AHR Signaling Interacting with Nutritional Factors Regulating the Expression of Markers in Vascular Inflammation and Atherogenesis

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
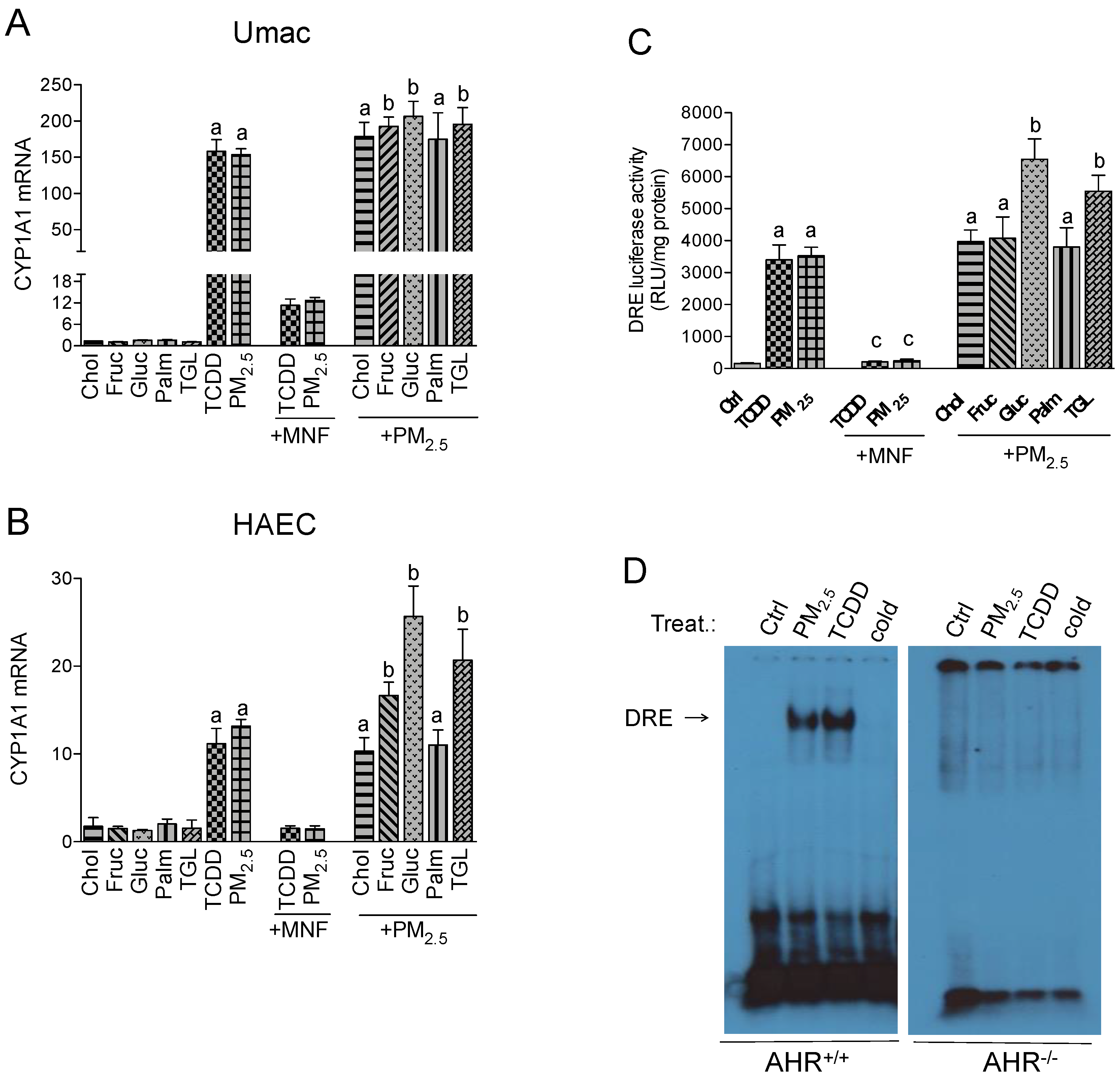
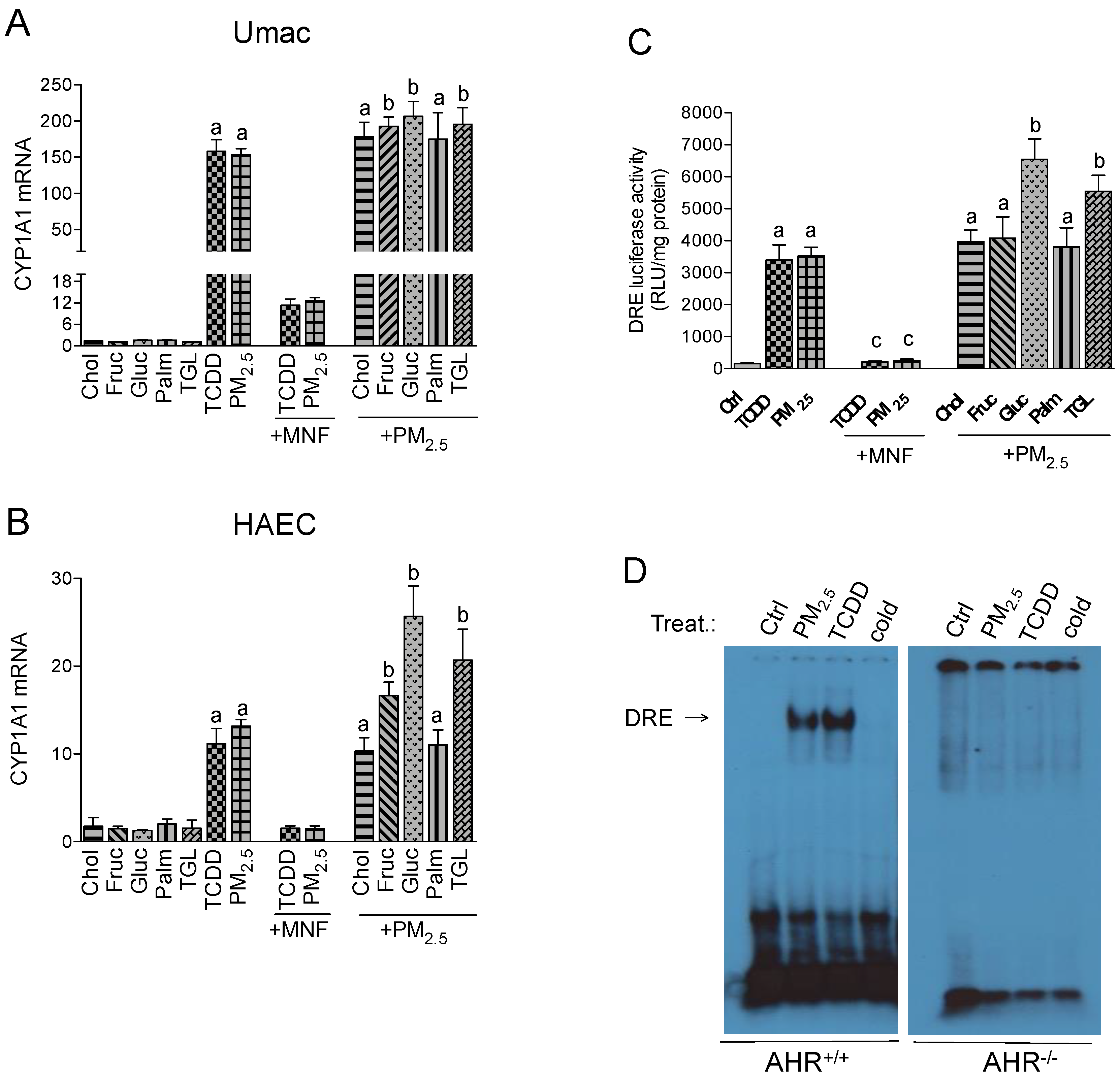
2.1. CYP1A1 Expression in Response to PM2.5 and Nutritional Factors
2.2. Effect of Nutritional Factors on PM2.5-Mediated AHR Activity
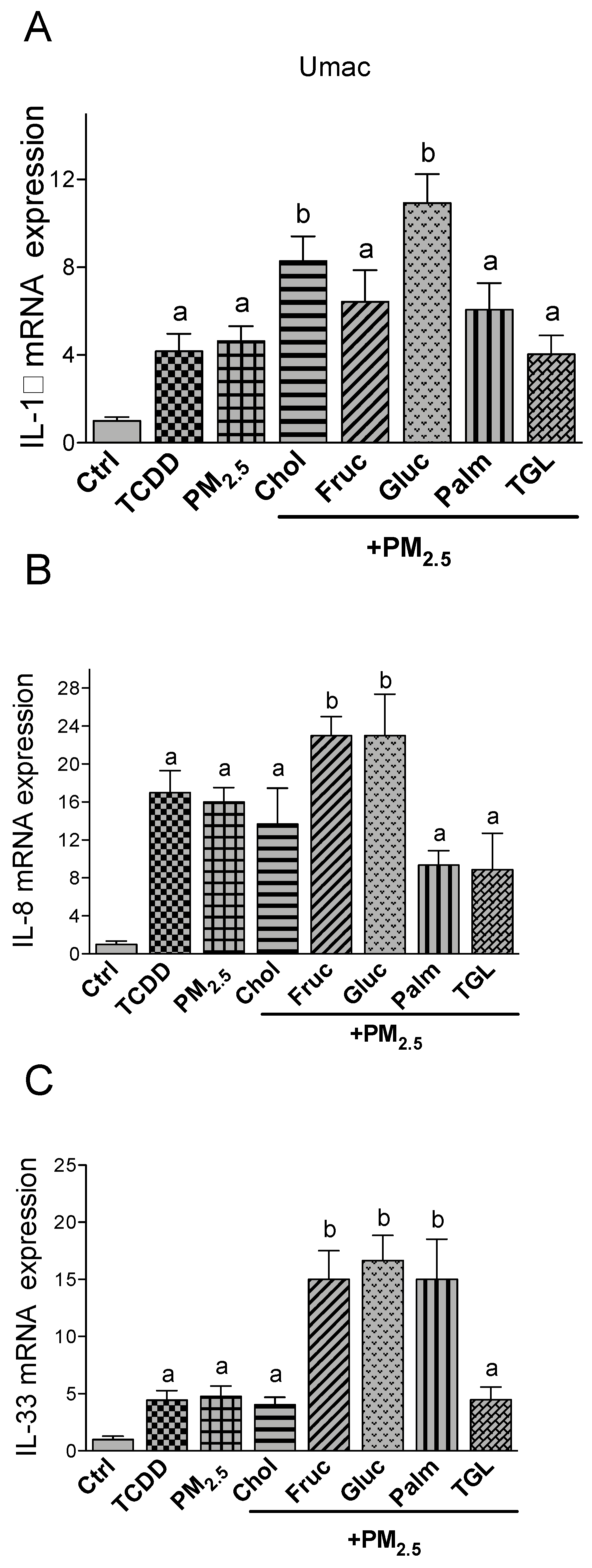
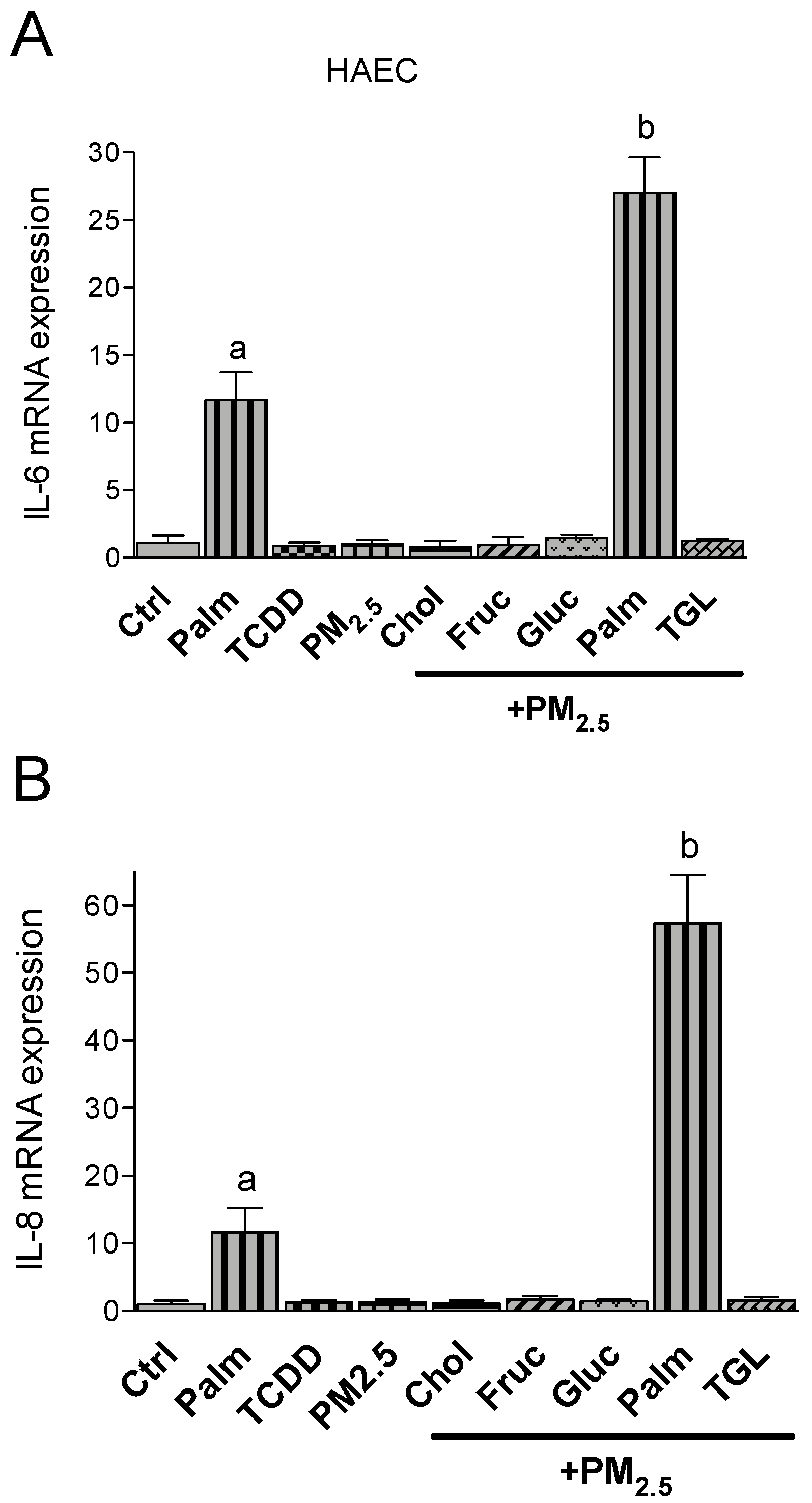
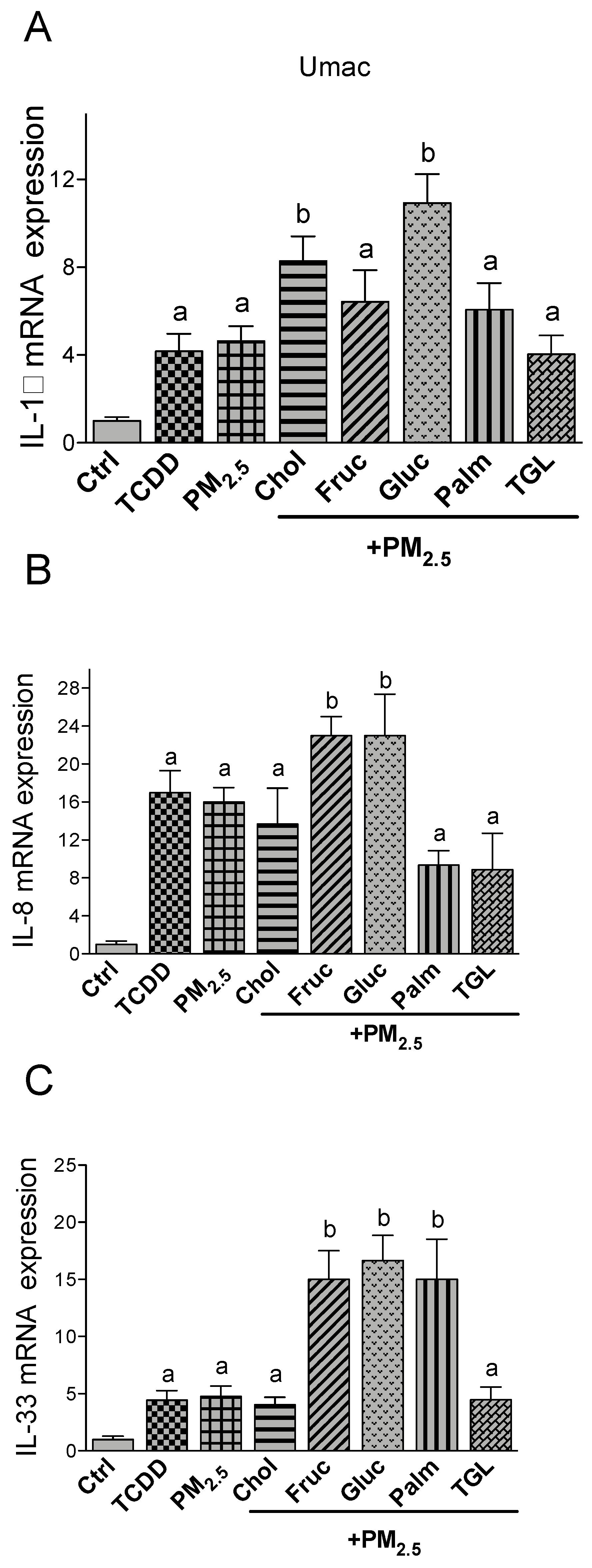
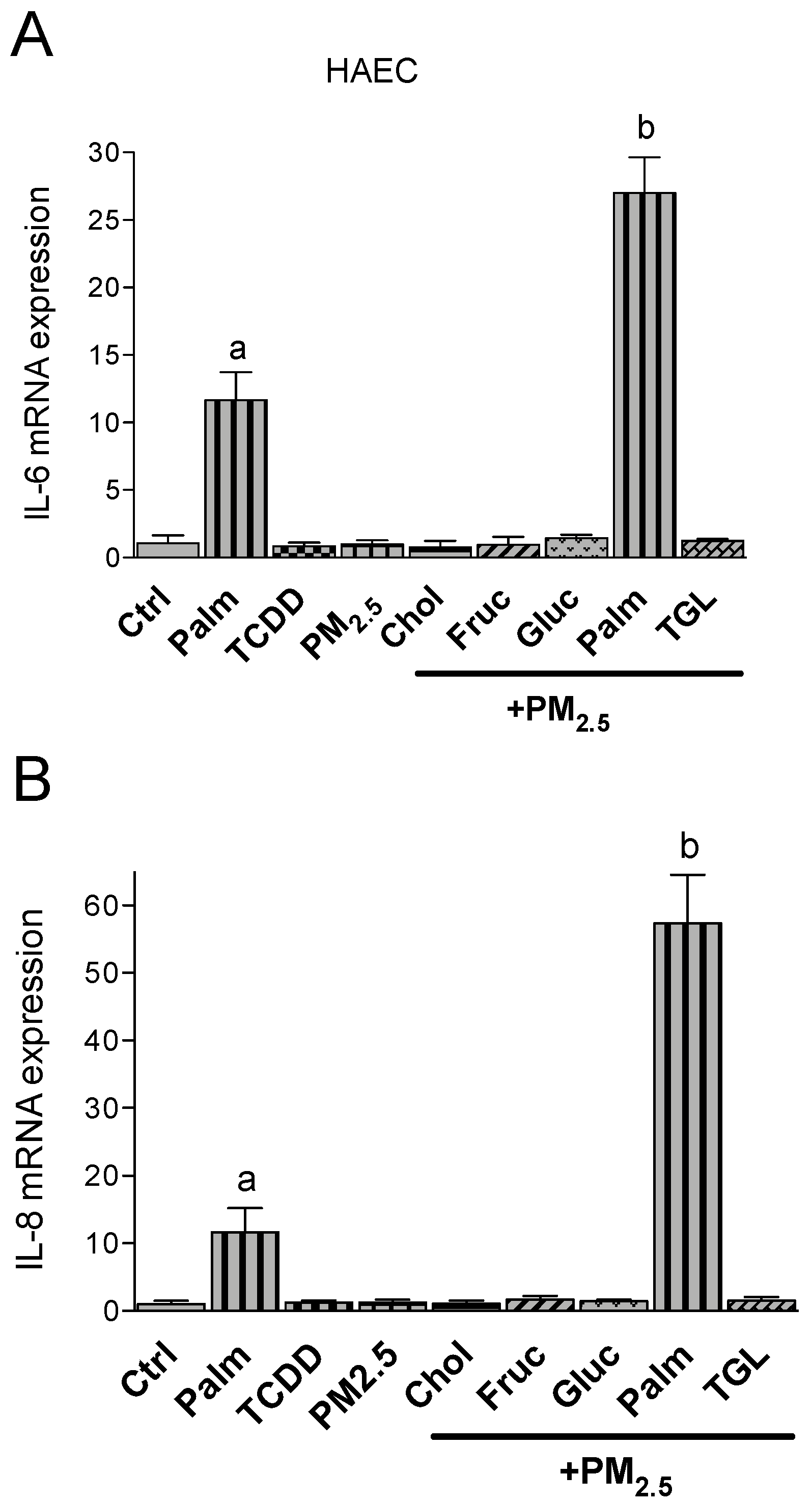
2.3. PM2.5 and Nutritional Factor Induced Expression of Pro-Inflammatory Cytokines
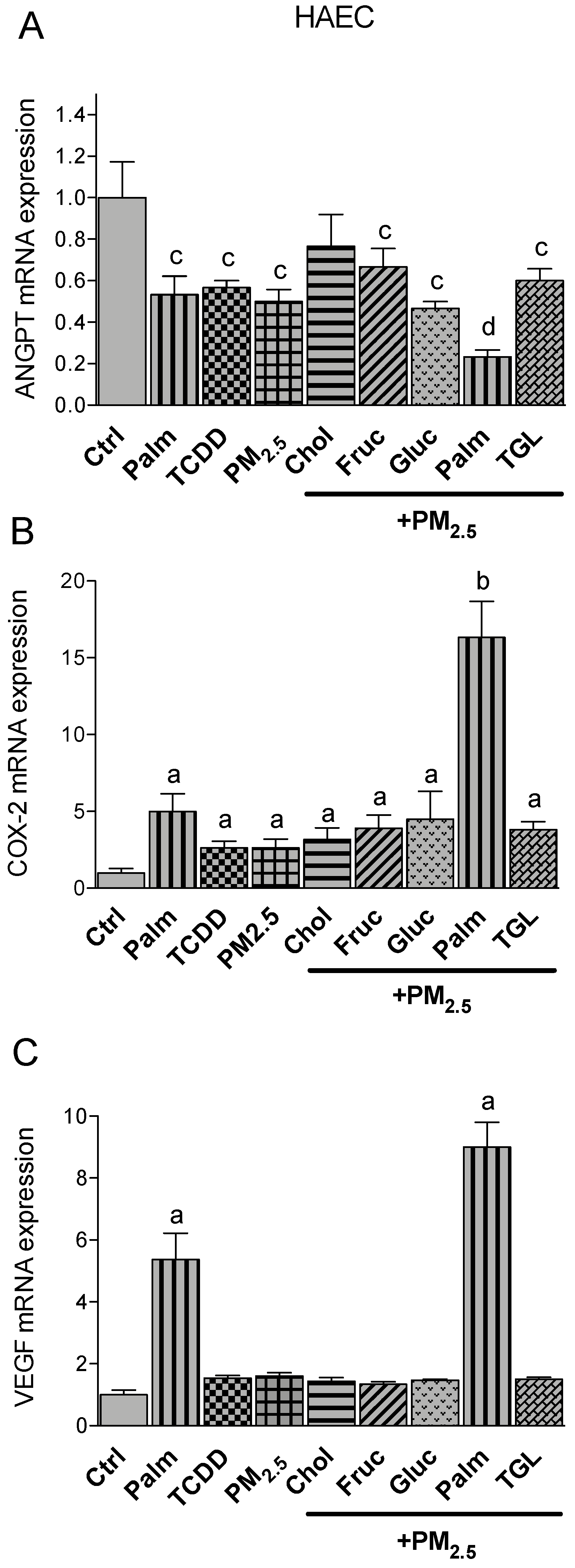
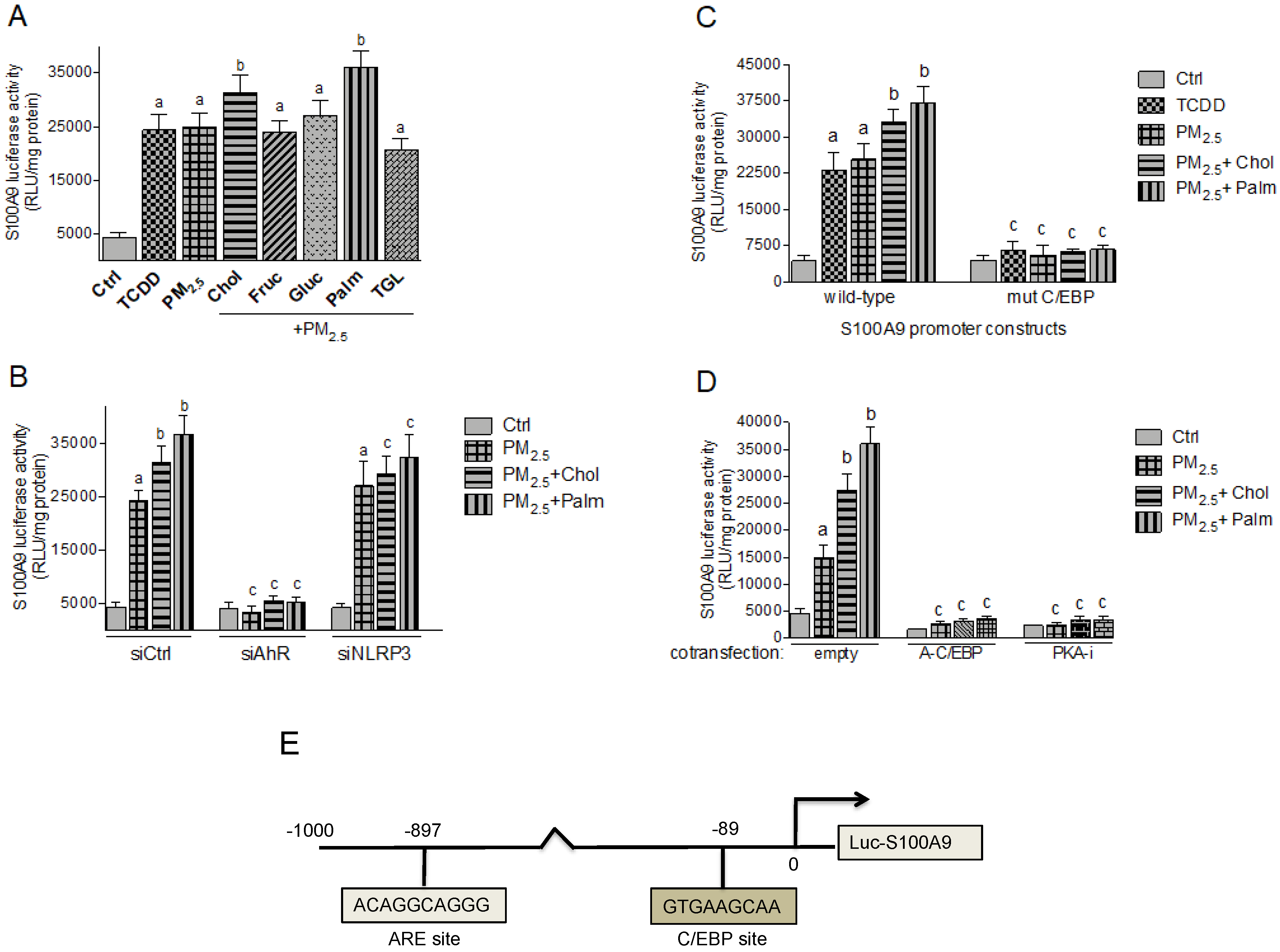
2.4. PM2.5 and Nutritional Factor Induced Expression of Atherogenic Markers
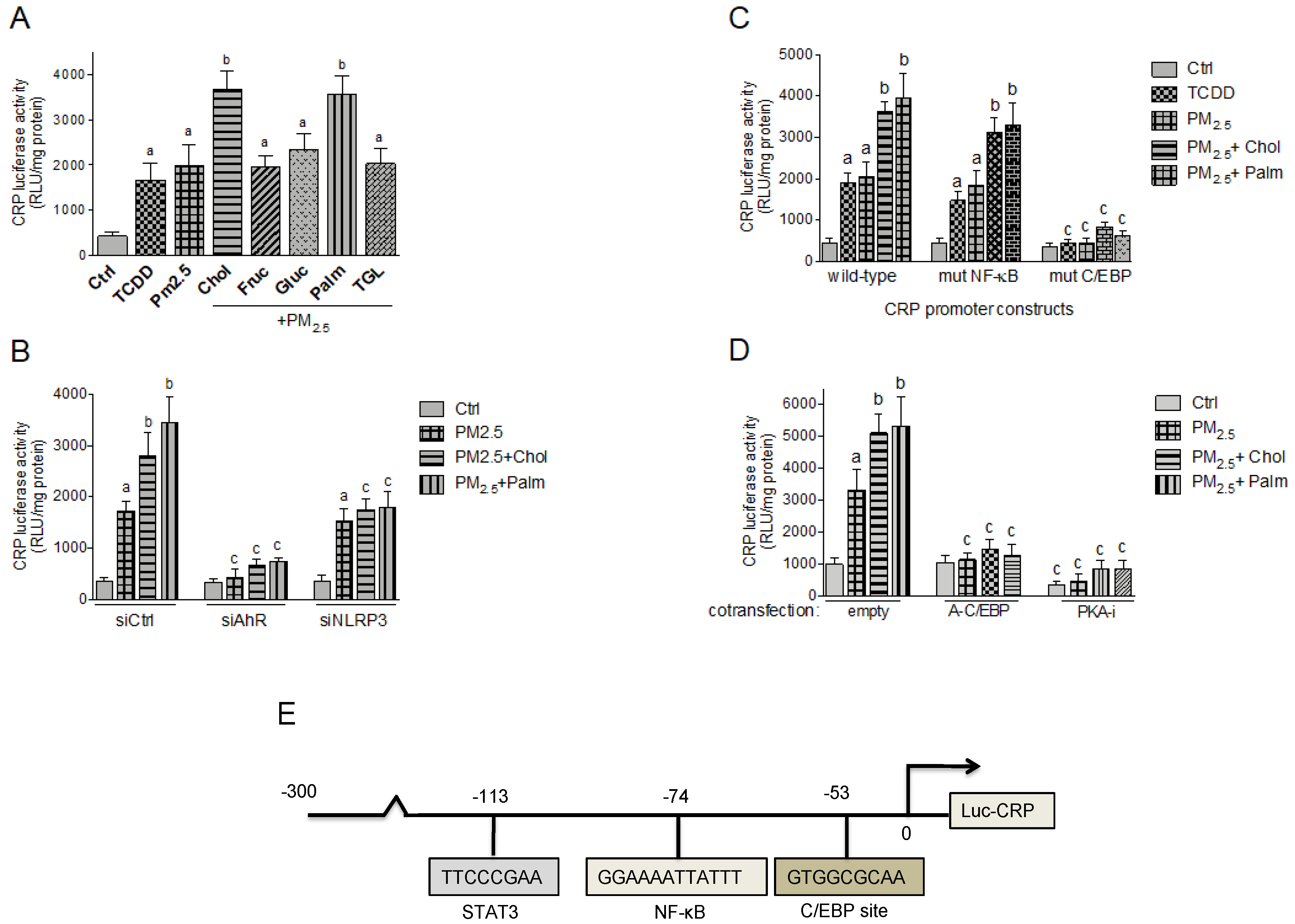
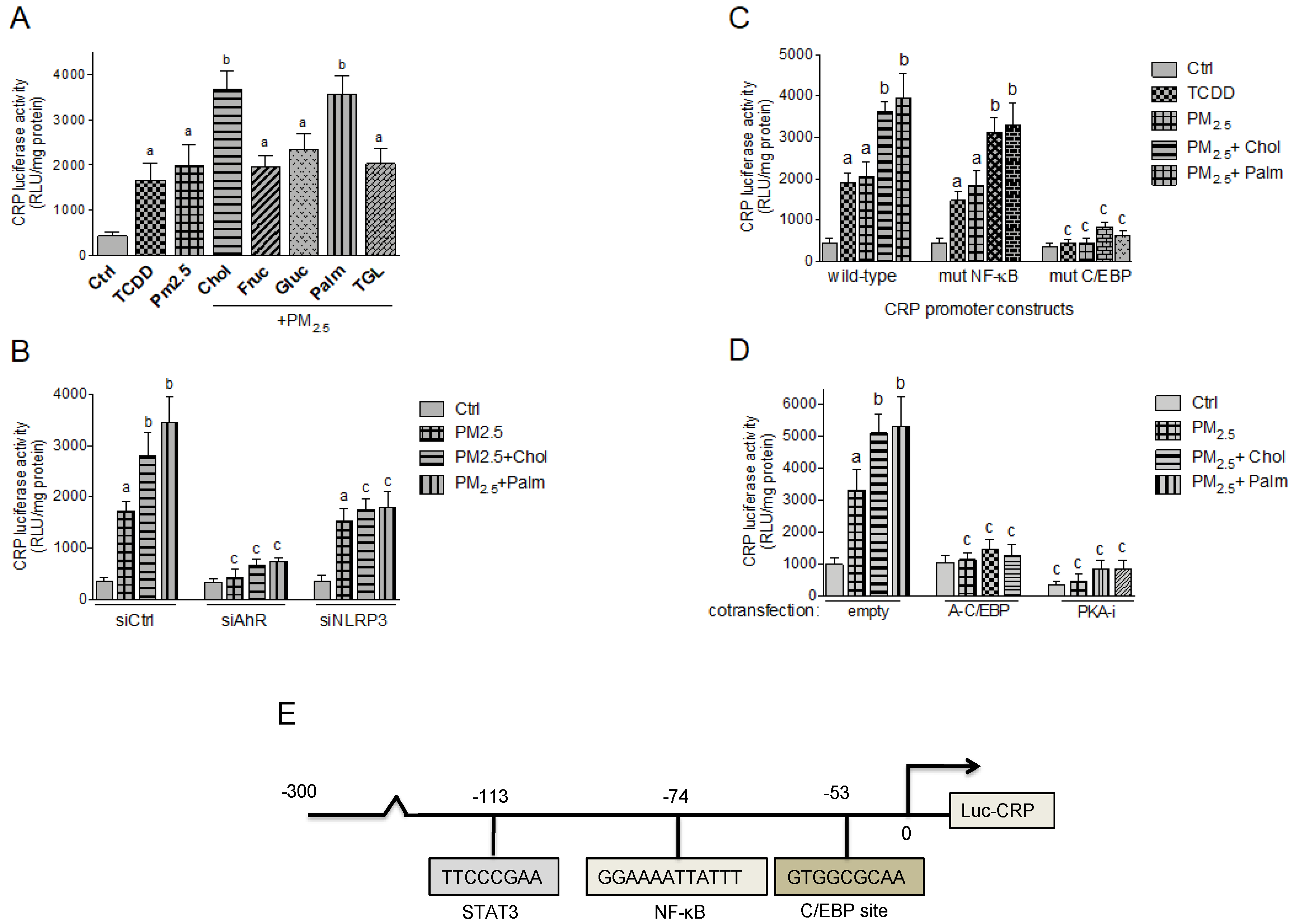
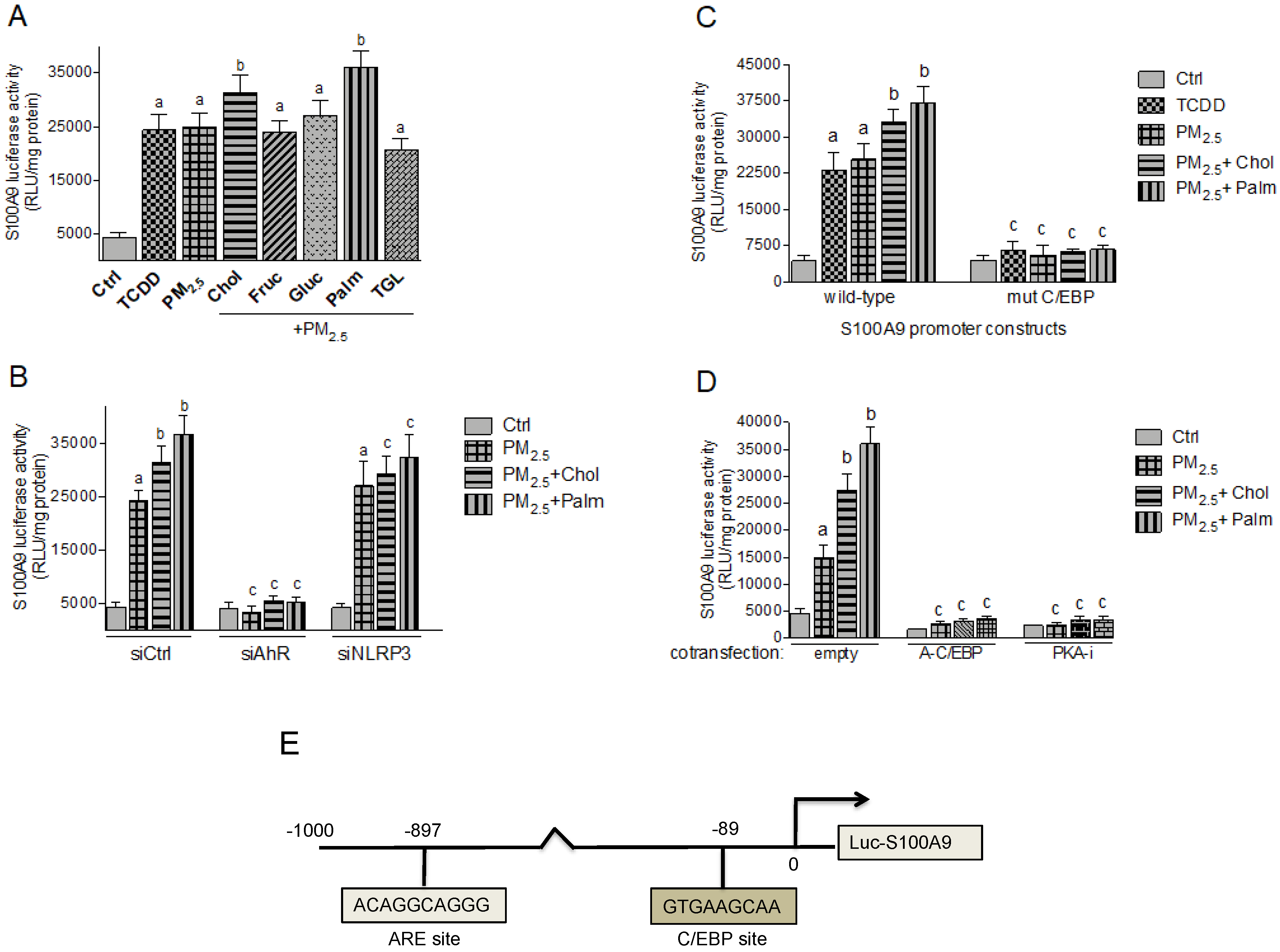
2.5. Effect of Nutritional Factors on PM2.5-Mediated CRP and S100A9 Promoter Activity
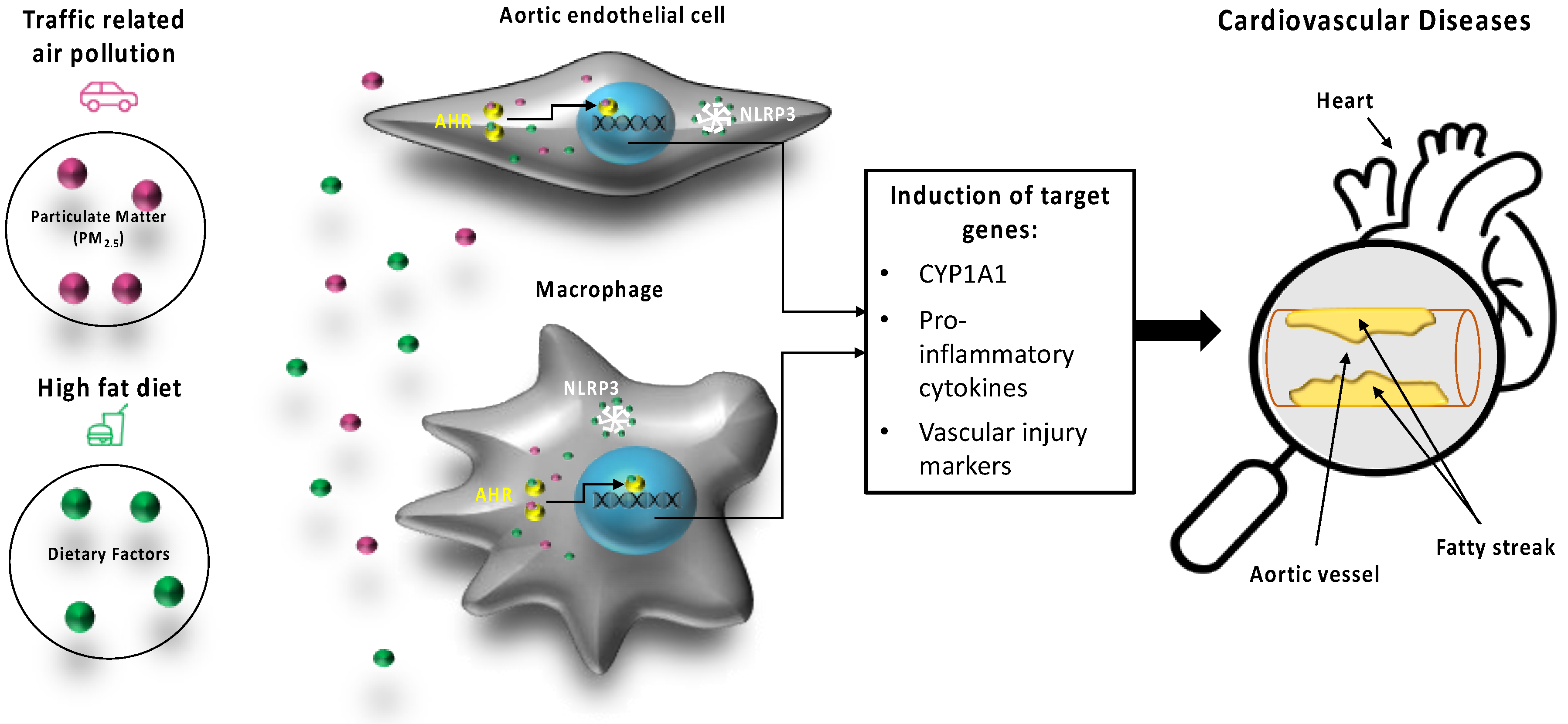
3. Discussion
4. Materials and Methods
4.1. Reagents and Preparation of PM
4.2. Cell Culture
4.3. RNA Isolation and Quantitative Real-Time RT-PCR (qPCR)
4.4. Transfection Experiments and Luciferase Assay
4.5. Gel-Mobility-Shift Assay (GMSA)
4.6. Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AHR | Aryl hydrocarbon receptor |
| ANGPT | Angiopoietin |
| ARE | antioxidant response element |
| ARNT | AHR nuclear translocator |
| BMM | Bone marrow-derived macrophages |
| C/EBPβ | CCAAT/enhancer-binding protein beta |
| CCL | C-C motif chemokine ligand |
| CXCL | C-X-C motif chemokine ligand |
| Chol | Cholesterol |
| COX-2 | Cyclooxygenase |
| CRP | C-reactive protein |
| CVD | Cardiovascular disease |
| CXCR | CXC-motive chemokine receptor |
| CYP DMSO DRE | Cytochrome P450 Dimethylsulfoxide Dioxin responsive element |
| Fruc | Fructose |
| Gluc | Glucose |
| GMSA | gel-mobility shift assay |
| HAEC | Human aortic endothelia cells |
| HFD | High-fat diet |
| IL | Interleukin |
| MNF | 3-methoxy-4-nitroflavone |
| NF-kB | Nuclear factor kappa-light-chain-enhancer of activated B-cells |
| NLRP3 | NLR family pyrin domain containing 3 |
| PAHs | Polycyclic aromatic hydrocarbons |
| PAI-2 | Plasminogen activator inhibitor 2 |
| Palm | Palmitic acid |
| PKA | Protein kinase A |
| PM | Particulate matter |
| qPCR | Quantitative Real-Time RT-PCR |
| S100A9 | Damage-associated molecular pattern molecule |
| siRNA | small interfering RNA |
| TCDD | 2,3,7,8-tetrachlorodibenzo-p-dioxin |
| TGL | Triglyceride |
| TPA | phorbol-12-myristate-13-acetate |
| TRAP | Traffic related air pollution |
| TSP-1 | Thrombospondin-1 |
| Umac | U937 derived macrophages (human) |
| VEGF | Vascular endothelia growth factor |
References
- Denison, M.S.; Nagy, S.R. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 309–334. [Google Scholar] [CrossRef] [PubMed]
- Bohonowych, J.E.; Denison, M.S. Persistent binding of ligands to the aryl hydrocarbon receptor. Toxicol. Sci. 2007, 98, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Rückerl, R.; Greven, S.; Ljungman, P.; Aalto, P.; Antoniades, C.; Bellander, T.; Berglind, N.; Chrysohoou, C.; Forastiere, F. Air pollution and inflammation (interleukin-6, C-reactive protein, fibrinogen) in myocardial infarction survivors. Environ. Health Perspect. 2007, 115, 1072–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunig, G.; Marsh, L.M.; Esmaeil, N.; Jackson, K.; Gordon, T.; Reibman, J.; Kwapiszewska, G.; Park, S.H. Perspective: Ambient air pollution: Inflammatory response and effects on the lung’s vasculature. Pulm. Circ. 2014, 4, 25–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef]
- Vogel, C.F.; Sciullo, E.; Wong, P.; Kuzmicky, P.; Kado, N.; Matsumura, F. Induction of proinflammatory cytokines and C-reactive protein in human macrophage cell line U937 exposed to air pollution particulates. Environ. Health Perspect. 2005, 113, 1536–1541. [Google Scholar] [CrossRef]
- Salcido-Neyoy, M.E.; Sánchez-Pérez, Y.; Osornio-Vargas, A.R.; Gonsebatt, M.E.; Meléndez-Zajgla, J.; Morales-Bárcenas, R.; Petrosyan, P.; Molina-Servin, E.D.; Vega, E.; Manzano-León, N.; et al. Induction of c-Jun by air particulate matter (PM(10)) of Mexico city: Participation of polycyclic aromatic hydrocarbons. Environ. Pollut. 2015, 203, 175–182. [Google Scholar] [CrossRef]
- Aung, H.H.; Lame, M.W.; Gohil, K.; He, G.; Denison, M.S.; Rutledge, J.C.; Wilson, D.W. Comparative gene responses to collected ambient particles in vitro: Endothelial responses. Physiol. Genomics 2011, 43, 917–929. [Google Scholar] [CrossRef]
- Gualtieri, M.; Ovrevik, J.; Mollerup, S.; Asare, N.; Longhin, E.; Dahlman, H.J.; Camatini, M.; Holme, J.A. Airborne urban particles (Milan winter-PM2.5) cause mitotic arrest and cell death: Effects on DNA, mitochondria, AhR binding and spindle organization. Mutat. Res. 2011, 713, 18–31. [Google Scholar] [CrossRef]
- Lauer, F.T.; Mitchell, L.A.; Bedrick, E.; McDonald, J.D.; Lee, W.Y.; Li, W.W.; Olvera, H.; Amaya, M.A.; Berwick, M.; Gonzales, M.; et al. Temporal-spatial analysis of U.S.-Mexico border environmental fine and coarse PM air sample extract activity in human bronchial epithelial cells. Toxicol. Appl. Pharmacol. 2009, 238, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, Y.; Ide, F.; Kishi, R.; Akutagawa, T.; Sakai, S.; Nakamura, M.; Ishikawa, T.; Fujii-Kuriyama, Y.; Nakatsuru, Y. Aryl hydrocarbon receptor plays a significant role in mediating airborne particulate-induced carcinogenesis in mice. Environ. Sci. Technol. 2007, 41, 3775–3780. [Google Scholar] [CrossRef]
- Ferecatu, I.; Borot, M.C.; Bossard, C.; Leroux, M.; Boggetto, N.; Marano, F.; Baeza-Squiban, A.; Andreau, K. Polycyclic aromatic hydrocarbon components contribute to the mitochondria-antiapoptotic effect of fine particulate matter on human bronchial epithelial cells via the aryl hydrocarbon receptor. Part. Fibre Toxicol. 2010, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Keebaugh, A.J.; Sioutas, C.; Pakbin, P.; Schauer, J.J.; Mendez, L.B.; Kleinman, M.T. Is atherosclerotic disease associated with organic components of ambient fine particles? Sci. Total Environ. 2015, 533, 69–75. [Google Scholar] [CrossRef]
- Kim, S.Y.; Sheppard, L.; Kaufman, J.D.; Bergen, S.; Szpiro, A.A.; Larson, T.V.; Adar, S.D.; Diez Roux, A.V.; Polak, J.F.; Vedal, S. Individual-level concentrations of fine particulate matter chemical components and subclinical atherosclerosis: A cross-sectional analysis based on 2 advanced exposure prediction models in the multi-ethnic study of atherosclerosis. Am. J. Epidemiol. 2014, 180, 718–728. [Google Scholar] [CrossRef] [Green Version]
- Andrysík, Z.; Vondráček, J.; Marvanová, S.; Ciganek, M.; Neča, J.; Pěnčíková, K.; Mahadevan, B.; Topinka, J.; Baird, W.M.; Kozubík, A.; et al. Activation of the aryl hydrocarbon receptor is the major toxic mode of action of an organic extract of a reference urban dust particulate matter mixture: The role of polycyclic aromatic hydrocarbons. Mutat. Res. 2011, 714, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Viera-Hutchins, L.; Garcia-Lloret, M.; Noval Rivas, M.; Wise, P.; McGhee, S.A.; Chatila, Z.K.; Daher, N.; Sioutas, C.; Chatila, T.A. Vehicular exhaust particles promote allergic airway inflammation through an aryl hydrocarbon receptor-notch signaling cascade. J. Allergy Clin. Immunol. 2015, 136, 441–453. [Google Scholar] [CrossRef] [Green Version]
- Korashy, H.M.; El-Kadi, A.O. The role of aryl hydrocarbon receptor in the pathogenesis of cardiovascular diseases. Drug Metab. Rev. 2006, 38, 411–450. [Google Scholar] [CrossRef] [PubMed]
- Puga, A. Perspectives on the potential involvement of the AH receptor-dioxin axis in cardiovascular disease. Toxicol. Sci. 2011, 120, 256–261. [Google Scholar] [CrossRef] [Green Version]
- O’Driscoll, C.A.; Gallo, M.E.; Fechner, J.H.; Schauer, J.J.; Mezrich, J.D. Real-world PM extracts differentially enhance Th17 differentiation and activate the aryl hydrocarbon receptor (AHR). Toxicology 2019, 414, 14–26. [Google Scholar]
- Castañeda, A.R.; Pinkerton, K.E.; Bein, K.J.; Magaña-Méndez, A.; Yang, H.T.; Ashwood, P.; Vogel, C.F. Ambient particulate matter activates the aryl hydrocarbon receptor (AHR) in dendritic cells and enhances Th17 polarization. Toxicol. Lett. 2018, 292, 85–96. [Google Scholar] [CrossRef]
- Vogel, C.F.; Sciullo, E.; Matsumura, F. Activation of inflammatory mediators and potential role of ah-receptor ligands in foam cell formation. Cardiovasc. Toxicol. 2004, 4, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Nishimura, N.; Kuo, V.; Fiehn, O.; Shahbaz, S.; Van-Winkle, L.; Matsumura, F.; Vogel, C.F. Activation of aryl hydrocarbon receptor induces vascular inflammation and promotes atherosclerosis in apolipoprotein E-/- mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1260–1267. [Google Scholar] [CrossRef] [Green Version]
- Zwaka, T.P.; Hombach, V.; Torzewski, J. C-reactive protein-mediated low density lipoprotein uptake by macrophages: Implications for atherosclerosis. Circulation 2001, 103, 1194–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Averill, M.M.; Kerkhoff, C.; Bornfeldt, K.E. S100A8 and S100A9 in cardiovascular biology and disease. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 223–229. [Google Scholar] [CrossRef] [Green Version]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Bornfeldt, K.E. Does elevated glucose promote atherosclerosis? pros and cons. Circ. Res. 2016, 119, 190–193. [Google Scholar] [CrossRef] [Green Version]
- Talayero, B.G.; Sacks, F.M. The role of triglycerides in atherosclerosis. Curr. Cardiol. Rep. 2011, 13, 544–552. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Liu, J.; Pang, X.; Wang, S.; Zhao, J.; Zhang, X.; Feng, L. Palmitic acid exerts pro-inflammatory effects on vascular smooth muscle cells by inducing the expression of C-reactive protein, inducible nitric oxide synthase and tumor necrosis factor-α. Int. J. Mol. Med. 2014, 34, 1706–1712. [Google Scholar] [CrossRef] [Green Version]
- Gidding, S.S.; Allen, N.B. Cholesterol and atherosclerotic cardiovascular disease: A lifelong problem. J. Am. Heart Assoc. 2019, 8, e012924. [Google Scholar] [CrossRef]
- Sheedy, F.J.; Moore, K.J. IL-1 signaling in atherosclerosis: Sibling rivalry. Nat. Immunol. 2013, 14, 1030–1032. [Google Scholar] [CrossRef]
- Buckley, M.L.; Williams, J.O.; Chan, Y.H.; Laubertová, L.; Gallagher, H.; Moss, J.W.E.; Ramji, D.P. The interleukin-33-mediated inhibition of expression of two key genes implicated in atherosclerosis in human macrophages requires MAP kinase, phosphoinositide 3-kinase and nuclear factor-κB signaling pathways. Sci. Rep. 2019, 9, 11317. [Google Scholar] [CrossRef]
- Vogel, C.; Boerboom, A.M.; Baechle, C.; El-Bahay, C.; Kahl, R.; Degen, G.H.; Abel, J. Regulation of prostaglandin endoperoxide H synthase-2 induction by dioxin in rat hepatocytes: Possible c-Src-mediated pathway. Carcinogenesis 2000, 21, 2267–2274. [Google Scholar] [CrossRef]
- Sutter, T.R.; Guzman, K.; Dold, K.M.; Greenlee, W.F. Targets for dioxin: Genes for plasminogen activator inhibitor-2 and interleukin-1 beta. Science 1991, 254, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, B.S.; Kujubu, D.A.; Perrin, D.M.; Herschman, H.R. Structure of the mitogen-inducible TIS10 gene and demonstration that the TIS10-encoded protein is a functional prostaglandin G/H synthase. J. Biol. Chem. 1992, 267, 4338–4344. [Google Scholar]
- Costelloe, E.O.; Stacey, K.J.; Antalis, T.M.; Hume, D.A. Regulation of the plasminogen activator inhibitor-2 (PAI-2) gene in murine macrophages. Demonstration of a novel pattern of responsiveness to bacterial endotoxin. J. Leukoc. Biol. 1999, 66, 172–182. [Google Scholar] [CrossRef]
- Cipollone, F.; Fazia, M.L. COX-2 and atherosclerosis. J. Cardiovasc. Pharmacol. 2006, 47, S26–S36. [Google Scholar] [CrossRef]
- Farris, S.D.; Hu, J.H.; Krishnan, R.; Emery, I.; Chu, T.; Du, L.; Kremen, M.; Dichek, H.L.; Gold, E.; Ramsey, S.A.; et al. Mechanisms of urokinase plasminogen activator (uPA)-mediated atherosclerosis: Role of the uPA receptor and S100A8/A9 proteins. J. Biol. Chem. 2011, 286, 22665–22677. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Mo, Y.; Gu, A.; Wan, R.; Zhang, Q.; Tollerud, D.J. Effects of urban particulate matter with high glucose on human monocytes U937. J. Appl. Toxicol. 2016, 36, 586–595. [Google Scholar] [CrossRef]
- Dabir, P.M.T.; Krukovets, I.; Stenina, O. Aryl hydrocarbon receptor is activated by glucose and regulates the thrombospondin-1 gene promoter in endothelial cells. Circ. Res. 2008, 102, 1558–1565. [Google Scholar] [CrossRef] [Green Version]
- Vogel, C.F.A.; Van Winkle, L.S.; Esser, C.; Haarmann-Stemmann, T. The aryl hydrocarbon receptor as a target of environmental stressors—Implications for pollution mediated stress and inflammatory responses. Redox Biol. 2020, 34, 101530. [Google Scholar] [CrossRef] [PubMed]
- Găman, M.A.; Epîngeac, M.E.; Diaconu, C.C.; Găman, A.M. Evaluation of oxidative stress levels in obesity and diabetes by the free oxygen radical test and free oxygen radical defence assays and correlations with anthropometric and laboratory parameters. World J. Diabetes 2020, 11, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Celletti, F.L.; Waugh, J.M.; Amabile, P.G.; Brendolan, A.; Hilfiker, P.A.; Dake, M.D. Vascular endothelial growth factor enhances atherosclerotic plaque progression. Nat. Med. 2001, 7, 425–429. [Google Scholar] [CrossRef]
- Lemstrom, K.B.; Krebs, R.; Nykanen, A.I.; Tikkanen, J.M.; Sihvola, R.K.; Aaltola, E.M.; Hayry, P.J.; Wood, J.; Alitalo, K.; Ylä-Herttuala, S.; et al. Vascular endothelial growth factor enhances cardiac allograft arteriosclerosis. Circulation 2002, 105, 2524–2530. [Google Scholar] [CrossRef] [Green Version]
- Nykänen, A.I.; Krebs, R.; Saaristo, A.; Turunen, P.; Alitalo, K.; Ylä-Herttuala, S.; Koskinen, P.K.; Lemström, K.B. Angiopoietin-1 protects against the development of cardiac allograft arteriosclerosis. Circulation. 2003, 107, 1308–1314. [Google Scholar] [CrossRef] [Green Version]
- Bobryshev, Y.V. Lord RS. S-100 positive cells in human arterial intima and in atherosclerotic lesions. Cardiovasc. Res. 1995, 29, 689–696. [Google Scholar] [CrossRef]
- Temchura, V.V.; Frericks, M.; Nacken, W.; Esser, C. Role of the aryl hydrocarbon receptor in thymocyte emigration in vivo. Eur. J. Immunol. 2005, 35, 2738–2747. [Google Scholar] [CrossRef]
- Sutter, C.H.; Bodreddigari, S.; Campion, C.; Wible, R.S.; Sutter, T.R. 2,3,7,8-Tetrachlorodibenzo-p-dioxin increases the expression of genes in the human epidermal differentiation complex and accelerates epidermal barrier formation. Toxicol. Sci. 2011, 124, 128–137. [Google Scholar] [CrossRef] [Green Version]
- Murray, I.A.; Morales, J.L.; Flaveny, C.A.; Dinatale, B.C.; Chiaro, C.; Gowdahalli, K.; Amin, S.; Perdew, G.H. Evidence for ligand-mediated selective modulation of aryl hydrocarbon receptor activity. Mol. Pharmacol. 2010, 77, 247–254. [Google Scholar] [CrossRef] [Green Version]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [Green Version]
- Snodgrass, R.G.; Huang, S.; Choi, I.W.; Rutledge, J.C.; Hwang, D.H. Inflammasome-mediated secretion of IL-1β in human monocytes through TLR2 activation; modulation by dietary fatty acids. J. Immunol. 2013, 191, 4337–4347. [Google Scholar] [CrossRef] [Green Version]
- Vogel, C.F.; Sciullo, E.; Park, S.; Liedtke, C.; Trautwein, C.; Matsumura, F. Dioxin increases C/EBPbeta transcription by activating cAMP/protein kinase A. J. Biol. Chem. 2004, 279, 8886–8894. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, A.; Cha-Molstad, H.; Samols, D.; Kushner, I. Transactivation of C-reactive protein by IL-6 requires synergistic interaction of CCAAT/enhancer binding protein beta (C/EBP beta) and Rel p50. J. Immunol. 2001, 166, 2378–2384. [Google Scholar] [CrossRef] [Green Version]
- Bando, M.; Zou, X.; Hiroshima, Y.; Kataoka, M.; Ross, K.F.; Shinohara, Y.; Nagata, T.; Herzberg, M.C.; Kido, J.I. Mechanism of interleukin-1α transcriptional regulation of S100A9 in a human epidermal keratinocyte cell line. Biochim. Biophys. Acta 2013, 1829, 954–962. [Google Scholar] [CrossRef] [Green Version]
- Vogel, C.F.A.; Ishihara, Y.; Campbell, C.E.; Kado, S.Y.; Nguyen-Chi, A.; Sweeney, C.; Pollet, M.; Haarmann-Stemmann, T.; Tuscano, J.M. A protective role of aryl hydrocarbon receptor repressor in inflammation and tumor growth. Cancers 2019, 11, 589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patten, K.T.; González, E.A.; Valenzuela, A.; Berg, E.; Wallis, C.; Garbow, J.R.; Silverman, J.L.; Bein, K.J.; Wexler, A.S.; Lein, P.J. Effects of early life exposure to traffic-related air pollution on brain development in juvenile Sprague-Dawley rats. Transl. Psychiatry 2020, 10, 166, PMCID:7264203. [Google Scholar] [CrossRef]
- Bein, K.J.; Wexler, A.S. A high-efficiency, low-bias method for extracting particulate matter from filter and impactor substrates. Atmos. Environ. 2014, 90, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Bein, K.J.; Wexler, A.S. Compositional variance in extracted particulate matter using different filter extraction techniques. Atmos. Environ. 2015, 107, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, Y.; Haarmann-Stemmann, T.; Kado, N.Y.; Vogel, C.F.A. Interleukin 33 expression induced by aryl hydrocarbon receptor in macrophages. Toxicol. Sci. 2019, 170, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.; Khan, E.M.; Leung, P.S.; Gershwin, M.E.; Chang, W.L.W.; Wu, D.; Haarmann-Stemmann, T.; Hoffmann, A.; Denison, M.S. Cross-talk between aryl hydrocarbon receptor and the inflammatory response: A role for nuclear factor-κB. J. Biol. Chem. 2014, 289, 1866–1875. [Google Scholar] [CrossRef] [Green Version]
- Vogel, C.F.; Sciullo, E.; Li, W.; Wong, P.; Lazennec, G.; Matsumura, F. RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol. Endocrinol. 2007, 21, 2941–2955. [Google Scholar] [CrossRef] [Green Version]
- Sumi, D.; Shimizu, Y.; Himeno, S. Involvement of Nrf2 activation in the upregulation of S100A9 by exposure to inorganic arsenite. Int. J. Mol. Med. 2013, 31, 259–264. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahlem, C.; Kado, S.Y.; He, Y.; Bein, K.; Wu, D.; Haarmann-Stemmann, T.; Kado, N.Y.; Vogel, C.F.A. AHR Signaling Interacting with Nutritional Factors Regulating the Expression of Markers in Vascular Inflammation and Atherogenesis. Int. J. Mol. Sci. 2020, 21, 8287. https://doi.org/10.3390/ijms21218287
Dahlem C, Kado SY, He Y, Bein K, Wu D, Haarmann-Stemmann T, Kado NY, Vogel CFA. AHR Signaling Interacting with Nutritional Factors Regulating the Expression of Markers in Vascular Inflammation and Atherogenesis. International Journal of Molecular Sciences. 2020; 21(21):8287. https://doi.org/10.3390/ijms21218287
Chicago/Turabian StyleDahlem, Carla, Sarah Y. Kado, Yi He, Keith Bein, Dalei Wu, Thomas Haarmann-Stemmann, Norman Y. Kado, and Christoph F. A. Vogel. 2020. "AHR Signaling Interacting with Nutritional Factors Regulating the Expression of Markers in Vascular Inflammation and Atherogenesis" International Journal of Molecular Sciences 21, no. 21: 8287. https://doi.org/10.3390/ijms21218287
APA StyleDahlem, C., Kado, S. Y., He, Y., Bein, K., Wu, D., Haarmann-Stemmann, T., Kado, N. Y., & Vogel, C. F. A. (2020). AHR Signaling Interacting with Nutritional Factors Regulating the Expression of Markers in Vascular Inflammation and Atherogenesis. International Journal of Molecular Sciences, 21(21), 8287. https://doi.org/10.3390/ijms21218287