An Overview of the Main Genetic, Epigenetic and Environmental Factors Involved in Autism Spectrum Disorder Focusing on Synaptic Activity



 ,
,  and
and 
Abstract
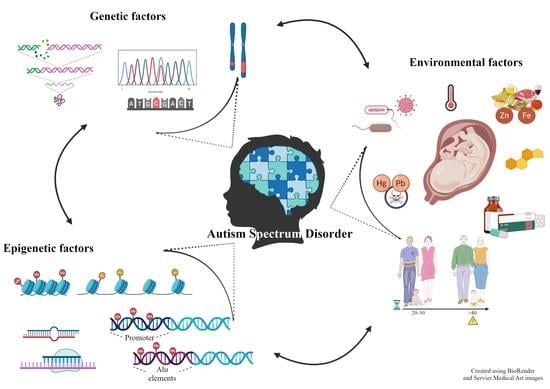
1. Introduction
2. Autism
2.1. Clinical Characteristics of ASD
2.2. Epidemiology
3. Genetic and Epigenetic Factors
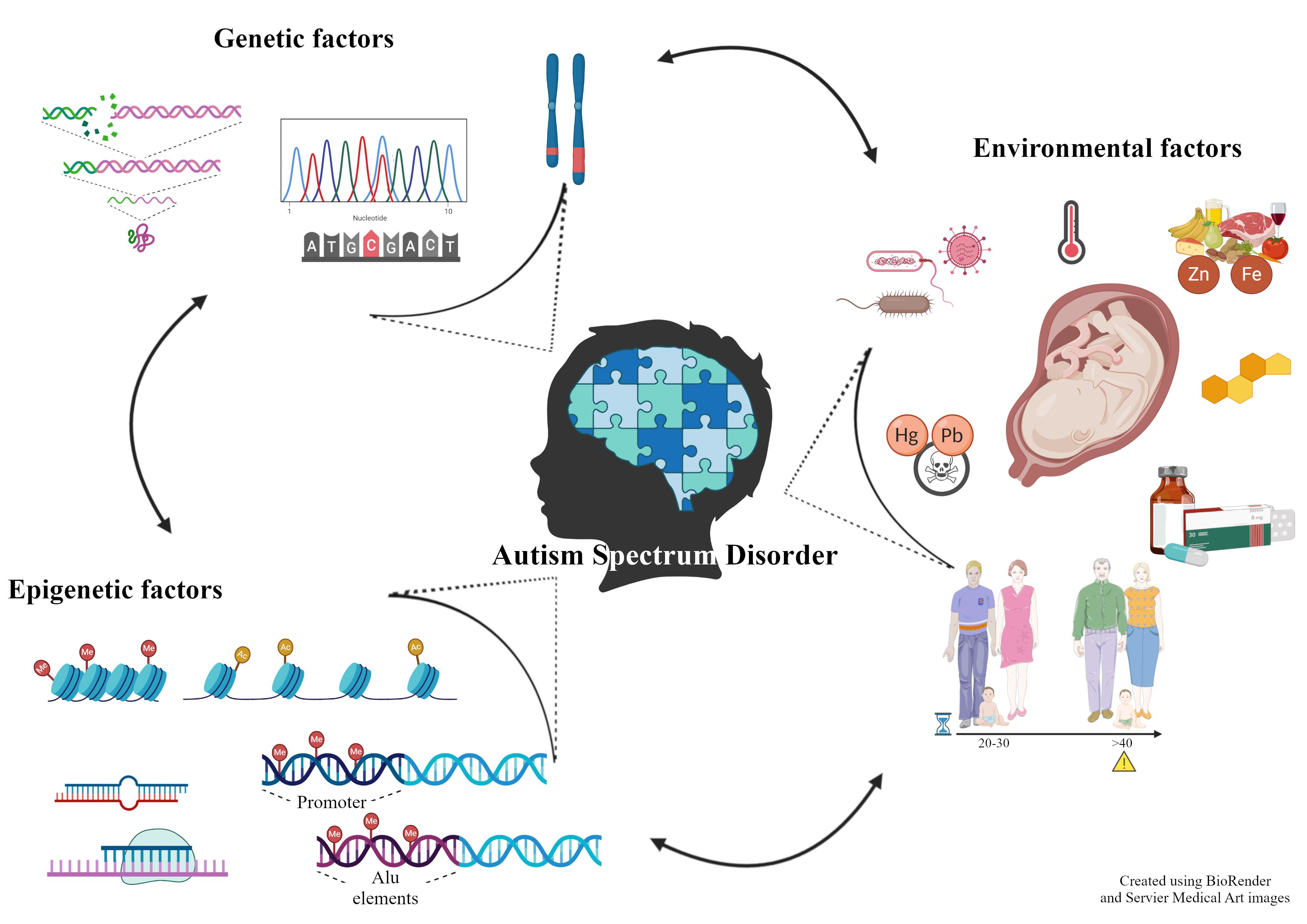
3.1. Relevant Candidate Genes
Synaptic Architecture and Functionality
- Cell adhesion molecules
- Scaffold proteins
- Voltage-gated ion channels
3.2. Epigenetic Factors
- DNA methylation
- miRNA
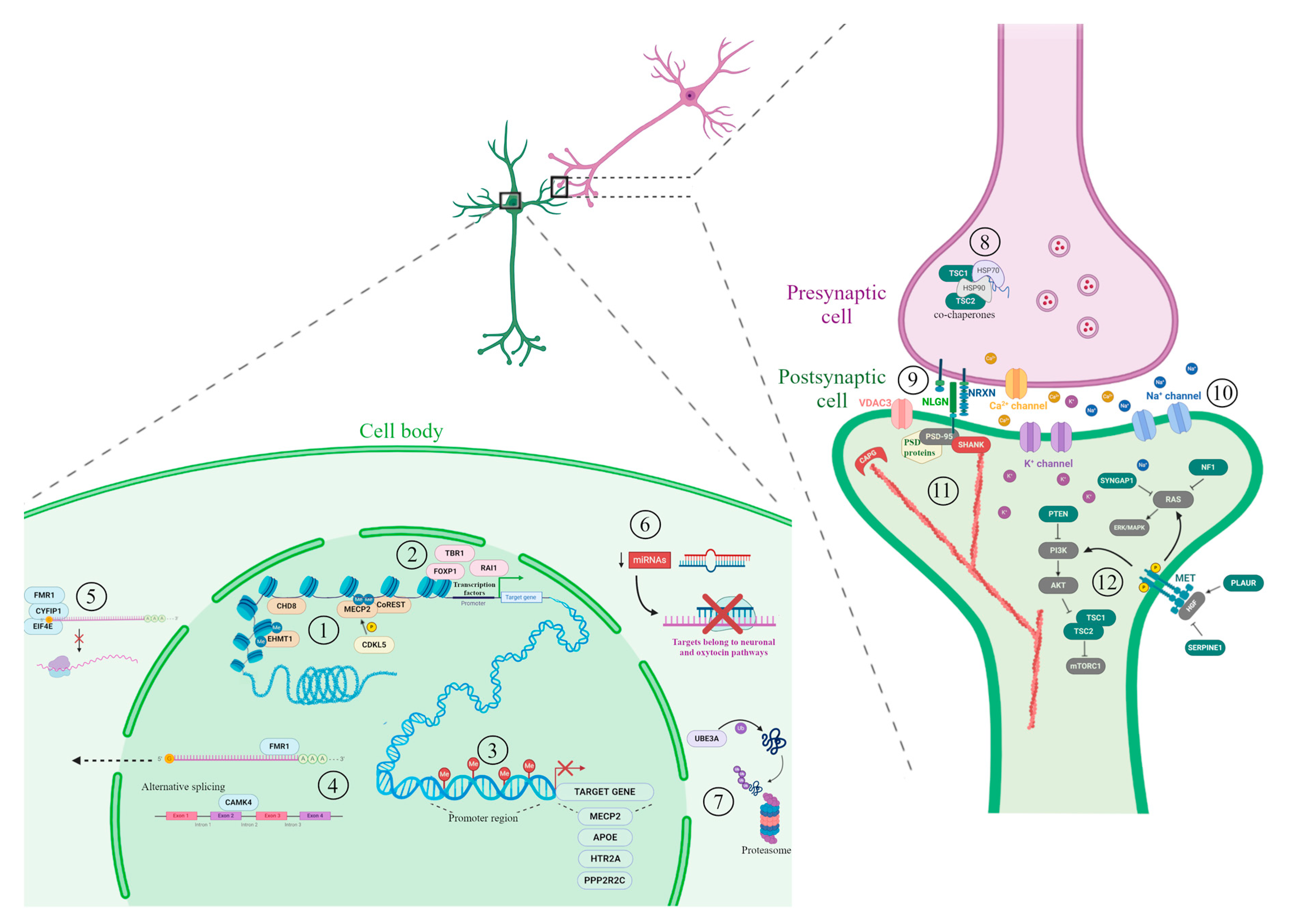
4. Environmental Factors
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kana, R.K.; Uddin, L.Q.; Ekenet, T.; Echugani, D.; Müller, R.-A. Brain connectivity in autism. Front. Hum. Neurosci. 2014, 8, 349. [Google Scholar] [CrossRef] [PubMed]
- Narzisi, A.; Muratori, F.; Calderoni, S.; Fabbro, F.; Urgesi, C. Neuropsychological Profile in High Functioning Autism Spectrum Disorders. J. Autism Dev. Disord. 2012, 43, 1895–1909. [Google Scholar] [CrossRef] [PubMed]
- Goin-Kochel, R.P.; Myers, B.J. Congenital Versus Regressive Onset of Autism Spectrum Disorders. Focus Autism Other Dev. Disabil. 2005, 20, 169–179. [Google Scholar] [CrossRef]
- Tamouza, R.; Fernell, E.; Eriksson, M.A.; Anderlid, B.; Manier, C.; Mariaselvam, C.M.; Boukouaci, W.; Leboyer, M.; Gillberg, C. HLA Polymorphism in Regressive and Non-Regressive Autism: A Preliminary Study. Autism Res. 2019, 13, 182–186. [Google Scholar] [CrossRef]
- Cohen, J.; Eyu, S.; Efu, Y.; Eli, X. Synaptic proteins and receptors defects in autism spectrum disorders. Front. Cell. Neurosci. 2014, 8, 276. [Google Scholar] [CrossRef]
- James, S.J.; Melnyk, S.; Jernigan, S.; Cleves, M.A.; Halsted, C.H.; Wong, D.H.; Cutler, P.; Bock, K.; Boris, M.; Bradstreet, J.J.; et al. Metabolic endophenotype and related genotypes are associated with oxidative stress in children with autism. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2006, 141, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Werling, D.M. The role of sex-differential biology in risk for autism spectrum disorder. Biol. Sex Differ. 2016, 7, 1–18. [Google Scholar] [CrossRef]
- Newschaffer, C.J.; Croen, L.A.; Daniels, J.; Giarelli, E.; Grether, J.K.; Levy, S.E.; Mandell, D.S.; Miller, L.A.; Pinto-Martin, J.; Reaven, J.; et al. The Epidemiology of Autism Spectrum Disorders. Annu. Rev. Public Health 2007, 28, 235–258. [Google Scholar] [CrossRef] [PubMed]
- Elsabbagh, M.; Mercure, E.; Hudry, K.; Chandler, S.; Pasco, G.; Charman, T.; Pickles, A.; Baron-Cohen, S.; Bolton, P.; Johnson, M.H. Infant Neural Sensitivity to Dynamic Eye Gaze Is Associated with Later Emerging Autism. Curr. Biol. 2012, 22, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Benvenuto, A.; Moavero, R.; Alessandrelli, R.; Manzi, B.; Curatolo, P. Syndromic autism: Causes and pathogenetic pathways. World J. Pediatr. 2009, 5, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Hu, V.W.; Devlin, C.A.; Debski, J.J. ASD Phenotype—Genotype Associations in Concordant and Discordant Monozygotic and Dizygotic Twins Stratified by Severity of Autistic Traits. Int. J. Mol. Sci. 2019, 20, 3804. [Google Scholar] [CrossRef]
- Colvert, E.; Tick, B.; McEwen, F.; Stewart, C.; Curran, S.R.; Woodhouse, E.; Gillan, N.; Hallett, V.; Lietz, S.; Garnett, T.; et al. Heritability of Autism Spectrum Disorder in a UK Population-Based Twin Sample. JAMA Psychiatry 2015, 72, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Larsson, H.; Hultman, C.M.; Reichenberg, A. The Familial Risk of Autism. JAMA 2014, 311, 1770–1777. [Google Scholar] [CrossRef]
- Eichler, E.E.; Flint, J.; Gibson, G.; Kong, A.; Leal, S.M.; Moore, J.H.; Nadeau, J.H. Missing heritability and strategies for finding the underlying causes of complex disease. Nat. Rev. Genet. 2010, 11, 446–450. [Google Scholar] [CrossRef]
- Génin, E. Missing heritability of complex diseases: Case solved? Hum. Genet. 2020, 139, 103–113. [Google Scholar] [CrossRef]
- Wiśniowiecka-Kowalnik, B.; Nowakowska, B.A. Genetics and epigenetics of autism spectrum disorder—Current evidence in the field. J. Appl. Genet. 2019, 60, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H. The Molecular Basis of Autism; Springer: New York, NY, USA, 2015; ISBN 9781493921904. [Google Scholar]
- Wang, K.; Zhang, H.; Ma, D.; Bucan, M.; Glessner, J.T.; Abrahams, B.S.; Salyakina, D.; Imielinski, M.; Bradfield, J.P.; Sleiman, P.M.A.; et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 2009, 459, 528–533. [Google Scholar] [CrossRef]
- Anney, R.; Klei, L.; Pinto, D.; Almeida, J.; Bacchelli, E.; Baird, G.; Bolshakova, N.; Bölte, S.; Bolton, P.F.; Bourgeron, T.; et al. Individual common variants exert weak effects on the risk for autism spectrum disorders. Hum. Mol. Genet. 2012, 21, 4781–4792. [Google Scholar] [CrossRef]
- Sener, E.F.; Canatan, H.; Ozkul, Y. Recent Advances in Autism Spectrum Disorders: Applications of Whole Exome Sequencing Technology. Psychiatry Investig. 2016, 13, 255–264. [Google Scholar] [CrossRef]
- Pinto, D.; Pagnamenta, A.T.; Klei, L.; Anney, R.; Merico, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010, 466, 368–372. [Google Scholar] [CrossRef]
- Marshall, C.R.; Noor, A.; Vincent, J.B.; Lionel, A.C.; Feuk, L.; Skaug, J.; Shago, M.; Moessner, R.; Pinto, D.; Ren, Y.; et al. Structural Variation of Chromosomes in Autism Spectrum Disorder. Am. J. Hum. Genet. 2008, 82, 477–488. [Google Scholar] [CrossRef]
- Lupski, J.R.; Stankiewicz, P. Genomic Disorders: Molecular Mechanisms for Rearrangements and Conveyed Phenotypes. PLoS Genet. 2005, 1, e49. [Google Scholar] [CrossRef] [PubMed]
- Devlin, B.; Scherer, S.W. Genetic architecture in autism spectrum disorder. Curr. Opin. Genet. Dev. 2012, 22, 229–237. [Google Scholar] [CrossRef]
- Napoli, E.; Russo, S.; Casula, L.; Alesi, V.; Amendola, F.A.; Angioni, A.; Novelli, A.; Valeri, G.; Menghini, D.; Vicari, S. Array-CGH Analysis in a Cohort of Phenotypically Well-Characterized Individuals with “Essential” Autism Spectrum Disorders. J. Autism Dev. Disord. 2018, 48, 442–449. [Google Scholar] [CrossRef]
- Beaudet, A.L. The utility of chromosomal microarray analysis in developmental and behavioral pediatrics. Child Dev. 2013, 84, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Jacquemont, M.-L.; Sanlaville, D.; Redon, R.; Raoul, O.; Cormier-Daire, V.; Lyonnet, S.; Amiel, J.; Le Merrer, M.; Heron, D.; De Blois, M.-C.; et al. Array-based comparative genomic hybridisation identifies high frequency of cryptic chromosomal rearrangements in patients with syndromic autism spectrum disorders. J. Med. Genet. 2006, 43, 843–849. [Google Scholar] [CrossRef]
- Holt, R.; Sykes, N.H.; Conceição, I.; Cazier, J.-B.; Anney, R.J.; Oliveira, G.G.; Gallagher, L.; Vicente, A.M.; Monaco, A.P.; Pagnamenta, A.T. CNVs leading to fusion transcripts in individuals with autism spectrum disorder. Eur. J. Hum. Genet. 2012, 20, 1141–1147. [Google Scholar] [CrossRef]
- Pagnamenta, A.T.; Bacchelli, E.; De Jonge, M.V.; Mirza, G.; Scerri, T.S.; Minopoli, F.; Chiocchetti, A.; Ludwig, K.U.; Hoffmann, P.; Paracchini, S.; et al. Characterization of a Family with Rare Deletions in CNTNAP5 and DOCK4 Suggests Novel Risk Loci for Autism and Dyslexia. Biol. Psychiatry 2010, 68, 320–328. [Google Scholar] [CrossRef]
- Ceroni, F.; Sagar, A.; Simpson, N.H.; Gawthrope, A.J.; Newbury, D.F.; Pinto, D.; Francis, S.M.; Tessman, D.C.; Cook, E.H.; Monaco, A.P.; et al. A Deletion Involving CD 38 and BST 1 Results in a Fusion Transcript in a Patient With Autism and Asthma. Autism Res. 2014, 7, 254–263. [Google Scholar] [CrossRef]
- Loi, E.; Moi, L.; Blois, S.; Bacchelli, E.; Benedetti, A.F.V.; Cameli, C.; Fadda, R.; Maestrini, E.; Carta, M.; Doneddu, G.; et al. ELMOD3—SH2D6 gene fusion as a possible co-star actor in autism spectrum disorder scenario. J. Cell. Mol. Med. 2019, 24, 2064–2069. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Schaaf, C.P. Autism genetics—An overview. Prenat. Diagn. 2016, 37, 14–30. [Google Scholar] [CrossRef]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.-Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef]
- Herrero, M.J.; Velmeshev, D.; Hernandez-Pineda, D.; Sethi, S.; Sorrells, S.; Banerjee, P.; Sullivan, C.; Gupta, A.R.; Kriegstein, A.R.; Corbin, J.G. Identification of amygdala-expressed genes associated with autism spectrum disorder. Mol. Autism 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hotulainen, P.; Hoogenraad, C. Actin in dendritic spines: Connecting dynamics to function. J. Cell Biol. 2010, 189, 619–629. [Google Scholar] [CrossRef]
- Van Spronsen, M.; Hoogenraad, C.C. Synapse Pathology in Psychiatric and Neurologic Disease. Curr. Neurol. Neurosci. Rep. 2010, 10, 207–214. [Google Scholar] [CrossRef]
- Kaizuka, T.; Takumi, T. Postsynaptic density proteins and their involvement in neurodevelopmental disorders. J. Biochem. 2018, 163, 447–455. [Google Scholar] [CrossRef]
- Südhof, T.C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 2008, 455, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-G.; Kishikawa, S.; Higgins, A.W.; Seong, I.-S.; Donovan, D.J.; Shen, Y.; Lally, E.; Weiss, L.A.; Najm, J.; Kutsche, K.; et al. Disruption of Neurexin 1 Associated with Autism Spectrum Disorder. Am. J. Hum. Genet. 2008, 82, 199–207. [Google Scholar] [CrossRef]
- Schaaf, C.P.; Boone, P.M.; Sampath, S.; Williams, C.; Bader, P.I.; Mueller, J.M.; Shchelochkov, O.A.; Brown, C.W.; Crawford, H.P.; Phalen, J.A.; et al. Phenotypic spectrum and genotype–phenotype correlations of NRXN1 exon deletions. Eur. J. Hum. Genet. 2012, 20, 1240–1247. [Google Scholar] [CrossRef]
- Radyushkin, K.; Hammerschmidt, K.; Boretius, S.; Varoqueaux, F.; El-Kordi, A.; Ronnenberg, A.; Winter, D.; Frahm, J.; Fischer, J.; Brose, N.; et al. Neuroligin-3-deficient mice: Model of a monogenic heritable form of autism with an olfactory deficit. Genes Brain Behav. 2009, 8, 416–425. [Google Scholar] [CrossRef]
- Liang, J.; Xu, W.; Hsu, Y.-T.; Yee, A.X.; Chen, L.; Südhof, T.C. Conditional neuroligin-2 knockout in adult medial prefrontal cortex links chronic changes in synaptic inhibition to cognitive impairments. Mol. Psychiatry 2015, 20, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Pak, C.; Danko, T.; Zhang, Y.; Aoto, J.; Anderson, G.R.; Maxeiner, S.; Yi, F.; Wernig, M.; Südhof, T.C. Human Neuropsychiatric Disease Modeling using Conditional Deletion Reveals Synaptic Transmission Defects Caused by Heterozygous Mutations in NRXN1. Cell Stem Cell 2015, 17, 316–328. [Google Scholar] [CrossRef]
- Arking, D.E.; Cutler, D.J.; Brune, C.W.; Teslovich, T.M.; West, K.; Ikeda, M.; Rea, A.; Guy, M.; Lin, S.; Cook, E.H.; et al. A Common Genetic Variant in the Neurexin Superfamily Member CNTNAP2 Increases Familial Risk of Autism. Am. J. Hum. Genet. 2008, 82, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Alarcón, M.; Abrahams, B.S.; Stone, J.L.; Duvall, J.A.; Perederiy, J.V.; Bomar, J.M.; Sebat, J.; Wigler, M.; Martin, C.L.; Ledbetter, D.H.; et al. Linkage, Association, and Gene-Expression Analyses Identify CNTNAP2 as an Autism-Susceptibility Gene. Am. J. Hum. Genet. 2008, 82, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Nord, A.S.; Network, S.P.; Roeb, W.; Dickel, D.E.; Walsh, T.; Kusenda, M.; O’Connor, K.L.; Malhotra, D.; McCarthy, S.E.; Stray, S.M.; et al. Reduced transcript expression of genes affected by inherited and de novo CNVs in autism. Eur. J. Hum. Genet. 2011, 19, 727–731. [Google Scholar] [CrossRef]
- Peñagarikano, O.; Abrahams, B.S.; Herman, E.I.; Winden, K.D.; Gdalyahu, A.; Dong, H.; Sonnenblick, L.I.; Gruver, R.; Almajano, J.; Bragin, A.; et al. Absence of CNTNAP2 Leads to Epilepsy, Neuronal Migration Abnormalities, and Core Autism-Related Deficits. Cell 2011, 147, 235–246. [Google Scholar] [CrossRef]
- Kreienkamp, H.-J. Scaffolding Proteins at the Postsynaptic Density: Shank as the Architectural Framework. In Protein-Protein Interactions as New Drug Targets; Springer: Berlin, Germany, 2008; Volume 186, pp. 365–380. [Google Scholar]
- Berkel, S.; Marshall, C.; Weiss, B.; Howe, J.; Roeth, R.; Moog, U.; Endris, V.; Roberts, W.; Szatmari, P.; Pinto, D.; et al. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat. Genet. 2010, 42, 489–491. [Google Scholar] [CrossRef]
- Jiang, Y.-H.; Ehlers, M.D. Modeling Autism by SHANK Gene Mutations in Mice. Neuron 2013, 78, 8–27. [Google Scholar] [CrossRef]
- Leblond, C.S.; Nava, C.; Polge, A.; Gauthier, J.; Huguet, G.; Lumbroso, S.; Giuliano, F.; Stordeur, C.; Depienne, C.; Mouzat, K.; et al. Meta-analysis of SHANK Mutations in Autism Spectrum Disorders: A Gradient of Severity in Cognitive Impairments. PLoS Genet. 2014, 10, e1004580. [Google Scholar] [CrossRef]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of Genes and Cellular Pathways Dysregulated in Autism Spectrum Disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef]
- Bacchelli, E.; Loi, E.; Cameli, C.; Moi, L.; Vega-Benedetti, A.F.; Blois, S.; Fadda, A.; Bonora, E.; Mattu, S.; Fadda, R.; et al. Analysis of a Sardinian Multiplex Family with Autism Spectrum Disorder Points to Post-Synaptic Density Gene Variants and Identifies CAPG as a Functionally Relevant Candidate Gene. J. Clin. Med. 2019, 8, 212. [Google Scholar] [CrossRef]
- Lahbib, S.; Leblond, C.S.; Hamza, M.; Regnault, B.; Lemée, L.; Mathieu, A.; Jaouadi, H.; Mkaouar, R.; Ben Youssef-Turki, I.; Belhadj, A.; et al. Homozygous 2p11.2 deletion supports the implication of ELMOD3 in hearing loss and reveals the potential association of CAPG with ASD/ID etiology. J. Appl. Genet. 2018, 60, 49–56. [Google Scholar] [CrossRef]
- Fan, Y.; Tang, X.; Vitriol, E.A.; Chen, G.; Zheng, J.Q. Actin capping protein is required for dendritic spine development and synapse formation. J. Neurosci. 2011, 31, 10228–11033. [Google Scholar] [CrossRef] [PubMed]
- Korobova, F.; Svitkina, T.M. Molecular Architecture of Synaptic Actin Cytoskeleton in Hippocampal Neurons Reveals a Mechanism of Dendritic Spine Morphogenesis. Mol. Biol. Cell 2010, 21, 165–176. [Google Scholar] [CrossRef]
- Schmunk, G.; Gargus, J.J. Channelopathy pathogenesis in autism spectrum disorders. Front. Genet. 2013, 4, 222. [Google Scholar] [CrossRef]
- Despang, P.; Salamon, S.; Breitenkamp, A.F.; Kuzmenkina, E.; Herzig, S.; Matthes, J. Autism-associated mutations in the CaVβ2 calcium-channel subunit increase Ba2+-currents and lead to differential modulation by the RGK-protein Gem. Neurobiol. Dis. 2020, 136, 104721. [Google Scholar] [CrossRef]
- Splawski, I.; Timothy, K.W.; Sharpe, L.M.; Decher, N.; Kumar, P.; Bloise, R.; Napolitano, C.; Schwartz, P.J.; Joseph, R.M.; Condouris, K.; et al. CaV1.2 Calcium Channel Dysfunction Causes a Multisystem Disorder Including Arrhythmia and Autism. Cell 2004, 119, 19–31. [Google Scholar] [CrossRef]
- Catterall, W.A.; Perez-Reyes, E.; Snutch, T.P.; Striessnig, J. International Union of Pharmacology. XLVIII. Nomenclature and Structure-Function Relationships of Voltage-Gated Calcium Channels. Pharmacol. Rev. 2005, 57, 411–425. [Google Scholar] [CrossRef]
- Barrett, C.F.; Tsien, R.W. The Timothy syndrome mutation differentially affects voltage- and calcium-dependent inactivation of CaV1.2 L-type calcium channels. Proc. Natl. Acad. Sci. USA 2008, 105, 2157–2162. [Google Scholar] [CrossRef]
- Hemara-Wahanui, A.; Berjukow, S.; Hope, C.I.; Dearden, P.K.; Wu, S.-B.; Wilson-Wheeler, J.; Sharp, D.M.; Lundon-Treweek, P.; Clover, G.M.; Hoda, J.-C.; et al. A CACNA1F mutation identified in an X-linked retinal disorder shifts the voltage dependence of Cav1.4 channel activation. Proc. Natl. Acad. Sci. USA 2005, 102, 7553–7558. [Google Scholar] [CrossRef]
- Pinggera, A.; Lieb, A.; Benedetti, B.; Lampert, M.; Monteleone, S.; Liedl, K.R.; Tuluc, P.; Striessnig, J. CACNA1D De Novo Mutations in Autism Spectrum Disorders Activate Cav1.3 L-Type Calcium Channels. Biol. Psychiatry 2015, 77, 816–822. [Google Scholar] [CrossRef]
- Pinggera, A.; Mackenroth, L.; Rump, A.; Schallner, J.; Beleggia, F.; Wollnik, B.; Striessnig, J. New gain-of-function mutation shows CACNA1D as recurrently mutated gene in autism spectrum disorders and epilepsy. Hum. Mol. Genet. 2017, 26, 2923–2932. [Google Scholar] [CrossRef]
- Boczek, N.J.; Miller, E.M.; Ye, D.; Nesterenko, V.V.; Tester, D.J.; Antzelevitch, C.; Czosek, R.J.; Ackerman, M.J.; Ware, S.M. Novel Timothy syndrome mutation leading to increase in CACNA1C window current. Heart Rhythm 2015, 12, 211–219. [Google Scholar] [CrossRef]
- Splawski, I.; Yoo, D.S.; Stotz, S.C.; Cherry, A.; Clapham, D.E.; Keating, M.T. CACNA1HMutations in Autism Spectrum Disorders. J. Biol. Chem. 2006, 281, 22085–22091. [Google Scholar] [CrossRef]
- Breitenkamp, A.F.S.; Matthes, J.; Nass, R.D.; Sinzig, J.; Lehmkuhl, G.; Nürnberg, P.; Herzig, S. Rare Mutations of CACNB2 Found in Autism Spectrum Disease-Affected Families Alter Calcium Channel Function. PLoS ONE 2014, 9, e95579. [Google Scholar] [CrossRef]
- Morrow, E.M.; Yoo, S.-Y.; Flavell, S.W.; Kim, T.-K.; Lin, Y.; Hill, R.S.; Mukaddes, N.M.; Balkhy, S.; Gascon, G.; Hashmi, A.; et al. Identifying Autism Loci and Genes by Tracing Recent Shared Ancestry. Science 2008, 321, 218–223. [Google Scholar] [CrossRef]
- Veeramah, K.R.; O’Brien, J.E.; Meisler, M.H.; Cheng, X.; Dib-Hajj, S.D.; Waxman, S.G.; Talwar, D.; Girirajan, S.; Eichler, E.E.; Restifo, L.L.; et al. De Novo Pathogenic SCN8A Mutation Identified by Whole-Genome Sequencing of a Family Quartet Affected by Infantile Epileptic Encephalopathy and SUDEP. Am. J. Hum. Genet. 2012, 90, 502–510. [Google Scholar] [CrossRef]
- Chen, C.-P.; Lin, S.-P.; Chern, S.-R.; Chen, Y.-J.; Tsai, F.-J.; Wu, P.-C.; Wang, W. Array-CGH detection of a de novo 2.8 Mb deletion in 2q24.2→q24.3 in a girl with autistic features and developmental delay. Eur. J. Med. Genet. 2010, 53, 217–220. [Google Scholar] [CrossRef]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.A.; Escayg, A.; Kearney, J.A.; Trudeau, M.; Macdonald, B.T.; Mori, M.; Reichert, J.; Buxbaum, J.D.; Meisler, M.H. Sodium channels SCN1A, SCN2A and SCN3A in familial autism. Mol. Psychiatry 2003, 8, 186–194. [Google Scholar] [CrossRef]
- Han, S.; Tai, C.; Westenbroek, R.E.; Yu, F.H.; Cheah, C.S.; Potter, G.B.; Rubenstein, J.L.; Scheuer, T.; De La Iglesia, H.O.; Catterall, W.A. Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature 2012, 489, 385–390. [Google Scholar] [CrossRef]
- Gilling, M.; Rasmussen, H.B.; Calloe, K.; Sequeira, A.F.; Baretto, M.; Oliveira, G.; Almeida, J.; Lauritsen, M.B.; Ullmann, R.; Boonen, S.E.; et al. Dysfunction of the Heteromeric KV7.3/KV7.5 Potassium Channel is Associated with Autism Spectrum Disorders. Front. Genet. 2013, 4, 54. [Google Scholar] [CrossRef]
- Gross, C.; Yao, X.; Pong, D.L.; Jeromin, A.; Bassell, G.J. Fragile X Mental Retardation Protein Regulates Protein Expression and mRNA Translation of the Potassium Channel Kv4.2. J. Neurosci. 2011, 31, 5693–5698. [Google Scholar] [CrossRef]
- Sicca, F.; Imbrici, P.; D’Adamo, M.C.; Moro, F.; Bonatti, F.; Brovedani, P.; Grottesi, A.; Guerrini, R.; Masi, G.; Santorelli, F.M.; et al. Autism with Seizures and Intellectual Disability: Possible Causative Role of Gain-of-function of the Inwardly-Rectifying K+ Channel Kir4.1. Neurobiol. Dis. 2011, 43, 239–247. [Google Scholar] [CrossRef]
- Laumonnier, F.; Eroger, S.; Guérin, P.; Molinari, F.; M’Rad, R.; Cahard, D.; Belhadj, A.; Halayem, M.; Persico, A.M.; Elia, M.; et al. Association of a Functional Deficit of the BKCa Channel, a Synaptic Regulator of Neuronal Excitability, With Autism and Mental Retardation. Am. J. Psychiatry 2006, 163, 1622. [Google Scholar] [CrossRef]
- Deng, P.Y.; Rotman, Z.; Blundon, J.A.; Cho, Y.; Cui, J.; Cavalli, V.; Zakharenko, S.S.; Klyachko, V.A. FMRP Regulates Neurotransmitter Release and Synaptic Information Transmission by Modulating Action Potential Duration via BK Channels. Neuron 2013, 77, 696–711. [Google Scholar] [CrossRef]
- Gonzalez–Gronow, M.; Cuchacovich, M.; Francos, R.; Cuchacovich, S.; Fernandez, M.D.P.; Blanco, A.; Bowers, E.V.; Kaczowka, S.; Pizzo, S.V. Antibodies against the voltage-dependent anion channel (VDAC) and its protective ligand hexokinase-I in children with autism. J. Neuroimmunol. 2010, 227, 153–161. [Google Scholar] [CrossRef]
- Gvozdjáková, A.; Löw, H.; Sun, I.; Navas, P.; Crane, F.L. Plasma membrane coenzyme Q: Evidence for a role in autism. Biol. Targets Ther. 2014, 8, 199. [Google Scholar] [CrossRef]
- Wong, C.C.Y.; Meaburn, E.L.; Ronald, A.; Price, T.S.; Jeffries, A.R.; Schalkwyk, L.C.; Plomin, R.; Mill, J. Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Mol. Psychiatry 2013, 19, 495–503. [Google Scholar] [CrossRef]
- Nguyen, A.; Rauch, T.A.; Pfeifer, G.P.; Hu, V.W. Global methylation profiling of lymphoblastoid cell lines reveals epigenetic contributions to autism spectrum disorders and a novel autism candidate gene, RORA, whose protein product is reduced in autistic brain. FASEB J. 2010, 24, 3036–3051. [Google Scholar] [CrossRef]
- Zhu, Y.; Mordaunt, C.E.; Yasui, D.H.; Marathe, R.; Coulson, R.L.; Dunaway, K.; Jianu, J.M.; Walker, C.K.; Ozonoff, S.; Hertz-Picciotto, I.; et al. Placental DNA methylation levels at CYP2E1 and IRS2 are associated with child outcome in a prospective autism study. Hum. Mol. Genet. 2019, 28, 2659–2674. [Google Scholar] [CrossRef]
- Andrews, S.V.; Sheppard, B.; Windham, G.C.; Schieve, L.A.; Schendel, D.E.; Croen, L.E.; Chopra, P.; Alisch, R.S.; Newschaffer, C.J.; Warren, S.T.; et al. Case-control meta-analysis of blood DNA methylation and autism spectrum disorder. Mol. Autism 2018, 9, 40. [Google Scholar] [CrossRef]
- Siu, M.T.; Butcher, D.T.; Turinsky, A.L.; Cytrynbaum, C.; Stavropoulos, D.J.; Walker, S.; Caluseriu, O.; Carter, M.; Lou, Y.; Nicolson, R.; et al. Functional DNA methylation signatures for autism spectrum disorder genomic risk loci: 16p11.2 deletions and CHD8 variants. Clin. Epigenetics 2019, 11, 1–19. [Google Scholar] [CrossRef]
- Kimura, R.; Nakata, M.; Funabiki, Y.; Suzuki, S.; Awaya, T.; Murai, T.; Hagiwara, M. An epigenetic biomarker for adult high-functioning autism spectrum disorder. Sci. Rep. 2019, 9, 13662–13667. [Google Scholar] [CrossRef] [PubMed]
- Saeliw, T.; Tangsuwansri, C.; Thongkorn, S.; Chonchaiya, W.; Suphapeetiporn, K.; Mutirangura, A.; Tencomnao, T.; Hu, V.W.; Sarachana, T. Integrated genome-wide Alu methylation and transcriptome profiling analyses reveal novel epigenetic regulatory networks associated with autism spectrum disorder. Mol. Autism 2018, 9, 27. [Google Scholar] [CrossRef]
- Wong, C.C.Y.; Smith, R.G.; Hannon, E.; Ramaswami, G.; Parikshak, N.N.; Assary, E.; Troakes, C.; Poschmann, J.; Schalkwyk, L.C.; Sun, W.; et al. Genome-wide DNA methylation profiling identifies convergent molecular signatures associated with idiopathic and syndromic autism in post-mortem human brain tissue. Hum. Mol. Genet. 2019, 28, 2201–2211. [Google Scholar] [CrossRef]
- Stathopoulos, S.; Gaujoux, R.; Lindeque, Z.; Mahony, C.; Van Der Colff, R.; Van Der Westhuizen, F.; O’Ryan, C. DNA Methylation Associated with Mitochondrial Dysfunction in a South African Autism Spectrum Disorder Cohort. Autism Res. 2020, 13, 1079–1093. [Google Scholar] [CrossRef]
- Hu, Z.; Yang, Y.; Zhao, Y.; Yu, H.; Ying, X.; Zhou, D.; Zhong, J.; Zheng, Z.; Liu, J.; Pan, R.; et al. APOE hypermethylation is associated with autism spectrum disorder in a Chinese population. Exp. Ther. Med. 2018, 15, 4749–4754. [Google Scholar] [CrossRef]
- Hranilović, D.; Blažević, S.; Stefulj, J.; Zill, P. DNA Methylation Analysis of HTR2A Regulatory Region in Leukocytes of Autistic Subjects. Autism Res. 2015, 9, 204–209. [Google Scholar] [CrossRef]
- Hu, Z.; Ying, X.; Huang, L.; Zhao, Y.; Zhou, D.; Liu, J.; Zhong, J.; Huang, T.; Zhang, W.; Cheng, F.; et al. Association of human serotonin receptor 4 promoter methylation with autism spectrum disorder. Medicine 2020, 99, e18838. [Google Scholar] [CrossRef]
- Lu, Z.; Liu, Z.; Mao, W.; Wang, X.; Zheng, X.; Chen, S.; Cao, B.; Huang, S.; Zhang, X.; Zhou, T.; et al. Locus-specific DNA methylation of Mecp2 promoter leads to autism-like phenotypes in mice. Cell Death Dis. 2020, 11, 85. [Google Scholar] [CrossRef]
- Hicks, S.D.; Middleton, F.A. A Comparative Review of microRNA Expression Patterns in Autism Spectrum Disorder. Front. Psychiatry 2016, 7, 176. [Google Scholar] [CrossRef]
- Hicks, S.D.; Carpenter, R.L.; Wagner, K.E.; Pauley, R.; Barros, M.; Tierney-Aves, C.; Barns, S.; Greene, C.D.; Middleton, F.A. Saliva MicroRNA Differentiates Children with Autism from Peers with Typical and Atypical Development. J. Am. Acad. Child. Adolesc. Psychiatry 2020, 59, 296–308. [Google Scholar] [CrossRef]
- Sehovic, E.; Spahic, L.; Smajlovic-Skenderagic, L.; Pistoljevic, N.; Dzanko, E.; Hajdarpasic, A. Identification of developmental disorders including autism spectrum disorder using salivary miRNAs in children from Bosnia and Herzegovina. PLoS ONE 2020, 15, e0232351. [Google Scholar] [CrossRef]
- Hagiwara, M.; Kimura, R.; Funabiki, Y.; Awaya, T.; Murai, T.; Hagiwara, M. MicroRNA profiling in adults with high-functioning autism spectrum disorder. Mol. Brain 2019, 12, 82–84. [Google Scholar] [CrossRef]
- Ozkul, Y.; Taheri, S.; Bayram, K.K.; Sener, E.F.; Mehmetbeyoglu, E.; Öztop, D.B.; Aybuga, F.; Tufan, E.; Bayram, A.; Dolu, N.; et al. A heritable profile of six miRNAs in autistic patients and mouse models. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Vasu, M.M.; Anitha, A.; Thanseem, I.; Suzuki, K.; Yamada, K.; Takahashi, T.; Wakuda, T.; Iwata, K.; Tsujii, M.; Sugiyama, T.; et al. Serum microRNA profiles in children with autism. Mol. Autism 2014, 5, 40. [Google Scholar] [CrossRef]
- Vatsa, N.; Kumar, V.; Singh, B.K.; Kumar, S.S.; Sharma, A.; Jana, N.R. Down-Regulation of miRNA-708 Promotes Aberrant Calcium Signaling by Targeting Neuronatin in a Mouse Model of Angelman Syndrome. Front. Mol. Neurosci. 2019, 12, 35. [Google Scholar] [CrossRef]
- Urdinguio, R.G.; Fernández, A.F.; López-Nieva, P.; Rossi, S.; Huertas, D.; Kulis, M.; Liu, C.-G.; Croce, C.M.; Calin, G.A.; Esteller, M. Disrupted microRNA expression caused by Mecp2 loss in a mouse model of Rett syndrome. Epigenetics 2010, 5, 656–663. [Google Scholar] [CrossRef]
- Edbauer, D.; Neilson, J.R.; Foster, K.A.; Wang, C.-F.; Seeburg, D.P.; Batterton, M.N.; Tada, T.; Dolan, B.M.; Sharp, P.A.; Sheng, M. Regulation of Synaptic Structure and Function by FMRP-Associated MicroRNAs miR-125b and miR-132. Neuron 2010, 65, 373–384. [Google Scholar] [CrossRef]
- Gaugler, T.; Klei, L.; Sanders, S.J.; Bodea, C.A.; Goldberg, A.P.; Lee, A.B.; Mahajan, M.C.; Manaa, D.; Pawitan, Y.; Reichert, J.G.; et al. Most genetic risk for autism resides with common variation. Nat. Genet. 2014, 46, 881–885. [Google Scholar] [CrossRef]
- Deng, W.; Zou, X.; Deng, H.; Li, J.; Tang, C.; Wang, X.; Guo, X. The Relationship Among Genetic Heritability, Environmental Effects, and Autism Spectrum Disorders. J. Child Neurol. 2015, 30, 1794–1799. [Google Scholar] [CrossRef]
- Edelson, L.R.; Saudino, K.J. Genetic and environmental influences on autistic-like behaviors in 2-year-old twins. Behav. Genet. 2009, 39, 255–264. [Google Scholar] [CrossRef]
- Bölte, S.; Girdler, S.; Marschik, P.B. The contribution of environmental exposure to the etiology of autism spectrum disorder. Cell. Mol. Life Sci. 2019, 76, 1275–1297. [Google Scholar] [CrossRef]
- Janecka, M.; Mill, J.; Basson, M.A.; Goriely, A.; Spiers, H.; Reichenberg, A.; Schalkwyk, L.; Fernandes, C. Advanced paternal age effects in neurodevelopmental disorders—Review of potential underlying mechanisms. Transl. Psychiatry 2017, 7, e1019. [Google Scholar] [CrossRef]
- Wu, S.; Wu, F.; Ding, Y.; Hou, J.; Bi, J.; Zhang, Z. Advanced parental age and autism risk in children: A systematic review and meta-analysis. Acta Psychiatr. Scand. 2016, 135, 29–41. [Google Scholar] [CrossRef]
- Atsem, S.; Reichenbach, J.; Potabattula, R.; Dittrich, M.; Nava, C.; Depienne, C.; Böhm, L.; Rost, S.; Hahn, T.; Schorsch, M.; et al. Paternal age effects on spermFOXK1andKCNA7methylation and transmission into the next generation. Hum. Mol. Genet. 2016, 25, 4996–5005. [Google Scholar] [CrossRef]
- Kojima, M.; Yassin, W.; Owada, K.; Aoki, Y.; Kuwabara, H.; Natsubori, T.; Iwashiro, N.; Gonoi, W.; Takao, H.; Kasai, K.; et al. Neuroanatomical Correlates of Advanced Paternal and Maternal Age at Birth in Autism Spectrum Disorder. Cereb. Cortex 2018, 29, 2524–2532. [Google Scholar] [CrossRef]
- Foldi, C.J.; Eyles, D.; McGrath, J.J.; Burne, T.H.J. Advanced paternal age is associated with alterations in discrete behavioural domains and cortical neuroanatomy of C57BL/6J mice. Eur. J. Neurosci. 2010, 31, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Sampino, S.; Juszczak, G.R.; Zacchini, F.; Swiergiel, A.H.; Modlinski, J.A.; Loi, P.; Ptak, G. Grand-paternal age and the development of autism-like symptoms in mice progeny. Transl. Psychiatry 2014, 4, e386. [Google Scholar] [CrossRef]
- Gardener, H.; Spiegelman, D.; Buka, S.L. Prenatal risk factors for autism: Comprehensive meta-analysis. Br. J. Psychiatry 2009, 195, 7–14. [Google Scholar] [CrossRef]
- Gardener, H.; Spiegelman, D.; Buka, S.L. Perinatal and Neonatal Risk Factors for Autism: A Comprehensive Meta-analysis. Pediatrics 2011, 128, 344–355. [Google Scholar] [CrossRef]
- Wang, C.; Geng, H.; Liu, W.; Zhang, G. Prenatal, perinatal, and postnatal factors associated with autism. Medicine 2017, 96, e6696. [Google Scholar] [CrossRef]
- Bejerot, S.; Eriksson, J.M.; Bonde, S.; Carlström, K.; Humble, M.B.; Eriksson, E. The extreme male brain revisited: Gender coherence in adults with autism spectrum disorder. Br. J. Psychiatry 2012, 201, 116–123. [Google Scholar] [CrossRef]
- Baron-Cohen, S.; Auyeung, B.; Nørgaard-Pedersen, B.; Hougaard, D.M.; Abdallah, M.W.; Melgaard, L.; Cohen, A.S.; Chakrabarti, B.; Ruta, L.; Lombardo, M.V. Elevated fetal steroidogenic activity in autism. Mol. Psychiatry 2015, 20, 369–376. [Google Scholar] [CrossRef]
- Baron-Cohen, S.; Tsompanidis, A.; Auyeung, B.; Nørgaard-Pedersen, B.; Hougaard, D.M.; Abdallah, M.; Cohen, A.; Pohl, A. Foetal oestrogens and autism. Mol. Psychiatry 2020, 25, 2970–2978. [Google Scholar] [CrossRef] [PubMed]
- Crider, A.; Thakkar, R.; Ahmed, A.O.; Pillai, A. Dysregulation of estrogen receptor beta (ERβ), aromatase (CYP19A1), and ER co-activators in the middle frontal gyrus of autism spectrum disorder subjects. Mol. Autism 2014, 5, 46. [Google Scholar] [CrossRef]
- Chakrabarti, B.; Dudbridge, F.; Kent, L.; Wheelwright, S.; Hill-Cawthorne, G.; Allison, C.; Banerjee-Basu, S.; Baron-Cohen, S. Genes related to sex steroids, neural growth, and social-emotional behavior are associated with autistic traits, empathy, and Asperger syndrome. Autism Res. 2009, 2, 157–177. [Google Scholar] [CrossRef]
- Jackson, A.A.; Robinson, S.M. Dietary guidelines for pregnancy: A review of current evidence. Public Health Nutr. 2001, 4, 625–630. [Google Scholar] [CrossRef]
- Van Eijsden, M.; Smits, L.J.M.; Van Der Wal, M.F.; Bonsel, G.J. Association between short interpregnancy intervals and term birth weight: The role of folate depletion. Am. J. Clin. Nutr. 2008, 88, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, C.; Kosidou, K.; Dalman, C.; Lundberg, M.; Lee, B.K.; Rai, D.; Karlsson, H.; Gardner, R.; Arver, S. Maternal vitamin D deficiency and the risk of autism spectrum disorders: Population-based study. BJPsych Open 2016, 2, 170–172. [Google Scholar] [CrossRef]
- Robea, M.; Luca, A.-C.; Ciobica, A. Relationship between Vitamin Deficiencies and Co-Occurring Symptoms in Autism Spectrum Disorder. Medicine 2020, 56, 245. [Google Scholar] [CrossRef]
- Saghazadeh, A.; Ahangari, N.; Hendi, K.; Saleh, F.; Rezaei, N. Status of essential elements in autism spectrum disorder: Systematic review and meta-analysis. Rev. Neurosci. 2017, 28, 783–809. [Google Scholar] [CrossRef]
- Schmidt, R.J.; Tancredi, D.J.; Krakowiak, P.; Hansen, R.L.; Ozonoff, S. Maternal Intake of Supplemental Iron and Risk of Autism Spectrum Disorder. Am. J. Epidemiol. 2014, 180, 890–900. [Google Scholar] [CrossRef]
- Hagmeyer, S.; Mangus, K.; Boeckers, T.M.; Grabrucker, A.M. Effects of Trace Metal Profiles Characteristic for Autism on Synapses in Cultured Neurons. Neural Plast. 2015, 2015, 1–16. [Google Scholar] [CrossRef]
- Goyal, D.K.; Neil, J.R.; Simmons, S.D.; Mansab, F.; Benjamin, S.; Pitfield, V.; Boulet, S.; Miyan, J.A. Zinc Deficiency in Autism: A Controlled Study. Insights Biomed. 2019, 4, 4. [Google Scholar] [CrossRef]
- Yasuda, H.; Yoshida, K.; Yasuda, Y.; Tsutsui, T. Infantile zinc deficiency: Association with autism spectrum disorders. Sci. Rep. 2011, 1, 129. [Google Scholar] [CrossRef]
- Li, S.-O.; Wang, J.-L.; Bjørklund, G.; Zhao, W.-N.; Yin, C.-H. Serum copper and zinc levels in individuals with autism spectrum disorders. NeuroReport 2014, 25, 1216–1220. [Google Scholar] [CrossRef]
- Grabrucker, S.; Boeckers, T.M.; Grabrucker, A.M. Gender Dependent Evaluation of Autism like Behavior in Mice Exposed to Prenatal Zinc Deficiency. Front. Behav. Neurosci. 2016, 10, 37. [Google Scholar] [CrossRef]
- Grabrucker, S.; Jannetti, L.; Eckert, M.; Gaub, S.; Chhabra, R.; Pfaender, S.; Mangus, K.; Reddy, P.P.; Rankovic, V.; Schmeisser, M.J.; et al. Zinc deficiency dysregulates the synaptic ProSAP/Shank scaffold and might contribute to autism spectrum disorders. Brain 2013, 137, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Grabrucker, A.M.; Knight, M.J.; Proepper, C.; Bockmann, J.; Joubert, M.; Rowan, M.; Nienhaus, G.U.; Garner, C.C.; Bowie, J.U.; Kreutz, M.R.; et al. Concerted action of zinc and ProSAP/Shank in synaptogenesis and synapse maturation. EMBO J. 2011, 30, 569–581. [Google Scholar] [CrossRef]
- Frederickson, C.J.; Koh, J.-Y.; Bush, A.I. The neurobiology of zinc in health and disease. Nat. Rev. Neurosci. 2005, 6, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Vyas, Y.; Lee, K.; Jung, Y.; Montgomery, J.M. Influence of maternal zinc supplementation on the development of autism-associated behavioural and synaptic deficits in offspring Shank3-knockout mice. Mol. Brain 2020, 13, 1–18. [Google Scholar] [CrossRef]
- Shih, P.-Y.; Hsieh, B.-Y.; Lin, M.-H.; Huang, T.-N.; Tsai, C.-Y.; Pong, W.-L.; Lee, S.-P.; Hsueh, Y.-P. CTTNBP2 Controls Synaptic Expression of Zinc-Related Autism-Associated Proteins and Regulates Synapse Formation and Autism-like Behaviors. Cell Rep. 2020, 31, 107700. [Google Scholar] [CrossRef]
- Velie, E.M.; Block, G.; Shaw, G.M.; Samuels, S.J.; Schaffer, D.M.; Kulldorff, M. Maternal supplemental and dietary zinc intake and the occurrence of neural tube defects in California. Am. J. Epidemiol. 1999, 150, 605–616. [Google Scholar] [CrossRef]
- Gardner, R.M.; Lee, B.K.; Magnusson, C.; Rai, D.; Frisell, T.; Karlsson, H.; Idring, S.; Dalman, C. Maternal body mass index during early pregnancy, gestational weight gain, and risk of autism spectrum disorders: Results from a Swedish total population and discordant sibling study. Int. J. Epidemiol. 2015, 44, 870–883. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, S.; Xu, S.; Weng, S.; Liu, Z. Maternal Body Mass Index and Risk of Autism Spectrum Disorders in Offspring: A Meta-analysis. Sci. Rep. 2016, 6, 34248. [Google Scholar] [CrossRef] [PubMed]
- Getz, K.D.; Anderka, M.T.; Werler, M.M.; Jick, S.S. Maternal Pre-pregnancy Body Mass Index and Autism Spectrum Disorder among Offspring: A Population-Based Case-Control Study. Paediatr. Périnat. Epidemiol. 2016, 30, 479–487. [Google Scholar] [CrossRef]
- Li, M.; Fallin, M.D.; Riley, A.; Landa, R.; Walker, S.O.; Silverstein, M.; Caruso, D.; Pearson, C.; Kiang, S.; Dahm, J.L.; et al. The Association of Maternal Obesity and Diabetes with Autism and Other Developmental Disabilities. Pediatrics 2016, 137, e20152206. [Google Scholar] [CrossRef]
- Tang, S.; Wang, Y.; Gong, X.; Wang, G.-H. A Meta-Analysis of Maternal Smoking during Pregnancy and Autism Spectrum Disorder Risk in Offspring. Int. J. Environ. Res. Public Health 2015, 12, 10418–10431. [Google Scholar] [CrossRef]
- Rosen, B.N.; Lee, B.K.; Lee, N.L.; Yang, Y.; Burstyn, I. Maternal Smoking and Autism Spectrum Disorder: A Meta-analysis. J. Autism Dev. Disord. 2014, 45, 1689–1698. [Google Scholar] [CrossRef]
- Visser, J.C.; Rommelse, N.; Vink, L.; Schrieken, M.; Oosterling, I.J.; Van Der Gaag, R.J.; Buitelaar, J.K. Narrowly Versus Broadly Defined Autism Spectrum Disorders: Differences in Pre- and Perinatal Risk Factors. J. Autism Dev. Disord. 2012, 43, 1505–1516. [Google Scholar] [CrossRef]
- Perrone-McGovern, K.; Simon-Dack, S.; Niccolai, L. Prenatal and Perinatal Factors Related to Autism, IQ, and Adaptive Functioning. J. Genet. Psychol. 2015, 176, 1–10. [Google Scholar] [CrossRef]
- Eliasen, M.H.; Tolstrup, J.S.; Andersen, A.-M.N.; Gronbaek, M.; Olsen, J.; Strandberg-Larsen, K.; Grønbæk, M. Prenatal alcohol exposure and autistic spectrum disorders—A population-based prospective study of 80 552 children and their mothers. Int. J. Epidemiol. 2010, 39, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, C.; McCarthy, F.P.; Ryan, R.M.; Khashan, A.S. Maternal Alcohol Consumption during Pregnancy and the Risk of Autism Spectrum Disorders in Offspring: A Retrospective Analysis of the Millennium Cohort Study. J. Autism Dev. Disord. 2018, 48, 3773–3782. [Google Scholar] [CrossRef]
- Meador, K.J. Effects of in Utero Antiepileptic Drug Exposure. Epilepsy Curr. 2008, 8, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Veroniki, A.A.; Cogo, E.; Rios, P.; Straus, S.E.; Finkelstein, Y.; Kealey, R.; Reynen, E.; Soobiah, C.; Thavorn, K.; Hutton, B.; et al. Comparative safety of anti-epileptic drugs during pregnancy: A systematic review and network meta-analysis of congenital malformations and prenatal outcomes. BMC Med. 2017, 15, 1–20. [Google Scholar] [CrossRef]
- Kobayashi, T.; Matsuyama, T.; Takeuchi, M.; Ito, S. Autism spectrum disorder and prenatal exposure to selective serotonin reuptake inhibitors: A systematic review and meta-analysis. Reprod. Toxicol. 2016, 65, 170–178. [Google Scholar] [CrossRef]
- Wan, H.; Zhang, C.; Li, H.; Luan, S.; Liu, C. Association of maternal diabetes with autism spectrum disorders in offspring. Medicine 2018, 97, e9438. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Jing, J.; Bowers, K.; Liu, B.; Bao, W. Maternal diabetes and the risk of autism spectrum disorders in the offspring: A systematic review and meta-analysis. J. Autism Dev. Disord. 2014, 44, 766–775. [Google Scholar] [CrossRef]
- Jiang, H.-Y.; Xu, L.-L.; Shao, L.; Xia, R.-M.; Yu, Z.-H.; Ling, Z.; Yang, F.; Deng, M.; Ruan, B. Maternal infection during pregnancy and risk of autism spectrum disorders: A systematic review and meta-analysis. Brain Behav. Immun. 2016, 58, 165–172. [Google Scholar] [CrossRef]
- Zerbo, O.; Qian, Y.; Yoshida, C.; Grether, J.K.; Van De Water, J.; Croen, L.A. Maternal Infection during Pregnancy and Autism Spectrum Disorders. J. Autism Dev. Disord. 2015, 45, 4015–4025. [Google Scholar] [CrossRef]
- Atladóttir, H.Ó.; Thorsen, P.; Østergaard, L.; Schendel, D.E.; Lemcke, S.; Abdallah, M.; Parner, E.T. Maternal Infection Requiring Hospitalization During Pregnancy and Autism Spectrum Disorders. J. Autism Dev. Disord. 2010, 40, 1423–1430. [Google Scholar] [CrossRef]
- Chen, S.-W.; Zhong, X.-S.; Jiang, L.-N.; Zheng, X.-Y.; Xiong, Y.-Q.; Ma, S.-J.; Qiu, M.; Huo, S.-T.; Ge, J.; Chen, Q. Maternal autoimmune diseases and the risk of autism spectrum disorders in offspring: A systematic review and meta-analysis. Behav. Brain Res. 2016, 296, 61–69. [Google Scholar] [CrossRef]
- Wu, S.; Ding, Y.; Wu, F.; Li, R.; Xie, G.; Hou, J.; Mao, P. Family history of autoimmune diseases is associated with an increased risk of autism in children: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2015, 55, 322–332. [Google Scholar] [CrossRef]
- Brown, A.S.; Sourander, A.; Hinkka-Yli-Salomäki, S.; McKeague, I.W.; Sundvall, J.; Surcel, H.-M. Elevated maternal C-reactive protein and autism in a national birth cohort. Mol. Psychiatry 2013, 19, 259–264. [Google Scholar] [CrossRef]
- Brown, A.S.; Surcel, H.-M.; Hinkka-Yli-Salomäki, S.; Cheslack-Postava, K.; Bao, Y.; Sourander, A. Maternal thyroid autoantibody and elevated risk of autism in a national birth cohort. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2015, 57, 86–92. [Google Scholar] [CrossRef]
- Meyer, U.; Feldon, J.; Fatemi, S.H. In-vivo rodent models for the experimental investigation of prenatal immune activation effects in neurodevelopmental brain disorders. Neurosci. Biobehav. Rev. 2009, 33, 1061–1079. [Google Scholar] [CrossRef]
- Comi, A.M.; Zimmerman, A.W.; Frye, V.H.; Law, P.A.; Peeden, J.N. Familial Clustering of Autoimmune Disorders and Evaluation of Medical Risk Factors in Autism. J. Child Neurol. 1999, 14, 388–394. [Google Scholar] [CrossRef]
- Atladóttir, H.O.; Pedersen, M.G.; Thorsen, P.; Mortensen, P.B.; Deleuran, B.; Eaton, W.W.; Parner, E.T.; Sutton, R.M.; Niles, D.; Nysaether, J.; et al. Association of Family History of Autoimmune Diseases and Autism Spectrum Disorders. Pediatrics 2009, 124, 687–694. [Google Scholar] [CrossRef]
- Sanna, A.; Firinu, D.; Zavattari, P.; Valera, P. Zinc Status and Autoimmunity: A Systematic Review and Meta-Analysis. Nutrients 2018, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Hung, C.; Wong, S.; Giulivi, C. Toxicity of the Flame-Retardant BDE-49 on Brain Mitochondria and Neuronal Progenitor Striatal Cells Enhanced by a PTEN-Deficient Background. Toxicol. Sci. 2013, 132, 196–210. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Frye, R.E. Mitochondrial dysfunction in autism spectrum disorders: A systematic review and meta-analysis. Mol. Psychiatry 2012, 17, 290–314. [Google Scholar] [CrossRef]
- Yoshimasu, K.; Kiyohara, C.; Takemura, S.; Nakai, K. A meta-analysis of the evidence on the impact of prenatal and early infancy exposures to mercury on autism and attention deficit/hyperactivity disorder in the childhood. NeuroToxicology 2014, 44, 121–131. [Google Scholar] [CrossRef]
- Shelton, J.F.; Geraghty, E.M.; Tancredi, D.; Delwiche, L.D.; Schmidt, R.J.; Ritz, B.; Hansen, R.L.; Hertz-Picciotto, I. Neurodevelopmental Disorders and Prenatal Residential Proximity to Agricultural Pesticides: The CHARGE Study. Environ. Health Perspect. 2014, 122, 1103–1109. [Google Scholar] [CrossRef]
- Jeddi, M.Z.; Janani, L.; Memari, A.H.; Akhondzadeh, S.; Yunesian, M. The role of phthalate esters in autism development: A systematic review. Environ. Res. 2016, 151, 493–504. [Google Scholar] [CrossRef]
- Dingemans, M.M.; Berg, M.V.D.; Westerink, R.H.S. Neurotoxicity of Brominated Flame Retardants: (In)direct Effects of Parent and Hydroxylated Polybrominated Diphenyl Ethers on the (Developing) Nervous System. Environ. Health Perspect. 2011, 119, 900–907. [Google Scholar] [CrossRef]
- Wayman, G.A.; Bose, D.D.; Yang, N.; Lesiak, A.; Bruun, N.; Impey, S.; LeDoux, V.; Pessah, I.N.; Lein, P.J. PCB-95 Modulates the Calcium-Dependent Signaling Pathway Responsible for Activity-Dependent Dendritic Growth. Environ. Health Perspect. 2012, 120, 1003–1009. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Category | Gene Symbol | Gene Name | Alterations | Associated Syndromes |
|---|---|---|---|---|
| Chromatin regulators | ANKRD11 | Ankyrin repeat domain 11 | Mutations; copy number loss | KBG syndrome; Cornelia de Lange syndrome |
| ARID1B | AT-rich interaction domain 1B | Mutations; copy number loss; copy number gain; translocation | Coffin–Siris syndrome | |
| ASXL3 | ASXL Transcriptional Regulator 3 | Mutations | Bainbridge-Ropers syndrome | |
| ATRX | ATRX Chromatin Remodeler | Mutations; copy number loss | ||
| AUTS2 | Autism susceptibility candidate 2 | Mutations; copy number loss; copy number gain; inversion; translocation | ||
| CHD2 | Chromodomain helicase DNA binding protein 2 | Mutations; copy number loss | Tourette syndrome | |
| CHD7 | Chromodomain helicase DNA binding protein 7 | Mutations; copy number loss; translocation | CHARGE syndrome | |
| CHD8 | Chromodomain helicase DNA binding protein 8 | Mutations; copy number loss; copy number gain; translocation | ||
| CREBBP | CREB-binding protein | Mutations; copy number loss | Rubinstein–Taybi syndrome, Menke-Hennekam syndrome 1, Tourette syndrome | |
| EHMT1 | Euchromatic histone-lysine N-methyltransferase 1 | Mutations; copy number loss; copy number gain; translocation | Kleefstra syndrome | |
| MBD5 | Methyl-CpG binding domain protein 5 | Mutations; copy number loss; copy number gain; inversion; translocation | 2q23.1 microdeletion syndrome, Kleefstra syndrome | |
| MECP2 | Methyl CpG binding protein 2 | Mutations; copy number loss; copy number gain; promoter methylation | Rett syndrome, X-linked intellectual disability, MECP2 duplication syndrome | |
| SETD5 | SET domain containing 5 | Mutations; copy number loss | ||
| Transcription factors/regulators | ADNP | Activity-dependent neuroprotector homeobox | Mutations; copy number loss | Helsmoortel-Van der Aa syndrome |
| FOXG1 | Forkhead box G1 | Mutations; copy number loss; copy number gain; translocation | Rett syndrome, FOXG1 syndrome, West syndrome, | |
| FOXP1 | Forkhead box P1 | Mutations; copy number loss; inversion; translocation | ||
| FOXP2 | Forkhead box P2 | Mutations; copy number loss; translocation | FOXP2-related speech and language disorder | |
| MED13L | Mediator complex subunit 13-like | Mutations; copy number loss; copy number gain | ||
| POGZ | Pogo transposable element with ZNF domain | Mutations; copy number loss; copy number gain | White–Sutton syndrome | |
| RAI1 | Retinoic Acid Induced 1 | Mutations; copy number loss; copy number gain | Smith–Magenis syndrome, Potocki–Lupski syndrome | |
| TBR1 | T-box, brain, 1 | Mutations; copy number loss | ||
| TCF4 | Transcription factor 4 | Mutations; copy number loss; translocation | Pitt–Hopkins syndrome | |
| ZBTB20 | Zinc finger and BTB domain containing 20 | Mutations; copy number loss; translocation | 3q13.31 microdeletion syndrome, Primrose syndrome, | |
| mRNA binding and trafficking regulator | FMR1 and its pathways | Fragile X mental retardation 1 | Mutations; copy number loss | Fragile X syndrome, Fragile X-associated tremor/ataxia syndrome |
| Protein degradation | UBE3A | Ubiquitin protein ligase E3A | Mutations; copy number gain | Angelman syndrome |
| Cell growth/proliferation | DYRK1A | Dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A | Mutations; copy number loss; inversion; translocation | |
| NF1 | Neurofibromin 1 | Mutations; copy number loss | ||
| PTEN and its pathways | Phosphatase and tensin homolog | Mutations; copy number loss | Cowden syndrome, Macrocephaly/autism syndrome, PTEN hamartoma tumor syndrome | |
| SYNGAP1 | Synaptic Ras GTPase activating protein 1 | Mutations; copy number loss; translocation | ||
| TSC1/TSC2 | Tuberous sclerosis 1/2 | Mutations | ||
| Protein modification | CDKL5 | Cyclin-dependent kinase-like 5 | Mutations; copy number loss; copy number gain translocation | Rett syndrome, Angelman syndrome |
| Factor | Evidence | References |
|---|---|---|
| Parental age | Association studies; meta-analyses; animal studies | [108,109,110,111,112,113] |
| Perinatal factors | Meta-analyses | [114,115,116] |
| Sex steroids | Association studies | [117,118,119,120,121] |
| Maternal nutrition | Association studies; meta-analyses; in vitro studies, animal studies | [123,124,125,126,127,128,129,130,131,132,133,134,136,137,138,139,140,141,142] |
| Fetal exposure to drugs, smoke, alcohol | Association studies; meta-analyses | [143,144,145,146,147,148,149,150,151] |
| Maternal diseases | Meta-analyses | [152,153] |
| Maternal infections | Association studies; meta-analyses; animal studies | [154,155,156,157,158,159,160,161] |
| Fetal exposure to toxic xenobiotics | Association studies; meta-analyses; in vitro studies; animal studies | [166,167,168,169,170,171] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masini, E.; Loi, E.; Vega-Benedetti, A.F.; Carta, M.; Doneddu, G.; Fadda, R.; Zavattari, P. An Overview of the Main Genetic, Epigenetic and Environmental Factors Involved in Autism Spectrum Disorder Focusing on Synaptic Activity. Int. J. Mol. Sci. 2020, 21, 8290. https://doi.org/10.3390/ijms21218290
Masini E, Loi E, Vega-Benedetti AF, Carta M, Doneddu G, Fadda R, Zavattari P. An Overview of the Main Genetic, Epigenetic and Environmental Factors Involved in Autism Spectrum Disorder Focusing on Synaptic Activity. International Journal of Molecular Sciences. 2020; 21(21):8290. https://doi.org/10.3390/ijms21218290
Chicago/Turabian StyleMasini, Elena, Eleonora Loi, Ana Florencia Vega-Benedetti, Marinella Carta, Giuseppe Doneddu, Roberta Fadda, and Patrizia Zavattari. 2020. "An Overview of the Main Genetic, Epigenetic and Environmental Factors Involved in Autism Spectrum Disorder Focusing on Synaptic Activity" International Journal of Molecular Sciences 21, no. 21: 8290. https://doi.org/10.3390/ijms21218290
APA StyleMasini, E., Loi, E., Vega-Benedetti, A. F., Carta, M., Doneddu, G., Fadda, R., & Zavattari, P. (2020). An Overview of the Main Genetic, Epigenetic and Environmental Factors Involved in Autism Spectrum Disorder Focusing on Synaptic Activity. International Journal of Molecular Sciences, 21(21), 8290. https://doi.org/10.3390/ijms21218290