Antigen Design for Successful Isolation of Highly Challenging Therapeutic Anti-GPCR Antibodies


Abstract
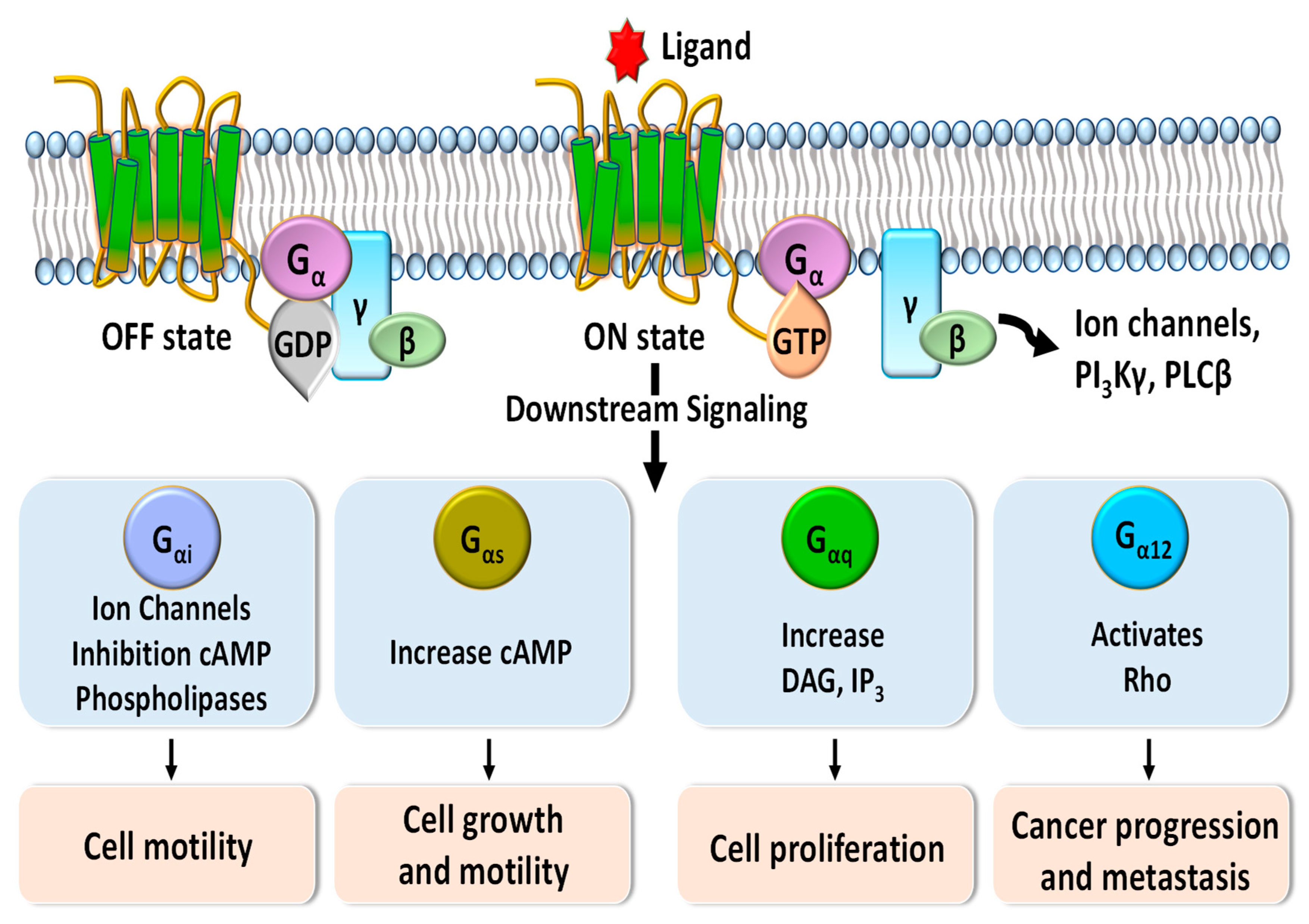
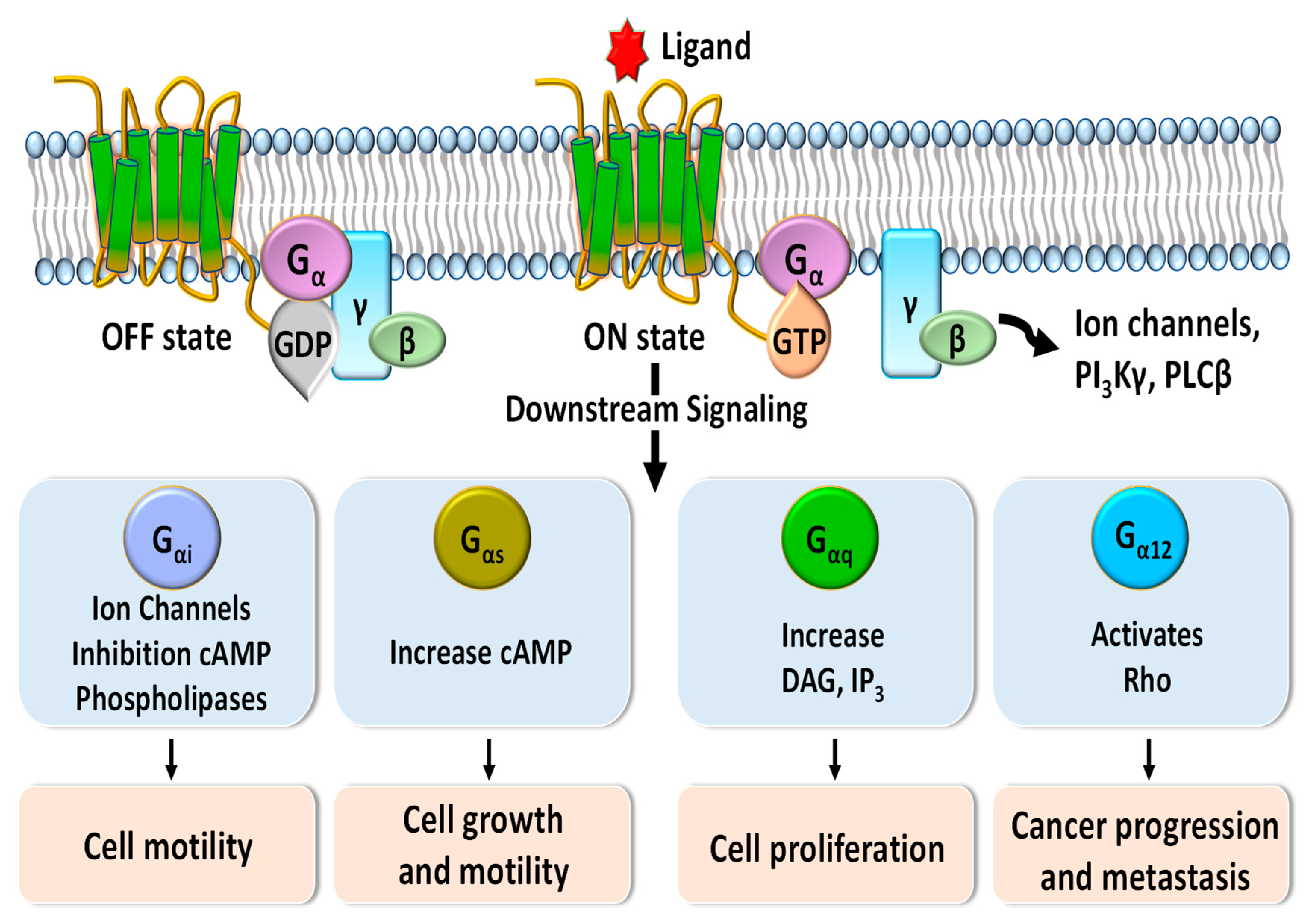
:1. Introduction
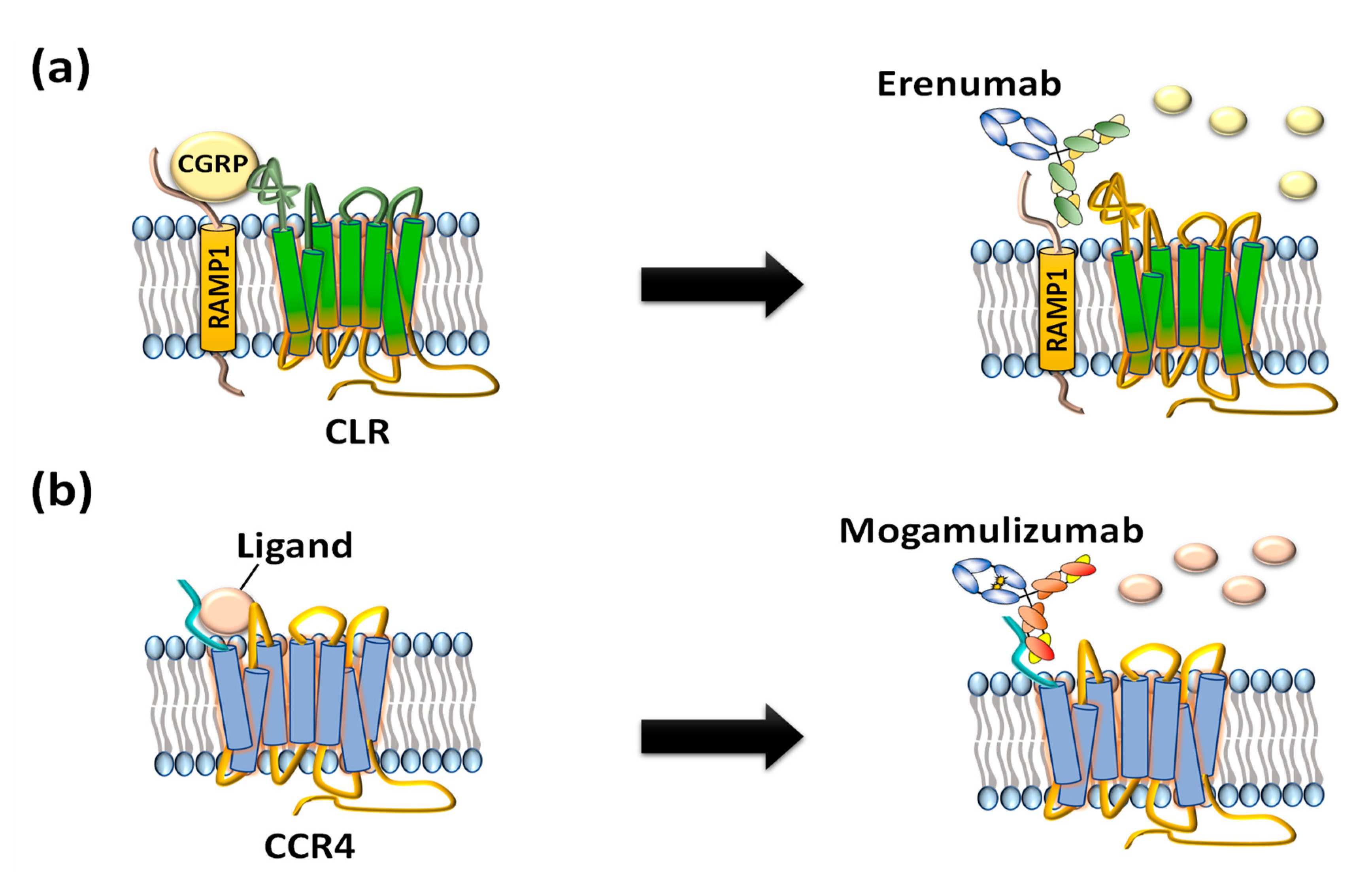
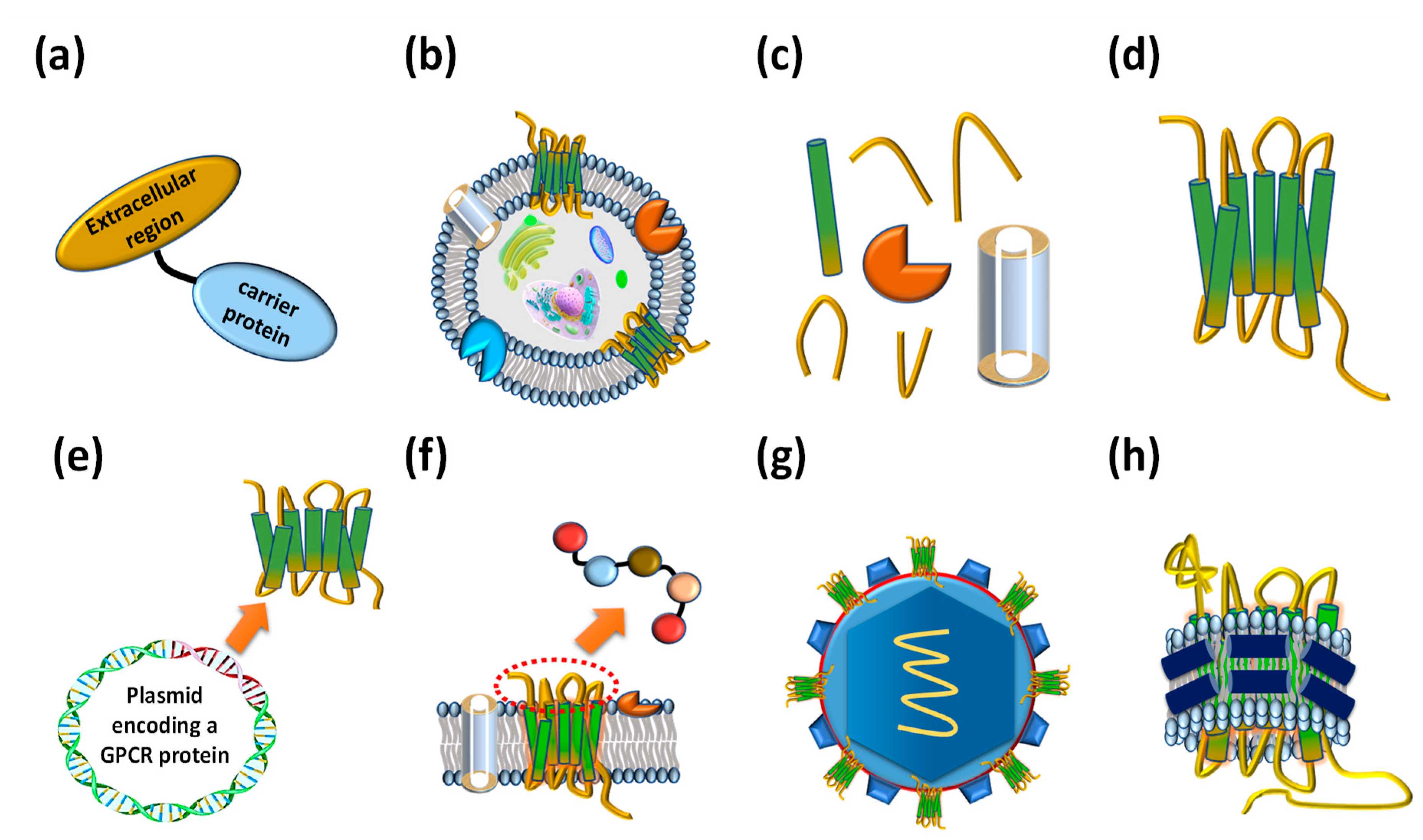
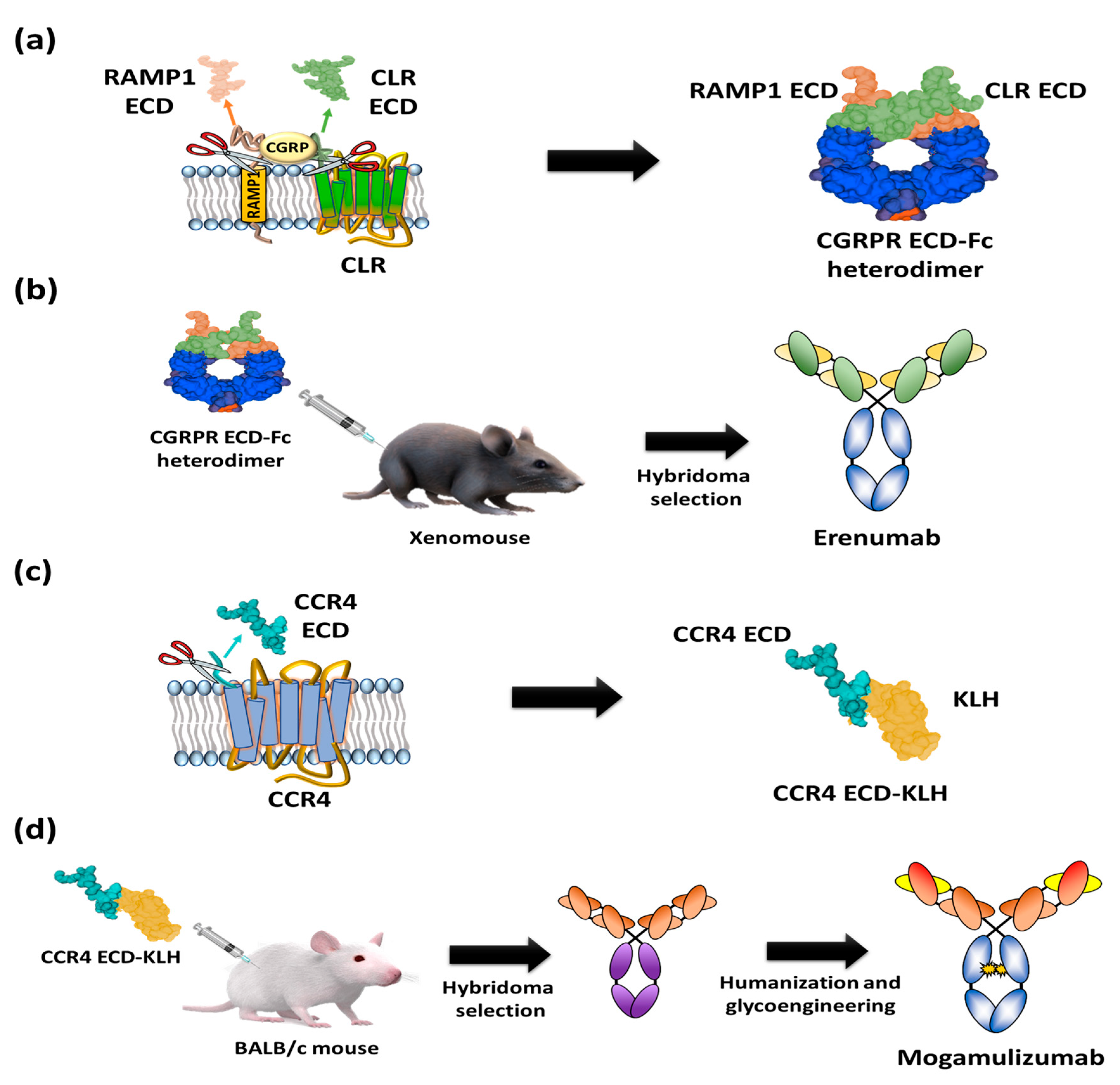
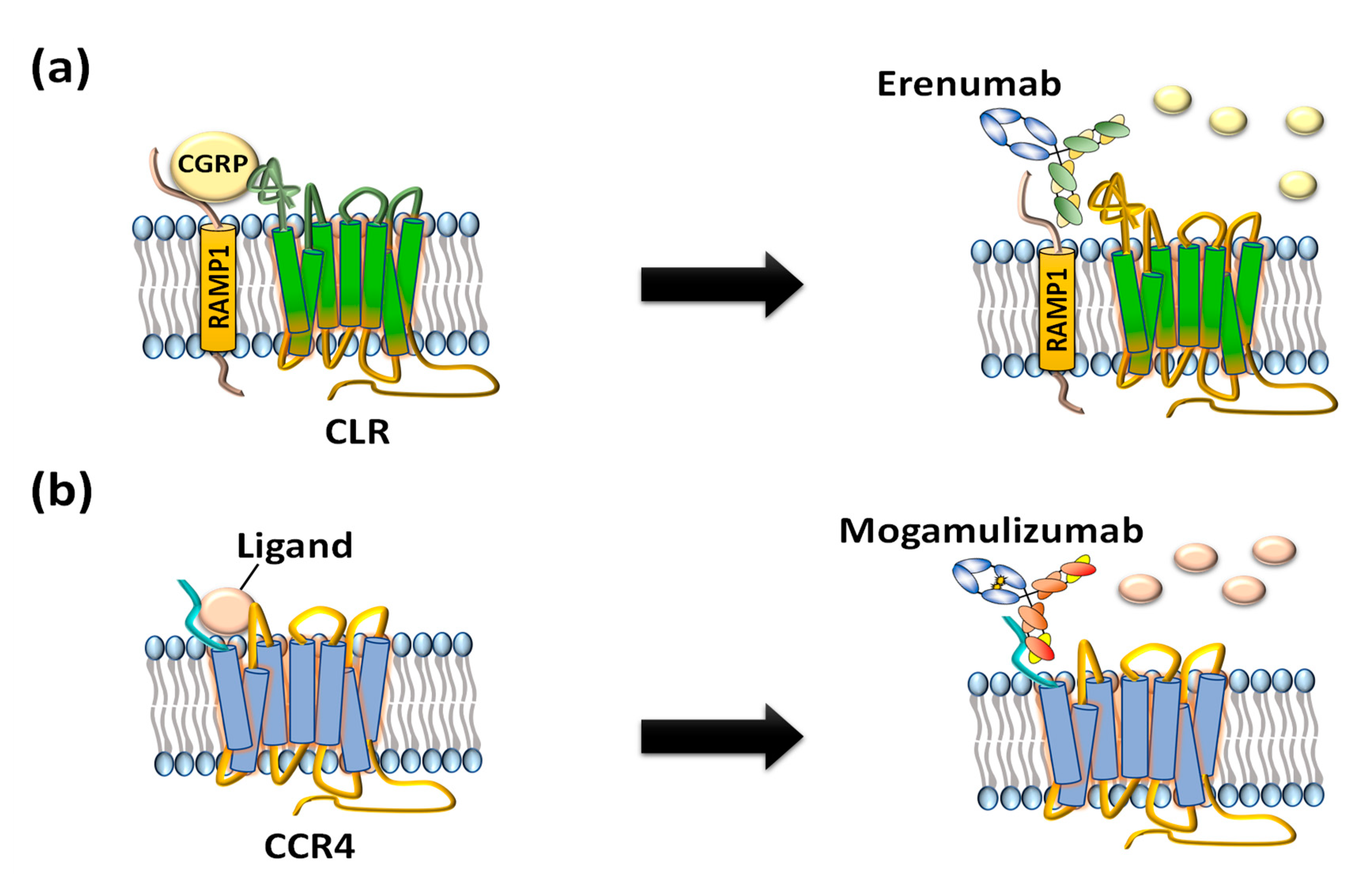
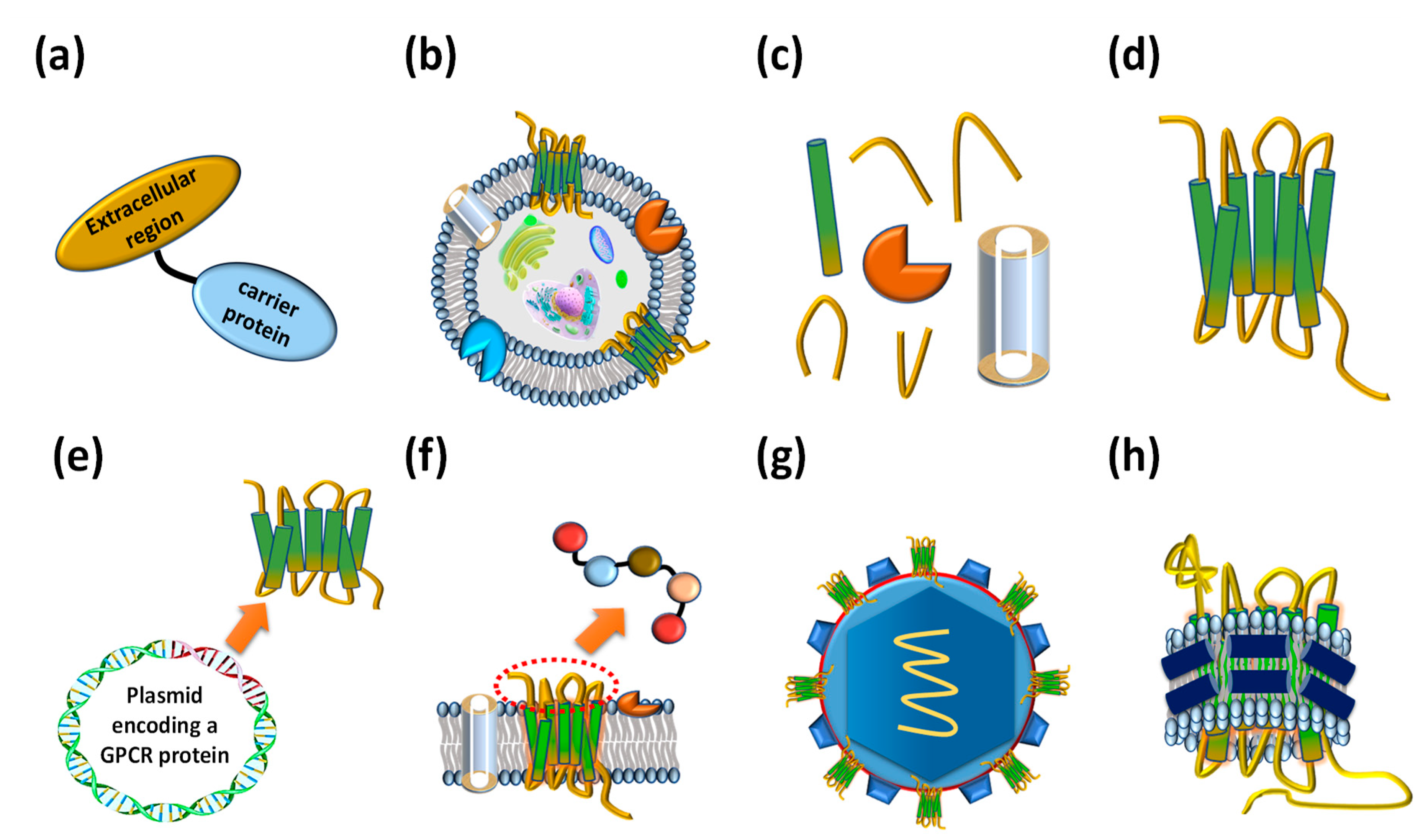
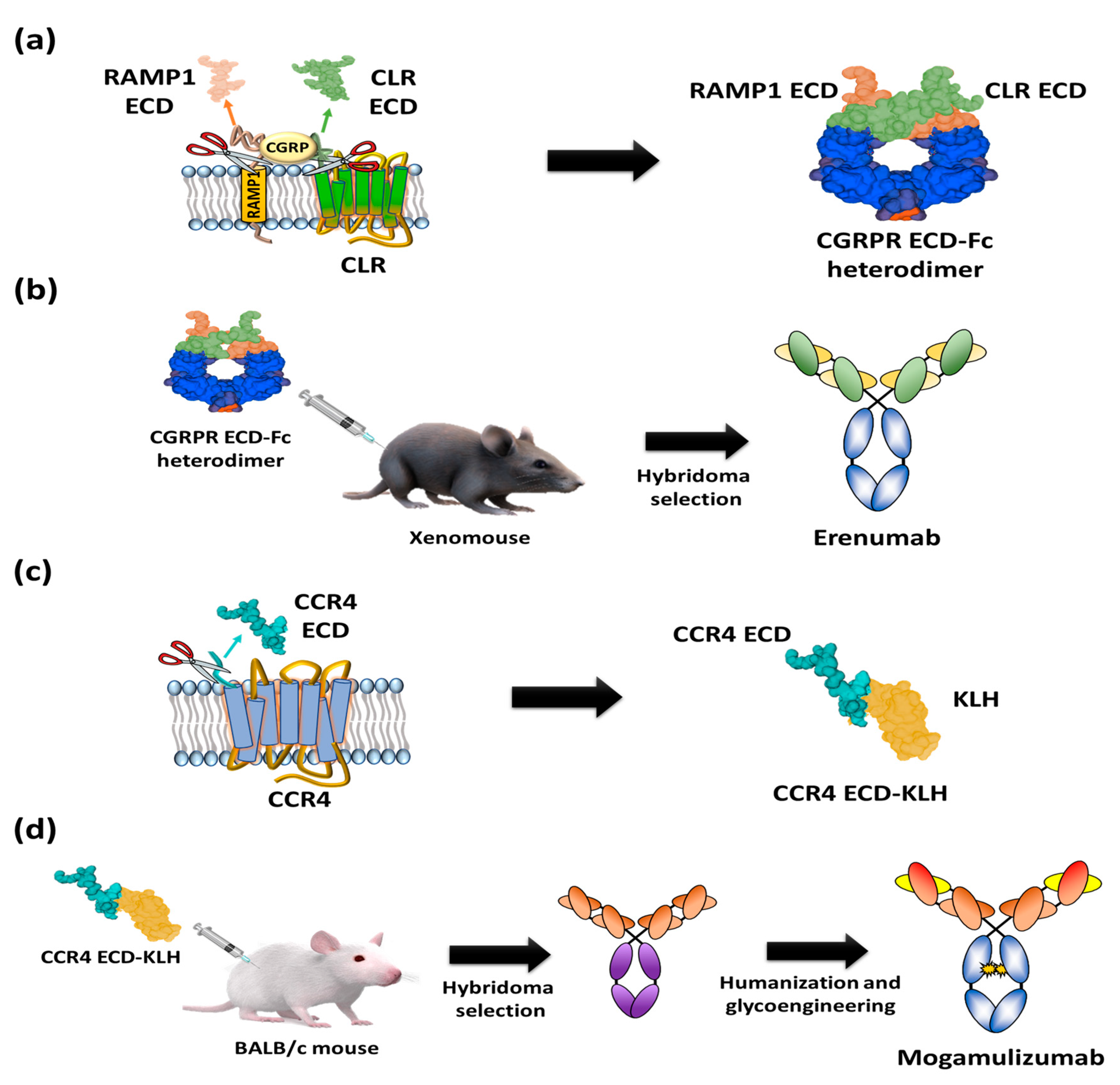
2. GPCR Extracellular Region Fusion Proteins as GPCR Antigens
3. GPCR-Expressing Cells or Membrane Fractions as GPCR Antigens
4. Purified Whole GPCR Proteins as GPCR Antigens
5. Other Types of Prepared GPCR Antigens
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wu, J.; Xie, N.; Zhao, X.; Nice, E.C.; Huang, C. Dissection of aberrant GPCR signaling in tumorigenesis—A systems biology approach. Cancer Genom Proteom. 2012, 9, 37–50. [Google Scholar]
- New, D.C.; Wong, Y.H. Molecular mechanisms mediating the G protein-coupled receptor regulation of cell cycle progression. J. Mol. Signal. 2007, 2, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zougman, A.; Hutchins, G.G.; Cairns, D.A.; Verghese, E.; Perry, S.L.; Jayne, D.G.; Selby, P.J.; Banks, R.E. Retinoic acid-induced protein 3: Identification and characterisation of a novel prognostic colon cancer biomarker. Eur. J. Cancer. 2013, 49, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Moyung, K.; Corriden, R.; Carter, H.; Insel, P.A. GPCRs show widespread differential mRNA expression and frequent mutation and copy number variation in solid tumors. PLoS Biol. 2019, 17, e3000434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feigin, M.E.; Xue, B.; Hammell, M.C.; Muthuswamy, S.K. G-protein–coupled receptor GPR161 is overexpressed in breast cancer and is a promoter of cell proliferation and invasion. Proc. Natl. Acad. Sci. USA 2014, 111, 4191–4196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef]
- Rask-Andersen, M.; Masuram, S.; Schioth, H.B. The druggable genome: Evaluation of drug targets in clinical trials suggests major shifts in molecular class and indication. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schioth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Chavali, S.; Masuho, I.; Jahn, L.J.; Martemyanov, K.A.; Gloriam, D.E.; Babu, M.M. Pharmacogenomics of GPCR Drug Targets. Cell 2018, 172, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Igawa, T.; Tsunoda, H.; Kuramochi, T.; Sampei, Z.; Ishii, S.; Hattori, K. Engineering the variable region of therapeutic IgG antibodies. In MAbs; Taylor & Francis: Abingdon, UK, 2011; Volume 3, pp. 243–252. [Google Scholar] [CrossRef] [Green Version]
- Dolgin, E. First GPCR-directed antibody passes approval milestone. Nat. Rev. Drug Discov. 2018, 17, 457–459. [Google Scholar] [CrossRef]
- Kasamon, Y.L.; Chen, H.; de Claro, R.A.; Nie, L.; Ye, J.; Blumenthal, G.M.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Mogamulizumab-kpkc for Mycosis Fungoides and Sezary Syndrome. Clin. Cancer Res. 2019, 25, 7275–7280. [Google Scholar] [CrossRef] [Green Version]
- Hoogenboom, H.R. Selecting and screening recombinant antibody libraries. Nat. Biotechnol. 2005, 23, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Amgen Inc. Erenumab (Aimovig™): US Prescribing Information. 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761077s000lbl.pdf (accessed on 13 October 2020).
- Kyowa Kirin Inc. Mogamulizumab (POTELIGEO®): US Prescribing Information. 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761051s000lbl.pdf (accessed on 13 October 2020).
- OncoMed Pharmaceuticals Inc. A Study of Vantictumab (OMP-18R5) in Combination with Docetaxel in Patients with Previously Treated NSCLC. 2013. Available online: https://clinicaltrials.gov/ct2/show/NCT01957007 (accessed on 13 October 2020).
- OncoMed Pharmaceuticals Inc. A Study of Vantictumab (OMP-18R5) in Combination with Paclitaxel in Locally Recurrent or Metastatic Breast Cancer. 2013. Available online: https://clinicaltrials.gov/ct2/show/NCT01973309 (accessed on 13 October 2020).
- OncoMed Pharmaceuticals Inc. A Study of Vantictumab (OMP-18R5) in Combination with Nab-Paclitaxel and Gemcitabine in Previously Untreated Stage IV Pancreatic Cancer. 2013. Available online: https://clinicaltrials.gov/ct2/show/NCT02005315 (accessed on 13 October 2020).
- CytoDyn Inc. PRO 140 in Treatment-Experienced HIV-1 Subjects. 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT03902522 (accessed on 13 October 2020).
- Millennium Pharmaceuticals Inc. Study of the Safety and Efficacy of MLN1202 in Patients in Multiple Sclerosis. 2010. Available online: https://clinicaltrials.gov/ct2/show/NCT01199640?cond=plozalizumab&draw=2&rank=3 (accessed on 13 October 2020).
- Bristol-Myers Squibb. An Investigational Immuno-therapy Study of Ulocuplumab in Combination with Low Dose Cytarabine in Patients with Newly Diagnosed Acute Myeloid Leukemia. 2014. Available online: https://clinicaltrials.gov/ct2/show/NCT02305563?cond=Ulocuplumab&draw=2&rank=4 (accessed on 13 October 2020).
- Innate Pharma. IPH5401 (Anti-C5aR) in Combination with Durvalumab in Patients with Advanced Solid Tumors (STELLAR-001). 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03665129 (accessed on 13 October 2020).
- Gmax Biopharm LLC. First Patient Dose of Glutazumab (GMA102) in the Phase II Clinical Trial for Treatment of Type 2 Diabetes. 2017. Available online: http://www.gmaxbiopharm.com/newng2_detail/id/15.html (accessed on 13 October 2020).
- REMD Biotherapeutics Inc. Multiple Dose Study to Evaluate the Efficacy, Safety and Pharmacodynamics of REMD-477 in Subjects with Type 1 Diabetes Mellitus. 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03117998 (accessed on 13 October 2020).
- Bird Rock Bio Inc. Safety tolerability, and PK of RYI-018 after Repeat Dosing in Subjects with Non-Alcoholic Fatty Liver Disease (NAFLD). 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03261739 (accessed on 13 October 2020).
- Lefkowitz, R.J.; Shenoy, S.K. Transduction of receptor signals by beta-arrestins. Science 2005, 308, 512–517. [Google Scholar] [CrossRef]
- Webb, D.R.; Handel, T.M.; Kretz-Rommel, A.; Stevens, R.C. Opportunities for functional selectivity in GPCR antibodies. Biochem. Pharmacol. 2013, 85, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Soto, A.G.; Smith, T.H.; Chen, B.; Bhattacharya, S.; Cordova, I.C.; Kenakin, T.; Vaidehi, N.; Trejo, J. N-linked glycosylation of protease-activated receptor-1 at extracellular loop 2 regulates G-protein signaling bias. Proc. Natl. Acad. Sci. USA 2015, 112, E3600–E3608. [Google Scholar] [CrossRef] [Green Version]
- Markham, A. Erenumab: First Global Approval. Drugs 2018, 78, 1157–1161. [Google Scholar] [CrossRef]
- Subramaniam, J.M.; Whiteside, G.; McKeage, K.; Croxtall, J.C. Mogamulizumab: First global approval. Drugs 2012, 72, 1293–1298. [Google Scholar] [CrossRef]
- Smith, D.C.; Rosen, L.S.; Chugh, R.; Goldman, J.W.; Xu, L.; Kapoun, A.; Brachmann, R.K.; Dupont, J.; Stagg, R.J.; Tolcher, A.W.; et al. First-in-human evaluation of the human monoclonal antibody vantictumab (OMP-18R5; anti-Frizzled) targeting the WNT pathway in a phase I study for patients with advanced solid tumors. J. Clin. Oncol. 2013, 31, 2540. [Google Scholar] [CrossRef]
- Brain, S.D.; Grant, A.D. Vascular actions of calcitonin gene-related peptide and adrenomedullin. Physiol. Rev. 2004, 84, 903–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, D.L.; Pioszak, A.A. Receptor Activity-Modifying Proteins (RAMPs): New Insights and Roles. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 469–487. [Google Scholar] [CrossRef] [Green Version]
- Kee, Z.; Kodji, X.; Brain, S.D. The Role of Calcitonin Gene Related Peptide (CGRP) in Neurogenic Vasodilation and Its Cardioprotective Effects. Front. Physiol. 2018, 9, 1249. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.L.; Khoshouei, M.; Deganutti, G.; Glukhova, A.; Koole, C.; Peat, T.S.; Radjainia, M.; Plitzko, J.M.; Baumeister, W.; Miller, L.J.; et al. Cryo-EM structure of the active, Gs-protein complexed, human CGRP receptor. Nature 2018, 561, 492–497. [Google Scholar] [CrossRef]
- King, C.T.; Gegg, C.V.; Hu, S.N.-Y.; Sen Lu, H.; Chan, B.M.; Berry, K.A.; Brankow, D.W.; Boone, T.J.; Kezunovic, N.; Kelley, M.R.; et al. Discovery of the Migraine Prevention Therapeutic Aimovig (Erenumab), the First FDA-Approved Antibody against a G-Protein-Coupled Receptor. ACS Pharmacol. Transl. Sci. 2019, 2, 485–490. [Google Scholar] [CrossRef] [Green Version]
- Booe, J.M.; Walker, C.S.; Barwell, J.; Kuteyi, G.; Simms, J.; Jamaluddin, M.A.; Warner, M.L.; Bill, R.M.; Harris, P.W.; Brimble, M.A.; et al. Structural Basis for Receptor Activity-Modifying Protein-Dependent Selective Peptide Recognition by a G Protein-Coupled Receptor. Mol. Cell 2015, 58, 1040–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Lehto, S.G.; Zhu, D.X.; Sun, H.; Zhang, J.; Smith, B.P.; Immke, D.C.; Wild, K.D.; Xu, C. Pharmacologic Characterization of AMG 334, a Potent and Selective Human Monoclonal Antibody against the Calcitonin Gene-Related Peptide Receptor. J. Pharmacol. Exp. Ther. 2016, 356, 223–231. [Google Scholar] [CrossRef] [Green Version]
- Garces, F.; Mohr, C.; Zhang, L.; Huang, C.S.; Chen, Q.; King, C.; Xu, C.; Wang, Z.L. Molecular Insight into Recognition of the CGRPR Complex by Migraine Prevention Therapy Aimovig (Erenumab). Cell Rep. 2020, 30, 1714–1723. [Google Scholar] [CrossRef] [Green Version]
- Goadsby, P.J.; Reuter, U.; Hallstrom, Y.; Broessner, G.; Bonner, J.H.; Zhang, F.; Sapra, S.; Picard, H.; Mikol, D.D.; Lenz, R.A. A Controlled Trial of Erenumab for Episodic Migraine. N. Engl. J. Med. 2017, 377, 2123–2132. [Google Scholar] [CrossRef]
- Ishida, T.; Ueda, R. CCR4 as a novel molecular target for immunotherapy of cancer. Cancer Sci. 2006, 97, 1139–1146. [Google Scholar] [CrossRef]
- Sugiyama, D.; Nishikawa, H.; Maeda, Y.; Nishioka, M.; Tanemura, A.; Katayama, I.; Ezoe, S.; Kanakura, Y.; Sato, E.; Fukumori, Y.; et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc. Natl. Acad. Sci. USA 2013, 110, 17945–17950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shitara, K.; Hanai, N.; Shoji, E.; Sakurada, M.; Furuya, A.; Nakamura, K.; Niwa, R.; Shibata, K.; Yamasaki, M. Method for producing recombinant antibody and antibody fragment thereof. U.S. Patent 8,632,996, 21 January 2014. [Google Scholar]
- Niwa, R.; Shoji-Hosaka, E.; Sakurada, M.; Shinkawa, T.; Uchida, K.; Nakamura, K.; Matsushima, K.; Ueda, R.; Hanai, N.; Shitara, K. Defucosylated chimeric anti-CC chemokine receptor 4 IgG1 with enhanced antibody-dependent cellular cytotoxicity shows potent therapeutic activity to T-cell leukemia and lymphoma. Cancer Res. 2004, 64, 2127–2133. [Google Scholar] [CrossRef] [Green Version]
- Niwa, R.; Sakurada, M.; Kobayashi, Y.; Uehara, A.; Matsushima, K.; Ueda, R.; Nakamura, K.; Shitara, K. Enhanced natural killer cell binding and activation by low-fucose IgG1 antibody results in potent antibody-dependent cellular cytotoxicity induction at lower antigen density. Clin. Cancer Res. 2005, 11, 2327–2336. [Google Scholar] [CrossRef] [Green Version]
- Yano, H.; Ishida, T.; Inagaki, A.; Ishii, T.; Ding, J.; Kusumoto, S.; Komatsu, H.; Iida, S.; Inagaki, H.; Ueda, R. Defucosylated anti CC chemokine receptor 4 monoclonal antibody combined with immunomodulatory cytokines: A novel immunotherapy for aggressive/refractory Mycosis fungoides and Sezary syndrome. Clin. Cancer Res. 2007, 13, 6494–6500. [Google Scholar] [CrossRef] [Green Version]
- Yoshie, O.; Matsushima, K. CCR4 and its ligands: From bench to bedside. Int. Immunol. 2014, 27, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Ishida, T.; Joh, T.; Uike, N.; Yamamoto, K.; Utsunomiya, A.; Yoshida, S.; Saburi, Y.; Miyamoto, T.; Takemoto, S.; Suzushima, H.; et al. Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: A multicenter phase II study. J. Clin. Oncol. 2012, 30, 837–842. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurney, A.; Axelrod, F.; Bond, C.J.; Cain, J.; Chartier, C.; Donigan, L.; Fischer, M.; Chaudhari, A.; Ji, M.; Kapoun, A.M.; et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 11717–11722. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Yang, M.; Wang, X.; Zhang, H.; Yao, C.; Sun, S.; Liu, Q.; Pan, H.; Liu, S.; Huan, Y.; et al. Glutazumab, a novel long-lasting GLP-1/anti-GLP-1R antibody fusion protein, exerts anti-diabetic effects through targeting dual receptor binding sites. Biochem. Pharmacol. 2018, 150, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Gu, W.; Yang, J.; Bi, V.; Shen, Y.; Lee, E.; Winters, K.A.; Komorowski, R.; Zhang, C.; Patel, J.J.; et al. Fully human monoclonal antibodies antagonizing the glucagon receptor improve glucose homeostasis in mice and monkeys. J. Pharmacol. Exp. Ther. 2009, 329, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Pettus, J.; Reeds, D.; Cavaiola, T.S.; Boeder, S.; Levin, M.; Tobin, G.; Cava, E.; Thai, D.; Shi, J.; Yan, H.; et al. Effect of a glucagon receptor antibody (REMD-477) in type 1 diabetes: A randomized controlled trial. Diabetes Obes. Metab. 2018, 20, 1302–1305. [Google Scholar] [CrossRef]
- Gilbert, J.; Lekstrom-Himes, J.; Donaldson, D.; Lee, Y.; Hu, M.; Xu, J.; Wyant, T.; Davidson, M.; MLN1202 Study Group. Effect of CC chemokine receptor 2 CCR2 blockade on serum C-reactive protein in individuals at atherosclerotic risk and with a single nucleotide polymorphism of the monocyte chemoattractant protein-1 promoter region. Am. J. Cardiol. 2011, 107, 906–911. [Google Scholar] [CrossRef]
- Olson, W.C.; Rabut, G.E.; Nagashima, K.A.; Tran, D.N.; Anselma, D.J.; Monard, S.P.; Segal, J.P.; Thompson, D.A.; Kajumo, F.; Guo, Y.; et al. Differential inhibition of human immunodeficiency virus type 1 fusion, gp120 binding, and CC-chemokine activity by monoclonal antibodies to CCR5. J. Virol. 1999, 73, 4145–4155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhne, M.R.; Mulvey, T.; Belanger, B.; Chen, S.; Pan, C.; Chong, C.; Cao, F.; Niekro, W.; Kempe, T.; Henning, K.A.; et al. BMS-936564/MDX-1338: A fully human anti-CXCR4 antibody induces apoptosis in vitro and shows antitumor activity in vivo in hematologic malignancies. Clin. Cancer Res. 2013, 19, 357–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massard, C.; Cassier, P.; Bendell, J.C.; Marie, D.B.; Blery, M.; Morehouse, C.; Ascierto, M.; Zerbib, R.; Mitry, E.; Tolcher, A.W. 1203P—Preliminary results of STELLAR-001, a dose escalation phase I study of the anti-C5aR, IPH5401, in combination with durvalumab in advanced solid tumours. Ann. Oncol. 2019, 30, v492. [Google Scholar] [CrossRef]
- Zhang, C.; Jing, S.; Zhang, H.; Wang, X.; Chenjiang, Y.A.O. Antibody Specifically Binding to GLP-1 R and Fusion Protein thereof with GLP-1 Patent. U.S. Patent 10,059,773, 28 August 2018. [Google Scholar]
- Trkola, A.; Ketas, T.J.; Nagashima, K.A.; Zhao, L.; Cilliers, T.; Morris, L.; Moore, J.P.; Maddon, P.J.; Olson, W.C. Potent, broad-spectrum inhibition of human immunodeficiency virus type 1 by the CCR5 monoclonal antibody PRO 140. J. Virol. 2001, 75, 579–588. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Jung, Y.; Lee, J.Y.; Lee, W.K.; Lim, D.; Yu, Y.G. Purification and characterization of recombinant human endothelin receptor type A. Protein Expr. Purif. 2012, 84, 14–18. [Google Scholar] [CrossRef]
- Corin, K.; Baaske, P.; Ravel, D.B.; Song, J.; Brown, E.; Wang, X.; Geissler, S.; Wienken, C.J.; Jerabek-Willemsen, M.; Duhr, S.; et al. A robust and rapid method of producing soluble, stable, and functional G-protein coupled receptors. PLoS ONE 2011, 6, e23036. [Google Scholar]
- Sarkar, C.A.; Dodevski, I.; Kenig, M.; Dudli, S.; Mohr, A.; Hermans, E.; Pluckthun, A. Directed evolution of a G protein-coupled receptor for expression, stability, and binding selectivity. Proc. Natl. Acad. Sci. USA 2008, 105, 14808–14813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Link, A.J.; Skretas, G.; Strauch, E.M.; Chari, N.S.; Georgiou, G. Efficient production of membrane-integrated and detergent-soluble G protein-coupled receptors in Escherichia coli. Protein Sci. 2008, 17, 1857–1863. [Google Scholar] [CrossRef] [Green Version]
- Vukoti, K.; Kimura, T.; Macke, L.; Gawrisch, K.; Yeliseev, A. Stabilization of functional recombinant cannabinoid receptor CB(2) in detergent micelles and lipid bilayers. PLoS ONE 2012, 7, e46290. [Google Scholar]
- Liu, S.; Wang, S.; Lu, S. DNA immunization as a technology platform for monoclonal antibody induction. Emerg. Microbes Infect. 2016, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Van der Woning, B.; De Boeck, G.; Blanchetot, C.; Bobkov, V.; Klarenbeek, A.; Saunders, M.; Waelbroeck, M.; Laeremans, T.; Steyaert, J.; Hultberg, A.; et al. DNA immunization combined with scFv phage display identifies antagonistic GCGR specific antibodies and reveals new epitopes on the small extracellular loops. In MAbs; Taylor & Francis: Abingdon, UK, 2016; Volume 8, pp. 1126–1135. [Google Scholar] [CrossRef] [Green Version]
- Heimann, A.S.; Gupta, A.; Gomes, I.; Rayees, R.; Schlessinger, A.; Ferro, E.S.; Unterwald, E.M.; Devi, L.A. Generation of G protein-coupled receptor antibodies differentially sensitive to conformational states. PLoS ONE 2017, 12, e0187306. [Google Scholar] [CrossRef] [Green Version]
- Boshuizen, R.S.; Marsden, C.; Turkstra, J.; Rossant, C.J.; Slootstra, J.; Copley, C.; Schwamborn, K. A combination of in vitro techniques for efficient discovery of functional monoclonal antibodies against human CXC chemokine receptor-2 (CXCR2). In MAbs; Taylor & Francis: Abingdon, UK, 2014; Volume 6, pp. 1415–1424. [Google Scholar] [CrossRef] [Green Version]
- Kushnir, N.; Streatfield, S.J.; Yusibov, V. Virus-like particles as a highly efficient vaccine platform: Diversity of targets and production systems and advances in clinical development. Vaccine 2012, 31, 58–83. [Google Scholar] [CrossRef]
- O’Rourke, J.P.; Peabody, D.S.; Chackerian, B. Affinity selection of epitope-based vaccines using a bacteriophage virus-like particle platform. Curr. Opin. Virol. 2015, 11, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Ho, T.T.; Nguyen, J.T.; Liu, J.; Stanczak, P.; Thompson, A.A.; Yan, Y.G.; Chen, J.; Allerston, C.K.; Dillard, C.L.; Xu, H.; et al. Method for rapid optimization of recombinant GPCR protein expression and stability using virus-like particles. Protein Expr. Purif. 2017, 133, 41–49. [Google Scholar] [CrossRef]
- Rouck, J.E.; Krapf, J.E.; Roy, J.; Huff, H.C.; Das, A. Recent advances in nanodisc technology for membrane protein studies (2012–2017). FEBS Lett. 2017, 591, 2057–2088. [Google Scholar] [CrossRef] [Green Version]
- Denisov, I.G.; Sligar, S.G. Nanodiscs in Membrane Biochemistry and Biophysics. Chem. Rev. 2017, 117, 4669–4713. [Google Scholar] [CrossRef]
- Kretz-Rommel, A.; Shi, L.; Ferrini, R.; Yang, T.; Xu, F.; Campion, B. Antibodies That Bind Human Cannabinoid 1 (CB1) Receptor. U.S. Patent 16/257,511, 17 October 2019. [Google Scholar]
- Mallat, A.; Lotersztajn, S. Endocannabinoids and liver disease. I. Endocannabinoids and their receptors in the liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G9–G12. [Google Scholar] [CrossRef] [Green Version]
- Di Marzo, V. Targeting the endocannabinoid system: To enhance or reduce? Nat. Rev. Drug Discov. 2008, 7, 438–455. [Google Scholar] [CrossRef] [PubMed]
- Osei-Hyiaman, D.; DePetrillo, M.; Pacher, P.; Liu, J.; Radaeva, S.; Bátkai, S.; Harvey-White, J.; Mackie, K.; Offertáler, L.; Wang, L.; et al. Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J. Clin. Investig. 2005, 115, 1298–1305. [Google Scholar] [CrossRef] [Green Version]
- Dodd, R.B.; Wilkinson, T.; Schofield, D.J. Therapeutic Monoclonal Antibodies to Complex Membrane Protein Targets: Antigen Generation and Antibody Discovery Strategies. Biodrugs 2018, 32, 339–355. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen Type | Antibody Screening Technology | Drug Name | Target | Indication | Status of Clinical Trial | Reference |
|---|---|---|---|---|---|---|
| RAMP1-Fc, CGRPR-Fc | Tg mouse immunization and hybridoma screening | AimovigTM (Erenumab) | CGRP receptor | Migraine | Approved | [14] |
| CCR4-KLH | Mouse immunization and hybridoma screening | Poteligeo® (Mogamulizumab) | CCR4 | Refractory mycosis fungoides and Sézary disease | Approved | [15] |
| FZD7-Fc | Synthetic human antibody phage library screening | Vantictumab (OMP-18R5) | FZD7 | Non-small cell lung cancer, pancreatic cancer, metastatic breast cancer | Phase I | [16,17,18] |
| CCR5 expressing cells | Mouse immunization and hybridoma screening | Leronlimab (Pro 140) | CCR5 | HIV infection | Phase III | [19] |
| CCR2 expressing cells | Mouse immunization and hybridoma screening | Plozalizumab (MLN-1202) | CCR2 | Relapsing-remitting multiple sclerosis | Phase II | [20] |
| CXCR4 expressing cells | Tg mouse immunization and phage library screening | Ulocuplumab (BMS-936564) | CXCR4 | Acute myeloid leukemia | Phase I/II | [21] |
| C5Aa1 expressing cells | Tg mouse immunization and hybridoma screening | Avdoralimab (IPH5401) | C5aR1 | Non-small cell lung cancer, liver cancer | Phase I | [22] |
| Membrane fraction of GLP1R expressing cells | Tg mouse immunization and hybridoma screening | Glutazumab (GMA102) | GLP1R | Type 2 diabetes | Phase II | [23] |
| Membrane fraction of GCGR expressing cells | Tg mouse immunization and hybridoma screening | Volagidemab (REMD-477) | GCGR | Diabetes mellitus | Phase II | [24] |
| Reconstituted CNR1 GLB complex | Mouse immunization and hybridoma/phage library screening | Namacizumab (RYI-018) | CNR1 | Non-alcoholic fatty liver disease | Phase I | [25] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ju, M.-S.; Jung, S.T. Antigen Design for Successful Isolation of Highly Challenging Therapeutic Anti-GPCR Antibodies. Int. J. Mol. Sci. 2020, 21, 8240. https://doi.org/10.3390/ijms21218240
Ju M-S, Jung ST. Antigen Design for Successful Isolation of Highly Challenging Therapeutic Anti-GPCR Antibodies. International Journal of Molecular Sciences. 2020; 21(21):8240. https://doi.org/10.3390/ijms21218240
Chicago/Turabian StyleJu, Man-Seok, and Sang Taek Jung. 2020. "Antigen Design for Successful Isolation of Highly Challenging Therapeutic Anti-GPCR Antibodies" International Journal of Molecular Sciences 21, no. 21: 8240. https://doi.org/10.3390/ijms21218240
APA StyleJu, M.-S., & Jung, S. T. (2020). Antigen Design for Successful Isolation of Highly Challenging Therapeutic Anti-GPCR Antibodies. International Journal of Molecular Sciences, 21(21), 8240. https://doi.org/10.3390/ijms21218240