AXL Receptor in Breast Cancer: Molecular Involvement and Therapeutic Limitations

 , , , , ,
, , , , , 
Abstract
1. Perspective Chapter
2. Introduction
3. AXL Receptor Tyrosine Kinase
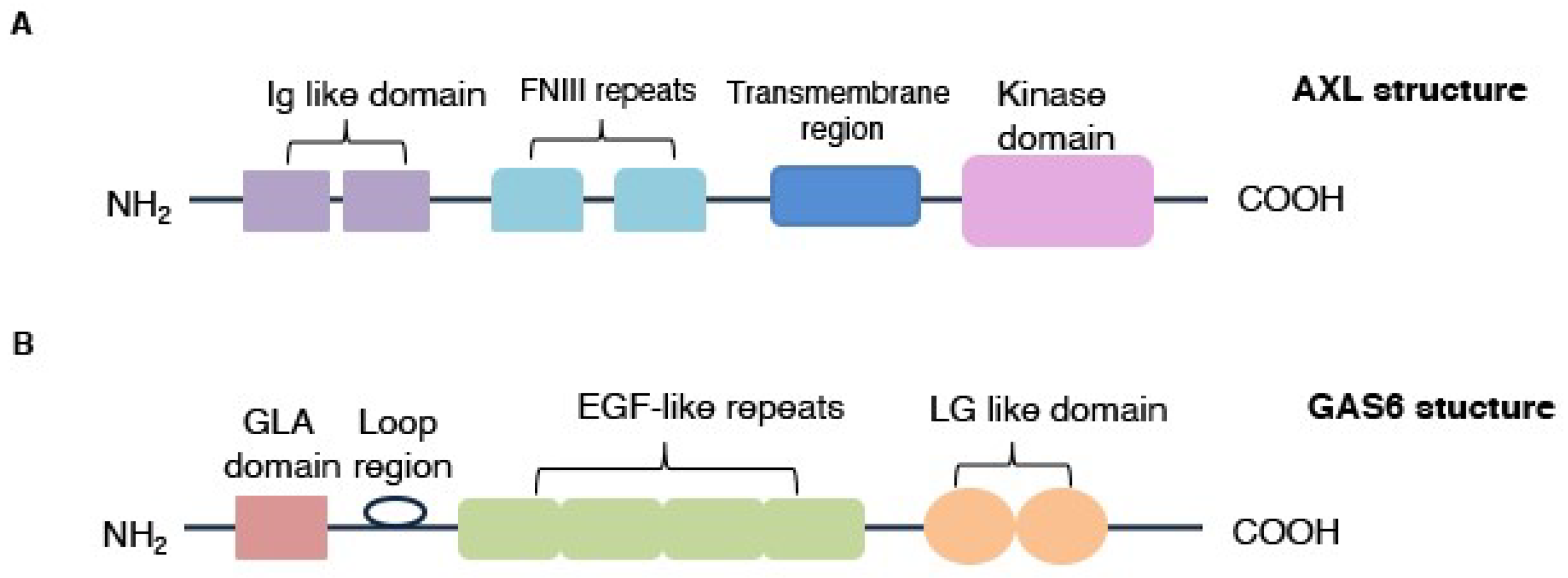
3.1. AXL Protein Structure
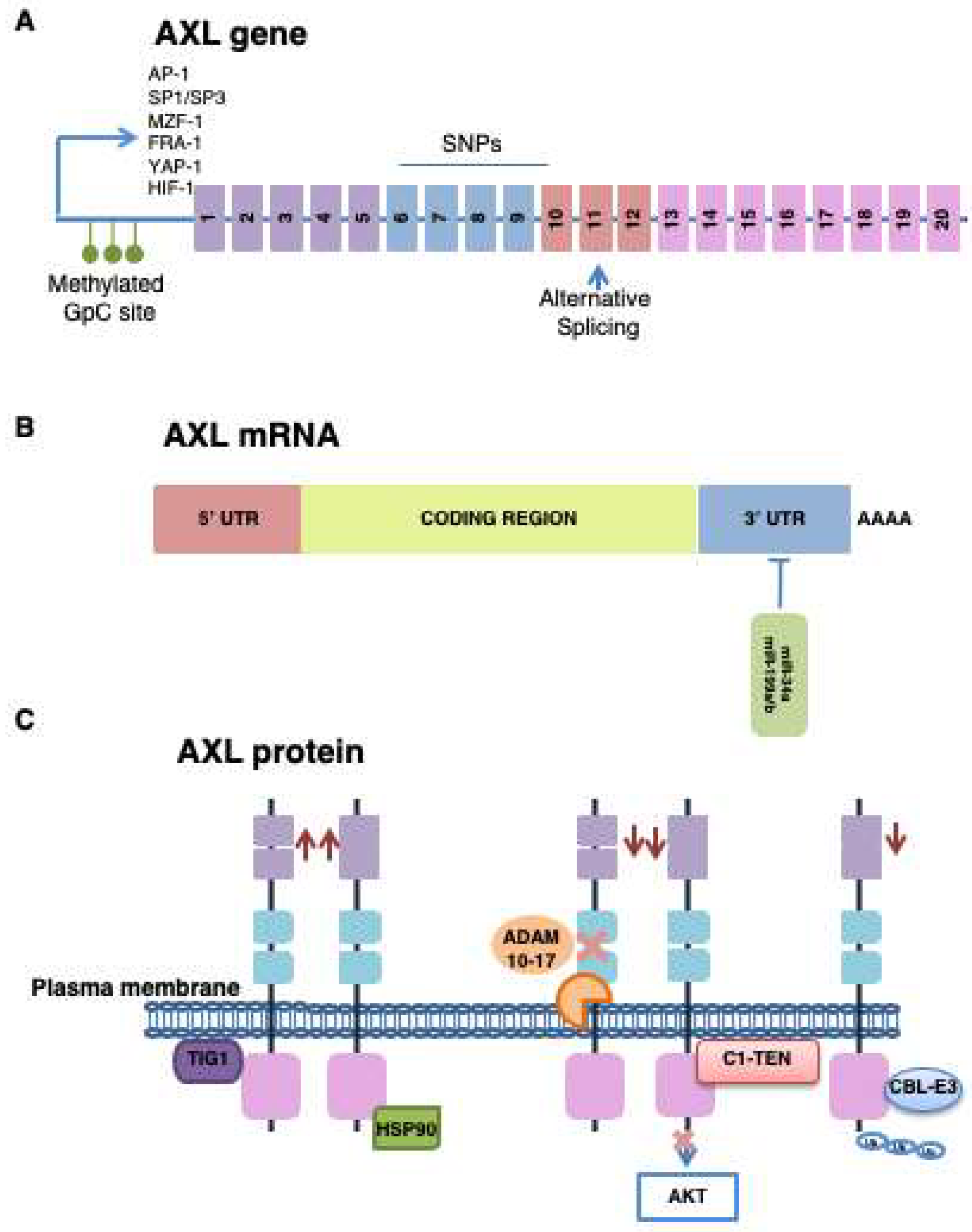
3.2. AXL Expression
3.2.1. Transcriptional Regulation
3.2.2. Post-Translational Modification
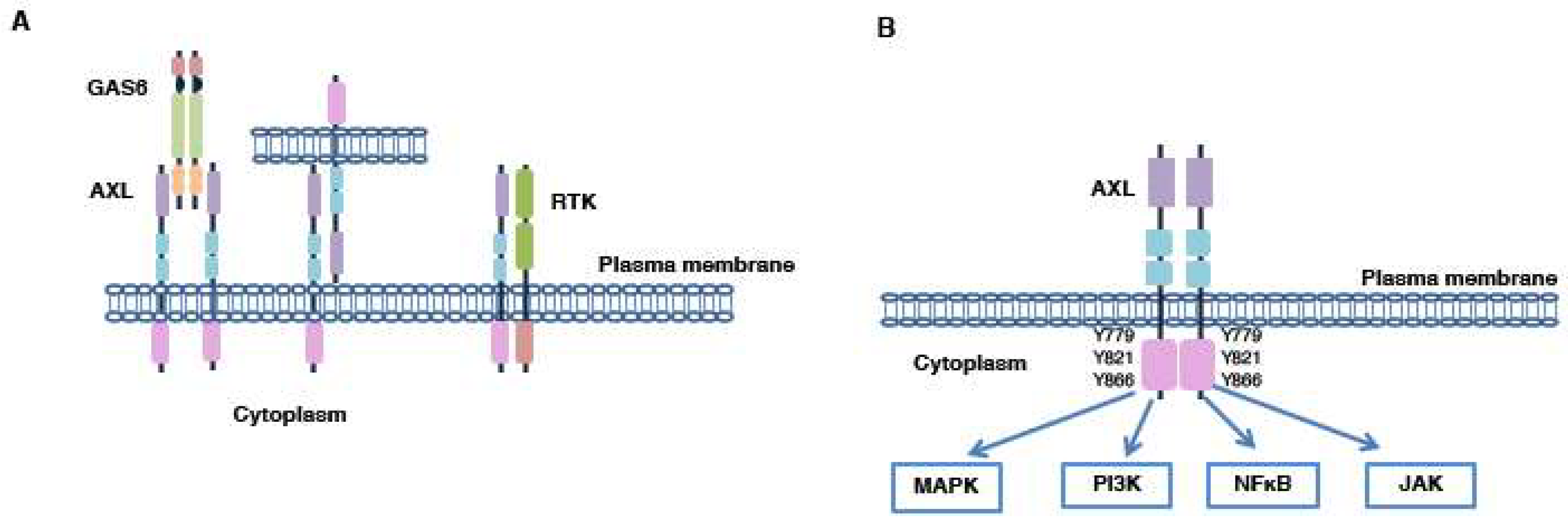
4. AXL Activation
4.1. GAS6/AXL Activation Mechanism
4.2. GAS6-Independent Activation Mechanisms
5. AXL Implications in Breast Cancers
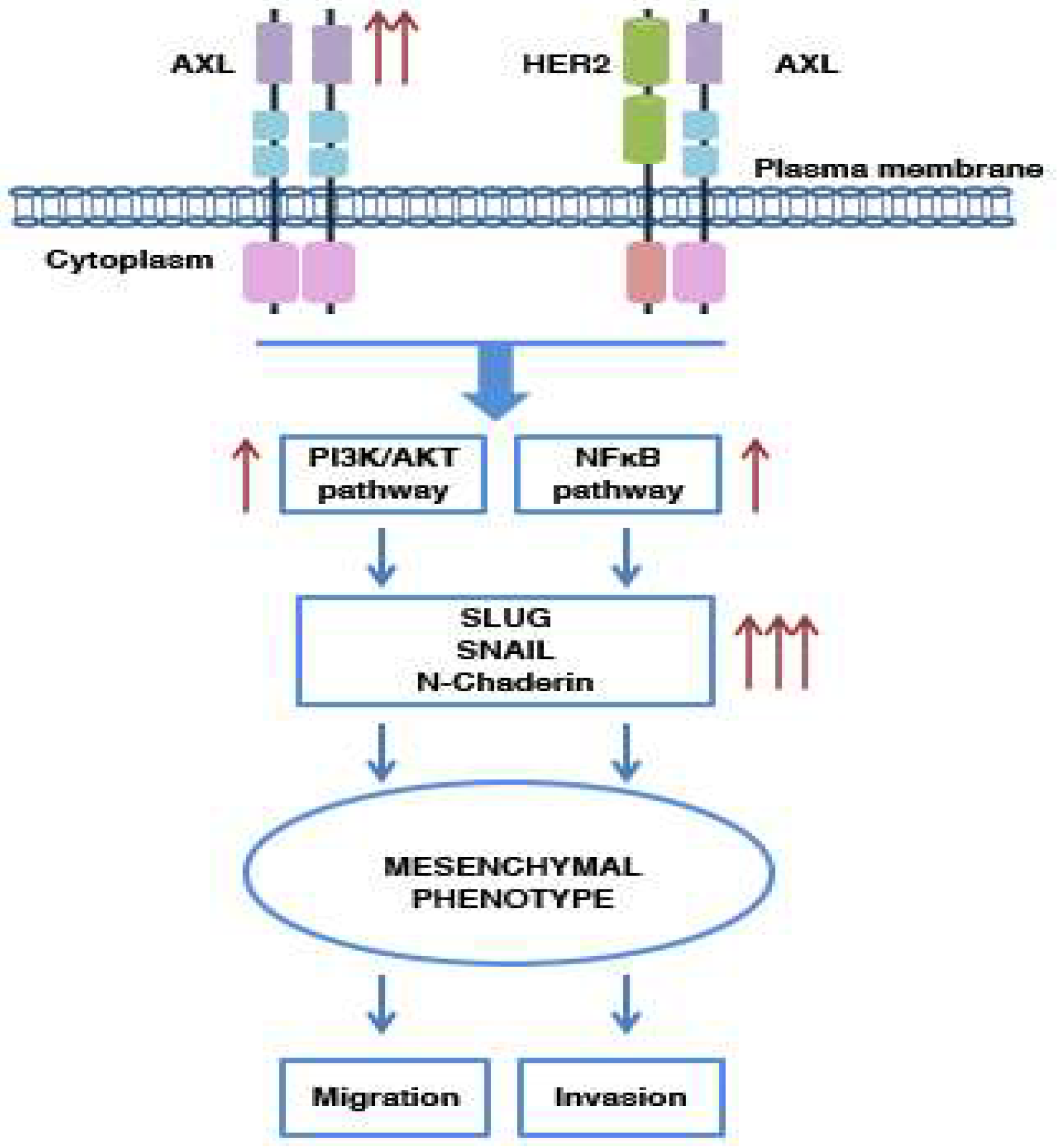
5.1. AXL and Metastasis: Its Involvement in Cell Migration and Invasion
5.2. AXL and EMT Process
5.3. AXL and Drug Resistance
6. AXL in BCSCs
7. AXL Inhibition in Breast Cancer Treatment
7.1. AXL Selective Inhibitors
7.2. Multi-Targets Inhibitors
8. AXL Clinical Implications: Limits and Future Hopes
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Abl | Abelson murine leukemia viral oncogene |
| ADAM | Disintegrin and metalloproteinase domain-containing protein |
| ALK | Anaplastic lymphoma receptor tyrosine kinase |
| AP-1 | Activator protein 1 |
| BAX | Bcl-2 Associated X-protein |
| BCSC | Breast cancer stem cell |
| C1-TEN | C1 domain-containing phosphatase and tensin homolog |
| CAFs | Cancer-associated fibroblasts |
| CAR-T | Chimeric antigen receptor T |
| CBL | Casitas B-lineage lymphoma |
| CCL | C-C motif chemokine ligand |
| CD | Cluster differentiation |
| ChK | Checkpoint kinases |
| CXCL | C-X-C Motif Chemokine Ligand |
| ECM | Extracellular Matrix |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial–mesenchymal transition |
| ER | Estrogen Receptor |
| FLT | fms-related tyrosine kinase |
| FNIII | Fibronectin type III |
| FRA-1 | Transcription factor Fos-related antigen 1 |
| GAS6 | Growth arrest-specific protein 6 |
| G-CSF | Granulocytic-colony-stimulating factor |
| G-MDSC | Granulocytic-myeloid-derived suppressor cell |
| GLA | Gamma-carboxy-glutamic acid |
| GRB2 | Growth factor receptor-bound protein 2 |
| HER2 | Human epidermal growth factor receptor 2 |
| HER3 | V-erb-b2 avian erythroblastic leukemia viral oncogene homolog3 |
| HGF | Hepatocyte growth factor |
| HIF-1 | hypoxia inducible factor 1 |
| HSP90 | Heat-shock protein 90 |
| Ig | Immunoglobulin |
| JAK | Janus Kinase |
| LG | Laminin G-like |
| MAPK | Mitogen-activated protein kinase |
| MEK | Mitogen-activated protein kinase kinase |
| MET | Hepatocyte growth factor receptor |
| MicroRNA | miRNA |
| MKNK1/2 | MAP kinase-interacting serine/threonine kinase 1 |
| MMP9 | Matrix metalloproteinase 9 |
| MST1R | Macrophage-stimulating protein receptor |
| MZF-1 | Myeloid Zinc Finger 1 |
| NFκB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NSCLC | Non-small-cell lung cancer |
| OS | Overall survival |
| PDGFR | Platelet-derived growth factor receptor |
| PI3K | PhosphoInositide3-Kinase |
| PLC γ | Phospholipase C γ |
| PR | Progesterone receptor |
| PROS1 | Protein S |
| PTEN | Phosphatase and tensin homolog on chromosome 10 |
| RET | Rearranged during Transfection |
| ROS1 | c-ros oncogene 1, receptor tyrosine kinase |
| RTKs | Receptor tyrosine kinases |
| sAXL | Soluble AXL |
| SNPs | Single polymorphisms |
| SP | Specificity protein |
| TAM | TYRO-3, AXL and MER |
| TGF-β | Transforming growth factor β |
| TIE-2 | Tyrosine kinase with immunoglobulin-like and EGF-like domains 2 |
| TIG1 | Tazarotene-induced gene 1 |
| TKs | Tyrosine kinases |
| TNBC | Triple-negative breast cancer |
| TULP-1 | Tubby-like protein 1 |
| VEGFR2 | Vascula Endothelial Growth Factor Receptor 2 |
| YAP1 | Yes-associated Protein 1 |
References
- Malhotra, G.K.; Zhao, X.; Band, H.; Band, V. Histological, molecular and functional subtypes of breast cancers. Cancer Biol. 2010, 10, 955–960. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Asmis, R.; Hawkins, G.A.; Howard, T.D.; Cox, L.A. The Need for Multi-Omics Biomarker Signatures in Precision Medicine. Int. J. Mol. Sci. 2019, 20, 4781. [Google Scholar] [CrossRef]
- Sun, C.; Bernards, R. Feedback and redundancy in receptor tyrosine kinase signaling: Relevance to cancer therapies. Trends Biochem. Sci. 2014, 39, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Paccez, J.D.; Vogelsang, M.; Parker, M.I.; Zerbini, L.F. The receptor tyrosine kinase Axl in cancer: Biological functions and therapeutic implications. Int. J. Cancer 2014, 134, 1024–1033. [Google Scholar] [CrossRef]
- Siveen, K.S.; Prabhu, K.S.; Achkar, I.W.; Kuttikrishnan, S.; Shyam, S.; Khan, A.Q.; Merhi, M.; Dermime, S.; Uddin, S. Role of Non Receptor Tyrosine Kinases in Hematological Malignances and its Targeting by Natural Products. Mol. Cancer 2018, 17, 31. [Google Scholar] [CrossRef]
- Meric, F.; Lee, W.P.; Sahin, A.; Zhang, H.; Kung, H.J.; Hung, M.C. Expression profile of tyrosine kinases in breast cancer. Clin. Cancer Res. 2002, 8, 361–367. [Google Scholar] [PubMed]
- Schenk, P.W.; Snaar-Jagalska, B.E. Signal perception and transduction: The role of protein kinases. Biochim. Biophys. Acta 1999, 1449, 1–24. [Google Scholar] [CrossRef]
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine kinase—Role and significance in Cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ye, X.; Tan, C.; Hongo, J.A.; Zha, J.; Liu, J.; Kallop, D.; Ludlam, M.J.; Pei, L. Axl as a potential therapeutic target in cancer: Role of Axl in tumor growth, metastasis and angiogenesis. Oncogene 2009, 28, 3442–3455. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Warner, S.L.; Vankayalapati, H.; Bearss, D.J.; Sharma, S. Targeting Axl and Mer kinases in cancer. Mol. Cancer 2011, 10, 1763–1773. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. The Receptor Tyrosine Kinase AXL in Cancer Progression. Cancers 2016, 8, 103. [Google Scholar] [CrossRef]
- Hafizi, S.; Dahlback, B. Signalling and functional diversity within the Axl subfamily of receptor tyrosine kinases. Cytokine Growth Factor Rev. 2006, 17, 295–304. [Google Scholar] [CrossRef]
- Wu, X.; Liu, X.; Koul, S.; Lee, C.Y.; Zhang, Z.; Halmos, B. AXL kinase as a novel target for cancer therapy. Oncotarget 2014, 5, 9546–9563. [Google Scholar] [CrossRef]
- Caberoy, N.B.; Alvarado, G.; Bigcas, J.L.; Li, W. Galectin-3 is a new MerTK-specific eat-me signal. J. Cell Physiol. 2012, 227, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM receptor tyrosine kinases: Biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008, 100, 35–83. [Google Scholar] [CrossRef]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef]
- Xie, S.; Li, Y.; Li, X.; Wang, L.; Yang, N.; Wang, Y.; Wei, H. Mer receptor tyrosine kinase is frequently overexpressed in human non-small cell lung cancer, confirming resistance to erlotinib. Oncotarget 2015, 6, 9206–9219. [Google Scholar] [CrossRef]
- Chien, C.W.; Hou, P.C.; Wu, H.C.; Chang, Y.L.; Lin, S.C.; Lin, S.C.; Lin, B.W.; Lee, J.C.; Chang, Y.J.; Sun, H.S.; et al. Targeting TYRO3 inhibits epithelial-mesenchymal transition and increases drug sensitivity in colon cancer. Oncogene 2016, 35, 5872–5881. [Google Scholar] [CrossRef]
- Antony, J.; Huang, R.Y. AXL-Driven EMT State as a Targetable Conduit in Cancer. Cancer Res. 2017, 77, 3725–3732. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov (accessed on 2 November 2020).
- Scaltriti, M.; Elkabets, M.; Baselga, J. Molecular Pathways: AXL, a Membrane Receptor Mediator of Resistance to Therapy. Clin. Cancer Res. 2016, 22, 1313–1317. [Google Scholar] [CrossRef]
- Cardone, C.; Blauensteiner, B.; Moreno-Viedma, V.; Martini, G.; Simeon, V.; Vitiello, P.P.; Ciardiello, D.; Belli, V.; Matrone, N.; Troiani, T.; et al. AXL is a predictor of poor survival and of resistance to anti-EGFR therapy in RAS wild-type metastatic colorectal cancer. Eur. J. Cancer 2020, 138, 1–10. [Google Scholar] [CrossRef]
- Yu, W.; Ge, X.; Lai, X.; Lv, J.; Wang, Y. The up-regulation of Axl is associated with a poor prognosis and promotes proliferation in pancreatic ductal adenocarcinoma. Int. J. Clin. Exp. Pathol. 2019, 12, 1626–1633. [Google Scholar]
- Ishikawa, M.; Sonobe, M.; Nakayama, E.; Kobayashi, M.; Kikuchi, R.; Kitamura, J.; Imamura, N.; Date, H. Higher expression of receptor tyrosine kinase Axl, and differential expression of its ligand, Gas6, predict poor survival in lung adenocarcinoma patients. Ann. Surg. Oncol. 2013, 20, S467. [Google Scholar] [CrossRef]
- Shen, Y.; Chen, X.; He, J.; Liao, D.; Zu, X. Axl inhibitors as novel cancer therapeutic agents. Life Sci. 2018, 198, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Gay, C.M.; Balaji, K.; Byers, L.A. Giving AXL the axe: Targeting AXL in human malignancy. Br. J. Cancer 2017, 116, 415–423. [Google Scholar] [CrossRef]
- Okimoto, R.A.; Bivona, T.G. AXL receptor tyrosine kinase as a therapeutic target in NSCLC. Lung Cancer 2015, 6, 27–34. [Google Scholar] [CrossRef]
- Varnum, B.C.; Young, C.; Elliott, G.; Garcia, A.; Bartley, T.D.; Fridell, Y.W.; Hunt, R.W.; Trail, G.; Clogston, C.; Toso, R.J.; et al. Axl receptor tyrosine kinase stimulated by the vitamin K-dependent protein encoded by growth-arrest-specific gene 6. Nature 1995, 373, 623–626. [Google Scholar] [CrossRef]
- Janssen, J.W.; Schulz, A.S.; Steenvoorden, A.C.; Schmidberger, M.; Strehl, S.; Ambros, P.F.; Bartram, C.R. A novel putative tyrosine kinase receptor with oncogenic potential. Oncogene 1991, 6, 2113–2120. [Google Scholar]
- O’Bryan, J.P.; Frye, R.A.; Cogswell, P.C.; Neubauer, A.; Kitch, B.; Prokop, C.; Espinosa, R., 3rd; Le Beau, M.M.; Earp, H.S.; Liu, E.T. axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol. Cell Biol. 1991, 11, 5016–5031. [Google Scholar] [CrossRef]
- Schulz, A.S.; Schleithoff, L.; Faust, M.; Bartram, C.R.; Janssen, J.W. The genomic structure of the human UFO receptor. Oncogene 1993, 8, 509–513. [Google Scholar]
- Faust, M.; Ebensperger, C.; Schulz, A.S.; Schleithoff, L.; Hameister, H.; Bartram, C.R.; Janssen, J.W. The murine ufo receptor: Molecular cloning, chromosomal localization and in situ expression analysis. Oncogene 1992, 7, 1287–1293. [Google Scholar] [PubMed]
- Hurtado, B.; Abasolo, N.; Munoz, X.; Garcia, N.; Benavente, Y.; Rubio, F.; Garcia de Frutos, P.; Krupinski, J.; Sala, N. Association study between polymorphims in GAS6-TAM genes and carotid atherosclerosis. Thromb. Haemost. 2010, 104, 592–598. [Google Scholar] [CrossRef]
- Brand, T.M.; Iida, M.; Stein, A.P.; Corrigan, K.L.; Braverman, C.M.; Luthar, N.; Toulany, M.; Gill, P.S.; Salgia, R.; Kimple, R.J.; et al. AXL mediates resistance to cetuximab therapy. Cancer Res. 2014, 74, 5152–5164. [Google Scholar] [CrossRef]
- Mudduluru, G.; Allgayer, H. The human receptor tyrosine kinase Axl gene--promoter characterization and regulation of constitutive expression by Sp1, Sp3 and CpG methylation. Biosci. Rep. 2008, 28, 161–176. [Google Scholar] [CrossRef]
- Mudduluru, G.; Vajkoczy, P.; Allgayer, H. Myeloid zinc finger 1 induces migration, invasion, and in vivo metastasis through Axl gene expression in solid cancer. Mol. Cancer Res. 2010, 8, 159–169. [Google Scholar] [CrossRef]
- Sayan, A.E.; Stanford, R.; Vickery, R.; Grigorenko, E.; Diesch, J.; Kulbicki, K.; Edwards, R.; Pal, R.; Greaves, P.; Jariel-Encontre, I.; et al. Fra-1 controls motility of bladder cancer cells via transcriptional upregulation of the receptor tyrosine kinase AXL. Oncogene 2012, 31, 1493–1503. [Google Scholar] [CrossRef]
- Nalwoga, H.; Ahmed, L.; Arnes, J.B.; Wabinga, H.; Akslen, L.A. Strong Expression of Hypoxia-Inducible Factor-1alpha (HIF-1alpha) Is Associated with Axl Expression and Features of Aggressive Tumors in African Breast Cancer. PLoS ONE 2016, 11, e0146823. [Google Scholar] [CrossRef]
- Manfioletti, G.; Brancolini, C.; Avanzi, G.; Schneider, C. The protein encoded by a growth arrest-specific gene (gas6) is a new member of the vitamin K-dependent proteins related to protein S, a negative coregulator in the blood coagulation cascade. Mol. Cell Biol. 1993, 13, 4976–4985. [Google Scholar] [CrossRef]
- Mudduluru, G.; Ceppi, P.; Kumarswamy, R.; Scagliotti, G.V.; Papotti, M.; Allgayer, H. Regulation of Axl receptor tyrosine kinase expression by miR-34a and miR-199a/b in solid cancer. Oncogene 2011, 30, 2888–2899. [Google Scholar] [CrossRef] [PubMed]
- Hajalirezay Yazdi, S.; Paryan, M.; Mohammadi-Yeganeh, S. An integrated approach of bioinformatic prediction and in vitro analysis identified that miR-34a targets MET and AXL in triple-negative breast cancer. Cell Mol. Biol. Lett. 2018, 23, 51. [Google Scholar] [CrossRef]
- Budagian, V.; Bulanova, E.; Orinska, Z.; Duitman, E.; Brandt, K.; Ludwig, A.; Hartmann, D.; Lemke, G.; Saftig, P.; Bulfone-Paus, S. Soluble Axl is generated by ADAM10-dependent cleavage and associates with Gas6 in mouse serum. Mol. Cell Biol. 2005, 25, 9324–9339. [Google Scholar] [CrossRef]
- Miller, M.A.; Oudin, M.J.; Sullivan, R.J.; Wang, S.J.; Meyer, A.S.; Im, H.; Frederick, D.T.; Tadros, J.; Griffith, L.G.; Lee, H.; et al. Reduced Proteolytic Shedding of Receptor Tyrosine Kinases Is a Post-Translational Mechanism of Kinase Inhibitor Resistance. Cancer Discov. 2016, 6, 382–399. [Google Scholar] [CrossRef]
- Flem Karlsen, K.; McFadden, E.; Florenes, V.A.; Davidson, B. Soluble AXL is ubiquitously present in malignant serous effusions. Gynecol. Oncol. 2019, 152, 408–415. [Google Scholar] [CrossRef]
- Flem-Karlsen, K.; Nyakas, M.; Farstad, I.N.; McFadden, E.; Wernhoff, P.; Jacobsen, K.D.; Florenes, V.A.; Maelandsmo, G.M. Soluble AXL as a marker of disease progression and survival in melanoma. PLoS ONE 2020, 15, e0227187. [Google Scholar] [CrossRef]
- Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef]
- Wu, Z.; Gholami, A.M.; Kuster, B. Systematic identification of the HSP90 candidate regulated proteome. Mol. Cell Proteom. 2012, 11, M111 016675. [Google Scholar] [CrossRef]
- Wang, X.; Saso, H.; Iwamoto, T.; Xia, W.; Gong, Y.; Pusztai, L.; Woodward, W.A.; Reuben, J.M.; Warner, S.L.; Bearss, D.J.; et al. TIG1 promotes the development and progression of inflammatory breast cancer through activation of Axl kinase. Cancer Res. 2013, 73, 6516–6525. [Google Scholar] [CrossRef]
- Valverde, P. Effects of Gas6 and hydrogen peroxide in Axl ubiquitination and downregulation. Biochem. Biophys. Res. Commun. 2005, 333, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, G.P.; Guida, T.; Alfano, L.; Avilla, E.; Santoro, M.; Carlomagno, F.; Melillo, R.M. Molecular mechanism of 17-allylamino-17-demethoxygeldanamycin (17-AAG)-induced AXL receptor tyrosine kinase degradation. J. Biol. Chem. 2013, 288, 17481–17494. [Google Scholar] [CrossRef]
- Hafizi, S.; Alindri, F.; Karlsson, R.; Dahlback, B. Interaction of Axl receptor tyrosine kinase with C1-TEN, a novel C1 domain-containing protein with homology to tensin. Biochem. Biophys. Res. Commun. 2002, 299, 793–800. [Google Scholar] [CrossRef]
- Hafizi, S.; Ibraimi, F.; Dahlback, B. C1-TEN is a negative regulator of the Akt/PKB signal transduction pathway and inhibits cell survival, proliferation, and migration. FASEB J. 2005, 19, 971–973. [Google Scholar] [CrossRef]
- Braunger, J.; Schleithoff, L.; Schulz, A.S.; Kessler, H.; Lammers, R.; Ullrich, A.; Bartram, C.R.; Janssen, J.W. Intracellular signaling of the Ufo/Axl receptor tyrosine kinase is mediated mainly by a multi-substrate docking-site. Oncogene 1997, 14, 2619–2631. [Google Scholar] [CrossRef]
- Abba, M.C.; Fabris, V.T.; Hu, Y.; Kittrell, F.S.; Cai, W.W.; Donehower, L.A.; Sahin, A.; Medina, D.; Aldaz, C.M. Identification of novel amplification gene targets in mouse and human breast cancer at a syntenic cluster mapping to mouse ch8A1 and human ch13q34. Cancer Res. 2007, 67, 4104–4112. [Google Scholar] [CrossRef][Green Version]
- Gomes, A.M.; Carron, E.C.; Mills, K.L.; Dow, A.M.; Gray, Z.; Fecca, C.R.; Lakey, M.A.; Carmeliet, P.; Kittrell, F.; Medina, D.; et al. Stromal Gas6 promotes the progression of premalignant mammary cells. Oncogene 2019, 38, 2437–2450. [Google Scholar] [CrossRef]
- Tanaka, M.; Siemann, D.W. Gas6/Axl Signaling Pathway in the Tumor Immune Microenvironment. Cancers 2020, 12, 1850. [Google Scholar] [CrossRef]
- Ibrahim, A.M.; Gray, Z.; Gomes, A.M.; Myers, L.; Behbod, F.; Machado, H.L. Gas6 expression is reduced in advanced breast cancers. NPJ Precis. Oncol. 2020, 4, 9. [Google Scholar] [CrossRef]
- Burchert, A.; Attar, E.C.; McCloskey, P.; Fridell, Y.W.; Liu, E.T. Determinants for transformation induced by the Axl receptor tyrosine kinase. Oncogene 1998, 16, 3177–3187. [Google Scholar] [CrossRef]
- Heiring, C.; Dahlback, B.; Muller, Y.A. Ligand recognition and homophilic interactions in Tyro3: Structural insights into the Axl/Tyro3 receptor tyrosine kinase family. J. Biol. Chem. 2004, 279, 6952–6958. [Google Scholar] [CrossRef]
- Bellosta, P.; Costa, M.; Lin, D.A.; Basilico, C. The receptor tyrosine kinase ARK mediates cell aggregation by homophilic binding. Mol. Cell Biol. 1995, 15, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G.; Rothlin, C.V. Immunobiology of the TAM receptors. Nat. Rev. Immunol. 2008, 8, 327–336. [Google Scholar] [CrossRef]
- Korshunov, V.A. Axl-dependent signalling: A clinical update. Clin. Sci. (Lond.) 2012, 122, 361–368. [Google Scholar] [CrossRef]
- Vouri, M.; Croucher, D.R.; Kennedy, S.P.; An, Q.; Pilkington, G.J.; Hafizi, S. Axl-EGFR receptor tyrosine kinase hetero-interaction provides EGFR with access to pro-invasive signalling in cancer cells. Oncogenesis 2016, 5, e266. [Google Scholar] [CrossRef]
- Namba, K.; Shien, K.; Takahashi, Y.; Torigoe, H.; Sato, H.; Yoshioka, T.; Takeda, T.; Kurihara, E.; Ogoshi, Y.; Yamamoto, H.; et al. Activation of AXL as a Preclinical Acquired Resistance Mechanism Against Osimertinib Treatment in EGFR-Mutant Non-Small Cell Lung Cancer Cells. Mol. Cancer Res. 2019, 17, 499–507. [Google Scholar] [CrossRef]
- Meyer, A.S.; Miller, M.A.; Gertler, F.B.; Lauffenburger, D.A. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Sci. Signal 2013, 6, ra66. [Google Scholar] [CrossRef]
- Li, W.; Xiong, X.; Abdalla, A.; Alejo, S.; Zhu, L.; Lu, F.; Sun, H. HGF-induced formation of the MET-AXL-ELMO2-DOCK180 complex promotes RAC1 activation, receptor clustering, and cancer cell migration and invasion. J. Biol. Chem. 2018, 293, 15397–15418. [Google Scholar] [CrossRef]
- Goyette, M.A.; Duhamel, S.; Aubert, L.; Pelletier, A.; Savage, P.; Thibault, M.P.; Johnson, R.M.; Carmeliet, P.; Basik, M.; Gaboury, L.; et al. The Receptor Tyrosine Kinase AXL Is Required at Multiple Steps of the Metastatic Cascade during HER2-Positive Breast Cancer Progression. Cell Rep. 2018, 23, 1476–1490. [Google Scholar] [CrossRef] [PubMed]
- Bilancio, A.; Migliaccio, A. Phosphoinositide 3-kinase assay in breast cancer cell extracts. Methods Mol. Biol. 2014, 1204, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef]
- Han, J.; Tian, R.; Yong, B.; Luo, C.; Tan, P.; Shen, J.; Peng, T. Gas6/Axl mediates tumor cell apoptosis, migration and invasion and predicts the clinical outcome of osteosarcoma patients. Biochem. Biophys. Res. Commun. 2013, 435, 493–500. [Google Scholar] [CrossRef]
- Lee, H.J.; Jeng, Y.M.; Chen, Y.L.; Chung, L.; Yuan, R.H. Gas6/Axl pathway promotes tumor invasion through the transcriptional activation of Slug in hepatocellular carcinoma. Carcinogenesis 2014, 35, 769–775. [Google Scholar] [CrossRef]
- Zajac, O.; Leclere, R.; Nicolas, A.; Meseure, D.; Marchio, C.; Vincent-Salomon, A.; Roman-Roman, S.; Schoumacher, M.; Dubois, T. AXL Controls Directed Migration of Mesenchymal Triple-Negative Breast Cancer Cells. Cells 2020, 9, 247. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Knyazev, P.G.; Cheburkin, Y.V.; Sharma, K.; Knyazev, Y.P.; Orfi, L.; Szabadkai, I.; Daub, H.; Keri, G.; Ullrich, A. AXL is a potential target for therapeutic intervention in breast cancer progression. Cancer Res. 2008, 68, 1905–1915. [Google Scholar] [CrossRef]
- Li, Y.; Jia, L.; Ren, D.; Liu, C.; Gong, Y.; Wang, N.; Zhang, X.; Zhao, Y. Axl mediates tumor invasion and chemosensitivity through PI3K/Akt signaling pathway and is transcriptionally regulated by slug in breast carcinoma. IUBMB Life 2014, 66, 507–518. [Google Scholar] [CrossRef]
- Ridley, A.J. Rho GTPase signalling in cell migration. Curr. Opin. Cell Biol. 2015, 36, 103–112. [Google Scholar] [CrossRef]
- Melchionna, R.; Spada, S.; Di Modugno, F.; D’Andrea, D.; Di Carlo, A.; Panetta, M.; Mileo, A.M.; Sperduti, I.; Antoniani, B.; Gallo, E.; et al. The actin modulator hMENA regulates GAS6-AXL axis and pro-tumor cancer/stromal cell cooperation. EMBO Rep. 2020, e50078. [Google Scholar] [CrossRef]
- Tai, K.Y.; Shieh, Y.S.; Lee, C.S.; Shiah, S.G.; Wu, C.W. Axl promotes cell invasion by inducing MMP-9 activity through activation of NF-kappaB and Brg-1. Oncogene 2008, 27, 4044–4055. [Google Scholar] [CrossRef]
- Berclaz, G.; Altermatt, H.J.; Rohrbach, V.; Kieffer, I.; Dreher, E.; Andres, A.C. Estrogen dependent expression of the receptor tyrosine kinase axl in normal and malignant human breast. Ann. Oncol. 2001, 12, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Gjerdrum, C.; Tiron, C.; Hoiby, T.; Stefansson, I.; Haugen, H.; Sandal, T.; Collett, K.; Li, S.; McCormack, E.; Gjertsen, B.T.; et al. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc. Natl. Acad. Sci. USA 2010, 107, 1124–1129. [Google Scholar] [CrossRef]
- Migliaccio, A.; Castoria, G.; de Falco, A.; Bilancio, A.; Giovannelli, P.; Di Donato, M.; Marino, I.; Yamaguchi, H.; Appella, E.; Auricchio, F. Polyproline and Tat transduction peptides in the study of the rapid actions of steroid receptors. Steroids 2012, 77, 974–978. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Q.; Wang, Q.; Cao, J.; Sun, J.; Zhu, Z. Mechanisms of resistance to estrogen receptor modulators in ER+/HER2- advanced breast cancer. Cell Mol. Life Sci. 2020, 77, 559–572. [Google Scholar] [CrossRef]
- D’Alfonso, T.M.; Hannah, J.; Chen, Z.; Liu, Y.; Zhou, P.; Shin, S.J. Axl receptor tyrosine kinase expression in breast cancer. J. Clin. Pathol. 2014, 67, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, L.; Nalwoga, H.; Arnes, J.B.; Wabinga, H.; Micklem, D.R.; Akslen, L.A. Increased tumor cell expression of Axl is a marker of aggressive features in breast cancer among African women. APMIS 2015, 123, 688–696. [Google Scholar] [CrossRef]
- Jin, G.; Wang, Z.; Wang, J.; Zhang, L.; Chen, Y.; Yuan, P.; Liu, D. Expression of Axl and its prognostic significance in human breast cancer. Oncol. Lett. 2017, 13, 621–628. [Google Scholar] [CrossRef]
- Chen, F.; Song, Q.; Yu, Q. Axl inhibitor R428 induces apoptosis of cancer cells by blocking lysosomal acidification and recycling independent of Axl inhibition. Am. J. Cancer Res. 2018, 8, 1466–1482. [Google Scholar]
- Goyette, M.A.; Cusseddu, R.; Elkholi, I.; Abu-Thuraia, A.; El-Hachem, N.; Haibe-Kains, B.; Gratton, J.P.; Cote, J.F. AXL knockdown gene signature reveals a drug repurposing opportunity for a class of antipsychotics to reduce growth and metastasis of triple-negative breast cancer. Oncotarget 2019, 10, 2055–2067. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Storci, G.; Sansone, P.; Trere, D.; Tavolari, S.; Taffurelli, M.; Ceccarelli, C.; Guarnieri, T.; Paterini, P.; Pariali, M.; Montanaro, L.; et al. The basal-like breast carcinoma phenotype is regulated by SLUG gene expression. J. Pathol. 2008, 214, 25–37. [Google Scholar] [CrossRef]
- Asiedu, M.K.; Beauchamp-Perez, F.D.; Ingle, J.N.; Behrens, M.D.; Radisky, D.C.; Knutson, K.L. AXL induces epithelial-to-mesenchymal transition and regulates the function of breast cancer stem cells. Oncogene 2014, 33, 1316–1324. [Google Scholar] [CrossRef]
- Antony, J.; Tan, T.Z.; Kelly, Z.; Low, J.; Choolani, M.; Recchi, C.; Gabra, H.; Thiery, J.P.; Huang, R.Y. The GAS6-AXL signaling network is a mesenchymal (Mes) molecular subtype-specific therapeutic target for ovarian cancer. Sci. Signal. 2016, 9, ra97. [Google Scholar] [CrossRef]
- Wilson, C.; Ye, X.; Pham, T.; Lin, E.; Chan, S.; McNamara, E.; Neve, R.M.; Belmont, L.; Koeppen, H.; Yauch, R.L.; et al. AXL inhibition sensitizes mesenchymal cancer cells to antimitotic drugs. Cancer Res. 2014, 74, 5878–5890. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.; Tong, P.; Diao, L.; Li, L.; Fan, Y.; Hoff, J.; Heymach, J.V.; Wang, J.; Byers, L.A. Targeting AXL and mTOR Pathway Overcomes Primary and Acquired Resistance to WEE1 Inhibition in Small-Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 6239–6253. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Kimura, A.; Miyazaki, T.; Tanaka, H.; Morimoto, M.; Nakai, K.; Soeda, J. Cabozantinib inhibits AXL- and MET-dependent cancer cell migration induced by growth-arrest-specific 6 and hepatocyte growth factor. Biochem. Biophys. Rep. 2020, 21, 100726. [Google Scholar] [CrossRef]
- Claas, A.M.; Atta, L.; Gordonov, S.; Meyer, A.S.; Lauffenburger, D.A. Systems Modeling Identifies Divergent Receptor Tyrosine Kinase Reprogramming to MAPK Pathway Inhibition. Cell Mol. Bioeng. 2018, 11, 451–469. [Google Scholar] [CrossRef]
- Liu, L.; Greger, J.; Shi, H.; Liu, Y.; Greshock, J.; Annan, R.; Halsey, W.; Sathe, G.M.; Martin, A.M.; Gilmer, T.M. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: Activation of AXL. Cancer Res. 2009, 69, 6871–6878. [Google Scholar] [CrossRef]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.; Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef]
- Guo, Z.; Li, Y.; Zhang, D.; Ma, J. Axl inhibition induces the antitumor immune response which can be further potentiated by PD-1 blockade in the mouse cancer models. Oncotarget 2017, 8, 89761–89774. [Google Scholar] [CrossRef]
- Schoumacher, M.; Burbridge, M. Key Roles of AXL and MER Receptor Tyrosine Kinases in Resistance to Multiple Anticancer Therapies. Curr. Oncol. Rep. 2017, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, T.A.; Rafat, M.; Castellini, L.; Shehade, H.; Kariolis, M.S.; Hui, A.B.; Stehr, H.; von Eyben, R.; Jiang, D.; Ellies, L.G.; et al. Reprogramming the immunological microenvironment through radiation and targeting Axl. Nat. Commun. 2016, 7, 13898. [Google Scholar] [CrossRef]
- Ludwig, K.F.; Du, W.; Sorrelle, N.B.; Wnuk-Lipinska, K.; Topalovski, M.; Toombs, J.E.; Cruz, V.H.; Yabuuchi, S.; Rajeshkumar, N.V.; Maitra, A.; et al. Small-Molecule Inhibition of Axl Targets Tumor Immune Suppression and Enhances Chemotherapy in Pancreatic Cancer. Cancer Res. 2018, 78, 246–255. [Google Scholar] [CrossRef]
- Haider, C.; Hnat, J.; Wagner, R.; Huber, H.; Timelthaler, G.; Grubinger, M.; Coulouarn, C.; Schreiner, W.; Schlangen, K.; Sieghart, W.; et al. Transforming Growth Factor-beta and Axl Induce CXCL5 and Neutrophil Recruitment in Hepatocellular Carcinoma. Hepatology 2019, 69, 222–236. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef]
- Kasikara, C.; Davra, V.; Calianese, D.; Geng, K.; Spires, T.E.; Quigley, M.; Wichroski, M.; Sriram, G.; Suarez-Lopez, L.; Yaffe, M.B.; et al. Pan-TAM Tyrosine Kinase Inhibitor BMS-777607 Enhances Anti-PD-1 mAb Efficacy in a Murine Model of Triple-Negative Breast Cancer. Cancer Res. 2019, 79, 2669–2683. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, Y.; Liu, W.; Li, X. Engineered IL-7 Receptor Enhances the Therapeutic Effect of AXL-CAR-T Cells on Triple-Negative Breast Cancer. Biomed. Res. Int. 2020, 2020, 4795171. [Google Scholar] [CrossRef]
- Sousa, B.; Ribeiro, A.S.; Paredes, J. Heterogeneity and Plasticity of Breast Cancer Stem Cells. Adv. Exp. Med. Biol. 2019, 1139, 83–103. [Google Scholar] [CrossRef]
- Wang, C.; Jin, H.; Wang, N.; Fan, S.; Wang, Y.; Zhang, Y.; Wei, L.; Tao, X.; Gu, D.; Zhao, F.; et al. Gas6/Axl Axis Contributes to Chemoresistance and Metastasis in Breast Cancer through Akt/GSK-3beta/beta-catenin Signaling. Theranostics 2016, 6, 1205–1219. [Google Scholar] [CrossRef]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef]
- Lee, J.E.; Kwon, Y.J.; Baek, H.S.; Ye, D.J.; Cho, E.; Choi, H.K.; Oh, K.S.; Chun, Y.J. Synergistic induction of apoptosis by combination treatment with mesupron and auranofin in human breast cancer cells. Arch. Pharm. Res. 2017, 40, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Ryu, Y.S.; Shin, S.; An, H.G.; Kwon, T.U.; Baek, H.S.; Kwon, Y.J.; Chun, Y.J. Synergistic Induction of Apoptosis by the Combination of an Axl Inhibitor and Auranofin in Human Breast Cancer Cells. Biomol. Ther. 2020, 28, 473–481. [Google Scholar] [CrossRef]
- Ye, X.; Li, Y.; Stawicki, S.; Couto, S.; Eastham-Anderson, J.; Kallop, D.; Weimer, R.; Wu, Y.; Pei, L. An anti-Axl monoclonal antibody attenuates xenograft tumor growth and enhances the effect of multiple anticancer therapies. Oncogene 2010, 29, 5254–5264. [Google Scholar] [CrossRef]
- Cerchia, L.; Esposito, C.L.; Camorani, S.; Rienzo, A.; Stasio, L.; Insabato, L.; Affuso, A.; de Franciscis, V. Targeting Axl with an high-affinity inhibitory aptamer. Mol. Ther. 2012, 20, 2291–2303. [Google Scholar] [CrossRef]
- Quirico, L.; Orso, F.; Esposito, C.L.; Bertone, S.; Coppo, R.; Conti, L.; Catuogno, S.; Cavallo, F.; de Franciscis, V.; Taverna, D. Axl-148b chimeric aptamers inhibit breast cancer and melanoma progression. Int. J. Biol. Sci. 2020, 16, 1238–1251. [Google Scholar] [CrossRef]
- Ma, Z.; Yao, G.; Zhou, B.; Fan, Y.; Gao, S.; Feng, X. The Chk1 inhibitor AZD7762 sensitises p53 mutant breast cancer cells to radiation in vitro and in vivo. Mol. Med. Rep. 2012, 6, 897–903. [Google Scholar] [CrossRef]
- Park, J.S.; Lee, C.; Kim, H.K.; Kim, D.; Son, J.B.; Ko, E.; Cho, J.H.; Kim, N.D.; Nan, H.Y.; Kim, C.Y.; et al. Suppression of the metastatic spread of breast cancer by DN10764 (AZD7762)-mediated inhibition of AXL signaling. Oncotarget 2016, 7, 83308–83318. [Google Scholar] [CrossRef][Green Version]
- Shen, Y.; Zhang, W.; Liu, J.; He, J.; Cao, R.; Chen, X.; Peng, X.; Xu, H.; Zhao, Q.; Zhong, J.; et al. Therapeutic activity of DCC-2036, a novel tyrosine kinase inhibitor, against triple-negative breast cancer patient-derived xenografts by targeting AXL/MET. Int. J. Cancer 2019, 144, 651–664. [Google Scholar] [CrossRef]
- Iacovelli, R.; Ciccarese, C.; Fornarini, G.; Massari, F.; Bimbatti, D.; Mosillo, C.; Rebuzzi, S.E.; Di Nunno, V.; Grassi, M.; Fantinel, E.; et al. Cabozantinib-related cardiotoxicity: A prospective analysis in a “real world” cohort of metastatic renal cell carcinoma patients. Br. J. Clin. Pharm. 2019. [Google Scholar] [CrossRef]
- Viola, D.; Elisei, R. Management of Medullary Thyroid Cancer. Endocrinol. Metab. Clin. N. Am. 2019, 48, 285–301. [Google Scholar] [CrossRef]
- Nokihara, H.; Nishio, M.; Yamamoto, N.; Fujiwara, Y.; Horinouchi, H.; Kanda, S.; Horiike, A.; Ohyanagi, F.; Yanagitani, N.; Nguyen, L.; et al. Phase 1 Study of Cabozantinib in Japanese Patients With Expansion Cohorts in Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2018. [Google Scholar] [CrossRef]
- Tolaney, S.M.; Ziehr, D.R.; Guo, H.; Ng, M.R.; Barry, W.T.; Higgins, M.J.; Isakoff, S.J.; Brock, J.E.; Ivanova, E.V.; Paweletz, C.P.; et al. Phase II and Biomarker Study of Cabozantinib in Metastatic Triple-Negative Breast Cancer Patients. Oncologist 2017, 22, 25–32. [Google Scholar] [CrossRef]
- Grullich, C. Cabozantinib: Multi-kinase Inhibitor of MET, AXL, RET, and VEGFR2. Recent Results Cancer Res. 2018, 211, 67–75. [Google Scholar] [CrossRef]
- Kawada, I.; Hasina, R.; Arif, Q.; Mueller, J.; Smithberger, E.; Husain, A.N.; Vokes, E.E.; Salgia, R. Dramatic antitumor effects of the dual MET/RON small-molecule inhibitor LY2801653 in non-small cell lung cancer. Cancer Res. 2014, 74, 884–895. [Google Scholar] [CrossRef]
- Konicek, B.W.; Capen, A.R.; Credille, K.M.; Ebert, P.J.; Falcon, B.L.; Heady, G.L.; Patel, B.K.R.; Peek, V.L.; Stephens, J.R.; Stewart, J.A.; et al. Merestinib (LY2801653) inhibits neurotrophic receptor kinase (NTRK) and suppresses growth of NTRK fusion bearing tumors. Oncotarget 2018, 9, 13796–13806. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Cortes, J.E.; Kim, D.W.; Khoury, H.J.; Brummendorf, T.H.; Porkka, K.; Martinelli, G.; Durrant, S.; Leip, E.; Kelly, V.; et al. Bosutinib safety and management of toxicity in leukemia patients with resistance or intolerance to imatinib and other tyrosine kinase inhibitors. Blood 2014, 123, 1309–1318. [Google Scholar] [CrossRef]
- Vultur, A.; Buettner, R.; Kowolik, C.; Liang, W.; Smith, D.; Boschelli, F.; Jove, R. SKI-606 (bosutinib), a novel Src kinase inhibitor, suppresses migration and invasion of human breast cancer cells. Mol. Cancer 2008, 7, 1185–1194. [Google Scholar] [CrossRef]
- Hebbard, L.; Cecena, G.; Golas, J.; Sawada, J.; Ellies, L.G.; Charbono, A.; Williams, R.; Jimenez, R.E.; Wankell, M.; Arndt, K.T.; et al. Control of mammary tumor differentiation by SKI-606 (bosutinib). Oncogene 2011, 30, 301–312. [Google Scholar] [CrossRef]
- Zou, H.Y.; Li, Q.; Lee, J.H.; Arango, M.E.; McDonnell, S.R.; Yamazaki, S.; Koudriakova, T.B.; Alton, G.; Cui, J.J.; Kung, P.P.; et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007, 67, 4408–4417. [Google Scholar] [CrossRef]
- Ayoub, N.M.; Al-Shami, K.M.; Alqudah, M.A.; Mhaidat, N.M. Crizotinib, a MET inhibitor, inhibits growth, migration, and invasion of breast cancer cells in vitro and synergizes with chemotherapeutic agents. Onco Targets Ther. 2017, 10, 4869–4883. [Google Scholar] [CrossRef]
- Rayson, D.; Lupichuk, S.; Potvin, K.; Dent, S.; Shenkier, T.; Dhesy-Thind, S.; Ellard, S.L.; Prady, C.; Salim, M.; Farmer, P.; et al. Canadian Cancer Trials Group IND197: A phase II study of foretinib in patients with estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2-negative recurrent or metastatic breast cancer. Breast Cancer Res. Treat. 2016, 157, 109–116. [Google Scholar] [CrossRef]
- Yan, S.; Vandewalle, N.; De Beule, N.; Faict, S.; Maes, K.; De Bruyne, E.; Menu, E.; Vanderkerken, K.; De Veirman, K. AXL Receptor Tyrosine Kinase as a Therapeutic Target in Hematological Malignancies: Focus on Multiple Myeloma. Cancers 2019, 11, 1727. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AXL Function(s) | |
|---|---|
| Tumor progression and metastasis | Rearrangement of the actin cytoskeleton Involvement in cell polarization Mediator between tumor cells and TME ECM degradation Involvement in intravasation and extravasation phenomena |
| EMT process | ↓E-cadherin and β-catenin ↑N-cadherin, Vimentin and Slug |
| Drug resistance | Transactivation with other members of RTK family Polarization of macrophages into M2-subtype Induction of a pro-tumoral immune microenvironment |
| Selective Inhibitors | ||||
|---|---|---|---|---|
| Target(s) | Function(s) | Field(s) | Reference(s) | |
| BGB324 (Bemcentinib or R428) | AXL | ATP-competitive AXL inhibitor that promotes apoptosis and reduces cell growth and metastasis | Preclinical and clinical | [87,93,108,109,110,111] |
| NA80X-1 | AXL | Selective AXL inhibitor that decreases cell motility and invasion | Preclinical | [75] |
| YW327.6S2 | AXL | Monoclonal antibody that inhibits GAS6/AXL interaction | Preclinical | [112] |
| GL21-T | AXL | RNA-based aptamer that blocks AXL’s catalytic activity and inhibits mobility and metastasis | Preclinical | [113,114] |
| DN10764 (AZD7762) | AXL ChKs | Selective AXL inhibitor that decreases cell proliferation, invasion and migration and induces apoptosis | Preclinical | [115,116] |
| SGI-7079 | AXL | Selective AXL inhibitor that decreases cell proliferation and metastasis | Preclinical | [50] |
| Non-Selective Inhibitors | ||||
| Rebastinib (DCC-2036) | MET VEGFR2 SRC AXL | Multi-target inhibitor that decreases cell proliferation, invasion, migration and EMT | Preclinical and Clinical | [27,117] |
| Cabozantinib (XL184) | RET VEGFR2 Flt 1-3-4 Tie2 MET AXL | Multi-target inhibitor that decreases cellular invasion and promotes immune system activation | Preclinical and clinical | [29,118,119,120,121,122] |
| Foretinib (XL880 or GSK-1363089) | MET RET VEGFR2 AXL | Multi-target inhibitor that restores the response to lapatinib in HER2+ context | Preclinical and clinical | [97] |
| Merestinib (LY2801653) | MET MST1R MKNK1/2 AXL | Multi-target agent that inhibits angiogenesis and mitosis | Preclinical and clinical | [123,124] |
| Bosutinib (SKI-606) | SRC Abl MEK BMX AXL | Multi-target inhibitors that decreases invasion, metastasis and tumor differentiation | Preclinical and clinical | [125,126,127] |
| Crizotinib (PF-02341066) | MET ALK ROS1 AXL | ATP-competitive agent that inhibits cell proliferation | Preclinical and clinical | [128,129] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falcone, I.; Conciatori, F.; Bazzichetto, C.; Bria, E.; Carbognin, L.; Malaguti, P.; Ferretti, G.; Cognetti, F.; Milella, M.; Ciuffreda, L. AXL Receptor in Breast Cancer: Molecular Involvement and Therapeutic Limitations. Int. J. Mol. Sci. 2020, 21, 8419. https://doi.org/10.3390/ijms21228419
Falcone I, Conciatori F, Bazzichetto C, Bria E, Carbognin L, Malaguti P, Ferretti G, Cognetti F, Milella M, Ciuffreda L. AXL Receptor in Breast Cancer: Molecular Involvement and Therapeutic Limitations. International Journal of Molecular Sciences. 2020; 21(22):8419. https://doi.org/10.3390/ijms21228419
Chicago/Turabian StyleFalcone, Italia, Fabiana Conciatori, Chiara Bazzichetto, Emilio Bria, Luisa Carbognin, Paola Malaguti, Gianluigi Ferretti, Francesco Cognetti, Michele Milella, and Ludovica Ciuffreda. 2020. "AXL Receptor in Breast Cancer: Molecular Involvement and Therapeutic Limitations" International Journal of Molecular Sciences 21, no. 22: 8419. https://doi.org/10.3390/ijms21228419
APA StyleFalcone, I., Conciatori, F., Bazzichetto, C., Bria, E., Carbognin, L., Malaguti, P., Ferretti, G., Cognetti, F., Milella, M., & Ciuffreda, L. (2020). AXL Receptor in Breast Cancer: Molecular Involvement and Therapeutic Limitations. International Journal of Molecular Sciences, 21(22), 8419. https://doi.org/10.3390/ijms21228419