2-Hydroxy-4-Methylbenzoic Anhydride Inhibits Neuroinflammation in Cellular and Experimental Animal Models of Parkinson’s Disease

 ,
,  ,
, 
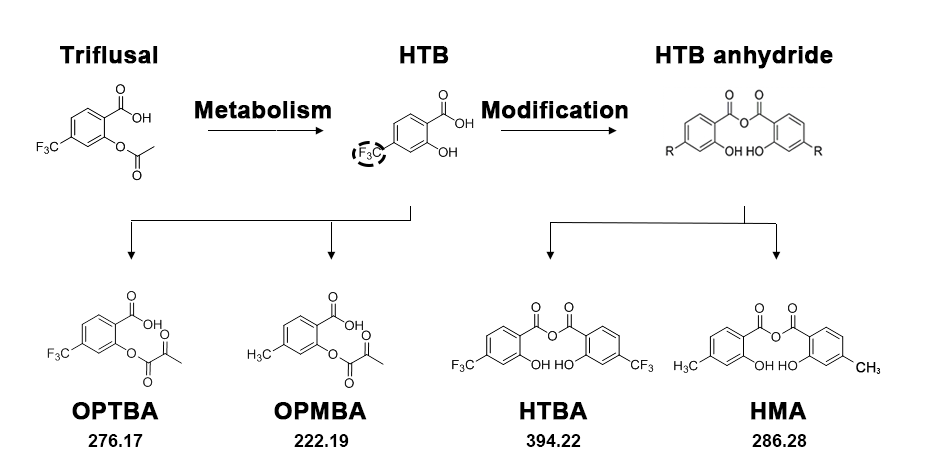
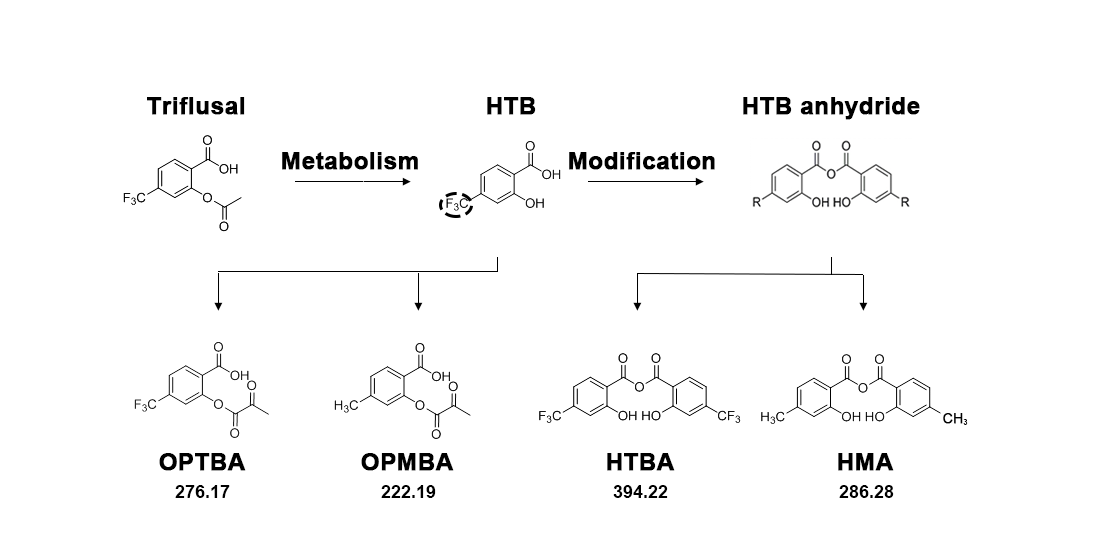{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract

1. Introduction
2. Results
2.1. Structure of HMA and Its Effect on Cell Viability and Nitric Oxide (NO) Production in Lipopolysaccharide (LPS)-Induced BV-2 Cells
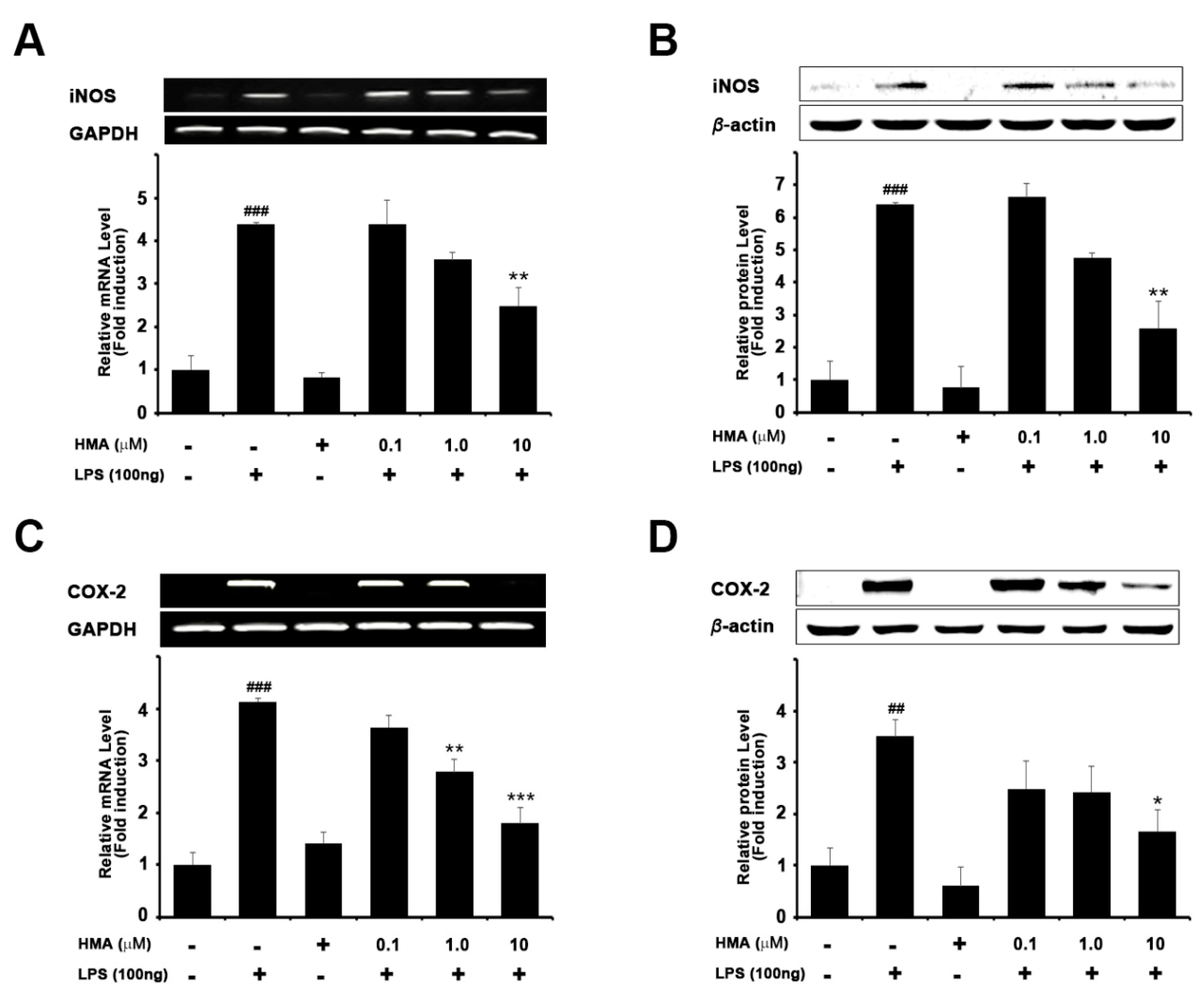
2.2. Effect of HMA on the Expression of iNOS and COX-2 on mRNA and Protein Levels in LPS-Induced Microglial BV-2 Cells Formatting of Mathematical Components
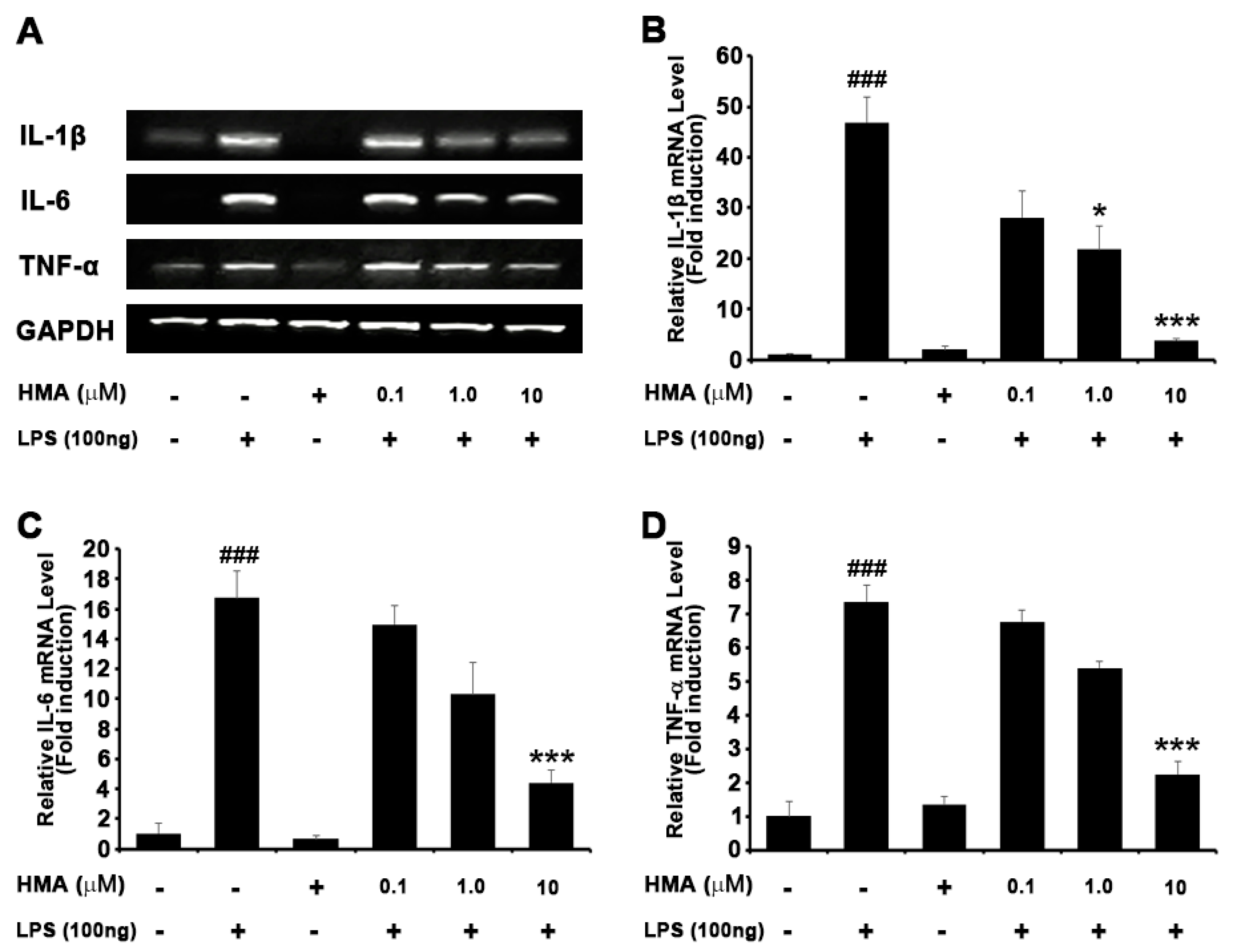
2.3. Effect of HMA on the Production of the LPS-Stimulated Proinflammatory Cytokines in BV-2 Cells
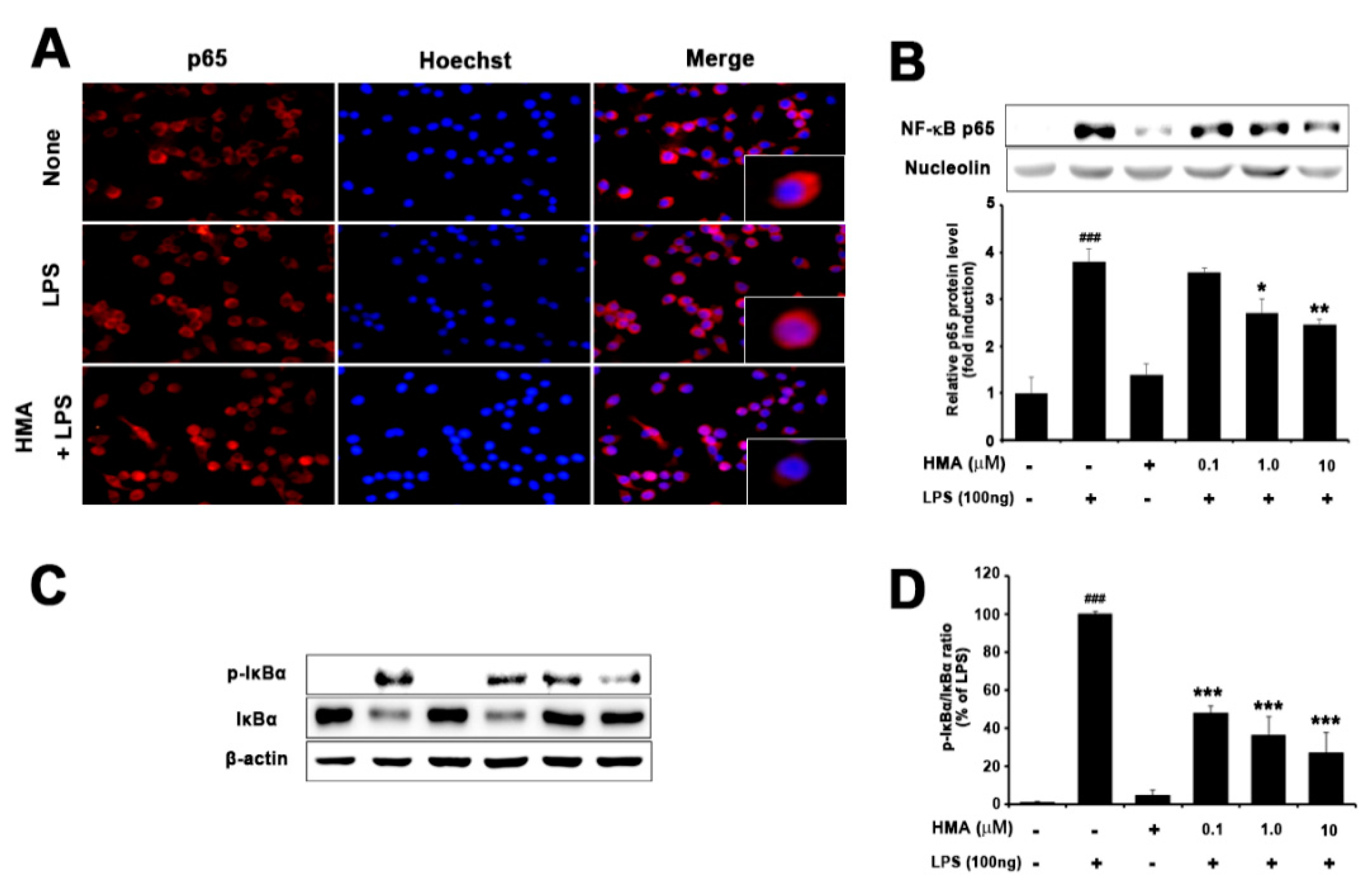
2.4. Effect of HMA on NF-κB (p65) Translocation and IκBα Phosphorylation and Degradation
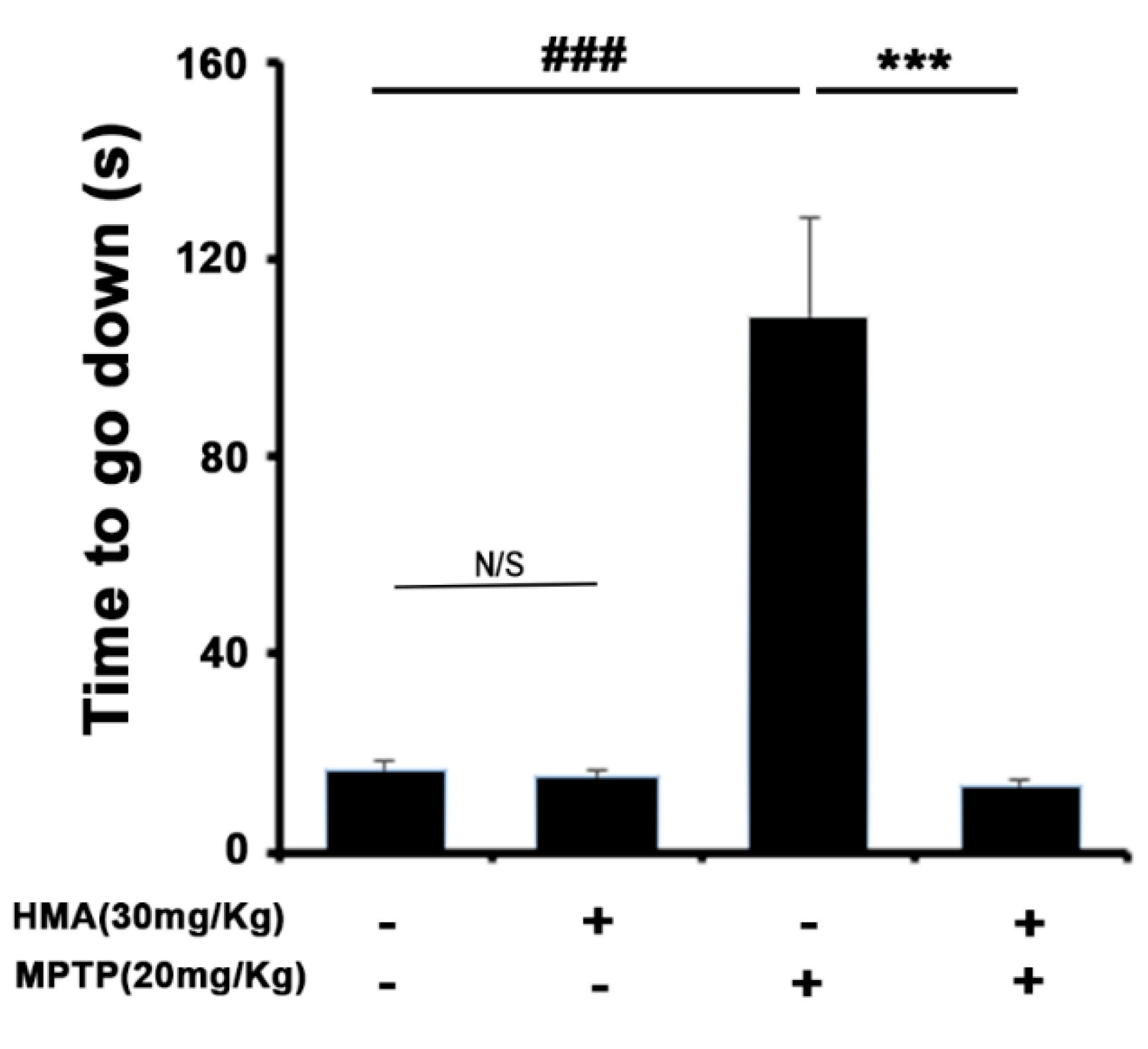
2.5. HMA Shows the Protective Effect of the Behavioral Deficit against MPTP Toxicity in a Mouse Model of Parkinson’s Disease (PD): The Pole Test
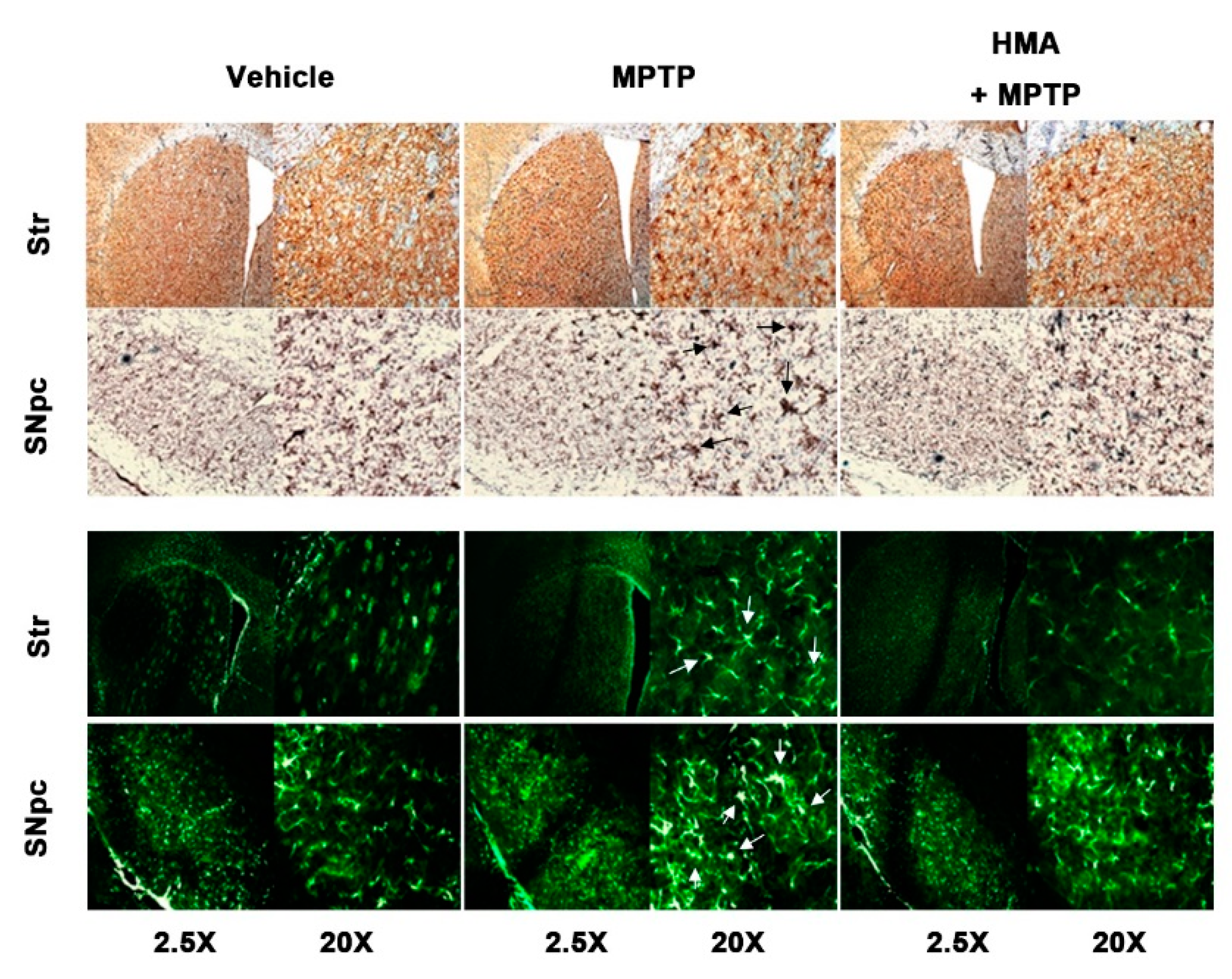
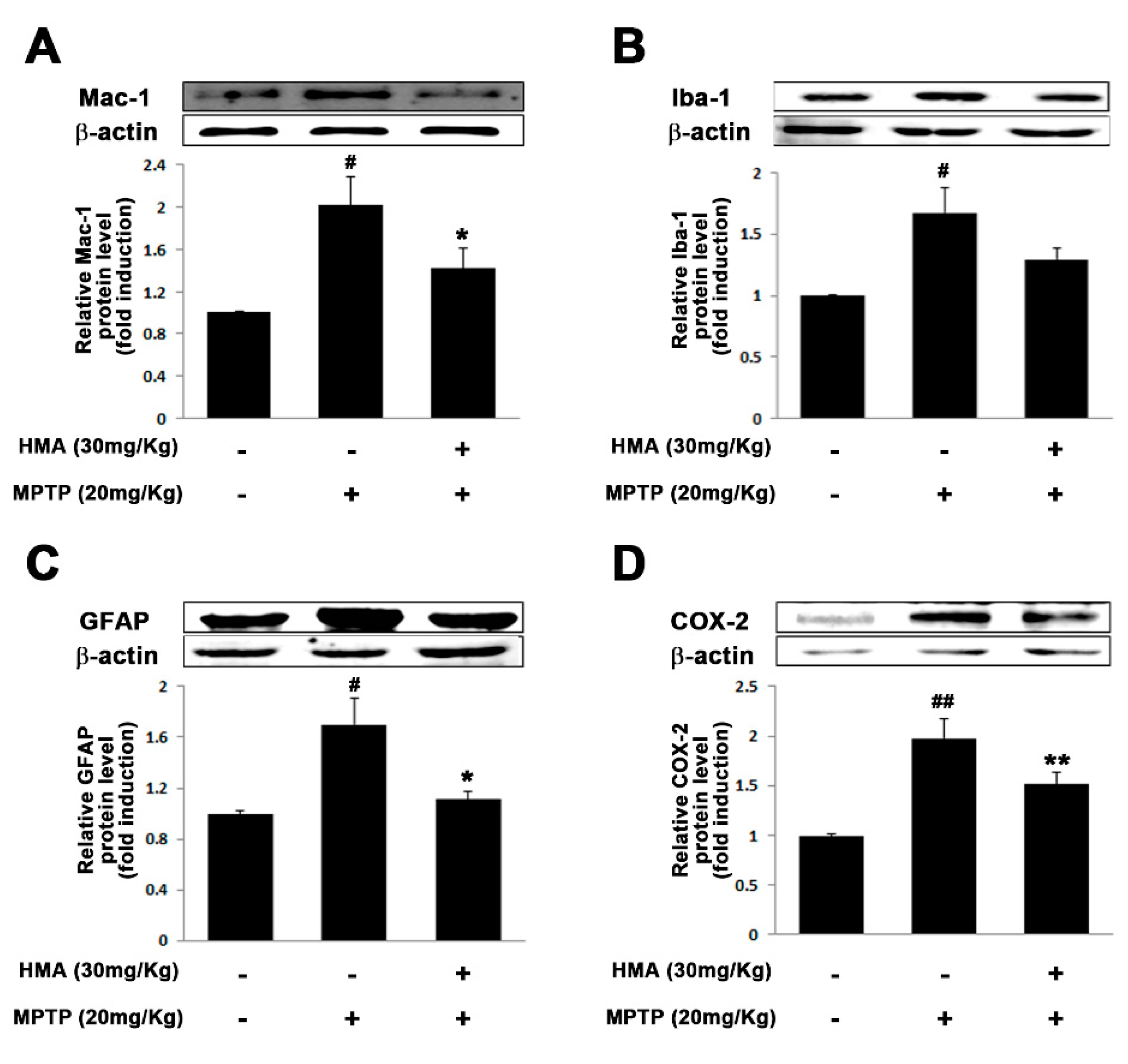
2.6. Effect of HMA on Microglial and Glial Activation in MPTP-Intoxicated Mouse Model of PD
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Synthesis of 2-Hydroxy-4-Methylbenzoic Anhydride (HMA)
4.3. Cell Culture and Treatment
4.4. Animals and Treatment
4.5. Cell Viability and NO Assay
4.6. Total RNA Extraction and RT-PCR
4.7. Western Blot Analysis
4.8. Immunocytochemistry
4.9. Immunohistochemistry
4.10. Pole Test
4.11. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Teeling:, J.L.; Perry, V.H. Systemic infection and inflammation in acute CNS injury and chronic neurodegeneration: Underlying mechanisms. Neuroscience 2009, 158, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H. Contribution of systemic inflammation to chronic neurodegeneration. Acta Neuropathol. 2010, 120, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef] [PubMed]
- Amor, S.; Woodroofe, M.N. Innate and adaptive immune responses in neurodegeneration and repair. Immunology 2014, 141, 287–291. [Google Scholar] [CrossRef]
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125 (Suppl. 2), S3–S23. [Google Scholar] [CrossRef]
- Wu, L.J.; Stevens, B.; Duan, S.; MacVicar, B.A. Microglia in neuronal circuits. Neural. Plast 2013, 2013, 586426. [Google Scholar] [CrossRef]
- Cunningham, C. Microglia and neurodegeneration: The role of systemic inflammation. Glia 2013, 61, 71–90. [Google Scholar] [CrossRef]
- Smith, J.A.; Das, A.; Ray, S.K.; Banik, N.L. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 2012, 87, 10–20. [Google Scholar] [CrossRef]
- Imamura, K.; Hishikawa, N.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 2003, 106, 518–526. [Google Scholar] [CrossRef]
- Alam, Q.; Alam, M.Z.; Mushtaq, G.; Damanhouri, G.A.; Rasool, M.; Kamal, M.A.; Haque, A. Inflammatory Process in Alzheimer’s and Parkinson’s Diseases: Central Role of Cytokines. Curr. Pharm. Des. 2016, 22, 541–548. [Google Scholar] [CrossRef]
- Murdoch, D.; Plosker, G.L. Triflusal: A review of its use in cerebral infarction and myocardial infarction, and as thromboprophylaxis in atrial fibrillation. Drugs 2006, 66, 671–692. [Google Scholar] [CrossRef] [PubMed]
- Fernandez de Arriba, A.; Cavalcanti, F.; Miralles, A.; Bayon, Y.; Alonso, A.; Merlos, M.; Garcia-Rafanell, J.; Forn, J. Inhibition of cyclooxygenase-2 expression by 4-trifluoromethyl derivatives of salicylate, triflusal, and its deacetylated metabolite, 2-hydroxy-4-trifluoromethylbenzoic acid. Mol. Pharm. 1999, 55, 753–760. [Google Scholar]
- Anninos, H.; Andrikopoulos, G.; Pastromas, S.; Sakellariou, D.; Theodorakis, G.; Vardas, P. Triflusal: An old drug in modern antiplatelet therapy. Review of its action, use, safety and effectiveness. Hell. J. Cardiol. 2009, 50, 199–207. [Google Scholar]
- Hernandez, M.; de Arriba, A.F.; Merlos, M.; Fuentes, L.; Crespo, M.S.; Nieto, M.L. Effect of 4-trifluoromethyl derivatives of salicylate on nuclear factor kappaB-dependent transcription in human astrocytoma cells. Br. J. Pharm. 2001, 132, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Mis, R.; Ramis, J.; Conte, L.; Forn, J. In-vitro protein binding interaction between a metabolite of triflusal, 2-hydroxy-4-trifluoromethylbenzoic acid and other drugs. J. Pharm. Pharmacol. 1992, 44, 935–937. [Google Scholar] [CrossRef]
- Mis, R.; Ramis, J.; Conte, L.; Forn, J. Binding of a metabolite of triflusal (2-hydroxy-4-trifluoromethylbenzoic acid) to serum proteins in rat and man. Eur. J. Clin. Pharm. 1992, 42, 175–179. [Google Scholar] [CrossRef]
- Moorcroft, M.J.; Davis, J.; Compton, R.G. Detection and determination of nitrate and nitrite: A review. Talanta 2001, 54, 785–803. [Google Scholar] [CrossRef]
- Scheiblich, H.; Roloff, F.; Singh, V.; Stangel, M.; Stern, M.; Bicker, G. Nitric oxide/cyclic GMP signaling regulates motility of a microglial cell line and primary microglia in vitro. Brain Res. 2014, 1564, 9–21. [Google Scholar] [CrossRef]
- Boje, K.M. Nitric oxide neurotoxicity in neurodegenerative diseases. Front. Biosci. 2004, 9, 763–776. [Google Scholar] [CrossRef]
- Kim, S.W.; Lee, H.K.; Kim, I.D.; Lee, H.; Luo, L.; Park, J.Y.; Yoon, S.H.; Lee, J.K. Robust neuroprotective effects of 2-((2-oxopropanoyl)oxy)-4-(trifluoromethyl)benzoic acid (OPTBA), a HTB/pyruvate ester, in the postischemic rat brain. Sci. Rep. 2016, 6, 31843. [Google Scholar] [CrossRef]
- Guha, M.; O’Connell, M.A.; Pawlinski, R.; Hollis, A.; McGovern, P.; Yan, S.F.; Stern, D.; Mackman, N. Lipopolysaccharide activation of the MEK-ERK1/2 pathway in human monocytic cells mediates tissue factor and tumor necrosis factor alpha expression by inducing Elk-1 phosphorylation and Egr-1 expression. Blood 2001, 98, 1429–1439. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.E.; Kim, I.S.; Jakaria, M.; Akther, M.; Choi, D.K. Importance of GPCR-Mediated Microglial Activation in Alzheimer’s Disease. Front. Cell Neurosci. 2018, 12, 258. [Google Scholar] [CrossRef]
- Orr, C.F.; Rowe, D.B.; Halliday, G.M. An inflammatory review of Parkinson’s disease. Prog. Neurobiol. 2002, 68, 325–340. [Google Scholar] [CrossRef]
- More, S.V.; Kumar, H.; Cho, D.Y.; Yun, Y.S.; Choi, D.K. Toxin-Induced Experimental Models of Learning and Memory Impairment. Int. J. Mol. Sci. 2016, 17, 1447. [Google Scholar] [CrossRef]
- Episcopo, F.L.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Marchetti, B. Reactive astrocytes are key players in nigrostriatal dopaminergic neurorepair in the MPTP mouse model of Parkinson’s disease: Focus on endogenous neurorestoration. Curr. Aging Sci. 2013, 6, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, C.P.; Tansey, M.G. A survey from 2012 of evidence for the role of neuroinflammation in neurotoxin animal models of Parkinson’s disease and potential molecular targets. Exp. Neurol. 2014, 256, 126–132. [Google Scholar] [CrossRef]
- Ye, J.; McGuinness, O.P. Inflammation during obesity is not all bad: Evidence from animal and human studies. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E466–E477. [Google Scholar] [CrossRef] [PubMed]
- Rock, R.B.; Gekker, G.; Hu, S.; Sheng, W.S.; Cheeran, M.; Lokensgard, J.R.; Peterson, P.K. Role of microglia in central nervous system infections. Clin. Microbiol. Rev. 2004, 17, 942–964. [Google Scholar] [CrossRef]
- Liang, X.; Wu, L.; Wang, Q.; Hand, T.; Bilak, M.; McCullough, L.; Andreasson, K. Function of COX-2 and prostaglandins in neurological disease. J. Mol. Neurosci. 2007, 33, 94–99. [Google Scholar] [CrossRef]
- Teismann, P.; Vila, M.; Choi, D.K.; Tieu, K.; Wu, D.C.; Jackson-Lewis, V.; Przedborski, S. COX-2 and neurodegeneration in Parkinson’s disease. Ann. N. Y. Acad. Sci. 2003, 991, 272–277. [Google Scholar] [CrossRef]
- Crews, F.T.; Sarkar, D.K.; Qin, L.; Zou, J.; Boyadjieva, N.; Vetreno, R.P. Neuroimmune Function and the Consequences of Alcohol Exposure. Alcohol. Res. 2015, 37, 331–341, 344–351. [Google Scholar]
- Tak, P.P.; Firestein, G.S. NF-kappaB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Simmons, L.J.; Surles-Zeigler, M.C.; Li, Y.; Ford, G.D.; Newman, G.D.; Ford, B.D. Regulation of inflammatory responses by neuregulin-1 in brain ischemia and microglial cells in vitro involves the NF-kappa B pathway. J. Neuroinflamm. 2016, 13, 237. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.W.; Koppula, S.; Hong, S.S.; Jeon, S.B.; Kwon, J.H.; Hwang, B.Y.; Park, E.J.; Choi, D.K. Regulation of microglia activity by glaucocalyxin-A: Attenuation of lipopolysaccharide-stimulated neuroinflammation through NF-kappaB and p38 MAPK signaling pathways. PLoS ONE 2013, 8, e55792. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.W.; Koppula, S.; Kumar, H.; Park, J.Y.; Kim, I.W.; More, S.V.; Kim, I.S.; Han, S.D.; Kim, S.K.; Yoon, S.H.; et al. alpha-Asarone attenuates microglia-mediated neuroinflammation by inhibiting NF kappa B activation and mitigates MPTP-induced behavioral deficits in a mouse model of Parkinson’s disease. Neuropharmacology 2015, 97, 46–57. [Google Scholar] [CrossRef]
- Kumar, H.; Kim, I.S.; More, S.V.; Kim, B.W.; Bahk, Y.Y.; Choi, D.K. Gastrodin protects apoptotic dopaminergic neurons in a toxin-induced Parkinson’s disease model. Evid. Based Complement. Altern. Med. 2013, 2013, 514095. [Google Scholar] [CrossRef]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- More, S.V.; Kumar, H.; Kim, I.S.; Koppulla, S.; Kim, B.W.; Choi, D.K. Strategic selection of neuroinflammatory models in Parkinson’s disease: Evidence from experimental studies. CNS Neurol. Disord. Drug Targets 2013, 12, 680–697. [Google Scholar] [CrossRef]
- Whitton, P.S. Inflammation as a causative factor in the aetiology of Parkinson’s disease. Br. J. Pharm. 2007, 150, 963–976. [Google Scholar] [CrossRef]
- Chung, Y.C.; Kim, S.R.; Jin, B.K. Paroxetine prevents loss of nigrostriatal dopaminergic neurons by inhibiting brain inflammation and oxidative stress in an experimental model of Parkinson’s disease. J. Immunol. 2010, 185, 1230–1237. [Google Scholar] [CrossRef]
- Lee, K.W.; Im, J.Y.; Woo, J.M.; Grosso, H.; Kim, Y.S.; Cristovao, A.C.; Sonsalla, P.K.; Schuster, D.S.; Jalbut, M.M.; Fernandez, J.R.; et al. Neuroprotective and anti-inflammatory properties of a coffee component in the MPTP model of Parkinson’s disease. Neurotherapeutics 2013, 10, 143–153. [Google Scholar] [CrossRef]
- Mohanasundari, M.; Sabesan, M. Modulating effect of Hypericum perforatum extract on astrocytes in MPTP induced Parkinson’s disease in mice. Eur. Rev. Med. Pharm. Sci. 2007, 11, 17–20. [Google Scholar]
- Jackson-Lewis, V.; Przedborski, S. Protocol for the MPTP mouse model of Parkinson’s disease. Nat. Protoc. 2007, 2, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.W.; Koppula, S.; Kim, J.W.; Lim, H.W.; Hwang, J.W.; Kim, I.S.; Park, P.J.; Choi, D.K. Modulation of LPS-stimulated neuroinflammation in BV-2 microglia by Gastrodia elata: 4-hydroxybenzyl alcohol is the bioactive candidate. J. Ethnopharmacol. 2012, 139, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Karthivashan, G.; Ko, H.M.; Cho, D.Y.; Kim, J.; Cho, D.J.; Ganesan, P.; Su-Kim, I.; Choi, D.K. Aqueous Extract of Dendropanax morbiferus Leaves Effectively Alleviated Neuroinflammation and Behavioral Impediments in MPTP-Induced Parkinson’s Mouse Model. Oxid. Med. Cell Longev. 2018, 2018, 3175214. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, S.-Y.; Kim, I.-S.; Koppula, S.; Park, J.-Y.; Kim, B.-W.; Yoon, S.-H.; Choi, D.-K. 2-Hydroxy-4-Methylbenzoic Anhydride Inhibits Neuroinflammation in Cellular and Experimental Animal Models of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8195. https://doi.org/10.3390/ijms21218195
Song S-Y, Kim I-S, Koppula S, Park J-Y, Kim B-W, Yoon S-H, Choi D-K. 2-Hydroxy-4-Methylbenzoic Anhydride Inhibits Neuroinflammation in Cellular and Experimental Animal Models of Parkinson’s Disease. International Journal of Molecular Sciences. 2020; 21(21):8195. https://doi.org/10.3390/ijms21218195
Chicago/Turabian StyleSong, Soo-Yeol, In-Su Kim, Sushruta Koppula, Ju-Young Park, Byung-Wook Kim, Sung-Hwa Yoon, and Dong-Kug Choi. 2020. "2-Hydroxy-4-Methylbenzoic Anhydride Inhibits Neuroinflammation in Cellular and Experimental Animal Models of Parkinson’s Disease" International Journal of Molecular Sciences 21, no. 21: 8195. https://doi.org/10.3390/ijms21218195
APA StyleSong, S.-Y., Kim, I.-S., Koppula, S., Park, J.-Y., Kim, B.-W., Yoon, S.-H., & Choi, D.-K. (2020). 2-Hydroxy-4-Methylbenzoic Anhydride Inhibits Neuroinflammation in Cellular and Experimental Animal Models of Parkinson’s Disease. International Journal of Molecular Sciences, 21(21), 8195. https://doi.org/10.3390/ijms21218195