Enhanced Collagen Deposition in the Duodenum of Patients with Hyaline Fibromatosis Syndrome and Protein Losing Enteropathy

 , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
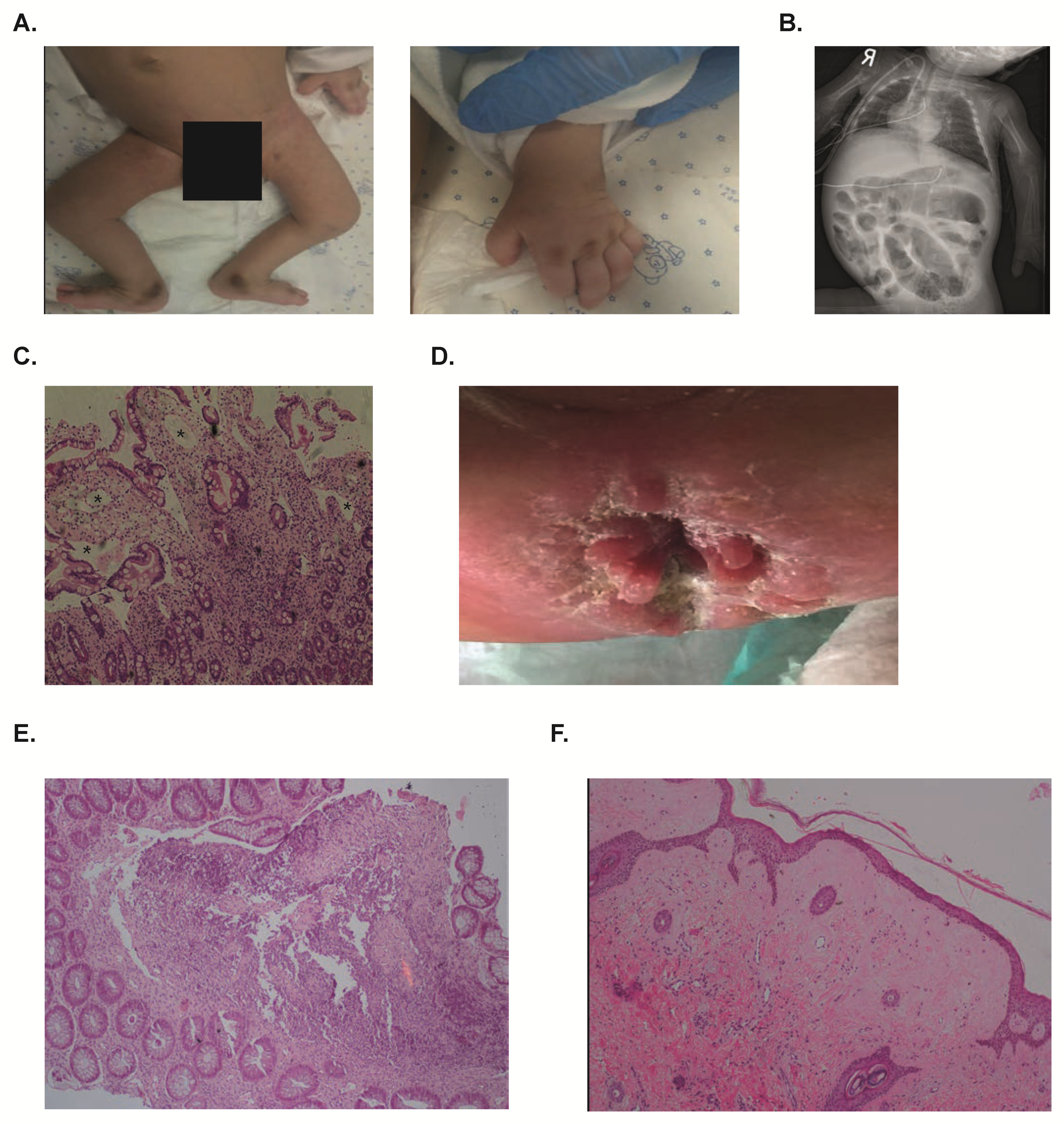
2.1. Clinical Phenotype
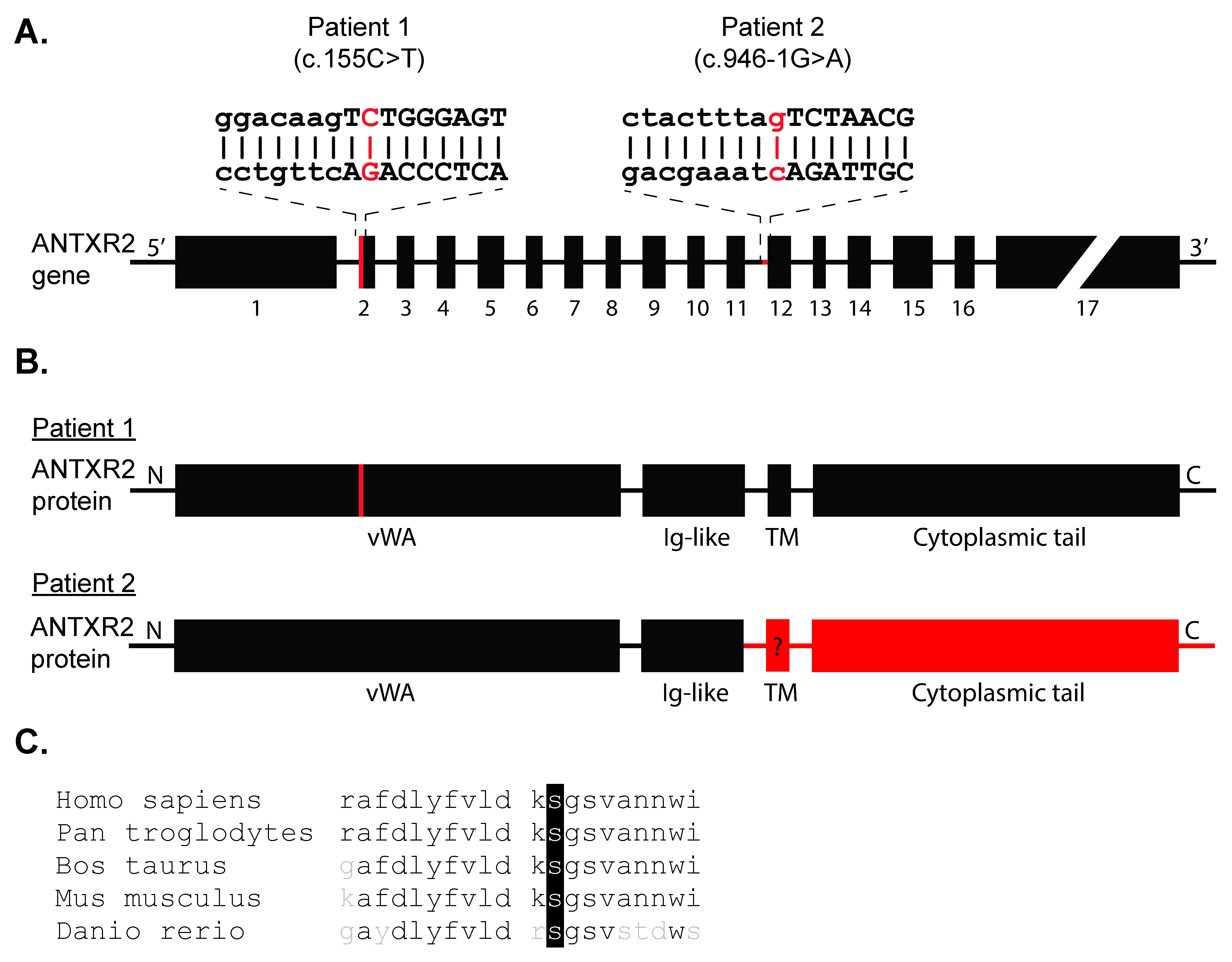
2.2. Genetic Studies
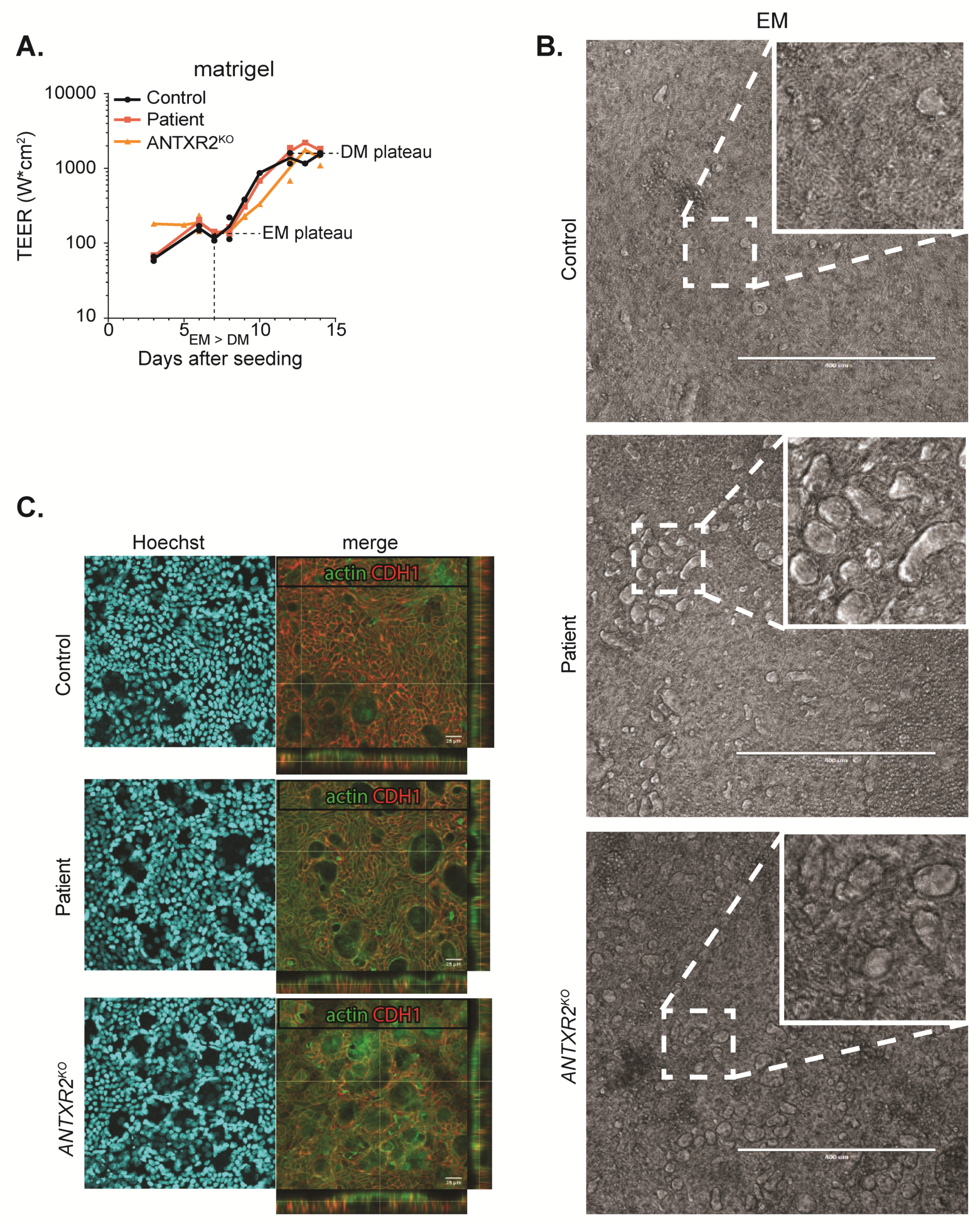
2.3. ANTXR2-deficient Organoids Are Not Affected in Growth and Polarization Potential
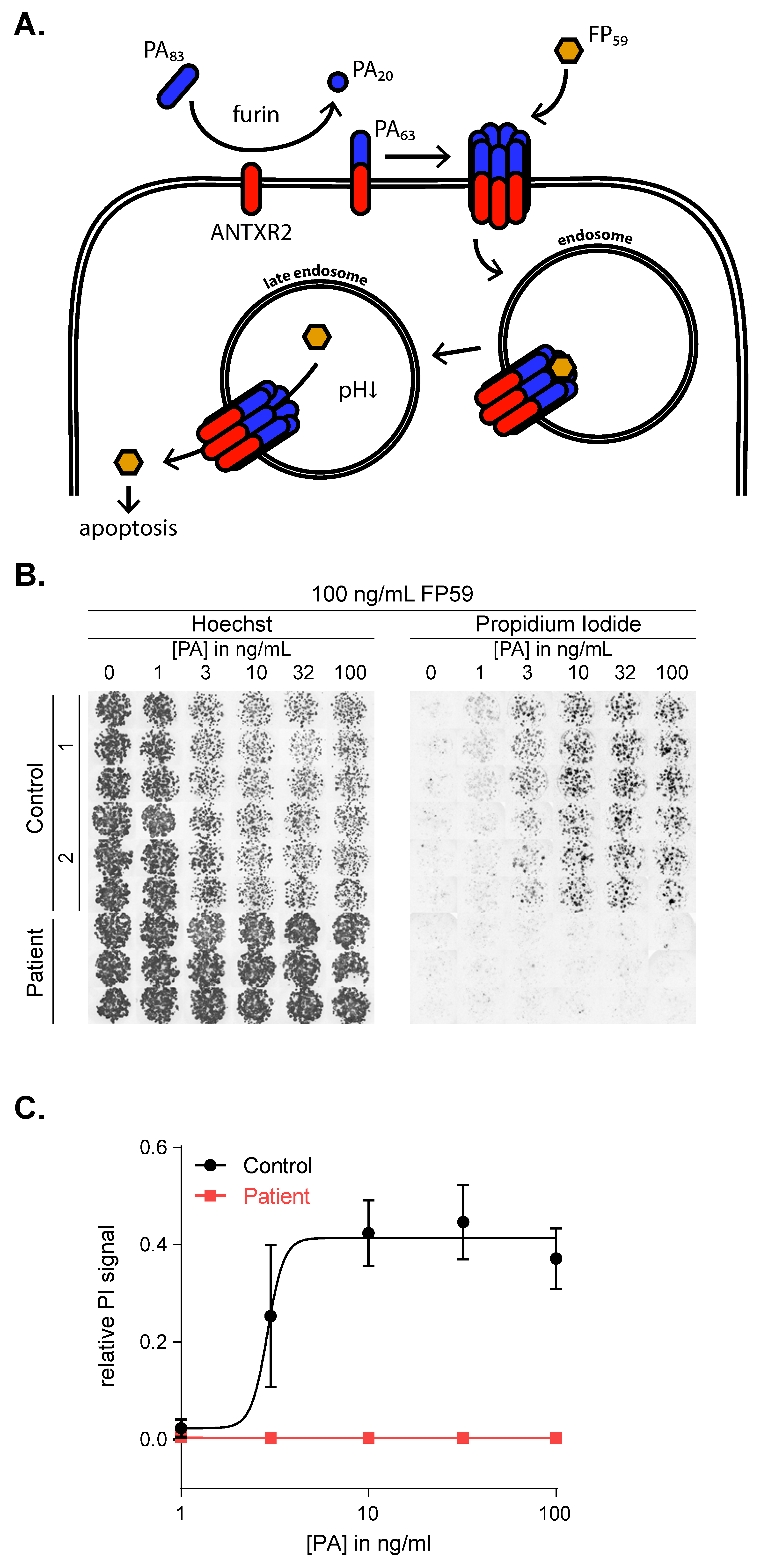
2.4. The c.155C>T Mutation Abolishes ANTXR2 Function
2.5. ANTXR2 Deficiency Does Not Influence Organoid Monolayer Formation
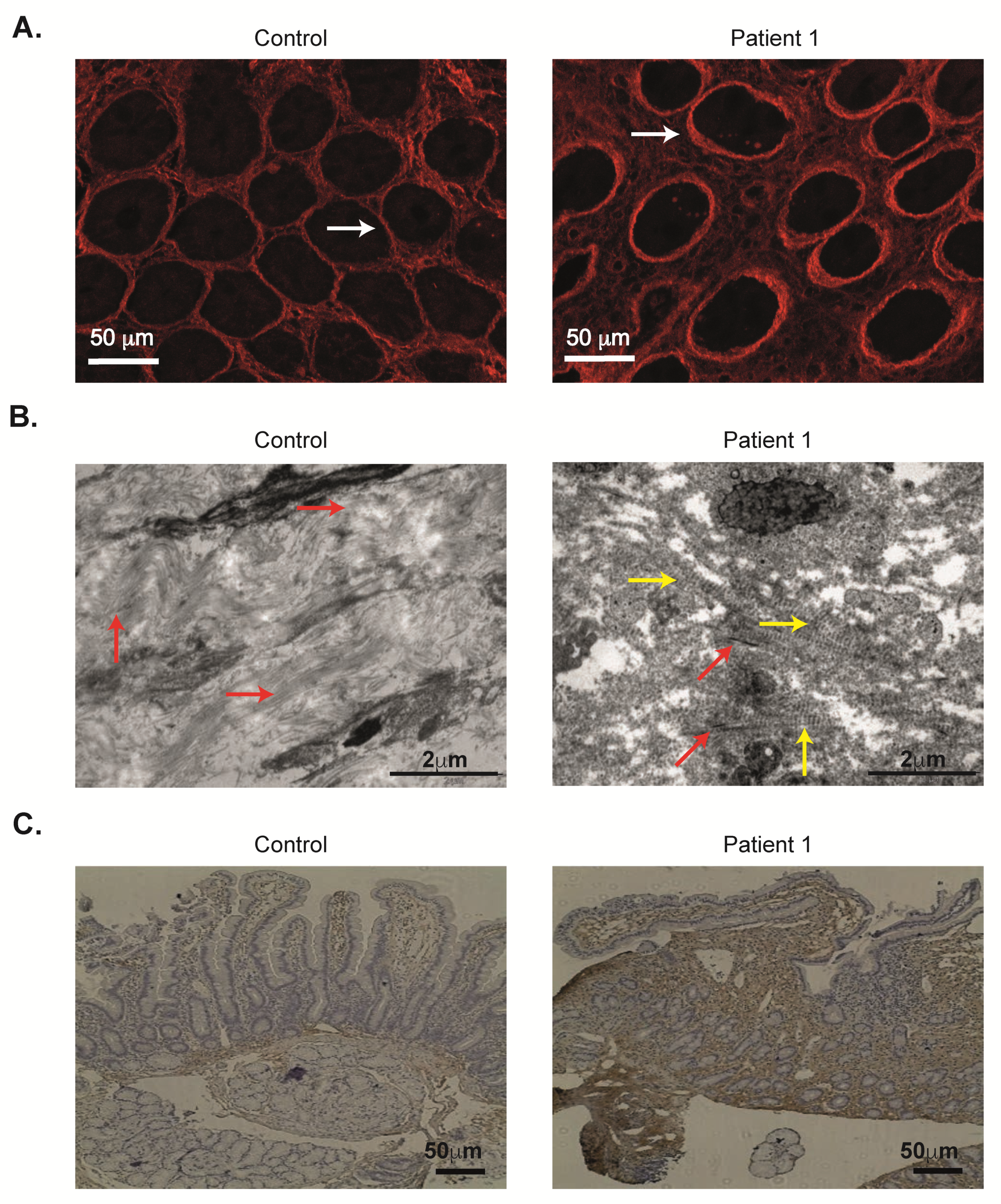
2.6. Abnormal Duodenal Collagen Deposition VI in ANTXR2 Deficiency
3. Discussion
4. Materials and Methods
4.1. Study Approval
4.2. DNA Sequencing
4.3. Organoid Culture
4.4. Quantitative Real-Time Polymerase Chain Reaction
4.5. Confocal Microscopy
4.6. Anthrax Toxin Cytotoxicity Assay
4.7. CRISPR-Cas9 Knockout of ANTXR2
4.8. Second Harmonic Generation Imaging
4.9. Electron Microscopy
4.10. Immunohistochemistry
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ANTXR2 | anthrax toxin receptor 2 |
| CMG2 | capillary morphogenesis protein 2 |
| DM | differentiation media |
| ECM | extracellular matrix |
| EGD | esophagogastroduodenoscopy |
| EF | edema factor |
| EM | expansion media |
| GF | growth factors |
| FFPE | formalin-fixed paraffin-embedded |
| H&E | hematoxylin and eosin |
| HFS | hyaline fibromatosis syndrome |
| ISH | infantile systemic hyalinosis |
| LF | lethal factor |
| MIDAS | metal ion-dependent adhesion site |
| MP | multiphoton |
| PA | protective antigen |
| PBS | phosphate buffered saline |
| PI | propidium iodide |
| PLE | protein losing enteropathy |
| ROCKi | ROCK inhibitor |
| RT | room temperature |
| SHG | second-harmonic generation |
| TEER | trans-epithelial electrical resistance |
| TPN | total parenteral nutrition |
| vWA | von Willebrand A |
References
- Burgi, J.; Kunz, B.; Abrami, L.; Deuquet, J.; Piersigilli, A.; Scholl-Burgi, S.; Lausch, E.; Unger, S.; Superti-Furga, A.; Bonaldo, P.; et al. CMG2/ANTXR2 regulates extracellular collagen VI which accumulates in hyaline fibromatosis syndrome. Nat. Commun. 2017, 8, 15861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeves, C.V.; Wang, X.; Charles-Horvath, P.C.; Vink, J.Y.; Borisenko, V.Y.; Young, J.A.; Kitajewski, J.K. Anthrax toxin receptor 2 functions in ECM homeostasis of the murine reproductive tract and promotes MMP activity. PLoS ONE 2012, 7, e34862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, K.; Ebihara, T.; Kusubata, M.; Adachi, E.; Arai, M.; Kawaguchi, N.; Utsunomiya, J.; Miki, Y.; Hiramoto, M.; Hattori, S.; et al. Abnormal collagen deposition in fibromas from patient with juvenile hyaline fibromatosis. J. Dermatol. Sci. 2009, 55, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Dowling, O.; Difeo, A.; Ramirez, M.C.; Tukel, T.; Narla, G.; Bonafe, L.; Kayserili, H.; Yuksel-Apak, M.; Paller, A.S.; Norton, K.; et al. Mutations in capillary morphogenesis gene-2 result in the allelic disorders juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am. J. Hum. Genet. 2003, 73, 957–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanks, S.; Adams, S.; Douglas, J.; Arbour, L.; Atherton, D.J.; Balci, S.; Bode, H.; Campbell, M.E.; Feingold, M.; Keser, G.; et al. Mutations in the gene encoding capillary morphogenesis protein 2 cause juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am. J. Hum. Genet. 2003, 73, 791–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denadai, R.; Raposo-Amaral, C.E.; Bertola, D.; Kim, C.; Alonso, N.; Hart, T.; Han, S.; Stelini, R.F.; Buzzo, C.L.; Raposo-Amaral, C.A.; et al. Identification of 2 novel ANTXR2 mutations in patients with hyaline fibromatosis syndrome and proposal of a modified grading system. Am. J. Med. Genet. A 2012, 158A, 732–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nofal, A.; Sanad, M.; Assaf, M.; Nofal, E.; Nassar, A.; Almokadem, S.; Attwa, E.; Elmosalamy, K. Juvenile hyaline fibromatosis and infantile systemic hyalinosis: A unifying term and a proposed grading system. J. Am. Acad. Dermatol. 2009, 61, 695–700. [Google Scholar] [CrossRef]
- Casas-Alba, D.; Martinez-Monseny, A.; Pino-Ramirez, R.M.; Alsina, L.; Castejon, E.; Navarro-Vilarrubi, S.; Perez-Duenas, B.; Serrano, M.; Palau, F.; Garcia-Alix, A. Hyaline fibromatosis syndrome: Clinical update and phenotype-genotype correlations. Hum. Mutat. 2018, 39, 1752–1763. [Google Scholar] [CrossRef]
- Bell, S.E.; Mavila, A.; Salazar, R.; Bayless, K.J.; Kanagala, S.; Maxwell, S.A.; Davis, G.E. Differential gene expression during capillary morphogenesis in 3D collagen matrices: Regulated expression of genes involved in basement membrane matrix assembly, cell cycle progression, cellular differentiation and G-protein signaling. J. Cell Sci. 2001, 114, 2755–2773. [Google Scholar]
- Glover, M.T.; Lake, B.D.; Atherton, D.J. Infantile systemic hyalinosis: Newly recognized disorder of collagen? Pediatrics 1991, 87, 228–234. [Google Scholar] [CrossRef]
- Alreheili, K.; AlMehaidib, A.; Alsaleem, K.; Banemi, M.; Aldekhail, W.; Al-Mayouf, S.M. Intestinal lymphangiectasia in a patient with infantile systemic hyalinosis syndrome: A rare cause of protein-losing enteropathy. Ann. Saudi Med. 2012, 32, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Buyukgebiz, B.; Ozturk, Y.; Arslan, N.; Ozer, E. A rare cause of protein-losing enteropathy and growth retardation in infancy: Infantile systemic hyalinosis. Turk. J. Pediatr. 2003, 45, 258–260. [Google Scholar] [PubMed]
- Schussler, E.; Linkner, R.V.; Levitt, J.; Mehta, L.; Martignetti, J.A.; Oishi, K. Protein-losing enteropathy and joint contractures caused by a novel homozygous ANTXR2 mutation. Adv. Genomics Genet. 2018, 8, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Braamskamp, M.J.; Dolman, K.M.; Tabbers, M.M. Clinical practice. Protein-losing enteropathy in children. Eur. J. Pediatr. 2010, 169, 1179–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneeberger, K.; Roth, S.; Nieuwenhuis, E.E.S.; Middendorp, S. Intestinal epithelial cell polarity defects in disease: Lessons from microvillus inclusion disease. Dis. Model Mech. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Vogel, G.F.; van Rijn, J.M.; Krainer, I.M.; Janecke, A.R.; Posovszky, C.; Cohen, M.; Searle, C.; Jantchou, P.; Escher, J.C.; Patey, N.; et al. Disrupted apical exocytosis of cargo vesicles causes enteropathy in FHL5 patients with Munc18-2 mutations. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Schneeberger, K.; Vogel, G.F.; Teunissen, H.; van Ommen, D.D.; Begthel, H.; El Bouazzaoui, L.; van Vugt, A.H.; Beekman, J.M.; Klumperman, J.; Muller, T.; et al. An inducible mouse model for microvillus inclusion disease reveals a role for myosin Vb in apical and basolateral trafficking. Proc. Natl. Acad. Sci. USA 2015, 112, 12408–12413. [Google Scholar] [CrossRef] [Green Version]
- Weis, V.G.; Knowles, B.C.; Choi, E.; Goldstein, A.E.; Williams, J.A.; Manning, E.H.; Roland, J.T.; Lapierre, L.A.; Goldenring, J.R. Loss of MYO5B in mice recapitulates Microvillus Inclusion Disease and reveals an apical trafficking pathway distinct to neonatal duodenum. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 131–157. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Zhang, Y.; Moayeri, M.; Liu, J.; Crown, D.; Fattah, R.J.; Wein, A.N.; Yu, Z.X.; Finkel, T.; Leppla, S.H. Key tissue targets responsible for anthrax-toxin-induced lethality. Nature 2013, 501, 63–68. [Google Scholar] [CrossRef] [Green Version]
- Deuquet, J.; Lausch, E.; Superti-Furga, A.; van der Goot, F.G. The dark sides of capillary morphogenesis gene 2. EMBO J. 2012, 31, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Friebe, S.; van der Goot, F.G.; Burgi, J. The Ins and Outs of Anthrax Toxin. Toxins (Basel) 2016, 8, 69. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Zhang, Y.; Hoover, B.; Leppla, S.H. The receptors that mediate the direct lethality of anthrax toxin. Toxins (Basel) 2012, 5, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. TEER measurement techniques for in vitro barrier model systems. J. Lab. Autom. 2015, 20, 107–126. [Google Scholar] [CrossRef] [Green Version]
- Shimshoni, E.; Afik, R.; Solomonov, I.; Adir, I.; Shenoy, A.; Adler, M.; Puricelli, L.; Ghini, V.; Mouhadeb, O.; Gluck, N.; et al. Unraveling the state-specific nature of the native extracellular matrix via multidimensional characterization of its material properties. bioRxic 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Shimshoni, E.; Yablecovitch, D.; Baram, L.; Dotan, I.; Sagi, I. ECM remodelling in IBD: Innocent bystander or partner in crime? The emerging role of extracellular molecular events in sustaining intestinal inflammation. Gut 2015, 64, 367–372. [Google Scholar] [CrossRef] [Green Version]
- Haas, J.T.; Winter, H.S.; Lim, E.; Kirby, A.; Blumenstiel, B.; DeFelice, M.; Gabriel, S.; Jalas, C.; Branski, D.; Grueter, C.A.; et al. DGAT1 mutation is linked to a congenital diarrheal disorder. J. Clin. Investig. 2012, 122, 4680–4684. [Google Scholar] [CrossRef] [Green Version]
- van Rijn, J.M.; Ardy, R.C.; Kuloglu, Z.; Harter, B.; van Haaften-Visser, D.Y.; van der Doef, H.P.J.; van Hoesel, M.; Kansu, A.; van Vugt, A.H.M.; Thian, M.; et al. Intestinal Failure and Aberrant Lipid Metabolism in Patients With DGAT1 Deficiency. Gastroenterology 2018, 155, 130–143.e15. [Google Scholar] [CrossRef] [Green Version]
- Elkadri, A.; Thoeni, C.; Deharvengt, S.J.; Murchie, R.; Guo, C.; Stavropoulos, J.D.; Marshall, C.R.; Wales, P.; Bandsma, R.; Cutz, E.; et al. Mutations in Plasmalemma Vesicle Associated Protein Result in Sieving Protein-Losing Enteropathy Characterized by Hypoproteinemia, Hypoalbuminemia, and Hypertriglyceridemia. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 381–394.e7. [Google Scholar] [CrossRef] [Green Version]
- Kurolap, A.; Eshach-Adiv, O.; Gonzaga-Jauregui, C.; Dolnikov, K.; Mory, A.; Paperna, T.; Hershkovitz, T.; Overton, J.D.; Kaplan, M.; Glaser, F.; et al. Establishing the role of PLVAP in protein-losing enteropathy: A homozygous missense variant leads to an attenuated phenotype. J. Med. Genet. 2018, 55, 779–784. [Google Scholar] [CrossRef]
- Kurolap, A.; Eshach-Adiv, O.; Hershkovitz, T.; Paperna, T.; Mory, A.; Oz-Levi, D.; Zohar, Y.; Mandel, H.; Chezar, J.; Azoulay, D.; et al. Loss of CD55 in Eculizumab-Responsive Protein-Losing Enteropathy. N. Engl. J. Med. 2017, 377, 87–89. [Google Scholar] [CrossRef]
- Ozen, A.; Comrie, W.A.; Ardy, R.C.; Dominguez Conde, C.; Dalgic, B.; Beser, O.F.; Morawski, A.R.; Karakoc-Aydiner, E.; Tutar, E.; Baris, S.; et al. CD55 Deficiency, Early-Onset Protein-Losing Enteropathy, and Thrombosis. N. Engl. J. Med. 2017, 377, 52–61. [Google Scholar] [CrossRef]
- Alders, M.; Hogan, B.M.; Gjini, E.; Salehi, F.; Al-Gazali, L.; Hennekam, E.A.; Holmberg, E.E.; Mannens, M.M.; Mulder, M.F.; Offerhaus, G.J.; et al. Mutations in CCBE1 cause generalized lymph vessel dysplasia in humans. Nat. Genet. 2009, 41, 1272–1274. [Google Scholar] [CrossRef]
- Kurolap, A.; Eshach Adiv, O.; Hershkovitz, T.; Tabib, A.; Karbian, N.; Paperna, T.; Mory, A.; Vachyan, A.; Slijper, N.; Steinberg, R.; et al. Eculizumab Is Safe and Effective as a Long-term Treatment for Protein-losing Enteropathy Due to CD55 Deficiency. J. Pediatr. Gastroenterol. Nutr. 2019, 68, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, K.; Ohno, A.; Kono, M.; Kitoh, H.; Itomi, K.; Akiyama, M. Hyperpigmentation over the metacarpophalangeal joints and the malleoli in a case of hyaline fibromatosis syndrome with ANTXR2 mutations. J. Eur. Acad. Dermatol. Venereol. 2016, 30, e44–e46. [Google Scholar] [CrossRef]
- Deuquet, J.; Lausch, E.; Guex, N.; Abrami, L.; Salvi, S.; Lakkaraju, A.; Ramirez, M.C.; Martignetti, J.A.; Rokicki, D.; Bonafe, L.; et al. Hyaline fibromatosis syndrome inducing mutations in the ectodomain of anthrax toxin receptor 2 can be rescued by proteasome inhibitors. EMBO Mol. Med. 2011, 3, 208–221. [Google Scholar] [CrossRef]
- Levitt, D.G.; Levitt, M.D. Protein losing enteropathy: Comprehensive review of the mechanistic association with clinical and subclinical disease states. Clin. Exp. Gastroenterol. 2017, 10, 147–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozen, A. CHAPLE syndrome uncovers the primary role of complement in a familial form of Waldmann’s disease. Immunol. Rev. 2019, 287, 20–32. [Google Scholar] [CrossRef]
- Alders, M.; Al-Gazali, L.; Cordeiro, I.; Dallapiccola, B.; Garavelli, L.; Tuysuz, B.; Salehi, F.; Haagmans, M.A.; Mook, O.R.; Majoie, C.B.; et al. Hennekam syndrome can be caused by FAT4 mutations and be allelic to Van Maldergem syndrome. Hum. Genet. 2014, 133, 1161–1167. [Google Scholar] [CrossRef] [PubMed]
- Brouillard, P.; Dupont, L.; Helaers, R.; Coulie, R.; Tiller, G.E.; Peeden, J.; Colige, A.; Vikkula, M. Loss of ADAMTS3 activity causes Hennekam lymphangiectasia-lymphedema syndrome 3. Hum. Mol. Genet. 2017, 26, 4095–4104. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, M.; Matano, M.; Nanki, K.; Sato, T. Efficient genetic engineering of human intestinal organoids using electroporation. Nat. Protoc. 2015, 10, 1474–1485. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Rijn, J.M.; Werner, L.; Aydemir, Y.; Spronck, J.M.A.; Pode-Shakked, B.; van Hoesel, M.; Shimshoni, E.; Polak-Charcon, S.; Talmi, L.; Eren, M.; et al. Enhanced Collagen Deposition in the Duodenum of Patients with Hyaline Fibromatosis Syndrome and Protein Losing Enteropathy. Int. J. Mol. Sci. 2020, 21, 8200. https://doi.org/10.3390/ijms21218200
van Rijn JM, Werner L, Aydemir Y, Spronck JMA, Pode-Shakked B, van Hoesel M, Shimshoni E, Polak-Charcon S, Talmi L, Eren M, et al. Enhanced Collagen Deposition in the Duodenum of Patients with Hyaline Fibromatosis Syndrome and Protein Losing Enteropathy. International Journal of Molecular Sciences. 2020; 21(21):8200. https://doi.org/10.3390/ijms21218200
Chicago/Turabian Stylevan Rijn, Jorik M., Lael Werner, Yusuf Aydemir, Joey M.A. Spronck, Ben Pode-Shakked, Marliek van Hoesel, Elee Shimshoni, Sylvie Polak-Charcon, Liron Talmi, Makbule Eren, and et al. 2020. "Enhanced Collagen Deposition in the Duodenum of Patients with Hyaline Fibromatosis Syndrome and Protein Losing Enteropathy" International Journal of Molecular Sciences 21, no. 21: 8200. https://doi.org/10.3390/ijms21218200
APA Stylevan Rijn, J. M., Werner, L., Aydemir, Y., Spronck, J. M. A., Pode-Shakked, B., van Hoesel, M., Shimshoni, E., Polak-Charcon, S., Talmi, L., Eren, M., Weiss, B., H.J. Houwen, R., Barshack, I., Somech, R., Nieuwenhuis, E. E. S., Sagi, I., Raas-Rothschild, A., Middendorp, S., & Shouval, D. S. (2020). Enhanced Collagen Deposition in the Duodenum of Patients with Hyaline Fibromatosis Syndrome and Protein Losing Enteropathy. International Journal of Molecular Sciences, 21(21), 8200. https://doi.org/10.3390/ijms21218200