Current Evidence and Future Perspectives on Pharmacological Treatment of Calcific Aortic Valve Stenosis

 ,
,  , ,
, ,
Abstract
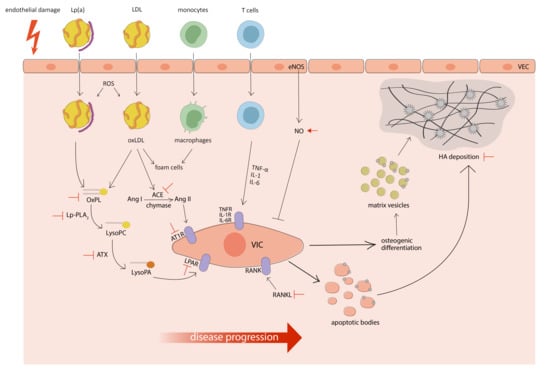
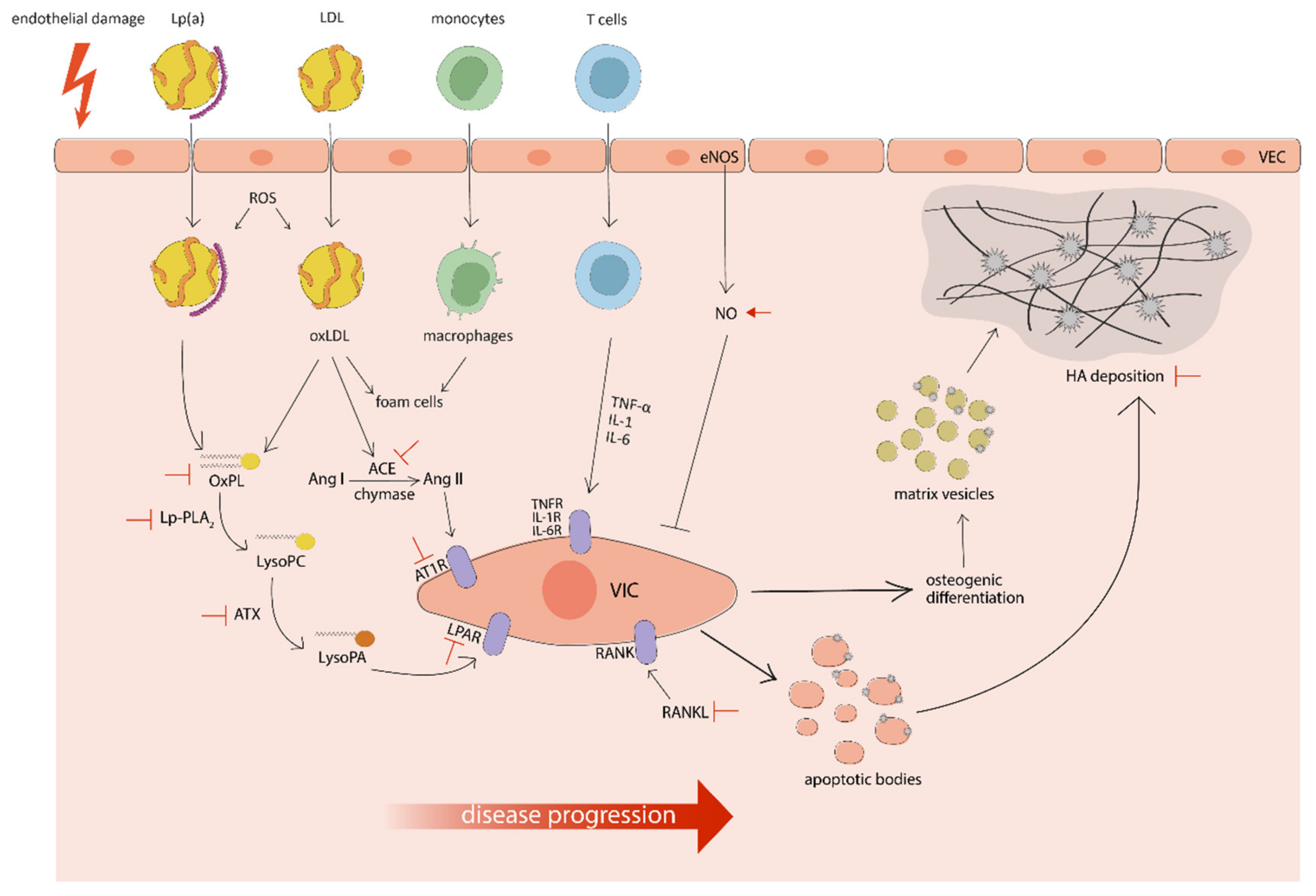
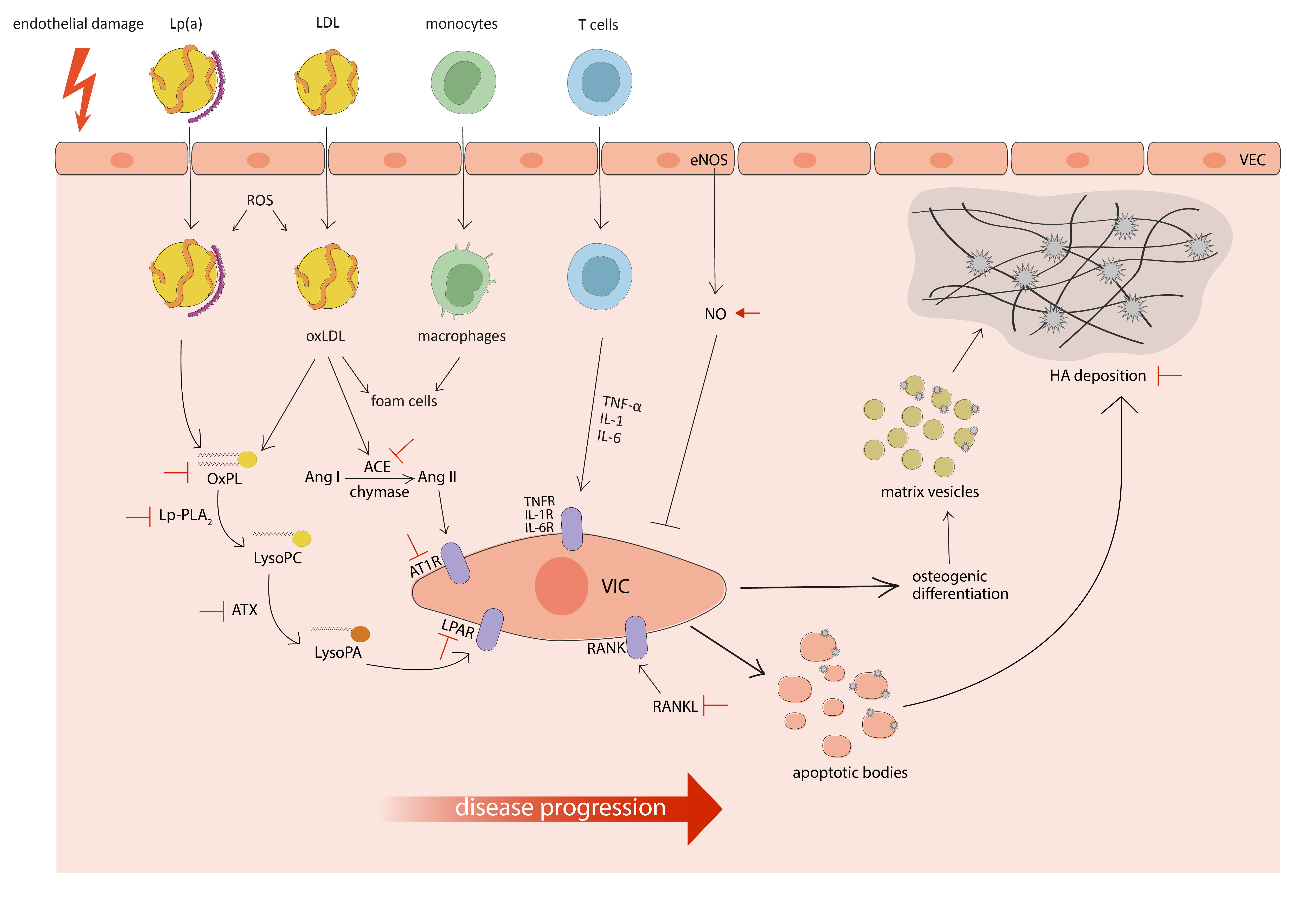
:
1. Introduction
2. Methods
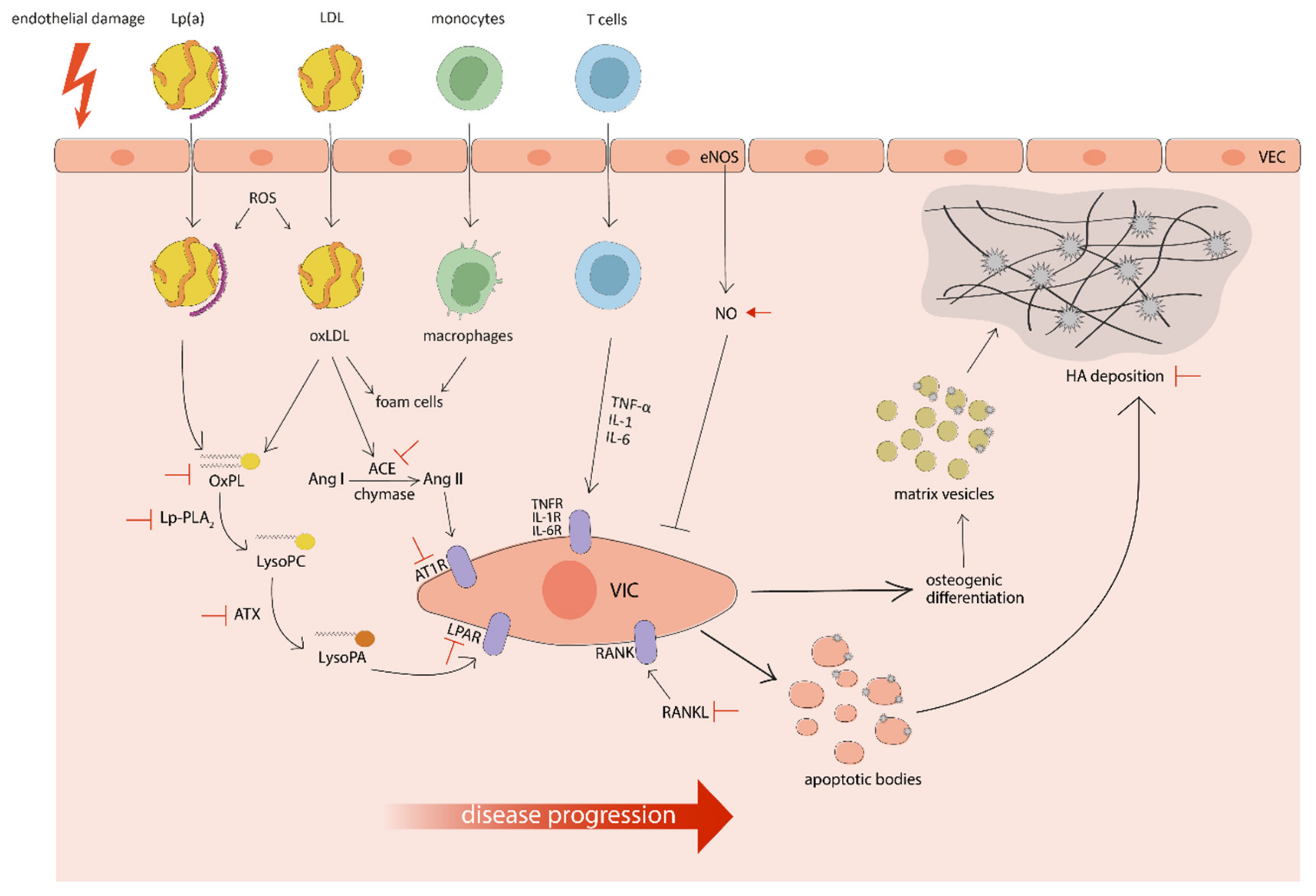
3. Pathophysiology of CAVS
4. Purpose
5. Lipid-Lowering Interventions
5.1. Statins
5.1.1. Current Evidence
5.1.2. Future Perspectives
5.2. Lp(a)-Lowering Therapies
5.2.1. Current Evidence
5.2.2. Future Perspectives
5.3. PCSK9 Inhibitors
5.3.1. Current Evidence
5.3.2. Future Perspectives
6. RAAS Blockade
6.1. ACE Inhibitors/ARBs
6.1.1. Current Evidence
6.1.2. Future Perspectives
6.2. Mineralocorticoid Receptor Antagonists
6.2.1. Current Evidence
6.2.2. Future Perspectives
7. Modulators of Nitric Oxide Pathway
7.1. Current Evidence
7.2. Future Perspectives
8. Anti-Calcific Agents
8.1. Inhibitors of Bone Resorption
8.1.1. Current Evidence
8.1.2. Future Perspectives
8.2. Vitamin K
8.2.1. Current Evidence
8.2.2. Future Perspectives
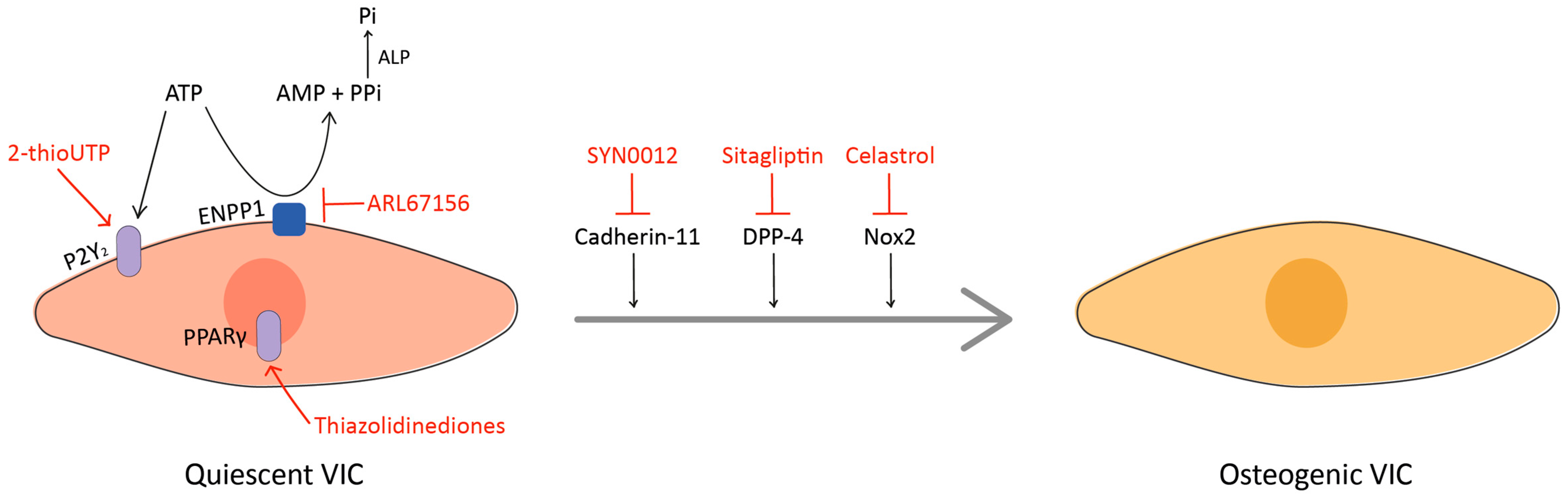
9. Emerging Targets
9.1. Nox2
9.2. ENPP1 and P2Y2
9.3. DPP-4
9.4. Cadherin-11
9.5. PPARγ
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lindman, B.R.; Clavel, M.A.; Mathieu, P.; Iung, B.; Lancellotti, P.; Otto, C.M.; Pibarot, P. Calcific aortic stenosis. Nat. Rev. Dis. Primers 2016, 2, 16006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osnabrugge, R.L.J.; Mylotte, D.; Head, S.J.; Nkomo, V.T.; LeReun, C.M.; Bogers, A.J.; Piazza, N.; Kappetein, A.P. Aortic stenosis in the elderly: Disease prevalence and number of candidates for transcatheter aortic valve replacement: A meta-analysis and modeling study. J. Am. Coll. Cardiol. 2013, 62, 1002–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nkomo, V.T.; Gardin, J.M.; Skelton, T.N.; Gottdiener, J.S.; Scott, C.G.; Enriquez-Sarano, M. Burden of valvular heart diseases: A population-based study. Lancet 2006, 368, 1005–1011. [Google Scholar] [CrossRef]
- Ancona, R.; Comenale Pint, S. Epidemiology of aortic valve stenosis (AS) and of aortic valve incompetence (AI): Is the prevalence of AS/AI similar in different parts of the world? Eur. J. Cardiol. Pract. 2020, 18, 10–12. [Google Scholar]
- Myasoedova, V.A.; Ravani, A.L.; Frigerio, B.; Valerio, V.; Moschetta, D.; Songia, P.; Poggio, P. Novel pharmacological targets for calcific aortic valve disease: Prevention and treatments. Pharmacol. Res. 2018, 136, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Head, S.J.; Çelik, M.; Kappetein, A.P. Mechanical versus bioprosthetic aortic valve replacement. Eur. Heart J. 2017. [Google Scholar] [CrossRef]
- Chen, H.Y.; Engert, J.C.; Thanassoulis, G. Risk factors for valvular calcification. Curr. Opin. Endocrinol. Diabetes Obes. 2019, 26, 96–102. [Google Scholar] [CrossRef]
- Hulin, A.; Hego, A.; Lancellotti, P.; Oury, C. Advances in Pathophysiology of Calcific Aortic Valve Disease Propose Novel Molecular Therapeutic Targets. Front. Cardiovasc. Med. 2018, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Peeters, F.E.C.M.; Meex, S.J.R.; Dweck, M.R.; Aikawa, E.; Crijns, H.J.G.M.; Schurgers, L.J.; Kietselaer, B.L.J.H. Calcific aortic valve stenosis: Hard disease in the heart: A biomolecular approach towards diagnosis and treatment. Eur. Heart J. 2018, 39, 2618–2624. [Google Scholar] [CrossRef] [Green Version]
- Perrucci, G.L.; Zanobini, M.; Gripari, P.; Songia, P.; Alshaikh, B.; Tremoli, E.; Poggio, P. Pathophysiology of aortic stenosis and mitral regurgitation. Compr. Physiol. 2017, 7, 799–818. [Google Scholar] [CrossRef]
- Tao, G.; Kotick, J.D.; Lincoln, J. Heart Valve Development, Maintenance, and Disease. The Role of Endothelial Cells; Elsevier Inc.: Amsterdam, The Netherlands, 2012; Volume 100. [Google Scholar] [CrossRef]
- Rutkovskiy, A.; Malashicheva, A.; Sullivan, G.; Bogdanova, M.; Kostareva, A.; Stensløkken, K.O.; Fiane, A.; Vaage, J. Valve interstitial cells: The key to understanding the pathophysiology of heart valve calcification. J. Am. Heart Assoc. 2017, 6, e006339. [Google Scholar] [CrossRef]
- Pawade, T.A.; Newby, D.E.; Dweck, M.R. Calcification in aortic stenosis: The skeleton key. J. Am. Coll. Cardiol. 2015, 66, 561–577. [Google Scholar] [CrossRef] [Green Version]
- Kaden, J.J.; Bickelhaupt, S.; Grobholz, R.; Haase, K.K.; Sarikoç, A.; Kiliç, R.; Brueckmann, M.; Lang, S.; Zahn, I.; Vahl, C.; et al. Receptor activator of nuclear factor κB ligand and osteoprotegerin regulate aortic valve calcification. J. Mol. Cell Cardiol. 2004, 36, 57–66. [Google Scholar] [CrossRef]
- Côté, N.; El Husseini, D.; Pépin, A.; Guauque-Olarte, S.; Ducharme, V.; Bouchard-Cannon, P.; Audet, A.; Fournier, D.; Gaudreault, N.; Derbali, H.; et al. ATP acts as a survival signal and prevents the mineralization of aortic valve. J. Mol. Cell Cardiol. 2012, 52, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, J.; Zhou, K.; Liao, X.; Zhou, X.; Shen, K. The methylation of Notch1 promoter mediates the osteogenesis differentiation in human aortic valve interstitial cells through Wnt/β-catenin signaling. J. Cell Physiol. 2019, 234, 20366–20376. [Google Scholar] [CrossRef]
- Rattazzi, M.; Iop, L.; Faggin, E.; Bertacco, E.; Zoppellaro, G.; Baesso, I.; Puato, M.; Torregrossa, G.; Fadini, G.P.; Agostini, C.; et al. Clones of interstitial cells from bovine aortic valve exhibit different calcifying potential when exposed to endotoxin and phosphate. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2165–2172. [Google Scholar] [CrossRef] [PubMed]
- Blaser, M.C.; Aikawa, E. Roles and Regulation of Extracellular Vesicles in Cardiovascular Mineral Metabolism. Front. Cardiovasc. Med. 2018, 5, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostyunin, A.E.; Yuzhalin, A.E.; Ovcharenko, E.A.; Kutikhin, A.G. Development of calcific aortic valve disease: Do we know enough for new clinical trials? J. Mol. Cell Cardiol. 2019, 132, 189–209. [Google Scholar] [CrossRef]
- Lim, S.Y. Role of statins in coronary artery disease. Chonnam Med. J. 2013, 49, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarrett, M.J.; Yao, Q.; Venardos, N.; Weyant, M.J.; Reece, T.B.; Meng, X.; Fullerton, D.A. Simvastatin down-regulates osteogenic response in cultured human aortic valve interstitial cells. J. Thorac. Cardiovasc. Surg. 2020. [Google Scholar] [CrossRef]
- Rajamannan, N.M.; Subramaniam, M.; Stock, S.R.; Stone, N.J.; Springett, M.; Ignatiev, K.I.; McConnell, J.P.; Singh, R.J.; Bonow, R.O.; Spelsberg, T.C. Atorvastatin inhibits calcification and enhances nitric oxide synthase production in the hypercholesterolaemic aortic valve. Heart 2005, 91, 806–810. [Google Scholar] [CrossRef] [Green Version]
- Rosenhek, R.; Rader, F.; Loho, N.; Gabriel, H.; Heger, M.; Klaar, U.; Schemper, M.; Binder, T.; Maurer, G.; Baumgartner, H. Statins but not angiotensin-converting enzyme inhibitors delay progression of aortic stenosis. Circulation 2004, 110, 1291–1295. [Google Scholar] [CrossRef] [Green Version]
- Bellamy, M.F.; Pellikka, P.A.; Klarich, K.W.; Tajik, A.J.; Enriquez-Sarano, M. Association of cholesterol levels, hydroxymethylglutaryl coenzyme-A reductase inhibitor treatment, and progression of aortic stenosis in the community. J. Am. Coll. Cardiol. 2002, 40, 1723–1730. [Google Scholar] [CrossRef] [Green Version]
- Moura, L.M.; Ramos, S.F.; Zamorano, J.L.; Barros, I.M.; Azevedo, L.F.; Rocha-Gonçalves, F.; Rajamannan, N.M. Rosuvastatin Affecting Aortic Valve Endothelium to Slow the Progression of Aortic Stenosis. J. Am. Coll. Cardiol. 2007, 49, 554–561. [Google Scholar] [CrossRef] [Green Version]
- Cowell, S.J.; Newby, D.E.; Prescott, R.J.; Bloomfield, P.; Reid, J.; Northridge, D.B.; Boon, N.A. A Randomized Trial of Intensive Lipid-Lowering Therapy in Calcific Aortic Stenosis. N. Engl. J. Med. 2005, 352, 2389–2397. [Google Scholar] [CrossRef]
- Dichtl, W.; Alber, H.F.; Feuchtner, G.M.; Hintringer, F.; Reinthaler, M.; Bartel, T.; Süssenbacher, A.; Grander, W.; Ulmer, H.; Pachinger, O.; et al. Prognosis and Risk Factors in Patients With Asymptomatic Aortic Stenosis and Their Modulation by Atorvastatin (20 mg). Am. J. Cardiol. 2008, 102, 743–748. [Google Scholar] [CrossRef]
- Chan, K.L.; Teo, K.; Dumesnil, J.G.; Ni, A.; Tam, J. Effect of Lipid Lowering With Rosuvastatin on Progression of Aortic Stenosis. Circulation 2010, 121, 306–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Linde, D.; Yap, S.C.; Van Dijk, A.P.J.; Budts, W.; Pieper, P.G.; van der Burgh, P.H.; Mulder, B.J.; Witsenburg, M.; Cuypers, J.A.; Lindemans, J.; et al. Effects of rosuvastatin on progression of stenosis in adult patients with congenital aortic stenosis (PROCAS Trial). Am. J. Cardiol. 2011, 108, 265–271. [Google Scholar] [CrossRef]
- Rossebø, A.B.; Pedersen, T.R.; Boman, K.; Brudi, P.; Chambers, J.B.; Egstrup, K.; Gerdts, E.; Gohlke-Bärwolf, C.; Holme, I.; Kesäniemi, Y.A.; et al. Intensive Lipid Lowering with Simvastatin and Ezetimibe in Aortic Stenosis. N. Engl. J. Med. 2008, 359, 1343–1356. [Google Scholar] [CrossRef] [Green Version]
- Teo, K.K.; Corsi, D.J.; Tam, J.W.; Dumesnil, J.G.; Chan, K.L. Lipid Lowering on Progression of Mild to Moderate Aortic Stenosis: Meta-analysis of the Randomized Placebo-Controlled Clinical Trials on 2344 Patients. Can. J. Cardiol. 2011, 27, 800–808. [Google Scholar] [CrossRef]
- Parolari, A.; Tremoli, E.; Cavallotti, L.; Trezzi, M.; Kassem, S.; Loardi, C.; Veglia, F.; Ferrari, G.; Pacini, D.; Alamanni, F. Do statins improve outcomes and delay the progression of non-rheumatic calcific aortic stenosis? Heart 2011, 97, 523–529. [Google Scholar] [CrossRef]
- Arsenault, B.J.; Boekholdt, S.M.; Mora, S.; DeMicco, D.A.; Bao, W.; Tardif, J.C.; Amarenco, P.; Pedersen, T.; Barter, P.; Waters, D.D. Impact of high-dose atorvastatin therapy and clinical risk factors on incident aortic valve stenosis in patients with cardiovascular disease (from TNT, IDEAL, and SPARCL). Am. J. Cardiol. 2014, 113, 1378–1382. [Google Scholar] [CrossRef]
- Nishimura, R.A.; Otto, C.M.; Bonow, R.O.; Carabello, B.A.; Erwin, J.P.; Guyton, R.A.; O’Gara, P.T.; Ruiz, C.E.; Skubas, N.J.; Sorajja, P.; et al. 2014 AHA/ACC Guideline for the Management of Patients with Valvular Heart Disease: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014, 129, 2440–2492. [Google Scholar] [CrossRef]
- Baumgartner, H.; Falk, V.; Bax, J.J.; De Bonis, M.; Hamm, C.; Holm, P.J.; Iung, B.; Lancellotti, P.; Lansac, E.; Rodriguez Muñoz, D.; et al. 2017 ESC/EACTS Guidelines for the Management of Valvular Heart Disease. Eur. Heart J. 2017, 38, 2739–2791. [Google Scholar] [CrossRef]
- Greve, A.M.; Bang, C.N.; Boman, K.; Egstrup, K.; Kesäniemi, Y.A.; Ray, S.; Pedersen, T.R.; Wachtell, K. Relation of Lipid-Lowering Therapy to Need for Aortic Valve Replacement in Patients With Asymptomatic Mild to Moderate Aortic Stenosis. Am. J. Cardiol. 2019, 124, 1736–1740. [Google Scholar] [CrossRef]
- Monzack, E.L.; Masters, K.S. A time course investigation of the statin paradox among valvular interstitial cell phenotypes. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, 903–909. [Google Scholar] [CrossRef] [Green Version]
- Thiago, L.; Tsuji, S.R.; Nyong, J.; Puga, M.E.; Gois, A.F.; Macedo, C.R.; Valente, O.; Atallah, Á.N. Statins for aortic valve stenosis. Cochrane Database Syst. Rev. 2016. [Google Scholar] [CrossRef] [Green Version]
- NCT02679261. Evaluating the Effectiveness of Atorvastatin on the Progression of Aortic Dilatation and Valvular Degeneration in Patients With Bicuspid Aortic Valve. Available online: https://clinicaltrials.gov/show/nct02679261 (accessed on 6 October 2020).
- Borrelli, M.J.; Youssef, A.; Boffa, M.B.; Koschinsky, M.L. New Frontiers in Lp(a)-Targeted Therapies. Trends Pharmacol. Sci. 2019, 40, 212–225. [Google Scholar] [CrossRef]
- Torzewski, M.; Ravandi, A.; Yeang, C.; Yeang, C.; Edel, A.; Bhindi, R.; Kath, S.; Twardowski, L.; Schmid, J.; Yang, X.; et al. Lipoprotein(a)-Associated Molecules Are Prominent Components in Plasma and Valve Leaflets in Calcific Aortic Valve Stenosis. JACC Basic Transl. Sci. 2017, 2, 229–240. [Google Scholar] [CrossRef]
- Zheng, K.H.; Tsimikas, S.; Pawade, T.; Kroon, J.; Jenkins, W.S.A.; Doris, M.K.; White, A.C.; Timmers, N.K.L.M.; Hjortnaes, J.; Rogers, M.A.; et al. Lipoprotein(a) and Oxidized Phospholipids Promote Valve Calcification in Patients With Aortic Stenosis. J. Am. Coll. Cardiol. 2019, 73, 2150–2162. [Google Scholar] [CrossRef]
- Ozkan, U.; Ozcelik, F.; Yildiz, M.; Budak, M. Lipoprotein(a) Gene Polymorphism Increases a Risk Factor for Aortic Valve Calcification. J. Cardiovasc. Dev. Dis. 2019, 6, 31. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Y.; Dufresne, L.; Burr, H.; Ambikkumar, A.; Yasui, N.; Luk, K.; Ranatunga, D.K.; Whitmer, R.A.; Lathrop, M.; Engert, J.C.; et al. Association of LPA variants with aortic stenosis a large-scale study using diagnostic and procedural codes from electronic health records. JAMA Cardiol. 2018, 3, 18–23. [Google Scholar] [CrossRef]
- Després, A.A.; Perrot, N.; Poulin, A.; Tastet, L.; Shen, M.; Chen, H.Y.; Bourgeois, R.; Trottier, M.; Tessier, M.; Guimond, J.; et al. Lipoprotein(a), Oxidized Phospholipids, and Aortic Valve Microcalcification Assessed by 18F-Sodium Fluoride Positron Emission Tomography and Computed Tomography. CJC Open 2019, 1, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Capoulade, R.; Yeang, C.; Chan, K.L.; Pibarot, P.; Tsimikas, S. Association of Mild to Moderate Aortic Valve Stenosis Progression with Higher Lipoprotein(a) and Oxidized Phospholipid Levels: Secondary Analysis of a Randomized Clinical Trial. JAMA Cardiol. 2018, 3, 1212–1217. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgrado, V.; Ference, B.A.; et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Atherosclerosis 2019, 290, 140–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, D.P.; Jacobson, T.A.; Jones, P.H.; Koschinsky, M.L.; McNeal, C.J.; Nordestgaard, B.G.; Orringer, C.E. Use of Lipoprotein(a) in clinical practice: A biomarker whose time has come. A scientific statement from the National Lipid Association. J. Clin. Lipidol. 2019, 13, 374–392. [Google Scholar] [CrossRef] [PubMed]
- Yeang, C.; Hung, M.Y.; Byun, Y.S.; Clopton, P.; Yang, X.; Witztum, J.L.; Tsimikas, S. Effect of therapeutic interventions on oxidized phospholipids on apolipoprotein B100 and lipoprotein(a). J. Clin. Lipidol. 2016, 10, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Reiner, Ž.; Simental-Mendía, L.E.; Ferretti, G.; Cicero, A.F.G. Effect of extended-release niacin on plasma lipoprotein(a) levels: A systematic review and meta-analysis of randomized placebo-controlled trials. Metabolism 2016, 65, 1664–1678. [Google Scholar] [CrossRef]
- Landray, M.J.; Haynes, R.; Hopewell, J.C.; Parish, S.; Aung, T.; Tomson, J.; Wallendszus, K.; Craig, M.; Jiang, L.; Collins, R.; et al. Effects of extended-release niacin with laropiprant in high-risk patients. N. Engl. J. Med. 2014, 371, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 371, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Langsted, A.; Nordestgaard, B.G. Antisense Oligonucleotides Targeting Lipoprotein(a). Curr. Atheroscler. Rep. 2019, 21, 30. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S. Potential Causality and Emerging Medical Therapies for Lipoprotein(a) and Its Associated Oxidized Phospholipids in Calcific Aortic Valve Stenosis. Circ. Res. 2019, 124, 405–415. [Google Scholar] [CrossRef]
- Leibundgut, G.; Scipione, C.; Yin, H.; Schneider, M.; Boffa, M.B.; Green, S.; Yang, X.; Dennis, E.; Witztum, J.L.; Koschinsky, M.L.; et al. Determinants of binding of oxidized phospholipids on apolipoprotein (a) and lipoprotein (a). J. Lipid Res. 2013, 54, 2815–2830. [Google Scholar] [CrossRef] [Green Version]
- Tsimikas, S.; Willeit, P.; Willeit, J.; Santer, P.; Mayr, M.; Xu, Q.; Mayr, A.; Witztum, J.L.; Kiechl, S. Oxidation-specific biomarkers, prospective 15-year cardiovascular and stroke outcomes, and net reclassification of cardiovascular events. J. Am. Coll. Cardiol. 2012, 60, 2218–2229. [Google Scholar] [CrossRef] [Green Version]
- Capoulade, R.; Chan, K.L.; Yeang, C.; Mathieu, P.; Bossé, Y.; Dumesnil, J.G.; Tam, J.W.; Teo, K.K.; Mahmut, A.; Yang, X.; et al. Oxidized phospholipids, lipoprotein(a), and progression of calcific aortic valve stenosis. J. Am. Coll. Cardiol. 2015, 66, 1236–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Que, X.; Hung, M.Y.; Yeang, C.; Gonen, A.; Prohaska, T.A.; Sun, X.; Diehl, C.; Määttä, A.; Gaddis, D.E.; Bowden, K.; et al. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature 2018, 558, 301–306. [Google Scholar] [CrossRef]
- Mahmut, A.; Boulanger, M.C.; El Husseini, D.; Fournier, D.; Bouchareb, R.; Després, J.P.; Pibarot, P.; Bossé, Y.; Mathieu, P. Elevated expression of lipoprotein-associated phospholipase A2 in calcific aortic valve disease: Implications for valve mineralization. J. Am. Coll. Cardiol. 2014, 63, 460–469. [Google Scholar] [CrossRef] [Green Version]
- Bouchareb, R.; Mahmut, A.; Nsaibia, M.J.; Boulanger, M.C.; Dahou, A.; Lépine, J.L.; Laflamme, M.H.; Hadji, F.; Couture, C.; Trahan, S.; et al. Autotaxin derived from lipoprotein(a) and valve interstitial cells promotes inflammation and mineralization of the aortic valve. Circulation 2015, 132, 677–690. [Google Scholar] [CrossRef]
- Nsaibia, M.J.; Mahmut, A.; Boulanger, M.C.; Arsenault, B.J.; Bouchareb, R.; Simard, S.; Witztum, J.L.; Clavel, M.A.; Pibarot, P.; Bossé, Y.; et al. Autotaxin interacts with lipoprotein(a) and oxidized phospholipids in predicting the risk of calcific aortic valve stenosis in patients with coronary artery disease. J. Intern. Med. 2016, 280, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Van der Aar, E.; Fagard, L.; Desrivot, J.; Dupont, S.; Heckmann, B.; Blanque, R.; Gheyle, L.; Ralic, J.; Vanhoutte, F. Favorable human safety, pharmacokinetics and pharmacodynamics of the autotaxin inhibitor GLPG1690, a potential new treatment in COPD. Eur. Respir. J. 2015, 304, OA484. [Google Scholar] [CrossRef]
- Maher, T.M.; van der Aar, E.M.; Van de Steen, O.; Allamassey, L.; Desrivot, J.; Dupont, S.; Fagard, L.; Ford, P.; Fieuw, A.; Wuyts, W. Safety, tolerability, pharmacokinetics, and pharmacodynamics of GLPG1690, a novel autotaxin inhibitor, to treat idiopathic pulmonary fibrosis (FLORA): A phase 2a randomised placebo-controlled trial. Lancet Respir. Med. 2018, 6, 627–635. [Google Scholar] [CrossRef]
- Mathieu, P.; Boulanger, M.C. Autotaxin and Lipoprotein Metabolism in Calcific Aortic Valve Disease. Front. Cardiovasc. Med. 2019, 6, 18. [Google Scholar] [CrossRef]
- Bouchareb, R.; Boulanger, M.C.; Tastet, L.; Mkannez, G.; Nsaibia, M.J.; Hadji, F.; Dahou, A.; Messadeq, Y.; Arsenault, B.J.; Pibarot, P.; et al. Activated platelets promote an osteogenic programme and the progression of calcific aortic valve stenosis. Eur. Heart J. 2019, 40, 1362–1373. [Google Scholar] [CrossRef]
- Capoulade, R.; Mahmut, A.; Tastet, L.; Arsenault, M.; Bédard, É.; Dumesnil, J.G.; Després, J.P.; Larose, É.; Arsenault, B.J.; Bossé, Y.; et al. Impact of plasma Lp-PLA2 activity on the progression of aortic stenosis: The PROGRESSA study. JACC Cardiovasc. Imaging 2015, 8, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Wihastuti, T.A.; Heriansyah, T.; Hanifa, H.; Andarini, S.; Sholichah, Z.; Sulfia, Y.H.; Adam, A.A.; Refialdinata, J.; Lutfiana, N.C. Darapladib inhibits atherosclerosis development in type 2 diabetes mellitus Sprague-Dawley rat model. Endocr. Regul. 2018, 52, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Choi, W.G.; Prasad, M.; Lennon, R.; Gulati, R.; Prasad, A.; Lerman, L.O.; Lerman, A. Long-term darapladib use does not affect coronary plaque composition assessed using multimodality intravascular imaging modalities: A randomized-controlled study. Coron Artery Dis. 2018, 29, 104. [Google Scholar] [CrossRef]
- NCT02109614. Early Aortic Valve Lipoprotein(a) Lowering Trial. Available online: https://clinicaltrials.gov/ct2/show/NCT02109614 (accessed on 6 October 2020).
- NCT03626662. Safety, Tolerability, Pharmacokinetics and Pharmacodynamics Study of AMG 890 in Subjects with Elevated Plasma Lipoprotein(a). Available online: https://clinicaltrials.gov/ct2/show/NCT03626662 (accessed on 6 October 2020).
- NCT04270760. Randomized Study to Evaluate Efficacy, Safety, and Tolerability of AMG 890 in Subjects With Elevated Lipoprotein(a). Available online: https://clinicaltrials.gov/ct2/show/NCT04270760 (accessed on 6 October 2020).
- Ferri, N.; Ruscica, M.; Coggi, D.; Bonomi, A.; Amato, M.; Frigerio, B.; Sansaro, D.; Ravani, A.; Veglia, F.; Capra, N.; et al. Sex-specific predictors of PCSK9 levels in a European population: The IMPROVE study. Atherosclerosis 2020, 309, 39–46. [Google Scholar] [CrossRef]
- Warden, B.A.; Fazio, S.; Shapiro, M.D. The PCSK9 revolution: Current status, controversies, and future directions: The PCSK9 revolution. Trends Cardiovasc. Med. 2020, 30, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.G.; He, Y.F.; Chen, Y.L.; Zhao, F.M.; Song, Y.Q.; Zhang, H.; Ma, Y.H.; Guan, X.; Zhang, W.Y.; Chen, X.L.; et al. Proprotein convertase subtilisin/kexin type 9 levels and aortic valve calcification: A prospective, cross sectional study. J. Int. Med. Res. 2016, 44, 865–874. [Google Scholar] [CrossRef] [Green Version]
- Perrot, N.; Valerio, V.; Moschetta, D.; Boekholdt, S.M.; Dina, C.; Chen, H.Y.; Abner, E.; Martinsson, A.; Manikpurage, H.D.; Rigade, S.; et al. Genetic and In Vitro Inhibition of PCSK9 and Calcific Aortic Valve Stenosis. JACC Basic Transl. Sci. 2020, 5, 649–661. [Google Scholar] [CrossRef]
- Poggio, P.; Songia, P.; Cavallotti, L.; Barbieri, S.S.; Zanotti, I.; Arsenault, B.J.; Valerio, V.; Ferri, N.; Capoulade, R.; Camera, M. PCSK9 Involvement in Aortic Valve Calcification. J. Am. Coll. Cardiol. 2018, 72, 3225–3227. [Google Scholar] [CrossRef]
- Nissen, S.E.; Stroes, E.; Dent-Acosta, R.E.; Rosenson, R.S.; Lehman, S.J.; Sattar, N.; Preiss, D.; Bruckert, E.; Ceška, R.; Lepor, N.; et al. Efficacy and tolerability of evolocumab vs ezetimibe in patients with muscle-related statin intolerance: The GAUSS-3 randomized clinical trial. JAMA J. Am. Med. Assoc. 2016, 315, 1580–1590. [Google Scholar] [CrossRef]
- Cho, L.; Dent, R.; Stroes, E.S.G.; Stein, E.A.; Sullivan, D.; Ruzza, A.; Flower, A.; Somaratne, R.; Rosenson, R.S. Persistent Safety and Efficacy of Evolocumab in Patients with Statin Intolerance: A Subset Analysis of the OSLER Open-Label Extension Studies. Cardiovasc. Drugs Ther. 2018, 32, 365–372. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, M.L.; Fazio, S.; Giugliano, R.P.; Stroes, E.S.G.; Kanevsky, E.; Gouni-Berthold, I.; Im, K.; Lira Pineda, A.; Wasserman, S.M.; Češka, R.; et al. Lipoprotein(a), PCSK9 Inhibition, and Cardiovascular Risk. Circulation 2019, 139, 1483–1492. [Google Scholar] [CrossRef]
- Qamar, A.; Giugliano, R.P.; Keech, A.C.; Kuder, J.F.; Murphy, S.A.; Kurtz, C.E.; Wasserman, S.M.; Sever, P.S.; Pedersen, T.R.; Sabatine, M.S. Interindividual Variation in Low-Density Lipoprotein Cholesterol Level Reduction with Evolocumab: An Analysis of FOURIER Trial Data. JAMA Cardiol. 2019, 4, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Watts, G.F.; Chan, D.C.; Somaratne, R.; Wasserman, S.M.; Scott, R.; Marcovina, S.M.; Barrett, P.H.R. Controlled study of the effect of proprotein convertase subtilisin-kexin type 9 inhibition with evolocumab on lipoprotein(a) particle kinetics. Eur. Heart J. 2018, 39, 2577–2585. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Puri, R.; Anderson, T.; Ballantyne, C.M.; Cho, L.; Kastelein, J.J.P.; Koenig, W.; Somaratne, R.; Kassahun, H.; Yang, J.; et al. Effect of Evolocumab on Coronary Plaque Composition. J. Am. Coll. Cardiol. 2018, 72, 2012–2021. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- NCT03051360. PCSK9 Inhibitors in the Progression of Aortic Stenosis. Available online: https://clinicaltrials.gov/show/NCT03051360 (accessed on 6 October 2020).
- Fitzgerald, K.; White, S.; Borodovsky, A.; Bettencourt, B.R.; Strahs, A.; Clausen, V.; Wijngaard, P.; Horton, J.D.; Taubel, J.; Brooks, A.; et al. A highly durable RNAi therapeutic inhibitor of PCSK9. N. Engl. J. Med. 2017, 376, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.J.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Antonini-Canterin, F.; Huang, G.; Cervesato, E.; Faggiano, P.; Pavan, D.; Piazza, R.; Nicolosi, G.L. Symptomatic aortic stenosis: Does systemic hypertension play an additional role? Hypertension 2003, 41, 1268–1272. [Google Scholar] [CrossRef] [Green Version]
- Arjunon, S.; Rathan, S.; Jo, H.; Yoganathan, A.P. Aortic valve: Mechanical environment and mechanobiology. Ann. Biomed. Eng. 2013, 41, 1331–1346. [Google Scholar] [CrossRef] [Green Version]
- Rassa, A.; Zahr, F. Hypertension and Aortic Stenosis: A Review. Curr. Hypertens Rev. 2018, 14, 6–14. [Google Scholar] [CrossRef]
- Capoulade, R.; Clavel, M.A.; Mathieu, P.; Côté, N.; Dumesnil, J.G.; Arsenault, M.; Bédard, E.; Pibarot, P. Impact of hypertension and renin-angiotensin system inhibitors in aortic stenosis. Eur. J. Clin. Investig. 2013, 43, 1262–1272. [Google Scholar] [CrossRef]
- Poggio, P.; Folesani, G.; Raffa, G.M.; Songia, P.; Valenti, V.; Myasoedova, V.; Parolari, A. Antihypertensive Treatments in Patients Affected by Aortic Valve Stenosis. Curr. Pharm. Des. 2017, 23, 1188–1194. [Google Scholar] [CrossRef]
- Lindman, B.R.; Otto, C.M. Time to treat hypertension in patients with aortic stenosis. Circulation 2013, 128, 1281–1283. [Google Scholar] [CrossRef] [Green Version]
- Balakumar, P.; Jagadeesh, G. A century old renin-angiotensin system still grows with endless possibilities: AT1 receptor signaling cascades in cardiovascular physiopathology. Cell Signal. 2014, 26, 2147–2160. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Shen, Y.; Hu, W.; Chen, Z.; Li, Y. Angiotensin II promotes an osteoblast-like phenotype in porcine aortic valve myofibroblasts. Aging Clin. Exp. Res. 2016, 28, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Li, Y.; Jia, L.; Han, Y.; Cheng, J.; Li, H.; Qi, Y.; Du, J. Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF β/Smad activation and cardiac fibrosis induced by angiotensin II. PLoS ONE 2012, 7, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helske, S.; Lindstedt, K.A.; Laine, M.; Mäyränpää, M.; Werkkala, K.; Lommi, J.; Turto, H.; Kupari, M.; Kovanen, P.T. Induction of local angiotensin II-producing systems in stenotic aortic valves. J. Am. Coll. Cardiol. 2004, 44, 1859–1866. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, K.D.; Shavelle, D.M.; Caulfield, M.T.; McDonald, T.O.; Olin-Lewis, K.; Otto, C.M.; Probstfield, J.L. Association of angiotensin-converting enzyme with low-density lipoprotein in aortic valvular lesions and in human plasma. Circulation 2002, 106, 2224–2230. [Google Scholar] [CrossRef] [Green Version]
- Chockalingam, A.; Venkatesan, S.; Subramaniam, T.; Jagannathan, V.; Elangovan, S.; Alagesan, R.; Gnanavelu, G.; Dorairajan, S.; Krishna, B.P.; Chockalingam, V. Safety and efficacy of angiotensin-converting enzyme inhibitors in symptomatic severe aortic stenosis: Symptomatic Cardiac Obstruction-Pilot Study of Enalapril in Aortic Stenosis (SCOPE-AS). Am. Heart J. 2004, 147, 740. [Google Scholar] [CrossRef]
- O’Brien, K.D.; Zhao, X.Q.; Shavelle, D.M.; Caulfield, M.T.; Letterer, R.A.; Kapadia, S.R.; Probstfield, J.L.; Otto, C.M. Hemodynamic effects of the angiotensin-converting enzyme inhibitor, ramipril, in patients with mild to moderate aortic stenosis and preserved left ventricular function. J. Investig. Med. 2004, 52, 185. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Tsujino, T.; Naito, Y.; Ezumi, A.; Lee-Kawabata, M.; Nakao, S.; Goda, A.; Sakata, Y.; Yamamoto, K.; Daimon, T.; et al. Administration of angiotensin-converting enzyme inhibitors is associated with slow progression of mild aortic stenosis in Japanese patients. Heart Vessels 2011, 26, 252–257. [Google Scholar] [CrossRef]
- Jiménez-Candil, J.; Bermejo, J.; Yotti, R.; Cortina, C.; Moreno, M.; Cantalapiedra, J.L.; García-Fernández, M.A. Effects of angiotensin converting enzyme inhibitors in hypertensive patients with aortic valve stenosis: A drug withdrawal study. Heart 2005, 91, 1311–1318. [Google Scholar] [CrossRef] [Green Version]
- Dalsgaard, M.; Iversen, K.; Kjaergaard, J.; Grande, P.; Goetze, J.P.; Clemmensen, P.; Hassager, C. Short-term hemodynamic effect of angiotensin-converting enzyme inhibition in patients with severe aortic stenosis: A placebo-controlled, randomized study. Am. Heart J. 2014, 167, 226–234. [Google Scholar] [CrossRef]
- Bull, S.; Loudon, M.; Francis, J.M.; Joseph, J.; Gerry, S.; Karamitsos, T.D.; Prendergast, B.D.; Banning, A.P.; Neubauer, S.; Myerson, S.G. A prospective, double-blind, randomized controlled trial of the angiotensin-converting enzyme inhibitor Ramipril in Aortic Stenosis (RIAS trial). Eur. Heart J. Cardiovasc. Imaging 2015, 16, 834–841. [Google Scholar] [CrossRef]
- Helske-Suihko, S.; Laine, M.; Lommi, J.; Kaartinen, M.; Werkkala, K.; Kovanen, P.T.; Kupari, M. Is blockade of the renin-angiotensin system able to reverse the structural and functional remodeling of the left ventricle in severe aortic stenosis? J. Cardiovasc. Pharmacol. 2015, 65, 233–240. [Google Scholar] [CrossRef]
- Côté, N.; Couture, C.; Pibarot, P.; Després, J.P.; Mathieu, P. Angiotensin receptor blockers are associated with a lower remodelling score of stenotic aortic valves. Eur. J. Clin. Investig. 2011, 41, 1172–1179. [Google Scholar] [CrossRef]
- Côté, N.; Mahmut, A.; Fournier, D.; Boulanger, M.C.; Couture, C.; Després, J.P.; Trahan, S.; Bossé, Y.; Pagé, S.; Pibarot, P.; et al. Angiotensin receptor blockers are associated with reduced fibrosis and interleukin-6 expression in calcific aortic valve disease. Pathobiology 2013, 81, 15–24. [Google Scholar] [CrossRef]
- Nadir, M.A.; Wei, L.; Elder, D.H.J.; Libianto, R.; Lim, T.K.; Pauriah, M.; Pringle, S.D.; Doney, A.D.; Choy, A.M.; Struthers, A.D.; et al. Impact of renin-angiotensin system blockade therapy on outcome in aortic stenosis. J. Am. Coll. Cardiol. 2011, 58, 570–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, C.N.; Greve, A.M.; Køber, L.; Rossebø, A.B.; Ray, S.; Boman, K.; Nienaber, C.A.; Devereux, R.B.; Wachtell, K. Renin-angiotensin system inhibition is not associated with increased sudden cardiac death, cardiovascular mortality or all-cause mortality in patients with aortic stenosis. Int. J. Cardiol. 2014, 175, 492–498. [Google Scholar] [CrossRef]
- Yamamoto, K.; Yamamoto, H.; Takeuchi, M.; Kisanuki, A.; Akasaka, T.; Ohte, N.; Hirano, Y.; Yoshida, K.; Nakatani, S.; Takeda, Y.; et al. Risk factors for progression of degenerative aortic valve disease in the Japanese—The Japanese aortic stenosis study (JASS) prospective analysis. Circ. J. 2015, 79, 2050–2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NCT01589380. A Randomized Trial of Angiotensin Receptor bLocker, Fimasartan, in Aortic Stenosis (ALFA Trial). Available online: https://clinicaltrials.gov/show/nct01589380 (accessed on 6 October 2020).
- NCT03666351. Study to Evaluate the Effect on Improvement of LVH by the Control of BP in Hypertension Patients With AV Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT03666351 (accessed on 6 October 2020).
- Andersson, C.; Abdulla, J. Is the use of renin-angiotensin systeminhibitors in patients with aortic valve stenosis safe and of prognostic benefit? A systematic review and meta-analysis. Eur. Heart J. Cardiovasc. Pharmacother. 2017, 3, 21–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magne, J.; Guinot, B.; Le Guyader, A.; Bégot, E.; Marsaud, J.P.; Mohty, D.; Aboyans, V. Relation Between Renin-Angiotensin System Blockers and Survival Following Isolated Aortic Valve Replacement for Aortic Stenosis. Am. J. Cardiol. 2018, 121, 455–460. [Google Scholar] [CrossRef]
- Rodriguez-Gabella, T.; Catalá, P.; Muñoz-García, A.J.; Nombela-Franco, L.; Del Valle, R.; Gutiérrez, E.; Regueiro, A.; Jimenez-Diaz, V.A.; Ribeiro, H.B.; Rivero, F.; et al. Renin-Angiotensin System Inhibition Following Transcatheter Aortic Valve Replacement. J. Am. Coll. Cardiol. 2019, 74, 631–641. [Google Scholar] [CrossRef]
- Chen, S.; Redfors, B.; Nazif, T.; Kirtane, A.; Crowley, A.; Ben-Yehuda, O.; Kapadia, S.; Finn, M.T.; Goel, S.; Lindman, B.R.; et al. Impact of renin-angiotensin systeminhibitors on clinical outcomes in patients with severe aortic stenosis undergoing transcatheter aortic valve replacement: An analysis of from the PARTNER 2 trial and registries. Eur. Heart J. 2020, 41, 943–954. [Google Scholar] [CrossRef]
- NCT03315832. Efficacy of Angiotensin Receptor Blocker Following aortIc Valve Intervention for Aortic STenOsis: A Randomized mulTi-cEntric Double-blind Phase II Study. Available online: https://clinicaltrials.gov/show/NCT03315832 (accessed on 6 October 2020).
- Wang, P.; Quan, Z.; Luo, D.; Chen, W.; Peng, D. Spironolactone dose-dependently alleviates the calcification of aortic rings cultured in hyperphosphatemic medium with or without hyperglycemia by suppressing phenotypic transition of VSMCs through downregulation of Pit-1. Mol. Med. Rep. 2019, 49, 3622–3632. [Google Scholar] [CrossRef]
- Zhu, D.; Rashdan, N.A.; Chapman, K.E.; Hadoke, P.W.; MacRae, V.E. A novel role for the mineralocorticoid receptor in glucocorticoid driven vascular calcification. Vasc. Pharmacol. 2016, 86, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Rajagopalan, S.; Duquaine, D.; King, S.; Pitt, B.; Patel, P. Mineralocorticoid receptor antagonism in experimental atherosclerosis. Circulation 2002, 105, 2212–2216. [Google Scholar] [CrossRef] [Green Version]
- Pitt, B.; Reichek, N.; Willenbrock, R.; Zannad, F.; Phillips, R.A.; Roniker, B.; Kleiman, J.; Krause, S.; Burns, D.; Williams, G.H. Effects of Eplerenone, Enalapril, and Eplerenone/Enalapril in Patients with Essential Hypertension and Left Ventricular Hypertrophy: The 4E-Left Ventricular Hypertrophy Study. Circulation 2003, 108, 1831–1838. [Google Scholar] [CrossRef]
- Jolobe, O.M.P. Evolving strategies for the use of spironolactone in cardiovascular disease. Eur. J. Intern. Med. 2013, 24, 303–309. [Google Scholar] [CrossRef]
- Gkizas, S.; Koumoundourou, D.; Sirinian, X.; Rokidi, S.; Mavrilas, D.; Koutsoukos, P.; Papalois, A.; Apostolakis, E.; Alexopoulos, D.; Papadaki, H. Aldosterone receptor blockade inhibits degenerative processes in the early stage of calcific aortic stenosis. Eur. J. Pharmacol. 2010, 642, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.A.H.; Kerr, A.J.; Cowan, B.R.; Young, A.A.; Occleshaw, C.; Richards, A.M.; Edwards, C.; Whalley, G.A.; Freidlander, D.; Williams, M.; et al. A randomized trial of the aldosterone-receptor antagonist eplerenone in asymptomatic moderate-severe aortic stenosis. Am. Heart J. 2008, 156, 348–355. [Google Scholar] [CrossRef]
- NCT03923530. Pressure Assessment to Improve Outcomes After TAVR: A Registry. Available online: https://clinicaltrials.gov/ct2/show/NCT03923530 (accessed on 6 October 2020).
- Bosse, K.; Hans, C.P.; Zhao, N.; Koenig, S.N.; Huang, N.; Guggilam, A.; LaHaye, S.; Tao, G.; Lucchesi, P.A.; Lincoln, J.; et al. Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J. Mol. Cell Cardiol. 2013, 60, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.D.; Chu, Y.; Brooks, R.M.; Richenbacher, W.E.; Peña-Silva, R.; Heistad, D.D. Dysregulation of Antioxidant Mechanisms Contributes to Increased Oxidative Stress in Calcific Aortic Valvular Stenosis in Humans. J. Am. Coll. Cardiol. 2008, 52, 843–850. [Google Scholar] [CrossRef] [Green Version]
- Bertacco, E.; Renato, M.; Arrigoni, G.; Faggin, E.; Iop, L.; Puato, M.; Tessari, P.; Pauletto, P.; Rattazzi, M. Proteomic analysis of clonal interstitial aortic valve cells acquiring a pro-calcific profile. J. Proteome Res. 2010, 9, 5913–5921. [Google Scholar] [CrossRef]
- Kennedy, J.A.; Hua, X.; Mishra, K.; Murphy, G.A.; Rosenkranz, A.C.; Horowitz, J.D. Inhibition of calcifying nodule formation in cultured porcine aortic valve cells by nitric oxide donors. Eur. J. Pharmacol. 2009, 602, 28–35. [Google Scholar] [CrossRef]
- Richards, J.; El-Hamamsy, I.; Chen, S.; Sarang, Z.; Sarathchandra, P.; Yacoub, M.H.; Chester, A.H.; Butcher, J.T. Side-specific endothelial-dependent regulation of aortic valve calcification: Interplay of hemodynamics and nitric oxide signaling. Am. J. Pathol. 2013, 182, 1922–1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rattazzi, M.; Donato, M.; Bertacco, E.; Millioni, R.; Franchin, C.; Mortarino, C.; Faggin, E.; Nardin, C.; Scarpa, R.; Cinetto, F.; et al. L-Arginine prevents inflammatory and pro-calcific differentiation of interstitial aortic valve cells. Atherosclerosis 2020, 298, 27–35. [Google Scholar] [CrossRef]
- Claveau, D.; Piha-Gossack, A.; Friedland, S.N.; Afilalo, J.; Rudski, L. Complications Associated with Nitrate Use in Patients Presenting with Acute Pulmonary Edema and Concomitant Moderate or Severe Aortic Stenosis. Ann. Emerg. Med. 2015, 66, 355–362.e1. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, J.W.; Nishimura, R.A.; Borlaug, B.A.; Eleid, M.F. Hemodynamic Response to Nitroprusside in Patients With Low-Gradient Severe Aortic Stenosis and Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2017, 70, 1339–1348. [Google Scholar] [CrossRef]
- Lewis, G.D.; Shah, R.; Shahzad, K.; Camuso, J.M.; Pappagianopoulos, P.P.; Hung, J.; Tawakol, A.; Gerszten, R.E.; Systrom, D.M.; Bloch, K.D.; et al. Sildenafil improves exercise capacity and quality of life in patients with systolic heart failure and secondary pulmonary hypertension. Circulation 2007, 116, 1555–1562. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, X.D.; Long, M.; Li, F.; Hu, X.; Liao, X.X.; Du, Z.M. PDE5 inhibitor sildenafil in the treatment of heart failure: A meta-analysis of randomized controlled trials. Int. J. Cardiol. 2014, 172, 581–587. [Google Scholar] [CrossRef]
- Vandenwijngaert, S.; Pokreisz, P.; Hermans, H.; Gillijns, H.; Pellens, M.; Bax, N.A.; Coppiello, G.; Oosterlinck, W.; Balogh, A.; Papp, Z.; et al. Increased Cardiac Myocyte PDE5 Levels in Human and Murine Pressure Overload Hypertrophy Contribute to Adverse LV Remodeling. PLoS ONE 2013, 8, e58841. [Google Scholar] [CrossRef] [Green Version]
- NCT01275339. Aortic Stenosis and PhosphodiEsterase Type 5 iNhibition (ASPEN): A Pilot Study. Available online: https://ClinicalTrials.gov/show/NCT01275339 (accessed on 6 October 2020).
- Lindman, B.R.; Zajarias, A.; Madrazo, J.A.; Shah, J.; Gage, B.F.; Novak, E.; Johnson, S.N.; Chakinala, M.M.; Hohn, T.A.; Saghir, M.; et al. Effects of phosphodiesterase type 5 inhibition on systemic and pulmonary hemodynamics and ventricular function in patients with severe symptomatic aortic stenosis. Circulation 2012, 125, 2353–2362. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, A.; Rodrigues, C.; Moraes, T.; Rodrigues, G. In Endothelial Cells, the Activation or Stimulation of Soluble Guanylyl Cyclase Induces the Nitric Oxide Production by a Mechanism Dependent of Nitric Oxide Synthase Activation. J. Pharm. Pharm. Sci. 2018, 21, 38–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schäfer, A.; Fraccarollo, D.; Werner, L.; Bauersachs, J. Guanylyl cyclase activator ataciguat improves vascular function and reduces platelet activation in heart failure. Pharmacol. Res. 2010, 62, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Weissmann, N.; Hackemack, S.; Dahal, B.K.; Pullamsetti, S.S.; Savai, R.; Mittal, M.; Fuchs, B.; Medebach, T.; Dumitrascu, R.; Eickels, M.V.; et al. The soluble guanylate cyclase activator HMR1766 reverses hypoxia-induced experimental pulmonary hypertension in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, 658–665. [Google Scholar] [CrossRef]
- NCT02049203. Safety of Ataciguat in Patients with Moderate Calcific Aortic Valve Stenosis. Available online: https://clinicaltrials.gov/show/nct02049203 (accessed on 6 October 2020).
- NCT02481258. A Study Evaluating the Effects of Ataciguat (HMR1766) on Aortic Valve Calcification. Available online: https://clinicaltrials.gov/show/nct02481258 (accessed on 6 October 2020).
- Weiss, R.M.; Lund, D.D.; Chu, Y.; Brooks, R.M.; Zimmerman, K.A.; El Accaoui, R.; Davis, M.K.; Hajj, G.P.; Zimmerman, M.B.; Heistad, D.D. Osteoprotegerin Inhibits Aortic Valve Calcification and Preserves Valve Function in Hypercholesterolemic Mice. PLoS ONE 2013, 8, e65201. [Google Scholar] [CrossRef] [Green Version]
- Bucay, N.; Sarosi, I.; Dunstan, C.R.; Morony, S.; Tarpley, J.; Capparelli, C.; Scully, S.; Tan, H.L.; Xu, W.; Lacey, D.L.; et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998, 12, 1260–1268. [Google Scholar] [CrossRef]
- Buso, G.; Faggin, E.; Pauletto, P.; Rattazzi, M. Osteoprotegerin in Cardiovascular Disease: Ally or Enemy? Curr. Pharm. Des. 2014, 20, 5862–5869. [Google Scholar] [CrossRef]
- Rattazzi, M.; Faggin, E.; Bertacco, E.; Buso, R.; Puato, M.; Plebani, M.; Zaninotto, M.; Condotta, D.; Zoppellaro, G.; Tarantini, G.; et al. RANKL Expression Is Increased in Circulating Mononuclear Cells of Patients with Calcific Aortic Stenosis. J. Cardiovasc. Transl. Res. 2018, 11, 329–338. [Google Scholar] [CrossRef]
- Persy, V.; D’Haese, P. Vascular calcification and bone disease: The calcification paradox. Trends Mol. Med. 2009, 15, 405–416. [Google Scholar] [CrossRef]
- Pfister, R.; Michels, G.; Sharp, S.J.; Luben, R.; Wareham, N.J.; Khaw, K.T. Inverse association between bone mineral density and risk of aortic stenosis in men and women in EPIC-Norfolk prospective study. Int. J. Cardiol. 2015, 178, 29–30. [Google Scholar] [CrossRef]
- Helas, S.; Goettsch, C.; Schoppet, M.; Zeitz, U.; Hempel, U.; Morawietz, H.; Kostenuik, P.J.; Erben, R.G.; Hofbauer, L.C. Inhibition of receptor activator of NF-κB ligand by denosumab attenuates vascular calcium deposition in mice. Am. J. Pathol. 2009, 175, 473–478. [Google Scholar] [CrossRef] [Green Version]
- Lerman, D.A.; Prasad, S.; Alotti, N. Denosumab could be a Potential Inhibitor of Valvular Interstitial Cells Calcification in vitro. Int. J. Cardiovasc. Res. 2015, 5, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Elmariah, S.; Cimmino, G.; Fuster, V.; Badimon, J.J. Bisphosphonates Prevent Oxidized Low-Density Lipoprotein-Induced Expression of Osteogenic Markers in Aortic Valve Myofibroblasts. J. Am. Coll. Cardiol. 2010, 55, A149–E1398. [Google Scholar] [CrossRef] [Green Version]
- Price, P.A.; Faus, S.A.; Williamson, M.K. Bisphosphonates alendronate and ibandronate inhibit artery calcification at doses comparable to those that inhibit bone resorption. Arterioscler. Thromb Vasc. Biol. 2001, 21, 817–824. [Google Scholar] [CrossRef] [Green Version]
- Synetos, A.; Toutouzas, K.; Drakopoulou, M.; Koutagiar, I.; Benetos, G.; Kotronias, R.; Anousakis-Vlachochristou, N.; Latsios, G.; Karanasos, A.; Agrogiannis, G.; et al. Inhibition of Aortic Valve Calcification by Local Delivery of Zoledronic Acid—An Experimental Study. J. Cardiovasc. Transl. Res. 2018, 11, 192–200. [Google Scholar] [CrossRef]
- Corrado, A.; Santoro, N.; Cantatore, F.P. Extra-skeletal effects of bisphosphonates. Jt. Bone Spine. 2007, 74, 32–38. [Google Scholar] [CrossRef]
- Hashiba, H.; Aizawa, S.; Tamura, K.; Kogo, H. Inhibition of the progression of aortic calcification by etidronate treatment in hemodialysis patients: Long-term effects. Ther. Apher. Dial. 2006, 10, 59–64. [Google Scholar] [CrossRef]
- Ariyoshi, T.; Eishi, K.; Sakamoto, I.; Matsukuma, S.; Odate, T. Effect of etidronic acid on arterial calcification in dialysis patients. Clin. Drug Investig. 2006, 26, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Elmariah, S.; Delaney, J.A.C.; O’Brien, K.D.; Budoff, M.J.; Vogel-Claussen, J.; Fuster, V.; Kronmal, R.A.; Halperin, J.L. Bisphosphonate use and prevalence of valvular and vascular calcification in women: MESA (The Multi-Ethnic Study of Atherosclerosis). J. Am. Coll. Cardiol. 2010, 56, 1752–1759. [Google Scholar] [CrossRef] [Green Version]
- Innasimuthu, A.L.; Katz, W.E. Effect of bisphosphonates on the progression of degenerative aortic stenosis. Echocardiography 2011, 28, 1–7. [Google Scholar] [CrossRef]
- Sterbakova, G.; Vyskocil, V.; Linhartova, K. Bisphosphonates in calcific aortic stenosis: Association with slower progression in mild disease—A pilot retrospective study. Cardiology 2011, 117, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, O.; Cam, A.; Goel, S.S.; Houghtaling, P.L.; Williams, S.; Ruiz-Rodriguez, E.; Menon, V.; Kapadia, S.R.; Tuzcu, E.M.; Blackstone, E.H.; et al. Do bisphosphonates slow the progression of aortic stenosis? J. Am. Coll. Cardiol. 2012, 59, 1452–1459. [Google Scholar] [CrossRef] [Green Version]
- Dweck, M.R.; Newby, D.E. Osteoporosis is a major confounder in observational studies investigating bisphosphonate therapyin aortic stenosis. J. Am. Coll. Cardiol. 2012, 60, 1027. [Google Scholar] [CrossRef] [Green Version]
- NCT02132026. Study Investigating the Effect of Drugs Used to Treat Osteoporosis on the Progression of Calcific Aortic Stenosis. Available online: https://clinicaltrials.gov/ct2/show/NCT02132026 (accessed on 6 October 2020).
- Schurgers, L.J.; Uitto, J.; Reutelingsperger, C.P. Vitamin K-dependent carboxylation of matrix Gla-protein: A crucial switch to control ectopic mineralization. Trends Mol. Med. 2013, 19, 217–226. [Google Scholar] [CrossRef]
- Zebboudj, A.F.; Imura, M.; Boström, K. Matrix GLA protein, a regulatory protein for bone morphogenetic protein-2. J. Biol. Chem. 2002, 277, 4388–4394. [Google Scholar] [CrossRef] [Green Version]
- Chiyoya, M.; Seya, K.; Yu, Z.; Daitoku, K.; Motomura, S.; Imaizumi, T.; Fukuda, I.; Furukawa, K.I. Matrix Gla protein negatively regulates calcification of human aortic valve interstitial cells isolated from calcified aortic valves. J. Pharmacol. Sci. 2018, 136, 257–265. [Google Scholar] [CrossRef]
- Luo, G.; Ducy, P.; McKee, M.D.; Pinero, G.J.; Loyer, E.; Behringer, R.R.; Karsenty, G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protien. Nature 1997, 386, 78–81. [Google Scholar] [CrossRef]
- Venardos, N.; Bennett, D.; Weyant, M.J.; Reece, T.B.; Meng, X.; Fullerton, D.A. Matrix Gla protein regulates calcification of the aortic valve. J. Surg. Res. 2015, 199, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Schurgers, L.J.; Cranenburg, E.C.M.; Vermeer, C. Matrix Gla-protein: The calcification inhibitor in need of vitamin K. Thromb. Haemost. 2008, 100, 593–603. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Koretsune, Y.; Akasaka, T.; Kisanuki, A.; Ohte, N.; Takenaka, T.; Takeuchi, M.; Yoshida, K.; Iwade, K.; Okuyama, Y.; et al. Effects of vitamin K antagonist on aortic valve degeneration in non-valvular atrial fibrillation patients: Prospective 4-year observational study. Thromb. Res. 2017, 160, 69–75. [Google Scholar] [CrossRef]
- Rattazzi, M.; Faggin, E.; Bertacco, E.; Nardin, C.; Pagliani, L.; Plebani, M.; Cinetto, F.; Guidolin, D.; Puato, M.; Pauletto, P. Warfarin, but not rivaroxaban, promotes the calcification of the aortic valve in ApoE−/− mice. Cardiovasc. Ther. 2018, 36, e12438. [Google Scholar] [CrossRef]
- Pan, M.H.; Maresz, K.; Lee, P.S.; Wu, J.C.; Ho, C.T.; Popko, J.; Mehta, D.S.; Stohs, S.J.; Badmaev, V. Inhibition of TNF-α, IL-1α, and IL-1β by Pretreatment of Human Monocyte-Derived Macrophages with Menaquinone-7 and Cell Activation with TLR Agonists in Vitro. J. Med. Food 2016, 19, 663–669. [Google Scholar] [CrossRef]
- Ohsaki, Y.; Shirakawa, H.; Hiwatashi, K.; Furukawa, Y.; Mizutani, T.; Komai, M. Vitamin K suppresses lipopolysaccharide-induced inflammation in the rat. Biosci. Biotechnol. Biochem. 2006, 70, 926–932. [Google Scholar] [CrossRef] [Green Version]
- Shea, M.K.; Cushman, M.; Booth, S.L.; Burke, G.L.; Chen, H.; Kritchevsky, S.B. Associations between vitamin k status and haemostatic and inflammatory biomarkers in community-dwelling adults: The multi-ethnic study of atherosclerosis. Thromb. Haemost. 2014, 112, 438–444. [Google Scholar] [CrossRef] [Green Version]
- Lupo, M.G.; Biancorosso, N.; Brilli, E.; Tarantino, G.; Adorni, M.P.; Vivian, G.; Salvalaio, M.; Dall’Acqua, S.; Sut, S.; Neutel, C.; et al. Cholesterol-lowering action of a novel nutraceutical combination in uremic rats: Insights into the molecular mechanism in a hepatoma cell line. Nutrients 2020, 12, 436. [Google Scholar] [CrossRef] [Green Version]
- Zwakenberg, S.R.; De Jong, P.A.; Bartstra, J.W.; van Asperen, R.; Westerink, J.; de Valk, H.; Slart, R.H.J.A.; Luurtsema, G.; Wolterink, J.M.; de Borst, G.J.; et al. The effect of menaquinone-7 supplementation on vascular calcification in patients with diabetes: A randomized, double-blind, placebo-controlled trial. Am. J. Clin. Nutr. 2019, 110, 883–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurnatowska, I.; Grzelak, P.; Masajtis-Zagajewska, A.; Kaczmarska, M.; Stefańczyk, L.; Vermeer, C.; Maresz, K.; Nowicki, M. Effect of Vitamin K2 on progression of atherosclerosis and vascular calcification in nondialyzed patients with chronic kidney disease stages 3-5. Pol. Arch. Med. Wewn. 2015, 125, 631–640. [Google Scholar] [CrossRef]
- Shea, M.K.; O’Donnell, C.J.; Hoffmann, U.; Dallal, G.E.; Dawson-Hughes, B.; Ordovas, J.M.; Price, P.A.; Williamson, M.K.; Booth, S.L. Vitamin K supplementation and progression of coronary artery calcium in older men and women. Am. J. Clin. Nutr. 2009, 89, 1799–1807. [Google Scholar] [CrossRef]
- Geleijnse, J.M.; Vermeer, C.; Grobbee, D.E.; Schurgers, L.; Knapen, M.H.; van der Meer, I.M.; Hofman, A.; Witteman, J.C. Dietary Intake of Menaquinone Is Associated with a Reduced Risk of Coronary Heart Disease: The Rotterdam Study. J. Nutr. 2004, 134, 3100–3105. [Google Scholar] [CrossRef] [PubMed]
- Gast, G.C.M.; de Roos, N.M.; Sluijs, I.; Bots, M.L.; Beulens, J.W.; Geleijnse, J.M.; Witteman, J.C.; Grobbee, D.E.; Peeters, P.H.; van der Schouw, Y.T. A high menaquinone intake reduces the incidence of coronary heart disease. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 504–510. [Google Scholar] [CrossRef]
- Holden, R.M.; Booth, S.L.; Day, A.G.; Clase, C.M.; Zimmerman, D.; Moist, L.; Shea, M.K.; McCabe, K.M.; Jamal, S.A.; Tobe, S.; et al. Inhibiting the progression of arterial calcification with vitamin K in HemoDialysis patients (iPACK-HD) trial: Rationale and study design for a randomized trial of vitamin K in patients with end stage kidney disease. Can. J. Kidney Health Dis. 2015, 2, 53. [Google Scholar] [CrossRef] [Green Version]
- Krueger, T.; Schlieper, G.; Schurgers, L.; Cornelis, T.; Cozzolino, M.; Jacobi, J.; Jadoul, M.; Ketteler, M.; Rump, L.C.; Stenvinkel, P.; et al. Vitamin K1 to slow vascular calcification in haemodialysis patients (VitaVasK trial): A rationale and study protocol. Nephrol. Dial. Transplant. 2014, 29, 1633–1638. [Google Scholar] [CrossRef] [PubMed]
- Vossen, L.M.; Schurgers, L.J.; van Varik, B.J.; Kietselaer, B.L.; Vermeer, C.; Meeder, J.G.; Rahel, B.M.; van Cauteren, Y.J.; Hoffland, G.A.; Rennenberg, R.J.; et al. Menaquinone-7 supplementation to reduce vascular calcification in patients with coronary artery disease: Rationale and study protocol (VitaK-CAC Trial). Nutrients 2015, 7, 8905–8915. [Google Scholar] [CrossRef] [Green Version]
- Brandenburg, V.M.; Reinartz, S.; Kaesler, N.; Krüger, T.; Dirrichs, T.; Kramann, R.; Peeters, F.; Floege, J.; Keszei, A.; Marx, N.; et al. Slower progress of aortic valve calcification with Vitamin K supplementation: Results from a prospective interventional proof-of-concept study. Circulation 2017, 135, 2081–2083. [Google Scholar] [CrossRef]
- Lindholt, J.S.; Frandsen, N.E.; Fredgart, M.H.; Øvrehus, K.A.; Dahl, J.S.; Møller, J.E.; Folkestad, L.; Urbonaviciene, G.; Becker, S.W.; Lambrechtsen, J.; et al. Effects of menaquinone-7 supplementation in patients with aortic valve calcification: Study protocol for a randomised controlled trial. BMJ Open 2018, 8, e22019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peeters, F.E.C.M.; Van Mourik, M.J.W.; Meex, S.J.R.; Bucerius, J.; Schalla, S.M.; Gerretsen, S.C.; Mihl, C.; Dweck, M.R.; Schurgers, L.J.; Wildberger, J.E.; et al. Bicuspid aortic valve stenosis and the effect of vitamin K2 on calcification using 18F-sodium fluoride positron emission tomography/magnetic resonance: The BASIK2 rationale and trial design. Nutrients 2018, 10, 386. [Google Scholar] [CrossRef] [Green Version]
- Perrucci, G.L.; Songia, P.; Moschetta, D.; Barbagallo, V.A.; Valerio, V.; Myasoedova, V.A.; Alfieri, V.; Massaiu, I.; Roberto, M.; Malešević, M.; et al. Cyclophilin A inhibition as potential treatment of human aortic valve calcification. Pharmacol. Res. 2020, 158, 104888. [Google Scholar] [CrossRef]
- Chen, Z.; Gordillo-Martinez, F.; Jiang, L.; He, P.; Hong, W.; Wei, X.; Staines, K.A.; Macrae, V.E.; Zhang, C.; Yu, D.; et al. Zinc ameliorates human aortic valve calcification through GPR39 mediated ERK1/2 signalling pathway. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef]
- Artiach, G.; Carracedo, M.; Seime, T.; Plunde, O.; Laguna-Fernandez, A.; Matic, L.; Franco-Cereceda, A.; Bäck, M. Proteoglycan 4 is Increased in Human Calcified Aortic Valves and Enhances Valvular Interstitial Cell Calcification. Cells 2020, 9, 684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Wang, L.; Pan, Y.; Wang, X.; Ding, Y.; Zhou, C.; Shah, A.M.; Zhao, G.; Zhang, M. Celastrol Alleviates Aortic Valve Calcification Via Inhibition of NADPH Oxidase 2 in Valvular Interstitial Cells. JACC Basic Transl. Sci. 2020, 5, 35–49. [Google Scholar] [CrossRef]
- Côté, N.; El Husseini, D.; Pépin, A.; Bouvet, C.; Gilbert, L.A.; Audet, A.; Fournier, D.; Pibarot, P.; Moreau, P.; Mathieu, P. Inhibition of ectonucleotidase with ARL67156 prevents the development of calcific aortic valve disease in warfarin-treated rats. Eur. J. Pharmacol. 2012, 689, 139–146. [Google Scholar] [CrossRef]
- Bouchareb, R.; Côté, N.; Boulanger, M.C.; Le Quang, K.; El Husseini, D.; Asselin, J.; Hadji, F.; Lachance, D.; Shayhidin, E.E.; Mahmut, A.; et al. Carbonic anhydrase XII in valve interstitial cells promotes the regression of calcific aortic valve stenosis. J. Mol. Cell Cardiol. 2015, 82, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.; Lee, S.; Kim, S.M.; Lee, E.J.; Lee, S.R.; Kim, D.H.; Jang, J.Y.; Kang, S.W.; Lee, K.U.; Chang, E.J.; et al. Dipeptidyl peptidase-4 induces aortic valve calcifcation by inhibiting insulin-like growth factor-1 signaling in valvular interstitial cells. Circulation 2017, 135, 1935–1950. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.R.; Bowler, M.A.; Snider, J.C.; David Merryman, W. Targeting Cadherin-11 Prevents Notch1-Mediated Calcific Aortic Valve Disease. Circulation 2017, 135, 2448–2450. [Google Scholar] [CrossRef]
- Li, F.; Cai, Z.; Chen, F.; Shi, X.; Zhang, Q.; Chen, S.; Shi, J.; Wang, D.W.; Dong, N. Pioglitazone attenuates progression of aortic valve calcification via down-regulating receptor for advanced glycation end products. Basic Res. Cardiol. 2012, 107. [Google Scholar] [CrossRef]
- Chu, Y.; Lund, D.D.; Weiss, R.M.; Brooks, R.M.; Doshi, H.; Hajj, G.P.; Sigmund, C.D.; Heistad, D.D. Pioglitazone attenuates valvular calcification induced by hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.R.; Dai, Y.; Zhao, J.; Lin, L.; Wang, Y.; Wang, Y. A mechanistic overview of triptolide and celastrol, natural products from Tripterygium wilfordii Hook F. Front. Pharmacol. 2018, 9, 104. [Google Scholar] [CrossRef] [Green Version]
- Cascão, R.; Fonseca, J.E.; Moita, L.F. Celastrol: A spectrum of treatment opportunities in chronic diseases. Front. Med. 2017, 4, 69. [Google Scholar] [CrossRef] [Green Version]
- Yeang, C.; Tsimikas, S. Ancient Remedy for a Modern Disease: Will Celastrol Become a Treatment for Aortic Valve Stenosis? JACC Basic Transl. Sci. 2020, 5, 50–52. [Google Scholar] [CrossRef] [PubMed]
- Rattazzi, M.; Bertacco, E.; Iop, L.; D’Andrea, S.; Puato, M.; Buso, G.; Causin, V.; Gerosa, G.; Faggin, E.; Pauletto, P. Extracellular pyrophosphate is reduced in aortic interstitial valve cells acquiring a calcifying profile: Implications for aortic valve calcification. Atherosclerosis 2014, 237, 568–576. [Google Scholar] [CrossRef]
- Villa-Bellosta, R. New insights into endogenous mechanisms of protection against arterial calcification. Atherosclerosis 2020, 306, 68–74. [Google Scholar] [CrossRef] [Green Version]
- El Husseini, D.; Boulanger, M.C.; Mahmut, A.; Bouchareb, R.; Laflamme, M.H.; Fournier, D.; Pibarot, P.; Bossé, Y.; Mathieu, P. P2Y2 receptor represses IL-6 expression by valve interstitial cells through Akt: Implication for calcific aortic valve disease. J. Mol. Cell Cardiol. 2014, 72, 146–156. [Google Scholar] [CrossRef]
- Mulvihill, E.E.; Drucker, D.J. Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr. Rev. 2014, 35, 992–1019. [Google Scholar] [CrossRef] [Green Version]
- Shah, Z.; Kampfrath, T.; Deiuliis, J.A.; Zhong, J.; Pineda, C.; Ying, Z.; Xu, X.; Lu, B.; Moffatt-Bruce, S.; Durairaj, R.; et al. Long-term dipeptidyl-peptidase 4 inhibition reduces atherosclerosis and inflammation via effects on monocyte recruitment and chemotaxis. Circulation 2011, 124, 2338–2349. [Google Scholar] [CrossRef] [Green Version]
- Varennes, O.; Mary, A.; Bricca, G.; Kamel, S.; Bellien, J. Dipeptidyl Peptidase-4 Inhibition Prevents Vascular Calcification by Potentiating the Insulin-Like Growth Factor-1 Signaling Pathway. JACC Basic Transl. Sci. 2019, 4, 113–115. [Google Scholar] [CrossRef]
- Radcliff, K.; Tang, T.B.; Lim, J.; Zhang, Z.; Abedin, M.; Demer, L.L.; Tintut, Y. Insulin-like growth factor-I regulates proliferation and osteoblastic differentiation of calcifying vascular cells via extracellular signal-regulated protein kinase and phosphatidylinositol 3-kinase pathways. Circ. Res. 2005, 96, 398–400. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Bowen, C.; Lu, G.; Knapp, C.; Recknagel, A.; Norris, R.A.; Butcher, J.T. Cadherin-11 expression patterns in heart valves associate with key functions during embryonic cushion formation, valve maturation and calcification. Cells Tissues Organs. 2014, 198, 300–310. [Google Scholar] [CrossRef] [Green Version]
- Hutcheson, J.D.; Chen, J.; Sewell-Loftin, M.K.; Ryzhova, L.M.; Fisher, C.I.; Su, Y.R.; Merryman, W.D. Cadherin-11 regulates cell-cell tension necessary for calcific nodule formation by valvular myofibroblasts. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 114–120. [Google Scholar] [CrossRef] [Green Version]
- Sung, D.C.; Bowen, C.J.; Vaidya, K.A.; Zhou, J.; Chapurin, N.; Recknagel, A.; Zhou, B.; Chen, J.; Kotlikoff, M.; Butcher, J.T. Cadherin-11 Overexpression Induces Extracellular Matrix Remodeling and Calcification in Mature Aortic Valves. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1627–1637. [Google Scholar] [CrossRef] [Green Version]
- Bowen, C.J.; Zhou, J.; Sung, D.C.; Butcher, J.T. Cadherin-11 coordinates cellular migration and extracellular matrix remodeling during aortic valve maturation. Dev. Biol. 2015, 407, 145–157. [Google Scholar] [CrossRef] [Green Version]
- Schroer, A.K.; Bersi, M.R.; Clark, C.R.; Zhang, Q.; Sanders, L.H.; Hatzopoulos, A.K.; Force, T.L.; Majka, S.M.; Lal, H.; Merryman, W.D. Cadherin-11 blockade reduces inflammation-driven fibrotic remodeling and improves outcomes after myocardial infarction. JCI Insight 2019, 4, e131545. [Google Scholar] [CrossRef]
- Liu, L.; Liu, Y.; Zhang, Y.; Bi, X.; Nie, L.; Liu, C.; Xiong, J.; He, T.; Xu, X.; Yu, Y.; et al. High phosphate-induced downregulation of PPARγ contributes to CKD-associated vascular calcification. J. Mol. Cell Cardiol. 2018, 114, 264–275. [Google Scholar] [CrossRef]
- Ivanova, E.A.; Parolari, A.; Myasoedova, V.; Melnichenko, A.A.; Bobryshev, Y.V.; Orekhov, A.N. Peroxisome proliferator-activated receptor (PPAR) gamma in cardiovascular disorders and cardiovascular surgery. J. Cardiol. 2015, 66, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Chen, T.; Wu, L.; Zhao, X.; Mao, H.; Xing, C. Effect of pioglitazone on the calcification of rat vascular smooth muscle cells through the downregulation of the Wnt/β-catenin signaling pathway. Mol. Med. Rep. 2017, 16, 6208–6213. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.V.; Inzucchi, S.E.; Echouffo-Tcheugui, J.B.; Tang, F.; Lam, C.S.P.; Sperling, L.S.; Kosiborod, M. Understanding contemporary use of thiazolidinediones an analysis from the diabetes collaborative registry. Circ. Heart Fail. 2019, 12, e005855. [Google Scholar] [CrossRef]
- Summerhill, V.I.; Moschetta, D.; Orekhov, A.N.; Poggio, P.; Myasoedova, V.A. Sex-specific features of calcific aortic valve disease. Int. J. Mol. Sci. 2020, 21, 5620. [Google Scholar] [CrossRef]
- Myasoedova, V.A.; Di Minno, A.; Songia, P.; Massaiu, I.; Alfieri, V.; Valerio, V.; Moschetta, D.; Andreini, D.; Alamanni, F.; Pepi, M.; et al. Sex-specific differences in age-related aortic valve calcium load: A systematic review and meta-analysis. Ageing Res. Rev. 2020, 61, 101077. [Google Scholar] [CrossRef] [PubMed]
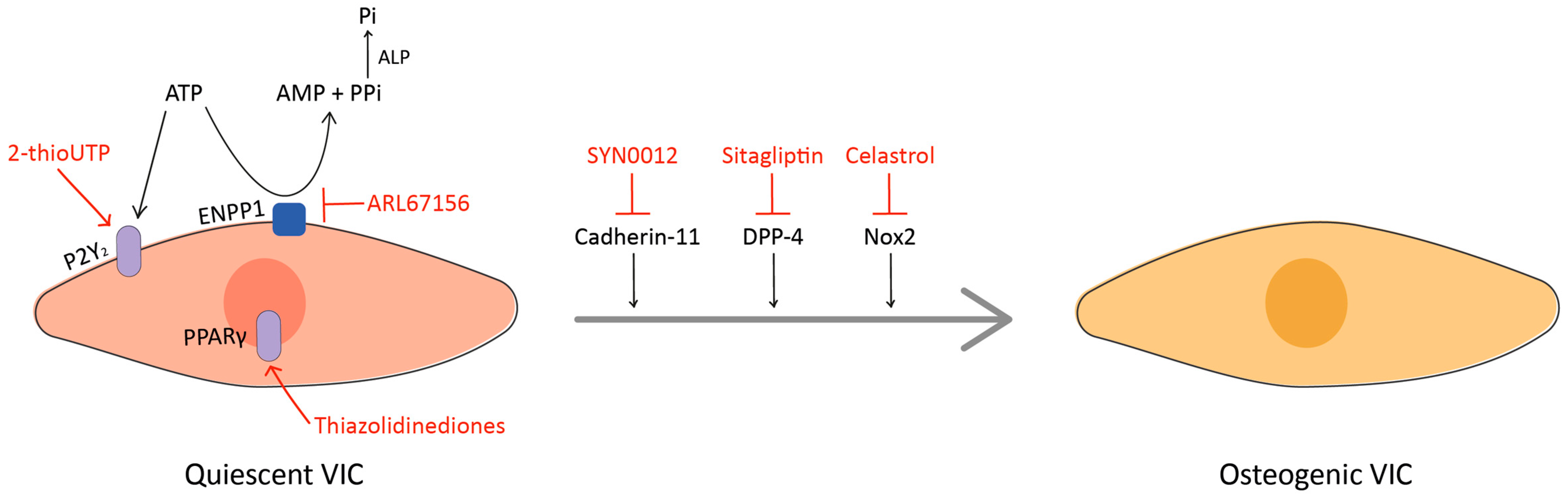

{kind=link}
{kind=link}
{kind=link}
| Trial Name | NCT | Treatment | Phase | Population | Enrolment (No. of Patients) | Primary Outcome |
|---|---|---|---|---|---|---|
| EAVaLL | NCT02109614 | Extended-release niacin vs. placebo | I | Aortic sclerosis or mild CAVS | 238 | Calcium score progression measured by cardiac CT at 2 years |
| NCT03051360 | PCSK9 inhibitor vs. placebo | II | Mild to moderate CAVS | 140 | Calcium score progression measured by cardiac CT and by NaF PET at 2 years | |
| NCT02481258 | Ataciguat vs. placebo | II | Moderate CAVS | 35 | Changes in AV calcium levels at 6 months | |
| SALTIRE II | NCT02132026 | Alendronate/denosumab vs. placebo | II | Peak aortic jet velocity >2.5 m/s and grade 2–4 calcification of the AV on echocardiography | 150 | Change in AV calcium score at 6 months and 2 years |
| AVADEC | NCT03243890 | Menaquinone-7 vs. placebo | - | AV calcification score above 300, but without clinical CAVS | 389 | Change in AV calcification at 2 years |
| BASIK2 | NCT02917525 | Vitamin K2 vs. placebo | II | Bicuspid AV and mild to moderate CAVS on prior echocardiography | 44 | Change in AV calcium metabolism measured by NaF PET at 6 months |
| Target | Treatment | Effects | Model | Ref. |
|---|---|---|---|---|
| Nox2 | Celastrol | Nox2 inhibition mitigates the severity of aortic valve fibrosis, calcification, and stenosis | rabbit | [190] |
| ENPP1 | ARL67156 | The inhibition of ENPP1 prevents the development of CAVS | rat | [191] |
| P2Y2 | 2-thioUTP | P2Y2 agonist promotes the regression of CAVS | mouse | [192] |
| DPP-4 | Sitagliptin | DPP-4 inhibition prevents CAVS development | rabbit | [193] |
| Cadherin-11 | SYN0012 | Cad-11-blocking antibody prevents Notch1-mediated CAVS | mouse | [194] |
| PPARγ | Pioglitazone | PPARγ agonist attenuates the progression of aortic valve calcification | rabbit | [195] |
| PPARγ agonist attenuates lipid deposition, calcification, and apoptosis in aortic valves | mouse | [196] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donato, M.; Ferri, N.; Lupo, M.G.; Faggin, E.; Rattazzi, M. Current Evidence and Future Perspectives on Pharmacological Treatment of Calcific Aortic Valve Stenosis. Int. J. Mol. Sci. 2020, 21, 8263. https://doi.org/10.3390/ijms21218263
Donato M, Ferri N, Lupo MG, Faggin E, Rattazzi M. Current Evidence and Future Perspectives on Pharmacological Treatment of Calcific Aortic Valve Stenosis. International Journal of Molecular Sciences. 2020; 21(21):8263. https://doi.org/10.3390/ijms21218263
Chicago/Turabian StyleDonato, Maristella, Nicola Ferri, Maria Giovanna Lupo, Elisabetta Faggin, and Marcello Rattazzi. 2020. "Current Evidence and Future Perspectives on Pharmacological Treatment of Calcific Aortic Valve Stenosis" International Journal of Molecular Sciences 21, no. 21: 8263. https://doi.org/10.3390/ijms21218263
APA StyleDonato, M., Ferri, N., Lupo, M. G., Faggin, E., & Rattazzi, M. (2020). Current Evidence and Future Perspectives on Pharmacological Treatment of Calcific Aortic Valve Stenosis. International Journal of Molecular Sciences, 21(21), 8263. https://doi.org/10.3390/ijms21218263