Novel Positive Allosteric Modulators of µ Opioid Receptor—Insight from In Silico and In Vivo Studies

 ,
,  , and
, and 
Abstract
1. Introduction
2. Results
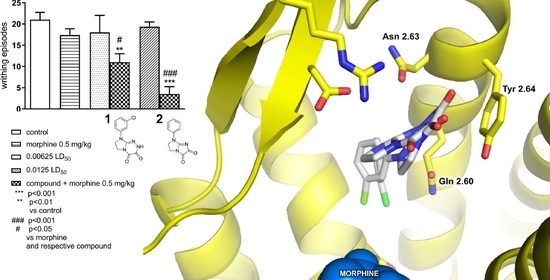
2.1. In Vivo Studies
2.1.1. Effect of Pre-Treatment with Cyprodime (CYP) on the Antinociceptive Effect of New Compounds 1 (0.0125 LD50) and 2 (0.05 LD50) in the ‘Writhing’ Test in Mice
2.1.2. Effect of Combined Administration of Morphine (0.5 mg/kg i.p.) and Tested Compounds 1 0.00625 LD50) and 2 (0.0125 LD50) in the ‘Writhing’ Test on Mice
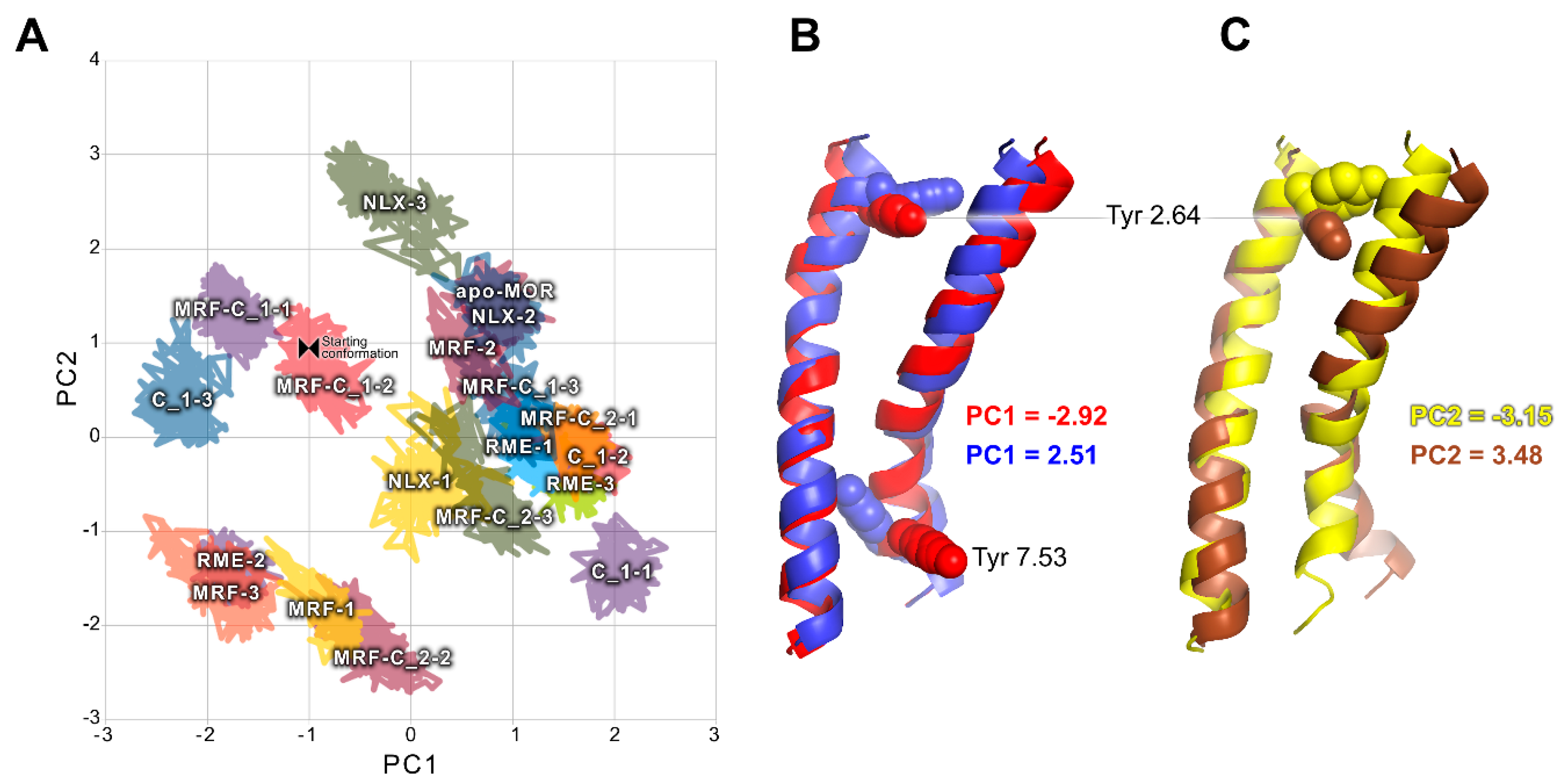
2.2. In Silico Studies
3. Discussion
4. Materials and Methods
4.1. Behavioral Studies
4.1.1. Animals
4.1.2. Drug Administration
4.1.3. Nociceptive Reactions
4.1.4. Statistics
4.2. In Silico Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BZD | Benzodiazepine |
| CYP | Cyprodime |
| DFT | Density functional theory |
| ECL | Extracellular loop |
| ESP | Electrostatic Potential |
| GPCR | G protein-coupled receptor |
| i.p. | Intraperitoneal |
| LD | Lethal dose |
| MD | Molecular dynamics |
| MOR | μ opioid receptor |
| PAM | Positive allosteric modulator |
| PC | Principal Component |
| PCA | Principal Component Analysis |
| POPC | 1-Palmitoyl-oleoyl-sn-glycero-phosphocholine |
| POPE | 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylethanolamine |
| s.c. | Subcutaneously |
| TM | Transmembrane |
| TRV-130 | Oliceridine |
References
- Ehrlich, A.T.; Kieffer, B.L.; Darcq, E. Current strategies toward safer mu opioid receptor drugs for pain management. Expert Opin. Ther. Targets 2019, 23, 315–326. [Google Scholar] [CrossRef]
- Fields, H.L.; Margolis, E.B. Understanding opioid reward. Trends Neurosci. 2015, 38, 217–225. [Google Scholar] [CrossRef]
- Benyamin, R.; Trescot, A.M.; Datta, S.; Buenaventura, R.; Adlaka, R.; Sehgal, N.; E Glaser, S.; Vallejo, R. Opioid complications and side effects. Pain Physician 2008, 11, 105–120. [Google Scholar]
- Urits, I.; Viswanath, O.; Orhurhu, V.; Gress, K.; Charipova, K.; Kaye, A.D.; Ngo, A. The Use of Mu-Opioid Receptor Biased Agonists: Oliceridine, an Opioid Analgesic with Reduced Adverse Effects. Curr. Pain Headache Rep. 2019, 23, 31. [Google Scholar] [CrossRef]
- Bartuzi, D.; Kaczor, A.A.; Matosiuk, D. Opportunities and Challenges in the Discovery of Allosteric Modulators of GPCRs. Methods Mol. Biol. 2018, 1705, 297–319. [Google Scholar] [CrossRef]
- Lazareno, S.; Doležal, V.; Popham, A.; Birdsall, N.J.M. Thiochrome Enhances Acetylcholine Affinity at Muscarinic M4 Receptors: Receptor Subtype Selectivity via Cooperativity Rather than Affinity. Mol. Pharmacol. 2004, 65, 257–266. [Google Scholar] [CrossRef]
- Christopoulos, A. Advances in G Protein-Coupled Receptor Allostery: From Function to Structure. Mol. Pharmacol. 2014, 86, 463–478. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Kenakin, T.P. Refining Efficacy: Allosterism and Bias in G Protein-Coupled Receptor Signaling. Methods Mol. Biol. 2011, 756, 3–35. [Google Scholar] [CrossRef]
- Bartuzi, D.; Kaczor, A.A.; Matosiuk, D. Signaling within Allosteric Machines: Signal Transmission Pathways Inside G Protein-Coupled Receptors. Molecules 2017, 22, 1188. [Google Scholar] [CrossRef]
- Conn, P.J.; Lindsley, C.W.; Meiler, J.; Niswender, C.M. Opportunities and challenges in the discovery of allosteric modulators of GPCRs for treating CNS disorders. Nat. Rev. Drug Discov. 2014, 13, 692–708. [Google Scholar] [CrossRef]
- Bartuzi, D.; Wróbel, T.M.; A Kaczor, A.; Matosiuk, D. Tuning Down the Pain - an Overview of Allosteric Modulation of Opioid Receptors: Mechanisms of Modulation, Allosteric Sites, Modulator Syntheses. Curr. Top. Med. Chem. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Burford, N.T.; Clark, M.J.; Wehrman, T.S.; Gerritz, S.W.; Banks, M.; O’Connell, J.; Traynor, J.R.; Alt, A. Discovery of positive allosteric modulators and silent allosteric modulators of the μ-opioid receptor. Proc. Natl. Acad. Sci. USA 2013, 110, 10830–10835. [Google Scholar] [CrossRef] [PubMed]
- Bisignano, P.; Burford, N.T.; Shang, Y.; Marlow, B.; Livingston, K.E.; Fenton, A.M.; Rockwell, K.; Budenholzer, L.; Traynor, J.R.; Gerritz, S.W.; et al. Ligand-Based Discovery of a New Scaffold for Allosteric Modulation of the μ-Opioid Receptor. J. Chem. Inf. Model. 2015, 55, 1836–1843. [Google Scholar] [CrossRef] [PubMed]
- Matosiuk, D.; Fidecka, S.; Antkiewicz-Michaluk, L.; Lipkowski, J.; Dybala, I.; Koziol, A.E. Synthesis and pharmacological activity of new carbonyl derivatives of 1-aryl-2-iminoimidazolidine: Part 2. Synthesis and pharmacological activity of 1,6-diaryl-5,7(1H)dioxo-2,3-dihydroimidazo[1,2-a][1,3,5]triazines. Eur. J. Med. Chem. 2002, 37, 761–772. [Google Scholar] [CrossRef]
- Matosiuk, D.; Fidecka, S.; Antkiewicz-Michaluk, L.; Dybała, I.; Kozioł, A.E. Synthesis and pharmacological activity of new carbonyl derivatives of 1-aryl-2-iminoimidazolidine. Part 1. Synthesis and pharmacological activity of chain derivatives of 1-aryl-2-iminoimidazolidine containing urea moiety. Part 1. Synthesis and pharmacological activity of chain derivatives of 1-aryl-2-iminoimidazolidine containing urea moiety. Eur. J. Med. Chem. 2001, 36, 783–797. [Google Scholar] [CrossRef] [PubMed]
- Matosiuk, D.; Fidecka, S.; Antkiewicz-Michaluk, L.; Dybala, I.; E Koziol, A. Synthesis and pharmacological activity of new carbonyl derivatives of 1-aryl-2-iminoimidazolidine. Part 3. Synthesis and pharmacological activity of 1-aryl-5,6(1H)dioxo-2,3-dihydroimidazo[1,2-a]imidazoles. Eur. J. Med. Chem. 2002, 37, 845–853. [Google Scholar] [CrossRef]
- Rządkowska, M.; Szacoń, E.; Kaczor, A.A.; Fidecka, S.; Kędzierska, E.; Matosiuk, D. Synthesis, central nervous system activity, and structure–activity relationship of 1-aryl-6-benzyl-7-hydroxy-2,3-dihydroimidazo[1,2-a]pyrimidine-5(1H)-ones. Med. Chem. Res. 2014, 23, 4221–4237. [Google Scholar] [CrossRef]
- Rzadkowska, M.; Szacon, E.; Kaczor, A.A.; Fidecka, S.; Kedzierska, E.; Matosiuk, D. Synthesis, pharmacological activity and molecular modeling of 1-aryl-7- hydroxy-2,3-dihydroimidazo[1,2-a]pyrimidine-5(1H)-ones and their 6-substituted derivatives. Med. Chem. 2014, 10, 460–475. [Google Scholar] [CrossRef]
- Szacoń, E.; Rządkowska, M.; Kaczor, A.A.; Kedzierska, E.; Orzelska-Górka, J.; Fidecka, S.; Matosiuk, D. Synthesis and Pharmacological Evaluation of Novel 1-(1,4-Alkylaryldisubstituted-4,5-dihydro-1H-imidazo)-3-substituted Urea Derivatives. Molecules 2016, 21, 582. [Google Scholar] [CrossRef]
- Szacoń, E.; Rządkowska, M.; Kaczor, A.A.; Kedzierska, E.; Mazur, A.; Fidecka, S.; Matosiuk, D. Synthesis, central nervous system activity and structure–activity relationship of N-substituted derivatives of 1-arylimidazolidyn-2-ylideneurea and products of their cyclization. J. Enzym. Inhib. Med. Chem. 2015, 30, 746–760. [Google Scholar] [CrossRef]
- Sztanke, K.; Fidecka, S.; Kedzierska, E.; Karczmarzyk, Z.; Pihlaja, K.; Matosiuk, D. Antinociceptive activity of new imidazolidine carbonyl derivatives. Eur. J. Med. Chem. 2005, 40, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Bartuzi, D.; Kaczor, A.A.; Matosiuk, D. Activation and Allosteric Modulation of Human μ Opioid Receptor in Molecular Dynamics. J. Chem. Inf. Model. 2015, 55, 2421–2434. [Google Scholar] [CrossRef] [PubMed]
- Bartuzi, D.; Kaczor, A.A.; Matosiuk, D. Interplay between Two Allosteric Sites and Their Influence on Agonist Binding in Human μ Opioid Receptor. J. Chem. Inf. Model. 2016, 56, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Bartuzi, D.; Kaczor, A.A.; Matosiuk, D. Molecular mechanisms of allosteric probe dependence in μ opioid receptor. J. Biomol. Struct. Dyn. 2019, 37, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Hothersall, J.D.; Torella, R.; Humphreys, S.; Hooley, M.; Brown, A.; McMurray, G.; Nickolls, S.A. Residues W320 and Y328 within the binding site of the μ-opioid receptor influence opiate ligand bias. Neuropharmacology 2017, 118, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Livingston, K.E.; Stanczyk, M.A.; Burford, N.T.; Alt, A.; Canals, M.; Traynor, J.R. Pharmacologic Evidence for a Putative Conserved Allosteric Site on Opioid Receptors. Mol. Pharmacol. 2018, 93, 157–167. [Google Scholar] [CrossRef]
- Shang, Y.; Yeatman, H.R.; Provasi, D.; Alt, A.; Christopoulos, A.; Canals, M.; Filizola, M. Proposed Mode of Binding and Action of Positive Allosteric Modulators at Opioid Receptors. ACS Chem. Biol. 2016, 11, 1220–1229. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Weinstein, H. [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Methods in Neurosciences; Sealfon, S.C., Ed.; Academic Press: San Diego, CA, USA, 1995; Volume 25, pp. 366–428. [Google Scholar]
- Le Bars, D.; Gozariu, M.; Cadden, S.W. Animal models of nociception. Pharmacol. Rev. 2001, 53, 597–652. [Google Scholar]
- Sierralta, F.; Miranda, H.F. Analgesic effect of benzodiazepines and flumazenil. Gen. Pharmacol. Vasc. Syst. 1992, 23, 739–742. [Google Scholar] [CrossRef]
- Pike, L.J.; Han, X.; Chung, K.-N.; Gross, R.W. Lipid Rafts Are Enriched in Arachidonic Acid and Plasmenylethanolamine and Their Composition Is Independent of Caveolin-1 Expression: A Quantitative Electrospray Ionization/Mass Spectrometric Analysis. Biochemistry 2002, 41, 2075–2088. [Google Scholar] [CrossRef]
- Żuk, J.; Bartuzi, D.; Matosiuk, D.; Kaczor, A.A. Preferential Coupling of Dopamine D2S and D2L Receptor Isoforms with Gi1 and Gi2 Proteins—In Silico Study. Int. J. Mol. Sci. 2020, 21, 436. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.H.; Beuming, T.; Banala, A.K.; Donthamsetti, P.; Pongetti, K.; LaBounty, A.; Levy, B.; Cao, J.; Michino, M.; Luedtke, R.R.; et al. Molecular Determinants of Selectivity and Efficacy at the Dopamine D3 Receptor. J. Med. Chem. 2012, 55, 6689–6699. [Google Scholar] [CrossRef] [PubMed]
- Männel, B.; Jaiteh, M.; Zeifman, A.; Randakova, A.; Möller, D.; Hübner, H.; Gmeiner, P.; Carlsson, J. Structure-Guided Screening for Functionally Selective D2 Dopamine Receptor Ligands from a Virtual Chemical Library. ACS Chem. Biol. 2017, 12, 2652–2661. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.K.; Abramyan, A.M.; Michino, M.; Free, R.B.; Sibley, D.R.; Javitch, J.A.; Lane, J.R.; Shi, L. The E2.65A mutation disrupts dynamic binding poses of SB269652 at the dopamine D2 and D3 receptors. PLoS Comput. Biol. 2018, 14, e1005948. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Salomon-Ferrer, R.; Lee, S.; Vaidehi, N. Conserved Mechanism of Conformational Stability and Dynamics in G-Protein-Coupled Receptors. J. Chem. Theory Comput. 2016, 12, 5575–5584. [Google Scholar] [CrossRef]
- Kedzierska, E.; Orzelska-Górka, J.; Perković, I.; Knežević, D.; Fidecka, S.; Kaiser, M.; Zorc, B. Pharmacological effects of primaquine ureas and semicarbazides on the central nervous system in mice and antimalarial activity in vitro. Fundam. Clin. Pharmacol. 2015, 30, 58–69. [Google Scholar] [CrossRef]
- Schmidhammer, H.; Burkard, W.P.; Eggstein-Aeppli, L.; Smith, C.F. Synthesis and biological evaluation of 14-alkoxymorphinans. 2. (-)-N-(Cyclopropylmethyl)-4,14-dimethoxymorphinan-6-one, a selective. mu. opioid receptor antagonist. J. Med. Chem. 1989, 32, 418–421. [Google Scholar] [CrossRef]
- Mendes, G.L.; Santos, A.R.; Malheiros, A.; Filho, V.C.; Yunes, R.A.; Calixto, J.B. Assessment of mechanisms involved in antinociception caused by sesquiterpene polygodial. J. Pharmacol. Exp. Ther. 2000, 292, 164–172. [Google Scholar]
- Allen, F.H. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 380–388. [Google Scholar] [CrossRef]
- Spartan ’10; Wavefunction, Inc.: Irvine, CA, USA, 2010.
- LigPrep; Schrödinger, LLC: New York, NY, USA, 2019.
- Epik; Schrödinger, LLC: New York, NY, USA, 2019.
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef]
- Jain, A.N. Surflex: Fully Automatic Flexible Molecular Docking Using a Molecular Similarity-Based Search Engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Small-Molecule Drug Discovery Suite 2019-3: Glide; Schrödinger, LLC: New York, NY, USA, 2019.
- Kaczor, A.A.; Bartuzi, D.; Matosiuk, D. Modeling the Active Conformation of Human µ Opioid Receptor. Lett. Drug Des. Discov. 2014, 11, 1053–1061. [Google Scholar] [CrossRef]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder Toward Realistic Biological Membrane Simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Case, D.A.; Berryman, J.T.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; et al. AMBER 2015; University of California: San Francisco, CA, USA, 2015. [Google Scholar]
- Jämbeck, J.P.M.; Lyubartsev, A.P. Another Piece of the Membrane Puzzle: Extending Slipids Further. J. Chem. Theory Comput. 2013, 9, 774–784. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Paloncýová, M.; Fabre, G.; DeVane, R.H.; Trouillas, P.; Berka, K.; Otyepka, M. Benchmarking of Force Fields for Molecule–Membrane Interactions. J. Chem. Theory Comput. 2014, 10, 4143–4151. [Google Scholar] [CrossRef]
- Vanquelef, E.; Simon, S.; Marquant, G.; Garcia, E.; Klimerak, G.; Delepine, J.C.; Cieplak, P.; Dupradeau, F.-Y. R.E.D. Server: A web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids Res. 2011, 39, W511–W517. [Google Scholar] [CrossRef]
- Da Silva, A.W.S.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System; Schrödinger, LLC: New York, NY, USA, 2012.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligands | Abbreviation | Source | Simulation Time | No. of Replicas |
|---|---|---|---|---|
| No ligands | Apo-MOR | [23] | 0.4 μs | 1 |
| (R)-methadone | RME | [23] | 0.4 μs | 3 |
| Morphine | MRF | New simulation | 0.4 μs | 3 |
| Morphine + compound 1 | MRF+C_1 | New simulation | 0.4 μs | 3 |
| Morphine + compound 2 | MRF+C_2 | New simulation | 0.4 μs | 3 |
| Compound 1 | C_1 | New simulation | 0.4 μs | 3 |
| Naloxone | NLX | New simulation | 0.4 μs | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartuzi, D.; Kędzierska, E.; Kaczor, A.A.; Schmidhammer, H.; Matosiuk, D. Novel Positive Allosteric Modulators of µ Opioid Receptor—Insight from In Silico and In Vivo Studies. Int. J. Mol. Sci. 2020, 21, 8463. https://doi.org/10.3390/ijms21228463
Bartuzi D, Kędzierska E, Kaczor AA, Schmidhammer H, Matosiuk D. Novel Positive Allosteric Modulators of µ Opioid Receptor—Insight from In Silico and In Vivo Studies. International Journal of Molecular Sciences. 2020; 21(22):8463. https://doi.org/10.3390/ijms21228463
Chicago/Turabian StyleBartuzi, Damian, Ewa Kędzierska, Agnieszka A. Kaczor, Helmut Schmidhammer, and Dariusz Matosiuk. 2020. "Novel Positive Allosteric Modulators of µ Opioid Receptor—Insight from In Silico and In Vivo Studies" International Journal of Molecular Sciences 21, no. 22: 8463. https://doi.org/10.3390/ijms21228463
APA StyleBartuzi, D., Kędzierska, E., Kaczor, A. A., Schmidhammer, H., & Matosiuk, D. (2020). Novel Positive Allosteric Modulators of µ Opioid Receptor—Insight from In Silico and In Vivo Studies. International Journal of Molecular Sciences, 21(22), 8463. https://doi.org/10.3390/ijms21228463