Viruses in Cancers of the Digestive System: Active Contributors or Idle Bystanders?



Abstract
:1. Introduction
2. Human Virome: Main Players
2.1. Phages
2.2. Viruses
3. Role of Viruses in the Development of Cancers of the Gastrointestinal System
3.1. Oral Cancer
3.2. Esophageal Cancer
3.3. Gastric Cancer
3.4. Liver Cancer
3.5. Bile Duct Cancer
3.6. Pancreatic Cancer
3.7. Colorectal Cancer
3.7.1. HBV and HCV
3.7.2. HPV
3.7.3. EBV
3.7.4. Cytomegalovirus
3.7.5. John Cunningham Virus
3.8. Anal Cancer
4. Discussion and Future Outlooks
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gillings, M.R.; Paulsen, I.T.; Tetu, S.G. Ecology and evolution of the human microbiota: Fire, farming and antibiotics. Genes 2015, 6, 841–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Yao, M.; Lv, L.; Ling, Z.; Li, L. The Human Microbiota in Health and Disease. Engineering 2017, 3, 71–82. [Google Scholar] [CrossRef]
- Columpsi, P.; Sacchi, P.; Zuccaro, V.; Cima, S.; Sarda, C.; Mariani, M.; Gori, A.; Bruno, R. Beyond the gut bacterial microbiota: The gut virome. J. Med. Virol. 2016, 88, 1467–1472. [Google Scholar] [CrossRef]
- Gregory, A.C.; Zablocki, O.; Zayed, A.A.; Howell, A.; Bolduc, B.; Sullivan, M.B. The Gut Virome Database Reveals Age-Dependent Patterns of Virome Diversity in the Human Gut. Cell Host Microbe 2020. [Google Scholar] [CrossRef] [PubMed]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoyles, L.; McCartney, A.L.; Neve, H.; Gibson, G.R.; Sanderson, J.D.; Heller, K.J.; van Sinderen, D. Characterization of virus-like particles associated with the human faecal and caecal microbiota. Res. Microbiol. 2014, 165, 803–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Mejía, J.L.; Muhammed, M.K.; Kot, W.; Neve, H.; Franz, C.M.A.P.; Hansen, L.H.; Vogensen, F.K.; Nielsen, D.S. Optimizing protocols for extraction of bacteriophages prior to metagenomic analyses of phage communities in the human gut. Microbiome 2015, 3, 64. [Google Scholar] [CrossRef] [Green Version]
- Neil, J.A.; Cadwell, K. The Intestinal Virome and Immunity. J. Immunol. 2018, 201, 1615–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M. Origins and evolution of viruses of eukaryotes: The ultimate modularity. Virology 2015, 479–480, 2–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norman, J.M.; Handley, S.A.; Virgin, H.W. Kingdom-agnostic metagenomics and the importance of complete characterization of enteric microbial communities. Gastroenterology 2014, 146, 1459–1469. [Google Scholar] [CrossRef]
- Kernbauer, E.; Ding, Y.; Cadwell, K. An enteric virus can replace the beneficial function of commensal bacteria. Nature 2014, 516, 94–98. [Google Scholar] [CrossRef]
- Yang, J.Y.; Kim, M.S.; Kim, E.; Cheon, J.H.; Lee, Y.S.; Kim, Y.; Lee, S.H.; Seo, S.U.; Shin, S.H.; Choi, S.S.; et al. Enteric Viruses Ameliorate Gut Inflammation via Toll-like Receptor 3 and Toll-like Receptor 7-Mediated Interferon-β Production. Immunity 2016, 44, 889–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krump, N.A.; You, J. Molecular mechanisms of viral oncogenesis in humans. Nat. Rev. Microbiol. 2018, 16, 684–698. [Google Scholar] [CrossRef]
- Galtier, M.; De Sordi, L.; Sivignon, A.; de Vallée, A.; Maura, D.; Neut, C.; Rahmouni, O.; Wannerberger, K.; Darfeuille-Michaud, A.; Desreumaux, P.; et al. Bacteriophages targeting adherent invasive Escherichia coli strains as a promising new treatment for Crohn’s disease. J. Crohn’s Colitis 2017, 11, 840–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, T.; Lu, X.-J.; Zhang, Y.; Cheung, C.P.; Lam, S.; Zhang, F.; Tang, W.; Ching, J.Y.L.; Zhao, R.; Chan, P.K.S.; et al. Gut mucosal virome alterations in ulcerative colitis. Gut 2019, 68, 1169–1179. [Google Scholar] [CrossRef] [Green Version]
- Emlet, C.; Ruffin, M.; Lamendella, R. Enteric Virome and Carcinogenesis in the Gut. Dig. Dis. Sci. 2020, 65, 852–864. [Google Scholar] [CrossRef] [Green Version]
- Gethings-Behncke, C.; Coleman, H.G.; Jordao, H.W.T.; Longley, D.B.; Crawford, N.; Murray, L.J.; Kunzmann, A.T. Fusobacterium nucleatum in the colorectum and its association with cancer risk and survival: A systematic review and meta-analysis. Cancer Epidemiol. Biomark. Prev. 2020, 29, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Gogokhia, L.; Buhrke, K.; Bell, R.; Hoffman, B.; Brown, D.G.; Hanke-Gogokhia, C.; Ajami, N.J.; Wong, M.C.; Ghazaryan, A.; Valentine, J.F.; et al. Expansion of Bacteriophages Is Linked to Aggravated Intestinal Inflammation and Colitis. Cell Host Microbe 2019, 25, 285–299.e8. [Google Scholar] [CrossRef] [Green Version]
- Brüssow, H.; Canchaya, C.; Hardt, W.-D. Phages and the Evolution of Bacterial Pathogens: From Genomic Rearrangements to Lysogenic Conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maronek, M.; Link, R.; Ambro, L.; Gardlik, R. Phages and Their Role in Gastrointestinal Disease: Focus on Inflammatory Bowel Disease. Cells 2020, 18, 1013. [Google Scholar] [CrossRef] [Green Version]
- Brown-Jaque, M.; Rodriguez Oyarzun, L.; Cornejo-Sánchez, T.; Martín-Gómez, M.T.; Gartner, S.; de Gracia, J.; Rovira, S.; Alvarez, A.; Jofre, J.; González-López, J.J.; et al. Detection of Bacteriophage Particles Containing Antibiotic Resistance Genes in the Sputum of Cystic Fibrosis Patients. Front. Microbiol. 2018, 9, 856. [Google Scholar] [CrossRef] [PubMed]
- Mirold, S.; Rabsch, W.; Rohde, M.; Stender, S.; Tschape, H.; Russmann, H.; Igwe, E.; Hardt, W.-D. Isolation of a temperate bacteriophage encoding the type III effector protein SopE from an epidemic Salmonella typhimurium strain. Proc. Natl. Acad. Sci. USA 1999, 96, 9845–9850. [Google Scholar] [CrossRef] [Green Version]
- Goh, S.; Chang, B.J.; Riley, T.V. Effect of phage infection on toxin production by Clostridium difficile. J. Med. Microbiol. 2005, 54, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, F.; Culp, W.D.; Massey, R.; Egevad, L.; Garland, D.; Persson, M.A.A.; Pisa, P. Tumor specific phage particles promote tumor regression in a mouse melanoma model. Cancer Immunol. Immunother. 2007, 56, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, F.; Tsagozis, P.; Lundberg, K.; Parsa, R.; Mangsbo, S.M.; Persson, M.A.A.; Harris, R.A.; Pisa, P. Tumor-Specific Bacteriophages Induce Tumor Destruction through Activation of Tumor-Associated Macrophages. J. Immunol. 2009, 182, 3105–3111. [Google Scholar] [CrossRef] [Green Version]
- Rampoldi, A.; Crooke, S.N.; Preininger, M.K.; Jha, R.; Maxwell, J.; Ding, L.; Spearman, P.; Finn, M.G.; Xu, C. Targeted Elimination of Tumorigenic Human Pluripotent Stem Cells Using Suicide-Inducing Virus-like Particles. ACS Chem. Biol. 2018, 13, 2329–2338. [Google Scholar] [CrossRef] [PubMed]
- Dabrowska, K.; Opolski, A.; Wietrzyk, J.; Switala-Jelen, K.; Boratynski, J.; Nasulewicz, A.; Lipinska, L.; Chybicka, A.; Kujawa, M.; Zabel, M.; et al. Antitumor activity of bacteriophages in murine experimental cancer models caused possibly by inhibition of β3 integrin signaling pathway. Acta Virol. 2004, 48, 241–248. [Google Scholar]
- Da̧browska, K.; Opolski, A.; Wietrzyk, J.; Nevozhay, D.; Szczaurska, K.; Świtała-Jeleń, K.; Boratyński, J.; Górski, A. Activity of bacteriophages in murine tumor models depends on the route of phage administration. Oncol. Res. 2005, 15, 183–187. [Google Scholar]
- Shan, J.; Ramachandran, A.; Thanki, A.M.; Vukusic, F.B.I.; Barylski, J.; Clokie, M.R.J. Bacteriophages are more virulent to bacteria with human cells than they are in bacterial culture; Insights from HT-29 cells. Sci. Rep. 2018, 8, 5091. [Google Scholar] [CrossRef]
- Zárate, S.; Taboada, B.; Yocupicio-Monroy, M.; Arias, C.F. Human Virome. Arch. Med. Res. 2017, 48, 701–716. [Google Scholar] [CrossRef]
- Landskron, G.; De La Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Simone, V.; Franzè, E.; Ronchetti, G.; Colantoni, A.; Fantini, M.C.; Di Fusco, D.; Sica, G.S.; Sileri, P.; MacDonald, T.T.; Pallone, F.; et al. Th17-type cytokines, IL-6 and TNF-α synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene 2015, 34, 3493–3503. [Google Scholar] [CrossRef]
- Morales-Sánchez, A.; Fuentes-Pananá, E.M. Human viruses and cancer. Viruses 2014, 6, 4047–4079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, M.; Broséus, J.; Feugier, P.; Thieblemont, C.; Laurent, B.; Danese, S.; Arnone, D.; Ndiaye, N.C.; Kokten, T.; Houlgatte, R.; et al. Characteristics Of Lymphoma In Patients With Inflammatory Bowel Disease: A Systematic Review. J. Crohn’s Colitis 2020. [Google Scholar] [CrossRef]
- Pal, A.; Kundu, R. Human Papillomavirus E6 and E7: The Cervical Cancer Hallmarks and Targets for Therapy. Front. Microbiol. 2020, 10, 3116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maden, C.; Beckmann, A.M.; Thomas, D.B.; Mcknight, B.; Sherman, K.J.; Ashley, R.L.; Corey, L.; Daling, J.R. Human papillomaviruses, herpes simplex viruses, and the risk of oral cancer in men. Am. J. Epidemiol. 1992, 135, 1093–1102. [Google Scholar] [CrossRef]
- Jalouli, J.; Jalouli, M.M.; Sapkota, D.; Ibrahim, S.O.; Larsson, P.A.; Sand, L. Human papilloma virus, herpes simplex virus and epstein barr virus in oral squamous cell carcinoma from eight different countries. Anticancer Res. 2012, 32, 571–580. [Google Scholar]
- Bjørge, T.; Hakulinen, T.; Engeland, A.; Jellum, E.; Koskela, P.; Lehtinen, M.; Luostarinen, T.; Paavonen, J.; Sapp, M.; Schiller, J.; et al. A prospective, seroepidemiological study of the role of human papillomavirus in esophageal cancer in norway. Cancer Res. 1997, 57, 3989–3992. [Google Scholar]
- Zhang, S.; Guo, L.; Chen, Q.; Zhang, M.; Liu, S.; Quan, P.; Lu, J. The association between human papillomavirus 16 and esophageal cancer in Chinese population: A meta-analysis. BMC Cancer 2015, 15, 99. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Tian, X.; Liu, F.; Zhao, Y.; Sun, M.; Chen, D.; Lu, C.; Wang, Z.; Shi, X.; Zhang, Q.; et al. Detection of HPV DNA in esophageal cancer specimens from different regions and ethnic groups: A descriptive study. BMC Cancer 2010, 10, 19. [Google Scholar] [CrossRef] [Green Version]
- Kirgan, D.; Manalo, P.; Hall, M.; Mcgregor, B. Association of Human Papillomavirus and Colon Neoplasms. Arch. Surg. 1990, 125, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Leu, S.; Chiang, H.; Fung, C.; Liu, W. Human Papillomavirus Type 18 in Colorectal Cancer. J. Microbiol. Immunol. Infect. 2001, 34, 87–91. [Google Scholar] [PubMed]
- Alexandrou, A.; Dimitriou, N.; Levidou, G.; Griniatsos, J.; Sougioultzis, S.; Korkolopoulou, P.; Felekouras, E.; Pikoulis, E.; Diamantis, T.; Tsigris, C.; et al. The Incidence of HPV Infection in Anal Cancer Patients in Greece. Acta Gastroenterol. Belg. 2014, 77, 213–216. [Google Scholar] [PubMed]
- Kabarriti, R.; Brodin, N.P.; Ohri, N.; Narang, R.; Huang, R.; Chuy, J.W.; Rajdev, L.N.; Kalnicki, S.; Guha, C.; Garg, M.K. Human papillomavirus, radiation dose and survival of patients with anal cancer. Acta Oncol. (Madr.) 2019, 58, 1745–1751. [Google Scholar] [CrossRef]
- Muresu, N.; Sotgiu, G.; Saderi, L.; Sechi, I.; Cossu, A.; Marras, V.; Meloni, M.; Martinelli, M.; Cocuzza, C.; Tanda, F.; et al. Distribution of hpv genotypes in patients with a diagnosis of anal cancer in an Italian region. Int. J. Environ. Res. Public Health 2020, 17, 4516. [Google Scholar] [CrossRef] [PubMed]
- Awerkiew, S.; Bollschweiler, E.; Metzger, R.; Schneider, P.M.; Hölscher, A.H.; Pfister, H. Esophageal cancer in germany is associated with Epstein-Barr-virus but not with papillomaviruses. Med. Microbiol. Immunol. 2003, 192, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Martínez-López, J.L.E.; Torres, J.; Camorlinga-Ponce, M.; Mantilla, A.; Leal, Y.A.; Fuentes-Pananá, E.M. Evidence of Epstein-Barr virus association with gastric cancer and non-atrophic gastritis. Viruses 2014, 6, 301–318. [Google Scholar] [CrossRef] [Green Version]
- Corallo, S.; Fucà, G.; Morano, F.; Salati, M.; Spallanzani, A.; Gloghini, A.; Volpi, C.C.; Trupia, D.V.; Lobefaro, R.; Guarini, V.; et al. Clinical Behavior and Treatment Response of Epstein-Barr Virus-Positive Metastatic Gastric Cancer: Implications for the Development of Future Trials. Oncologist 2020, 25, 780–786. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Wu, B.A.; Zeng, Y.M.; Chen, G.C.; Li, X.X.; Chen, J.T.; Guo, Y.W.; Li, M.H.; Zeng, Y. Epstein-Barr virus in hepatocellular carcinogenesis. World J. Gastroenterol. 2004, 10, 3409–3413. [Google Scholar] [CrossRef]
- Song, L.-B.; Zhang, X.; Zhang, C.-Q.; Zhang, Y.; Pan, Z.-Z.; Liao, W.-T.; Li, M.-Z.; Zeng, M.-S. Infection of Epstein-Barr Virus in Colorectal Cancer in Chinese. Chin. J. Cancer 2006, 25, 1356–1360. [Google Scholar]
- Fiorina, L.; Ricotti, M.; Vanoli, A.; Luinetti, O.; Dallera, E.; Riboni, R.; Paolucci, S.; Brugnatelli, S.; Paulli, M.; Pedrazzoli, P.; et al. Systematic analysis of human oncogenic viruses in colon cancer revealed EBV latency in lymphoid infiltrates. Infect. Agent. Cancer 2014, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- Laghi, L.; Randolph, A.E.; Chauhan, D.P.; Marra, G.; Major, E.O.; Neel, J.V.; Boland, C.R. JC virus DNA is present in the mucosa of the human colon and in colorectal cancers. Proc. Natl. Acad. Sci. USA 1999, 96, 7484–7489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, R.; Murai, Y.; Tsuneyama, K.; Abdel-Aziz, H.O.; Nomoto, K.; Takahashi, H.; Cheng, C.M.; Kuchina, T.; Harman, B.V.; Takano, Y. Detection of JC virus DNA sequences in colorectal cancers in Japan. Virchows Arch. 2005, 447, 723–730. [Google Scholar] [CrossRef]
- Jung, W.T.; Li, M.S.; Goel, A.; Boland, C.R. JC virus T-antigen expression in sporadic adenomatous polyps of the colon. Cancer 2008, 112, 1028–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokita, H.; Murai, S.; Kamitsukasa, H.; Yagura, M.; Harada, H.; Takahashi, M.; Okamoto, H. High TT virus load as an independent factor associated with the occurrence of hepatocellular carcinoma among patients with hepatitis C virus-related chronic liver disease. J. Med. Virol. 2002, 67, 501–509. [Google Scholar] [CrossRef]
- Iloeje, U.H.; Yang, H.I.; Jen, C.L.; Su, J.; Wang, L.Y.; You, S.L.; Lu, S.N.; Chen, C.J. Risk of pancreatic cancer in chronic hepatitis B virus infection: Data from the REVEAL-HBV cohort study. Liver Int. 2010, 30, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhao, Y.; Li, B.; Huang, J.; Wu, L.; Xu, D.; Yang, J.; He, J. Hepatitis viruses infection and risk of intrahepatic cholangiocarcinoma: Evidence from a meta-analysis. BMC Cancer 2012, 12, 289. [Google Scholar] [CrossRef] [Green Version]
- Hassan, M.M.; Li, D.; El-Deeb, A.S.; Wolff, R.A.; Bondy, M.L.; Davila, M.; Abbruzzese, J.L. Association between hepatitis B virus and pancreatic cancer. J. Clin. Oncol. 2008, 26, 4557–4562. [Google Scholar] [CrossRef]
- Su, F.H.; Le, T.N.; Muo, C.H.; Te, S.A.; Sung, F.C.; Yeh, C.C. Chronic hepatitis B virus infection associated with increased colorectal cancer risk in Taiwanese population. Viruses 2020, 12, 97. [Google Scholar] [CrossRef] [Green Version]
- Dimberg, J.; Hong, T.; Skarstedt, M.; Löfgren, S.; Zar, N.; Matussek, A. Detection of Cytomegalovirus DNA in Colorectal Tissue From Swedish and Vietnamese Patients With Colorectal Cancer. Anticancer Res. 2013, 33, 4947–4950. [Google Scholar]
- Chen, H.P.; Jiang, J.K.; Chen, C.Y.; Yang, C.Y.; Chen, Y.C.; Lin, C.H.; Chou, T.Y.; Cho, W.L.; Chan, Y.J. Identification of human cytomegalovirus in tumour tissues of colorectal cancer and its association with the outcome of non-elderly patients. J. Gen. Virol. 2016, 97, 2411–2420. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.P.; Jiang, J.K.; Chan, C.H.; Teo, W.H.; Yang, C.Y.; Chen, Y.C.; Chou, T.Y.; Lin, C.H.; Chan, Y.J. Genetic polymorphisms of the human cytomegalovirus UL144 gene in colorectal cancer and its association with clinical outcome. J. Gen. Virol. 2015, 96, 3613–3623. [Google Scholar] [CrossRef]
- Jin, F.; Vajdic, C.M.; Law, M.; Amin, J.; Van Leeuwen, M.; McGregor, S.; Poynten, I.M.; Templeton, D.J.; Grulich, A.E. Incidence and time trends of anal cancer among people living with HIV in Australia. AIDS 2019, 33, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Colón-López, V.; Shiels, M.S.; Machin, M.; Ortiz, A.P.; Strickler, H.; Castle, P.E.; Pfeiffer, R.M.; Engels, E.A. Anal cancer risk among people with HIV infection in the United States. J. Clin. Oncol. 2018, 36, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Grew, D.; Bitterman, D.; Leichman, C.G.; Leichman, L.; Sanfilippo, N.; Moore, H.G.; Du, K. HIV infection is associated with poor outcomes for patients with anal cancer in the highly active antiretroviral therapy era. Dis. Colon Rectum 2015, 58, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [Green Version]
- Gupta, B.; Johnson, N.W. Systematic review and meta-analysis of association of smokeless tobacco and of betel quid without tobacco with incidence of oral cancer in south asia and the pacific. PLoS ONE 2014, 9, e113385. [Google Scholar] [CrossRef]
- Conway, D.I.; Purkayastha, M.; Chestnutt, I.G. The changing epidemiology of oral cancer: Definitions, trends, and risk factors. Br. Dent. J. 2018, 225, 867. [Google Scholar] [CrossRef] [Green Version]
- Sand, L.; Jalouli, J. Viruses and oral cancer. Is there a link? Microbes Infect. 2014, 16, 371–378. [Google Scholar] [CrossRef]
- Kouvousi, M.; Xesfyngi, D.; Tsimplaki, E.; Argyri, E.; Ioannidou, G.; Ploxorou, M.; Lazaris, A.; Patsouris, E.; Panotopoulou, E. Prevalence of human papillomavirus in 45 greek patients with oral cancer. J. Oncol. 2013, 2013, 756510. [Google Scholar] [CrossRef] [Green Version]
- González-Ramírez, I.; Irigoyen-Camacho, M.E.; Ramírez-Amador, V.; Lizano-Soberón, M.; Carrillo-García, A.; García-Carrancá, A.; Sánchez-Pérez, Y.; Méndez-Martínez, R.; Granados-García, M.; Ruíz-Godoy, L.M.; et al. Association between age and high-risk human papilloma virus in Mexican oral cancer patients. Oral Dis. 2013, 19, 796–804. [Google Scholar] [CrossRef]
- Chaitanya, N.; Allam, N.; Gandhi Babu, D.; Waghray, S.; Badam, R.; Lavanya, R. Systematic meta-analysis on association of human papilloma virus and oral cancer. J. Cancer Res. Ther. 2016, 12, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Kane, S.; Patil, V.M.; Noronha, V.; Joshi, A.; Dhumal, S.; D’Cruz, A.; Bhattacharjee, A.; Prabhash, K. Predictivity of human papillomavirus positivity in advanced oral cancer. Indian J. Cancer 2015, 52, 403–405. [Google Scholar]
- Sritippho, T.; Pongsiriwet, S.; Lertprasertsuke, N.; Buddhachat, K.; Sastraruji, T.; Iamaroon, A. p16—A Possible Surrogate Marker for High-Risk Human Papillomaviruses in Oral Cancer? Asian Pac. J. Cancer Prev. 2016, 17, 4049–4057. [Google Scholar]
- Chiba, I.; Shindoh, M.; Yasuda, M.; Yamazaki, Y.; Amemiya, A.; Sato, Y.; Fujinaga, K.; Notani, K.; Fukuda, H. Mutations in the p53 gene and human papillomavirus infection as significant prognostic factors in squamous cell carcinomas of the oral cavity. Oncogene 1996, 12, 1663–1668. [Google Scholar] [PubMed]
- Cutilli, T.; Leocata, P.; Dolo, V.; Altobelli, E. Association between p53 status, human papillomavirus infection, and overall survival in advanced oral cancer after resection and combination systemic treatment. Br. J. Oral Maxillofac. Surg. 2016, 54, 198–202. [Google Scholar] [CrossRef]
- Mundi, N.; Prokopec, S.D.; Ghasemi, F.; Warner, A.; Patel, K.; MacNeil, D.; Howlett, C.; Stecho, W.; Plantinga, P.; Pinto, N.; et al. Genomic and human papillomavirus profiling of an oral cancer cohort identifies TP53 as a predictor of overall survival. Cancers Head Neck 2019, 4, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rustgi, A.K.; El-Serag, H.B. Esophageal carcinoma. N. Engl. J. Med. 2014, 371, 2499–2509. [Google Scholar] [CrossRef]
- Syrjänen, K.; Pyrhönen, S.; Aukee, S.; Koskela, E. Squamous cell papilloma of the esophagus: A tumour probably caused by human papilloma virus (HPV). Diagn. Histopathol. 1982, 5, 291–296. [Google Scholar]
- Lagergren, J.; Wang, Z.; Bergstro, R.; Dillner, J.; Nyre, O. Human Papillomavirus Infection and Esophageal Cancer: A Nationwide Seroepidemiologic Case—Control Study in Sweden. J. Natl. Cancer Inst. 1999, 91, 156–162. [Google Scholar] [CrossRef] [Green Version]
- Dąbrowski, A.; Kwaśniewski, W.; Skoczylas, T.; Bednarek, W.; Kuźma, D.; Goździcka-józefiak, A. Incidence of human papilloma virus in esophageal squamous cell carcinoma in patients from the Lublin region. World J. Gastroenterol. WJG 2012, 18, 5739–5744. [Google Scholar]
- Li, T.; Lu, Z.; Chen, K.; Guo, M.; Xing, H.; Mei, Q.; Yang, H.; Lechner, J.F.; Ke, Y. Human papillomavirus type 16 is an important infectious factor in the high incidence of esophageal cancer in Anyang area of China (HPV) infection in the pathogenesis of esophageal carcin-. Carcinogenesis 2001, 22, 929–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadpour, B.; Rouhi, S.; Khodabandehloo, M. Prevalence and Association of Human Papillomavirus with Esophageal Squamous Cell Carcinoma in Iran: A Systematic Review and Meta-Analysis. Iran. J. Public Health 2019, 48, 1215–1226. [Google Scholar]
- Baş, Y.; Aker, V.; Aylin, G. Effect of high-risk human papillomavirus in esophageal squamous cell carcinoma in Somalian and Turkish cases. Pathog. Dis. 2019, 77, ftz047. [Google Scholar] [CrossRef]
- Castillo, A.; Aguayo, F.; Koriyama, C.; Torres, M.; Carrascal, E.; Corvalan, A.; Roblero, J.P.; Naquira, C.; Palma, M.; Backhouse, C.; et al. Human papillomavirus in esophageal squamous cell carcinoma in Colombia and Chile. World J Gastroenterol. WJG 2006, 12, 6188–6192. [Google Scholar] [CrossRef]
- Da Costa, A.; Fregnani, J.; Pastrez, P.; Mariano, V.; Neto, C.; Guimarães, D.; de Oliveira, K.; Neto, S.; Nunes, E.; Ferreira, S.; et al. Prevalence of high risk HPV DNA in esophagus is high in Brazil but not related to esophageal squamous cell carcinoma. Histol. Histopathol. 2018, 33, 357–363. [Google Scholar]
- Koshiol, J.; Kreimer, A.R. Lessons from Australia: HPV is not a major risk factor foresophageal squamous cell carcinoma. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1889–1892. [Google Scholar] [CrossRef] [Green Version]
- Antunes, L.; Prolla, J.C.; Lopes, A.D.B.; Pires, M.; Carlos, L.; Antunes, M.; Prolla, J.C. No evidence of HPV DNA in esophageal squamous cell carcinoma in a population of Southern Brazil. World J. Gastroenterol. 2013, 19, 6598–6603. [Google Scholar] [CrossRef]
- Antonsson, A.; Nancarrow, D.J.; Brown, I.S.; Green, A.C.; Drew, P.A.; Watson, D.I.; Hayward, N.K.; Whiteman, D.C. High-risk human papillomavirus in esophageal squamous cell carcinoma. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2080–2087. [Google Scholar] [CrossRef] [Green Version]
- Chang, F.; Syrjänen, S.; Shen, Q.; Cintorino, M.; Santopietro, R.; Tosi, P.; Syrjänen, K. Evaluation of HPV, CMV, HSV and EBV in Esophageal Squamous Cell Carcinomas From a High-Incidence Area of China. Anticancer Res. 2000, 20, 3935–3940. [Google Scholar]
- Xi, R.; Zhang, X.; Chen, X.; Pan, S.; Hui, B.; Zhang, L.; Fu, S.; Li, X.; Zhang, X.; Gong, T.; et al. Human papillomavirus 16 infection predicts poor outcome in patients with esophageal squamous cell carcinoma. OncoTargets Ther. 2015, 8, 573–581. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.S.; Chow, K.C.; Wu, Y.C.; Li, W.Y.; Huang, M.H. Detection of Epstein-Barr virus in esophageal squamous cell carcinoma in Taiwan. Am. J. Gastroenterol. 1999, 94, 2834–2839. [Google Scholar] [CrossRef] [PubMed]
- Hong, T.; Shimada, Y.; Kano, M.; Kaganoi, J.; Uchida, S.; Komoto, I.; Yamabe, H.; Imamura, M. The Epstein-Barr virus is rarely associated with esophageal cancer. Int. J. Mol. Med. 2000, 5, 363–368. [Google Scholar] [CrossRef]
- Yanai, H.; Hirano, A.; Matsusaki, K.; Kawano, T.; Miura, O.; Yoshida, T.; Okita, K.; Shimizu, N. Epstein-Barr Virus Association Is Rare in Esophageal Squamous Cell Carcinoma. Int. J. Gastrointest. Cancer 2003, 33, 165–170. [Google Scholar] [CrossRef]
- Awerkiew, S.; Zur Hausen, A.; Baldus, S.E.; Hölscher, A.H.; Sidorenko, S.I.; Kutsev, S.I.; Pfister, H.J. Presence of Epstein-Barr virus in esophageal cancer is restricted to tumor infiltrating lymphocytes. Med. Microbiol. Immunol. 2005, 194, 187–191. [Google Scholar] [CrossRef]
- Sarbia, M.; Zur Hausen, A.; Feith, M.; Geddert, H.; Von Rahden, B.H.A.; Langer, R.; Von Weyhern, C.; Siewert, J.R.; Höfler, H.; Stein, H.J. Esophageal (Barrett’s) adenocarcinoma is not associated with Epstein-Barr virus infection: An analysis of 162 cases. Int. J. Cancer 2005, 117, 698–700. [Google Scholar] [CrossRef]
- Sunpaweravong, S.; Mitarnun, W.; Puttawibul, P. Absence of Epstein-Barr virus in esophageal squamous cell carcinoma. Dis. Esophagus 2005, 18, 398–399. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Awuti, I.; Deng, Y.; Li, D.; Niyazi, M.; Aniwar, J.; Sheyhidin, I.; Lu, G.; Li, G.; Zhang, L. Human papillomavirus promotes esophageal squamous cell carcinoma by regulating DNA methylation and expression of HLA-DQB1. Asia. Pac. J. Clin. Oncol. 2014, 10, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Zang, B.; Huang, G.; Wang, X.; Zheng, S. HPV-16 E6 promotes cell growth of esophageal cancer via downregulation of miR-125b and activation of Wnt/β-catenin signaling pathway. Int. J. Clin. Exp. Pathol. 2015, 8, 13687–13694. [Google Scholar]
- Rampias, T.; Boutati, E.; Pectasides, E.; Sasaki, C.; Kountourakis, P.; Weinberger, P.; Psyrri, A. Activation of Wnt signaling pathway by human papillomavirus E6 and E7 oncogenes in HPV16-positive oropharyngeal squamous carcinoma cells. Mol. Cancer Res. 2010, 8, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Rajendra, S.; Yang, T.; Xuan, W.; Sharma, P.; Pavey, D.; Lee, C.S.; Le, S.; Collins, J.; Wang, B. Active human papillomavirus involvement in Barrett’s dysplasia and oesophageal adenocarcinoma is characterized by wild-type p53 and aberrations of the retinoblastoma protein pathway. Int. J. Cancer 2017, 141, 2037–2049. [Google Scholar] [CrossRef]
- Yang, J.; Wu, H.; Wei, S.; Xiong, H.; Fu, X.; Qi, Z.; Jiang, Q.; Li, W.; Hu, G.; Yuan, X.; et al. HPV seropositivity joints with susceptibility loci identified in GWASs at apoptosis associated genes to increase the risk of Esophageal Squamous Cell Carcinoma (ESCC). BMC Cancer 2014, 14, 501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Liu, B.; Li, W.; Xiong, H.; Qiu, H.; Fu, Q.; Chen, B.; Hu, G.; Yuan, X. Association of p53 and MDM2 polymorphisms with risk of human papillomavirus (HPV)-related esophageal squamous cell carcinoma (ESCC). Cancer Epidemiol. 2013, 37, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, W.; Liu, W.; Mao, Y.; Fu, Z.; Liu, J.; Huang, W.; Zhang, Z.; An, D.; Li, B. Human papillomavirus infection increases the chemoradiation response of esophageal squamous cell carcinoma based on P53 mutation. Radiother. Oncol. 2017, 124, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, A.M.; Fregnani, J.H.T.G.; Pastrez, P.R.A.; Mariano, V.S.; Silva, E.M.; Neto, C.S.; Guimarães, D.P.; Villa, L.L.; Sichero, L.; Syrjanen, K.J.; et al. HPV infection and p53 and p16 expression in esophageal cancer: Are they prognostic factors? Infect. Agent. Cancer 2017, 12, 54. [Google Scholar] [CrossRef]
- Zhang, D.H.; Chen, J.Y.; Hong, C.Q.; Yi, D.Q.; Wang, F.; Cui, W. High-risk human papillomavirus infection associated with telomere elongation in patients with esophageal squamous cell carcinoma with poor prognosis. Cancer 2014, 120, 2673–2683. [Google Scholar] [CrossRef]
- Snietura, M.; Waniczek, D.; Piglowski, W.; Kopec, A.; Nowakowska-Zajdel, E.; Lorenc, Z.; Muc-Wierzgon, M. Potential role of human papilloma virus in the pathogenesis of gastric cancer. World J. Gastroenterol. 2014, 20, 6632–6637. [Google Scholar] [CrossRef]
- Zeng, Z.-M.; Luo, F.-F.; Zou, L.-X.; He, R.-Q.; Pan, D.-H.; Chen, X.; Xie, T.-T.; Li, Y.-Q.; Peng, Z.-G.; Chen, G. Human papillomavirus as a potential risk factor for gastric cancer: A meta-analysis of 1917 cases. OncoTargets Ther. 2016, 9, 7105–7114. [Google Scholar] [CrossRef] [Green Version]
- Pembrey, L.; Raynor, P.; Griffiths, P.; Chaytor, S.; Wright, J.; Hall, A.J. Seroprevalence of cytomegalovirus, Epstein Barr virus and varicella zoster virus among pregnant women in Bradford: A cohort study. PLoS ONE 2013, 8, e81881. [Google Scholar] [CrossRef] [Green Version]
- Dowd, J.B.; Palermo, T.; Brite, J.; McDade, T.W.; Aiello, A. Seroprevalence of Epstein-Barr Virus Infection in U.S. Children Ages 6–19, 2003–2010. PLoS ONE 2013, 8, e64921. [Google Scholar] [CrossRef] [Green Version]
- Rodriquenz, M.G.; Roviello, G.; D’Angelo, A.; Lavacchi, D.; Roviello, F.; Polom, K. MSI and EBV Positive Gastric Cancer’s Subgroups and Their Link With Novel Immunotherapy. J. Clin. Med. 2020, 9, 1427. [Google Scholar] [CrossRef]
- Aversa, J.G.; Song, M.; Hu, N.; Goldstein, A.M.; Hewitt, S.M.; Gulley, M.L.; Dawsey, S.; Camargo, M.C.; Taylor, P.R.; Rabkin, C.S. Low epstein–barr virus prevalence in cardia gastric cancer among a high-incidence Chinese population. Dig. Dis. Sci. 2020. [Google Scholar] [CrossRef]
- Yuan, X.Y.; Wang, M.Y.; Wang, X.Y.; Chang, A.Y.; Li, J. Non-detection of Epstein-Barr virus and Human Papillomavirus in a region of high gastric cancer risk indicates a lack of a role for these viruses in gastric carcinomas. Genet. Mol. Biol. 2013, 36, 183–184. [Google Scholar] [CrossRef]
- De Souza, C.R.T.; Almeida, M.C.A.; Khayat, A.S.; Da Silva, E.L.; Soares, P.C.; Chaves, L.C.; Burbano, R.M.R. Association between Helicobacter pylori, Epstein-Barr virus, human papillomavirus and gastric adenocarcinomas. World J. Gastroenterol. 2018, 24, 4928–4938. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Zhao, X.X.; Tan, C.; Zhang, Z.G.; Jiang, Y.; Chen, J.N.; Wei, H.B.; Xue, L.; Li, H.G.; Du, H.; et al. Epstein-Barr virus latent membrane protein 2A suppresses the expression of HER2 via a pathway involving TWIST and YB-1 in Epstein-Barr virus-associated gastric carcinomas. Oncotarget 2015, 6, 207–220. [Google Scholar] [CrossRef]
- Yoon, H.H.; Shi, Q.; Sukov, W.R.; Wiktor, A.E.; Khan, M.; Sattler, C.A.; Grothey, A.; Wu, T.T.; Diasio, R.B.; Jenkins, R.B.; et al. Association of HER2/ErbB2 expression and gene amplification with pathologic features and prognosis in esophageal adenocarcinomas. Clin. Cancer Res. 2012, 18, 546–554. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, M.; Stoss, O.; Shi, D.; Büttner, R.; Van De Vijver, M.; Kim, W.; Ochiai, A.; Rüschoff, J.; Henkel, T. Assessment of a HER2 scoring system for gastric cancer: Results from a validation study. Histopathology 2008, 52, 797–805. [Google Scholar] [CrossRef]
- Kreutzfeldt, J.; Rozeboom, B.; Dey, N.; De, P. The Trastuzumab Era: Current and Upcoming Targeted HER2+ Breast Cancer Therapies. Am. J. Cancer Res. 2020, 10, 1045–1067. [Google Scholar]
- Constanza Camargo, M.; Kim, W.H.; Chiaravalli, A.M.; Kim, K.M.; Corvalan, A.H.; Matsuo, K.; Yu, J.; Sung, J.J.Y.; Herrera-Goepfert, R.; Meneses-Gonzalez, F.; et al. Improved survival of gastric cancer with tumour Epstein-Barr virus positivity: An international pooled analysis. Gut 2014, 63, 236–243. [Google Scholar] [CrossRef]
- Wang, J.; Zheng, X.; Qin, Z.; Wei, L.; Lu, Y.; Peng, Q.; Gao, Y.; Zhang, X.; Zhang, X.; Li, Z.; et al. Epstein–Barr virus miR-BART3-3p promotes tumorigenesis by regulating the senescence pathway in gastric cancer. J. Biol. Chem. 2019, 294, 4854–4866. [Google Scholar] [CrossRef] [Green Version]
- Maucort-Boulch, D.; de Martel, C.; Franceschi, S.; Plummer, M. Fraction and incidence of liver cancer attributable to hepatitis B and C viruses worldwide. Int. J. Cancer 2018, 142, 2471–2477. [Google Scholar] [CrossRef] [Green Version]
- Bosetti, C.; Turati, F.; La Vecchia, C. Hepatocellular carcinoma epidemiology. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 753–770. [Google Scholar] [CrossRef]
- Xia, J.; Jiang, S.C.; Peng, H.J. Association between liver fluke infection and hepatobiliary pathological changes: A systematic review and meta-analysis. PLoS ONE 2015, 10, e0132673. [Google Scholar] [CrossRef] [Green Version]
- Arzumanyan, A.; Reis, H.M.G.P.V.; Feitelson, M.A. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat. Rev. Cancer 2013, 13, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Zemel, R.; Issachar, A.; Tur-Kaspa, R. The Role of Oncogenic Viruses in the Pathogenesis of Hepatocellular Carcinoma. Clin. Liver Dis. 2011, 15, 261–279. [Google Scholar] [CrossRef]
- Scinicariello, F.; Sato, T.; Lee, C.S.; Hsu, H.C.; Chan, T.S.; Tyring, S.K. Detection of human papillomavirus in primary hepatocellular carcinoma. Anticancer Res. 1992, 12, 763–766. [Google Scholar]
- Sugawara, Y.; Makuuchi, M.; Takada, K. Detection of Epstein-Barr virus DNA in hepatocellular carcinoma tissues from hepatitis C-positive patients. Scand. J. Gastroenterol. 2000, 35, 981–984. [Google Scholar]
- Chu, P.G.; Chen, Y.Y.; Chen, W.G.; Weiss, L.M. No direct role for epstein-barr virus in American hepatocellular carcinoma. Am. J. Pathol. 2001, 159, 1287–1292. [Google Scholar] [CrossRef] [Green Version]
- Akhter, S.; Liu, H.; Prabhu, R.; DeLucca, C.; Bastian, F.; Garry, R.F.; Schwartz, M.; Thung, S.N.; Dash, S. Epstein-Barr virus and human hepatocellular carcinoma. Cancer Lett. 2003, 192, 49–57. [Google Scholar] [CrossRef]
- Junying, J.; Herrmann, K.; Davies, G.; Lissauer, D.; Bell, A.; Timms, J.; Reynolds, G.M.; Hubscher, S.G.; Young, L.S.; Niedobitek, G.; et al. Absence of Epstein-Barr virus DNA in the tumor cells of European hepatocellular carcinoma. Virology 2003, 306, 236–243. [Google Scholar] [CrossRef] [Green Version]
- Zur Hausen, A.; van Beek, J.; Bloemena, E.; ten Kate, F.J.; Meijer, C.J.L.M.; van den Brule, A.J.C. No role for Epstein-Barr virus in Dutch hepatocellular carcinoma: A study at the DNA, RNA and protein levels. J. Gen. Virol. 2003, 84, 1863–1869. [Google Scholar] [CrossRef] [PubMed]
- Nishino, R.; Takano, A.; Oshita, H.; Ishikawa, N.; Akiyama, H.; Ito, H.; Nakayama, H.; Miyagi, Y.; Tsuchiya, E.; Kohno, N.; et al. Identification of Epstein-Barr virus-induced gene 3 as a novel serum and tissue biomarker and a therapeutic target for lung cancer. Clin. Cancer Res. 2011, 17, 6272–6286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaud, D.; Mirlekar, B.; Bischoff, S.; Cowley, D.O.; Vignali, D.A.A.; Pylayeva-Gupta, Y. Pancreatic cancer-associated inflammation drives dynamic regulation of p35 and Ebi3. Cytokine 2020, 125, 154817. [Google Scholar] [CrossRef]
- Hou, Y.M.; Dong, J.; Liu, M.Y.; Yu, S. Expression of Epstein-Barr virus-induced gene 3 in cervical cancer: Association with clinicopathological parameters and prognosis. Oncol. Lett. 2016, 11, 330–334. [Google Scholar] [CrossRef] [Green Version]
- Song, Q.; Chen, X.; Hu, W.; Mei, G.; Yang, X.; Wu, H. Downregulation of epstein-barr virus-induced gene 3 is associated with poor prognosis of hepatocellular carcinoma after curative resection. Oncol. Lett. 2018, 15, 7751–7759. [Google Scholar] [PubMed]
- Kang, H.J.; Oh, J.H.; Chun, S.M.; Kim, D.; Ryu, Y.M.; Hwang, H.S.; Kim, S.Y.; An, J.; Cho, E.J.; Lee, H.; et al. Immunogenomic landscape of hepatocellular carcinoma with immune cell stroma and EBV-positive tumor-infiltrating lymphocytes. J. Hepatol. 2019, 71, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, T.; Okamoto, H.; Konishi, K.; Yoshizawa, H.; Miyakawa, Y.; Mayumi, M. A novel DNA virus (TTV) associated with elevated transaminase levels in posttransfusion hepatitis of unknown etiology. Biochem. Biophys. Res. Commun. 1997, 241, 92–97. [Google Scholar] [CrossRef]
- Pineau, P.; Meddeb, M.; Raselli, R.; Qin, L.X.; Terris, B.; Tang, Z.Y.; Tiollais, P.; Mazzaferro, V.; Dejean, A. Effect of TT virus infection on hepatocellular carcinoma development: Results of a Euro-Asian survey. J. Infect. Dis. 2000, 181, 1138–1142. [Google Scholar] [CrossRef] [Green Version]
- Tagger, A.; Donato, F.; Ribero, M.L.; Binelli, G.; Gelatti, U.; Portera, G.; Albertini, A.; Fasola, M.; Chiesa, R.; Nardi, G. A case-control study on a novel DNA virus (TT virus) infection and hepatocellular carcinoma. Hepatology 1999, 30, 294–299. [Google Scholar] [CrossRef]
- Yoshida, H.; Kato, N.; Shiratori, Y.; Lan, K.H.; Ono-Nita, S.K.; Feng, Z.; Shiina, S.; Omata, M. Poor association of TT virus viremia with hepatocellular carcinoma. Liver 2000, 20, 247–252. [Google Scholar] [CrossRef]
- Hassoba, H.; Mahmoud, M.; Fahmy, H.; Leheta, O.; Sayed, A.; Fathy, A.; Serwah, A.; Abbas, A.; Nooman, Z.; Attia, F.; et al. TT Virus Infection Among Egyptian Patients With Hepatocellular Carcinoma. Egypt. J. Immunol. 2003, 10, 9–16. [Google Scholar]
- Ahn, D.H.; Bekaii-Saab, T. Biliary cancer: Intrahepatic cholangiocarcinoma vs. extrahepatic cholangiocarcinoma vs. gallbladder cancers: Classification and therapeutic implications. J. Gastrointest. Oncol. 2017, 8, 239–301. [Google Scholar] [CrossRef] [Green Version]
- Navas, M.C.; Glaser, S.; Dhruv, H.; Celinski, S.; Alpini, G.; Meng, F. Hepatitis C Virus Infection and Cholangiocarcinoma: An Insight into Epidemiologic Evidences and Hypothetical Mechanisms of Oncogenesis. Am. J. Pathol. 2019, 189, 1122–1132. [Google Scholar] [CrossRef] [Green Version]
- Qu, Z.; Cui, N.; Qin, M.; Wu, X. Epidemiological survey of biomarkers of hepatitis virus in patients with extrahepatic cholangiocarcinomas. Asia. Pac. J. Clin. Oncol. 2012, 8, 83–87. [Google Scholar] [CrossRef]
- Srivatanakul, P.; Honjo, S.; Kittiwatanachot, P.; Jedpiyawongse, A.; Khuhaprema, T.; Miwa, M. Hepatitis viruses and risk of cholangiocarcinoma in northeast Thailand. Asian Pac. J. Cancer Prev. 2010, 11, 985–988. [Google Scholar] [PubMed]
- Perumal, V.; Wang, J.; Thuluvath, P.; Choti, M.; Torbenson, M. Hepatitis C and hepatitis B nucleic acids are present in intrahepatic cholangiocarcinomas from the United States. Hum. Pathol. 2006, 37, 1211–1216. [Google Scholar] [CrossRef]
- Tan, J.H.; Zhou, W.Y.; Zhou, L.; Cao, R.C.; Zhang, G.W. Viral hepatitis B and C infections increase the risks of intrahepatic and extrahepatic cholangiocarcinoma: Evidence from a systematic review and meta-analysis. Turk. J. Gastroenterol. 2020, 31, 246–256. [Google Scholar] [CrossRef]
- Ahn, C.S.; Hwang, S.; Lee, Y.J.; Kim, K.H.; Moon, D.B.; Ha, T.Y.; Song, G.W.; Lee, S.G. Prognostic impact of hepatitis B virus infection in patients with intrahepatic cholangiocarcinoma. ANZ J. Surg. 2018, 88, 212–217. [Google Scholar] [CrossRef]
- Jeong, S.; Gao, L.; Tong, Y.; Xia, L.; Xu, N.; Sha, M.; Zhang, J.; Kong, X.; Gu, J.; Xia, Q. Prognostic Impact of Cirrhosis in Patients With Intrahepatic Cholangiocarcinoma Following Hepatic Resection. Can. J. Gastroenterol. Hepatol. 2017, 2017, 6543423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, J.W.; Kwan, B.S.; Cheon, Y.K.; Lee, T.Y.; Shim, C.S.; Kwon, S.Y.; Choe, W.H.; Yoo, B.C.; Yoon, J.M.; Lee, J.H. Prognostic impact of hepatitis B or C on intrahepatic cholangiocarcinoma. Korean J. Intern. Med. 2020, 35, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.Y.; He, X.D.; Xiu, D.R. Hepatitis B virus is associated with the clinical features and survival rate of patients with intrahepatic cholangiocarcinoma. Clin. Res. Hepatol. Gastroenterol. 2016, 40, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Klufah, F.; Mobaraki, G.; Chteinberg, E.; Alharbi, R.A.; Winnepenninckx, V.; Speel, E.J.M.; Rennspiess, D.; Damink, S.W.O.; Neumann, U.P.; Kurz, A.K.; et al. High prevalence of human polyomavirus 7 in cholangiocarcinomas and adjacent peritumoral hepatocytes: Preliminary findings. Microorganisms 2020, 8, 1125. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, S.; Song, F.; Cao, S.; Yin, X.; Xie, J.; Tu, X.; Xu, J.; Xu, X.; Dong, X.; et al. Hepatitis B virus status and the risk of pancreatic cancer: A meta-analysis. Eur. J. Cancer Prev. 2013, 22, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Abe, S.K.; Inoue, M.; Sawada, N.; Iwasaki, M.; Shimazu, T.; Yamaji, T.; Sasazuki, S.; Saito, E.; Tanaka, Y.; Mizokami, M.; et al. Hepatitis B and C virus infection and risk of pancreatic cancer: A population-based cohort study (JPHC study cohort II). Cancer Epidemiol. Biomarkers Prev. 2016, 25, 555–557. [Google Scholar] [CrossRef] [Green Version]
- Zhu, F.; Li, H.; Du, G.; Chen, J.; Cai, S. Chronic Hepatitis B Virus Infection and Pancreatic Cancer: A Case-Control Study in Southern China. Asian Pac. J. Cancer Prev. 2011, 12, 1405–1408. [Google Scholar]
- Chang, M.C.; Chen, C.H.; Liang, J.D.; Tien, Y.W.; Hsu, C.; Wong, J.M.; Chang, Y.T. Hepatitis B and C viruses are not risks for pancreatic adenocarcinoma. World J. Gastroenterol. 2014, 20, 5060–5065. [Google Scholar] [CrossRef]
- Huang, J.; Magnusson, M.; Törner, A.; Ye, W.; Duberg, A.S. Risk of pancreatic cancer among individuals with hepatitis C or hepatitis B virus infection: A nationwide study in Sweden. Br. J. Cancer 2013, 109, 2917–2923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.H.; Fu, J.J.; Wang, X.L.; Zhu, J.Y.; Ye, X.H.; Chen, S.D. Hepatitis B or C viral infection and risk of pancreatic cancer: A meta-analysis of observational studies. World J. Gastroenterol. 2013, 19, 4234–4241. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Sharma, R.; Lamerato, L.; Sheehan, M.; Krajenta, R.; Gordon, S.C. Is previous exposure to hepatitis B a risk factor for pancreatic cancer or hepatocellular carcinoma? J. Clin. Gastroenterol. 2014, 48, 729–733. [Google Scholar] [CrossRef]
- Andersen, E.S.; Omland, L.H.; Jepsen, P.; Krarup, H.; Christensen, P.B.; Obel, N.; Weis, N. Risk of all-type cancer, hepatocellular carcinoma, non-Hodgkin lymphoma and pancreatic cancer in patients infected with hepatitis B virus. J. Viral Hepat. 2015, 22, 828–834. [Google Scholar] [CrossRef]
- Chen, Y.; Bai, X.; Zhang, Q.; Wen, L.; Su, W.; Fu, Q.; Sun, X.; Lou, Y.; Yang, J.; Zhang, J.; et al. The hepatitis B virus X protein promotes pancreatic cancer through modulation of the PI3K/AKT signaling pathway. Cancer Lett. 2016, 380, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Tomasiewicz, K.; Modrzewska, R.; Lyczak, A.; Krawczuk, G. TT virus infection and pancreatic cancer: Relationship or accidental coexistence. World J. Gastroenterol. 2005, 11, 2847–2849. [Google Scholar] [CrossRef]
- Minton, K. Commensal viruses contribute to gut health. Nat. Rev. Immunol. 2019, 19, 721. [Google Scholar] [CrossRef]
- Santiago-Rodriguez, T.M.; Hollister, E.B. Human virome and disease: High-throughput sequencing for virus discovery, identification of phage-bacteria dysbiosis and development of therapeutic approaches with emphasis on the human gut. Viruses 2019, 11, 656. [Google Scholar] [CrossRef] [Green Version]
- Qiu, H.B.; Zhang, L.Y.; Zeng, Z.L.; Wang, Z.Q.; Luo, H.Y.; Keshari, R.P.; Zhou, Z.W.; Xu, R.H. HBV infection decreases risk of liver metastasis in patients with colorectal cancer: A cohort study. World J. Gastroenterol. 2011, 17, 804–808. [Google Scholar] [CrossRef]
- Wang, F.S.; Shao, Z.G.; Zhang, J.L.; Liu, Y.F. Colorectal liver metastases rarely occur in patients with chronic hepatitis virus infection. Hepatogastroenterology 2012, 59, 1390–1392. [Google Scholar] [CrossRef] [PubMed]
- Augustin, G.; Bruketa, T.; Korolija, D.; Milosevic, M. Lower incidence of hepatic metastases of colorectal cancer in patients with chronic liver diseases: Meta-analysis. Hepatogastroenterology. 2013, 60, 1164–1168. [Google Scholar] [PubMed]
- Li Destri, G.; Castaing, M.; Ferlito, F.; Minutolo, V.; Di Cataldo, A.; Puleo, S. Rare hepatic metastases of colorectal cancer in livers with symptomatic HBV and HCV hepatitis. Ann. Ital. Chir. 2013, 84, 323–327. [Google Scholar]
- García-Alonso, F.J.; Bonillo-Cambrodón, D.; Bermejo, A.; García-Martínez, J.; Hernández-Tejero, M.; Valer López Fando, P.; Piqueras, B.; Bermejo, F. Acceptance, yield and feasibility of attaching HCV birth cohort screening to colorectal cancer screening in Spain. Dig. Liver Dis. 2016, 48, 1237–1242. [Google Scholar] [CrossRef]
- McGregor, B.; Byrne, P.; Kirgan, D.; Albright, J.; Manalo, P.; Hall, M. Confirmation of the association of human papillomavirus with human colon cancer. Am. J. Surg. 1993, 166, 738–742. [Google Scholar] [CrossRef]
- Damin, D.C.; Caetano, M.B.; Rosito, M.A.; Schwartsmann, G.; Damin, A.S.; Frazzon, A.P.; Ruppenthal, R.D.; Alexandre, C.O.P. Evidence for an association of human papillomavirus infection and colorectal cancer. Eur. J. Surg. Oncol. 2007, 33, 569–574. [Google Scholar]
- Giuliani, L.; Ronci, C.; Bonifacio, D.; Di Bonito, L.; Favalli, C.; Perno, C.; Syrjänen, K.; Ciotti, M. Detection of oncogenic DNA viruses in colorectal cancer. Anticancer Res. 2008, 28, 1405–1410. [Google Scholar] [PubMed]
- Gornick, M.C.; Castellsague, X.; Sanchez, G.; Giordano, T.J.; Vinco, M.; Greenson, J.K.; Capella, G.; Raskin, L.; Rennert, G.; Gruber, S.B.; et al. Human papillomavirus is not associated with colorectal cancer in a large international study. Cancer Causes Control 2010, 21, 737–743. [Google Scholar] [PubMed]
- Burnett-Hartman, A.N.; Feng, Q.; Popov, V.; Kalidindi, A.; Newcomb, P.A. Human papillomavirus DNA is rarely detected in colorectal carcinomas and not associated with microsatellite instability: The seattle colon cancer family registry. Cancer Epidemiol. Biomark. Prev. 2013, 22, 317–319. [Google Scholar]
- Gazzaz, F.; Mosli, M.H.; Jawa, H.; Sibiany, A. Detection of human papillomavirus infection by molecular tests and its relation to colonic polyps and colorectal cancer. Saudi Med. J. 2016, 37, 256–261. [Google Scholar] [PubMed]
- Mahmoudvand, S.; Safaei, A.; Erfani, N.; Sarvari, J. Presence of human papillomavirus DNA in colorectal cancer tissues in Shiraz, Southwest Iran. Asian Pac. J. Cancer Prev. 2015, 16, 7883–7887. [Google Scholar]
- Qiu, Q.; Li, Y.; Fan, Z.; Yao, F.; Shen, W.; Sun, J.; Yuan, Y.; Chen, J.; Cai, L.; Xie, Y.; et al. Gene Expression Analysis of Human Papillomavirus-Associated Colorectal Carcinoma. Biomed. Res. Int. 2020, 2020, 5201587. [Google Scholar]
- Asem, M.S.; Buechler, S.; Wates, R.B.; Miller, D.L.; Stack, M.S. Wnt5a signaling in cancer. Cancers 2016, 8, 79. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.R.; Myint, T.; Goh, H.S. Over-expression of the c-myc proto-oncogene in colorectal carcinoma. Br. J. Cancer 1993, 68, 407–413. [Google Scholar]
- Spandidos, D.; Yiagnisis, M.; Papadimitriou, K.; Field, J. ras, c-myc and c-erbB-2 oncoproteins in human breast cancer. Anticancer Res. 1989, 9, 1385–1393. [Google Scholar]
- Little, C.D.; Nau, M.M.; Carney, D.N.; Gazdar, A.F.; Minna, J.D. Amplification and expression of the c-myc oncogene in human lung cancer cell lines. Nature 1983, 306, 194–196. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, K.; Hirohashi, S.; Sekiya, T. Increased expression of the c-myc gene without gene amplification in human lung cancer and colon cancer cell lines. Jpn. J. Cancer Res. GANN 1986, 77, 540–545. [Google Scholar] [PubMed]
- Sun, D.W.; Zhang, Y.Y.; Qi, Y.; Zhou, X.T.; Lv, G.Y. Prognostic significance of MMP-7 expression in colorectal cancer: A meta-analysis. Cancer Epidemiol. 2015, 39, 135–142. [Google Scholar]
- Otero, L.; Lacunza, E.; Vasquez, V.; Arbelaez, V.; Cardier, F.; González, F. Variations in AXIN2 predict risk and prognosis of colorectal cancer. BDJ Open 2019, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.X.; Zhang, L.; Simayi, D.; Zhang, N.; Tao, L.; Yang, L.; Zhao, J.; Chen, Y.Z.; Li, F.; Zhang, W.J. Human papillomavirus infection correlates with inflammatory stat3 signaling activity and IL-17 level in patients with colorectal cancer. PLoS ONE 2015, 10, e0118391. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Tait, S.W.G. Apoptosis and Cancer: Force Awakens, Phantom Menace, or Both? Int. Rev. Cell Mol. Biol. 2018, 337, 135–152. [Google Scholar]
- Karbasi, A.; Borhani, N.; Daliri, K.; Kazemi, B.; Manoochehri, M. Downregulation of external death receptor genes FAS and DR5 in colorectal cancer samples positive for human papillomavirus infection. Pathol. Res. Pract. 2015, 211, 444–448. [Google Scholar] [CrossRef]
- Buyru, N.; Tezol, A.; Dalay, N. Coexistence of K-ras mutations and HPV infection in colon cancer. BMC Cancer 2006, 6, 115. [Google Scholar] [CrossRef] [Green Version]
- Sayhan, N.; Yazici, H.; Budak, M.; Bitisik, O.; Dalay, N. P53 Codon 72 Genotypes in Colon Cancer. Association With Human Papillomavirus Infection. Res. Commun. Mol. Pathol. Pharmacol. 2001, 109, 25–34. [Google Scholar]
- Buyru, N.; Budak, M.; Yazici, H.; Dalay, N. p53 Gene Mutations Are Rare in Human Papillomavirus-Associated Colon Cancer. Oncol. Rep. 2003, 10, 2089–2092. [Google Scholar] [CrossRef]
- Sarvari, J.; Mahmoudvand, S.; Pirbonyeh, N.; Safaei, A.; Hosseini, S.Y. The very low frequency of Epstein-Barr JC and BK viruses DNA in colorectal cancer tissues in Shiraz, Southwest Iran. Polish J. Microbiol. 2018, 67, 73–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malki, M.I.; Gupta, I.; Fernandes, Q.; Aboulkassim, T.; Yasmeen, A.; Vranic, S.; Al Moustafa, A.E.; Al-Thawadi, H.A. Co-presence of Epstein–Barr virus and high-risk human papillomaviruses in Syrian colorectal cancer samples. Hum. Vaccines Immunother. 2020, 1. [Google Scholar] [CrossRef] [PubMed]
- Gupta, I.; Al Farsi, H.; Jabeen, A.; Skenderi, F.; Al-Thawadi, H.; Alahmad, Y.M.; Al Moustafa, A.E.; Vranic, S. High-risk human papillomaviruses and epstein–barr virus in colorectal cancer and their association with clinicopathological status. Pathogens 2020, 9, 456. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Chen, Q.; Du, W.; Chen, C.; Li, F.; Yang, J.; Peng, J.; Kang, D.; Lin, B.; Chai, X.; et al. Epstein-Barr Virus-Induced Gene 3 (EBI3) Blocking Leads to Induce Antitumor Cytotoxic T Lymphocyte Response and Suppress Tumor Growth in Colorectal Cancer by Bidirectional Reciprocal-Regulation STAT3 Signaling Pathway. Mediat. Inflamm. 2016, 2016, 3214105. [Google Scholar]
- Chen, H.P.; Jiang, J.K.; Chen, C.Y.; Chou, T.Y.; Chen, Y.C.; Chang, Y.T.; Lin, S.F.; Chan, C.H.; Yang, C.Y.; Lin, C.H.; et al. Human cytomegalovirus preferentially infects the neoplastic epithelium of colorectal cancer: A quantitative and histological analysis. J. Clin. Virol. 2012, 54, 240–244. [Google Scholar] [CrossRef]
- Bai, B.; Wang, X.; Chen, E.; Zhu, H. Human cytomegalovirus infection and colorectal cancer risk: A meta-analysis. Oncotarget 2016, 7, 76735–76742. [Google Scholar] [CrossRef]
- Chen, H.P.; Jiang, J.K.; Lai, P.Y.; Chen, C.Y.; Chou, T.Y.; Chen, Y.C.; Chan, C.H.; Lin, S.F.; Yang, C.Y.; Chen, C.Y.; et al. Tumoral presence of human cytomegalovirus is associated with shorter disease-free survival in elderly patients with colorectal cancer and higher levels of intratumoral interleukin-17. Clin. Microbiol. Infect. 2014, 20, 664–671. [Google Scholar] [CrossRef] [Green Version]
- Knösel, T.; Schewe, C.; Dietel, M.; Petersen, I. Cytomegalovirus is not associated with progression and metastasis of colorectal cancer. Cancer Lett. 2004, 211, 243–247. [Google Scholar]
- Mehrabani-Khasraghi, S.; Ameli, M.; Khalily, F. Demonstration of herpes simplex virus, cytomegalovirus, and Epstein-Barr virus in colorectal cancer. Iran. Biomed. J. 2016, 20, 302–306. [Google Scholar]
- Li, X.; Qian, D.; Ju, F.; Wang, B. Upregulation of Toll-like receptor 2 expression in colorectal cancer infected by human cytomegalovirus. Oncol. Lett. 2015, 9, 365–370. [Google Scholar] [CrossRef]
- Harkins, L.; Volk, A.L.; Samanta, M.; Mikolaenko, I.; Britt, W.J.; Bland, K.I.; Cobbs, C.S. Specific localisation of human cytomegalovirus nucleic acids and proteins in human colorectal cancer. Lancet 2002, 360, 1557–1563. [Google Scholar] [CrossRef]
- Sheng, J.; Sun, H.; Yu, F.-B.; Li, B.; Zhang, Y.; Zhu, Y.-T. The Role of Cyclooxygenase-2 in Colorectal Cancer. Int. J. Med. Sci. 2020, 17, 1095–1101. [Google Scholar] [CrossRef]
- Teo, W.H.; Chen, H.P.; Huang, J.C.; Chan, Y.J. Human cytomegalovirus infection enhances cell proliferation, migration and upregulation of EMT markers in colorectal cancer-derived stem cell-like cells. Int. J. Oncol. 2017, 51, 1415–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishna, B.A.; Poole, E.L.; Jackson, S.E.; Smit, M.J.; Wills, M.R.; Sinclair, J.H. Latency-associated expression of human cytomegalovirus US28 attenuates cell signaling pathways to maintain latent infection. MBio 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.Z.; Xu, J.G.; Zhou, Y.H.; Zheng, J.H.; Lin, K.Z.; Zheng, S.Z.; Ye, M.S.; He, Y.; Liu, C.B.; Xue, Z.X. Human cytomegalovirus-encoded US28 may act as a tumor promoter in colorectal cancer. World J. Gastroenterol. 2016, 22, 2789–2798. [Google Scholar] [CrossRef]
- Langemeijer, E.V.; Slinger, E.; de Munnik, S.; Schreiber, A.; Maussang, D.; Vischer, H.; Verkaar, F.; Leurs, R.; Siderius, M.; Smit, M.J. Constitutive ß-Catenin Signaling by the Viral Chemokine Receptor US28. PLoS ONE 2012, 7, e48935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maussang, D.; Langemeijer, E.; Fitzsimons, C.P.; Stigter-van Walsům, M.; Dijkman, R.; Borg, M.K.; Slinger, E.; Schreiber, A.; Michel, D.; Tensen, C.P.; et al. The human cytomegalovirus-encoded chemokine receptor US28 promotes angiogenesis and tumor formation via cyclooxygenase-2. Cancer Res. 2009, 69, 2861–2869. [Google Scholar] [CrossRef] [Green Version]
- Zu Rhein, G.M.; Chou, S.M. Particles resembling papova viruses in human cerebral demyelinating disease. Science (80-.) 1965, 148, 1477–1479. [Google Scholar] [CrossRef]
- Coelho, T.R.; Almeida, L.; Lazo, P.A. JC virus in the pathogenesis of colorectal cancer, an etiological agent or another component in a multistep process? Virol. J. 2010, 7, 42. [Google Scholar] [CrossRef] [Green Version]
- Mou, X.; Chen, L.; Liu, F.; Lin, J.; Diao, P.; Wang, H.; Li, Y.; Lin, J.; Teng, L.; Xiang, C. Prevalence of JC virus in chinese patients with colorectal cancer. PLoS ONE 2012, 7, e35900. [Google Scholar] [CrossRef] [Green Version]
- Shavaleh, R.; Kamandi, M.; Feiz Disfani, H.; Mansori, K.; Naseri, S.N.; Rahmani, K.; Ahmadi Kanrash, F. Association between JC virus and colorectal cancer: Systematic review and meta-analysis. Infect. Dis. (Auckl.) 2020, 52, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Newcomb, P.A.; Bush, A.; Stoner, G.; Lampe, J.; Potter, J.; Bigler, J. No Evidence of an Association of JC Virus and Colon Neoplasia. Cancer Epidemiol. Biomark. Prev. 2004, 13, 662–666. [Google Scholar]
- Lundstig, A.; Stattin, P.; Persson, K.; Sasnauskas, K.; Viscidi, R.P.; Gislefoss, R.E.; Dillner, J. No excess risk for colorectal cancer among subjects seropositive for the JC polyomavirus. Int. J. Cancer 2007, 121, 1098–1102. [Google Scholar] [CrossRef] [Green Version]
- Hampras, S.S.; Viscidi, R.P.; Helzlsouer, K.J.; Lee, J.H.; Fulp, W.J.; Giuliano, A.R.; Platz, E.A.; Rollison, D.E. Prospective study of seroreactivity to JC virus T-antigen and risk of colorectal cancers and adenomas. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2591–2596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esmailzadeh, N.; Ranaee, M.; Alizadeh, A.; Khademian, A.; Saber Amoli, S.; Sadeghi, F. Presence of JC Polyomavirus in Nonneoplastic Inflamed Colon Mucosa and Primary and Metastatic Colorectal Cancer. Gastrointest. Tumors 2020, 7, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Li, M.S.; Nagasaka, T.; Shin, S.K.; Fuerst, F.; Ricciardiello, L.; Wasserman, L.; Boland, C.R. Association of JC Virus T-Antigen Expression With the Methylator Phenotype in Sporadic Colorectal Cancers. Gastroenterology 2006, 130, 1950–1961. [Google Scholar] [CrossRef]
- Ricciardiello, L.; Baglioni, M.; Giovannini, C.; Pariali, M.; Cenacchi, G.; Ripalti, A.; Landini, M.; Sawa, H.; Nagashima, K.; Frisque, R.; et al. Induction of chromosomal instability in colonic cells by the human polyomavirus JC virus. Cancer Res. 2003, 63, 7256–7262. [Google Scholar]
- Niv, Y.; Vilkin, A.; Brenner, B.; Kendel, Y.; Morgenstern, S.; Levi, Z. HMLH1 promoter methylation and JC virus T antigen presence in the tumor tissue of colorectal cancer Israeli patients of different ethnic groups. Eur. J. Gastroenterol. Hepatol. 2010, 22, 938–941. [Google Scholar] [CrossRef]
- Vilkin, A.; Ronen, Z.; Levi, Z.; Morgenstern, S.; Halpern, M.; Niv, Y. Presence of JC virus DNA in the tumor tissue and normal mucosa of patients with sporadic colorectal cancer (CRC) or with positive family history and Bethesda criteria. Dig. Dis. Sci. 2012, 57, 79–84. [Google Scholar] [CrossRef]
- Coelho, T.R.; Gaspar, R.; Figueiredo, P.; Mendonça, C.; Lazo, P.A.; Almeida, L. Human JC polyomavirus in normal colorectal mucosa, hyperplastic polyps, sporadic adenomas, and adenocarcinomas in Portugal. J. Med. Virol. 2013, 85, 2119–2127. [Google Scholar] [CrossRef]
- Staib, C.; Pesch, J.; Gerwig, R.; Gerber, J.K.; Brehm, U.; Stangl, A.; Grummt, F. p53 inhibits JC virus DNA replication in vivo and interacts with JC virus large T-antigen. Virology 1996, 219, 237–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Link, A.; Shin, S.K.; Nagasaka, T.; Balaguer, F.; Koi, M.; Jung, B.; Richard Boland, C.; Goel, A. JC virus mediates invasion and migration in colorectal metastasis. PLoS ONE 2009, 4, e8146. [Google Scholar] [CrossRef]
- Enam, S.; Del Valle, L.; Lara, C.; Gan, D.-D.; Ortiz-Hidalgo, C.; Palazzo, J.P.; Khalili, K. Association of Human Polyomavirus JCV With Colon Cancer: Evidence for Interaction of Viral T-antigen and Beta-Catenin. Cancer Res. 2002, 62, 7093–7101. [Google Scholar] [PubMed]
- Enam, S.; Gan, D.-D.; White, M.K.; Del Valle, L.; Khalili, K. Regulation of Human Neurotropic JCV in Colon Cancer Cells. Anticancer Res. 2006, 26, 833–841. [Google Scholar] [PubMed]
- Ripple, M.J.; Struckhoff, A.P.; Trillo-Tinoco, J.; Li, L.; Margolin, D.A.; McGoey, R.; Valle, L. Del Activation of c-Myc and cyclin D1 by JCV T-Antigen and β-Catenin in colon cancer. PLoS ONE 2014, 9, e106257. [Google Scholar] [CrossRef] [Green Version]
- Nosho, K.; Shima, K.; Kure, S.; Irahara, N.; Baba, Y.; Chen, L.; Kirkner, G.J.; Fuchs, C.S.; Ogino, S. JC virus T-antigen in colorectal cancer is associated with p53 expression and chromosomal instability, independent of CpG island methylator phenotype. Neoplasia 2009, 11, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Hoff, P.M.; Coudry, R.; Moniz, C.M.V. Pathology of Anal Cancer. Surg. Oncol. Clin. N. Am. 2017, 26, 57–71. [Google Scholar] [CrossRef]
- Kang, Y.J.; Smith, M.; Canfell, K. Anal cancer in high-income countries: Increasing burden of disease. PLoS ONE 2018, 13, e0205105. [Google Scholar] [CrossRef]
- Frisch, M.; Fenger, C.; van den Brule, A.; Sørensen, P.; Meijer, C.; Walboomers, J.; Adami, H.; Melbye, M.; Glimelius, B. Variants of squamous cell carcinoma of the anal canal and perianal skin and their relation to human papillomaviruses. Cancer Res. 1999, 59, 753–757. [Google Scholar]
- Silva Dalla Libera, L.; Almeida de Carvalho, K.; Enocencio Porto Ramos, J.; Oliveira Cabral, L.; de Cassia Goncalves de Alencar, R.; Villa, L.; Alves, R.; Rabelo Santos, S.; Aparecida Dos Santos Carneiro, M.; Saddi, V. Human Papillomavirus and Anal Cancer: Prevalence, Genotype Distribution, and Prognosis Aspects From Midwestern Region of Brazil. J. Oncol. 2019, 2019, 6018269. [Google Scholar] [CrossRef] [Green Version]
- Urbute, A.; Rasmussen, C.L.; Belmonte, F.; Obermueller, T.; Prigge, E.S.; Arbyn, M.; Verdoodt, F.; Kjaer, S.K. Prognostic significance of HPV DNA and p16INK4a in anal cancer: A systematic review and meta-analysis. Cancer Epidemiol. Biomark. Prev. 2020, 29, 703–710. [Google Scholar] [CrossRef] [Green Version]
- Koerber, S.A.; Schoneweg, C.; Slynko, A.; Krug, D.; Haefner, M.F.; Herfarth, K.; Debus, J.; Sterzing, F.; Von Knebel Doeberitz, M.; Prigge, E.S.; et al. Influence of human papillomavirus and p16INK4a on treatment outcome of patients with anal cancer. Radiother. Oncol. 2014, 113, 331–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rödel, F.; Wieland, U.; Fraunholz, I.; Kitz, J.; Rave-Fränk, M.; Wolff, H.A.; Weiss, C.; Wirtz, R.; Balermpas, P.; Fokas, E.; et al. Human papillomavirus DNA load and p16INK4a expression predict for local control in patients with anal squamous cell carcinoma treated with chemoradiotherapy. Int. J. Cancer 2015, 136, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Welzel, G.; Ottstadt, M.; Lohr, F.; Severa, S.; Prigge, E.S.; Wentzensen, N.; Trunk, M.J.; Wenz, F.; Von Knebel-Doeberitz, M.; et al. Prognostic relevance of HPV infection and p16 overexpression in squamous cell anal cancer. Int. J. Radiat. Oncol. Biol. Phys. 2015, 93, 819–827. [Google Scholar] [CrossRef]
- Małusecka, E.; Chmielik, E.; Suwiński, R.; Giglok, M.; Lange, D.; Rutkowski, T.; Mazurek, A.M. Significance of HPV16 Viral Load Testing in Anal Cancer. Pathol. Oncol. Res. 2020, 26, 2191–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rademacher, B.L.; Matkowskyj, K.A.; Meske, L.M.; Romero, A.; Sleiman, H.; Carchman, E.H. The role of pharmacologic modulation of autophagy on anal cancer development in an HPV mouse model of carcinogenesis. Virology 2017, 507, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Rademacher, B.L.; Matkowskyj, K.A.; Lacount, E.D.; Carchman, E.H. Topical application of a dual PI3K/mTOR inhibitor prevents anal carcinogenesis in a human papillomavirus mouse model of anal cancer. Eur. J. Cancer Prev. 2019, 28, 483–491. [Google Scholar] [CrossRef]
- Wechsler, E.I.; Tugizov, S.; Herrera, R.; Da Costa, M.; Palefsky, J.M. E5 can be expressed in anal cancer and leads to epidermal growth factor receptor-induced invasion in a human papillomavirus 16-transformed anal epithelial cell line. J. Gen. Virol. 2018, 99, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Salit, I.E.; Tinmouth, J.; Chong, S.; Raboud, J.; Diong, C.; Su, D.S.; Sano, M.; Lytwyn, A.; Chapman, W.; Mahony, J. Screening for HIV-associated anal cancer: Correlation of HPV genotypes, p16, and E6 transcripts with anal pathology. Cancer Epidemiol. Biomark. Prev. 2009, 18, 1986–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.K.; Pitot, H.C.; Liem, A.; Lambert, P.F. Dominant role of HPV16 E7 in anal carcinogenesis. Virology 2011, 421, 114–118. [Google Scholar] [CrossRef] [Green Version]
- Da Silva Neto, M.M.; Brites, C.; Borges, Á.H. Cancer during HIV infection. APMIS 2020, 128, 121–128. [Google Scholar] [CrossRef]
- Silverberg, M.; Lau, B.; Justice, A.; Engels, E.; Gill, M.; Goedert, J.; Kirk, G.; D’Souza, G.; Bosch, R.; Brooks, J.; et al. Risk of anal cancer in HIV-infected and HIV-uninfected individuals in North America. Clin. Infect. Dis. 2012, 54, 1026–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wentzensen, N.; Follansbee, S.; Borgonovo, S.; Tokugawa, D.; Schwartz, L.; Lorey, T.S.; Sahasrabuddhe, V.V.; Lamere, B.; Gage, J.C.; Fetterman, B.; et al. Human papillomavirus genotyping, human papillomavirus mRNA expression, and p16/Ki-67 cytology to detect anal cancer precursors in HIV-infected MSM. AIDS 2012, 26, 2185–2192. [Google Scholar] [CrossRef] [PubMed]
- Combes, J.D.; Clifford, G.M.; Günthard, H.F.; Hauser, C.; Darling, K.E.A.; Valladares, P.; Battegay, M.; Waldeck, F.; Bernasconi, E.; Bertisch, B.; et al. Antibodies against HPV16E6 oncoprotein in the Swiss HIV cohort study: Kinetics and anal cancer risk prediction. Int. J. Cancer 2020, 147, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Bertisch, B.; Franceschi, S.; Lise, M.; Vernazza, P.; Keiser, O.; Schöni-Affolter, F.; Bouchardy, C.; Dehler, S.; Levi, F.; Jundt, G.; et al. Risk factors for anal cancer in persons infected with HIV: A nested case-control study in the Swiss HIV Cohort Study. Am. J. Epidemiol. 2013, 178, 877–884. [Google Scholar] [CrossRef]
- Meyer, J.E.; Panico, V.J.A.; Marconato, H.M.F.; Sherr, D.L.; Christos, P.; Pirog, E.C. HIV positivity but not HPV/p16 status is associated with higher recurrence rate in anal cancer. J. Gastrointest. Cancer 2013, 44, 450–455. [Google Scholar] [CrossRef]
- Liu, Y.; Sigel, K.M.; Westra, W.; Gitman, M.R.; Zheng, W.; Gaisa, M.M. HIV-Infected Patients With Anal Cancer Precursors: Clinicopathological Characteristics and Human Papillomavirus Subtype Distribution. Dis. Colon Rectum 2020, 63, 890–896. [Google Scholar] [CrossRef]
- De Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef] [Green Version]
- Kannen, V.; Parry, L.; Martin, F.L. Phages Enter the Fight against Colorectal Cancer. Trends Cancer 2019, 5, 577–579. [Google Scholar] [CrossRef] [PubMed]

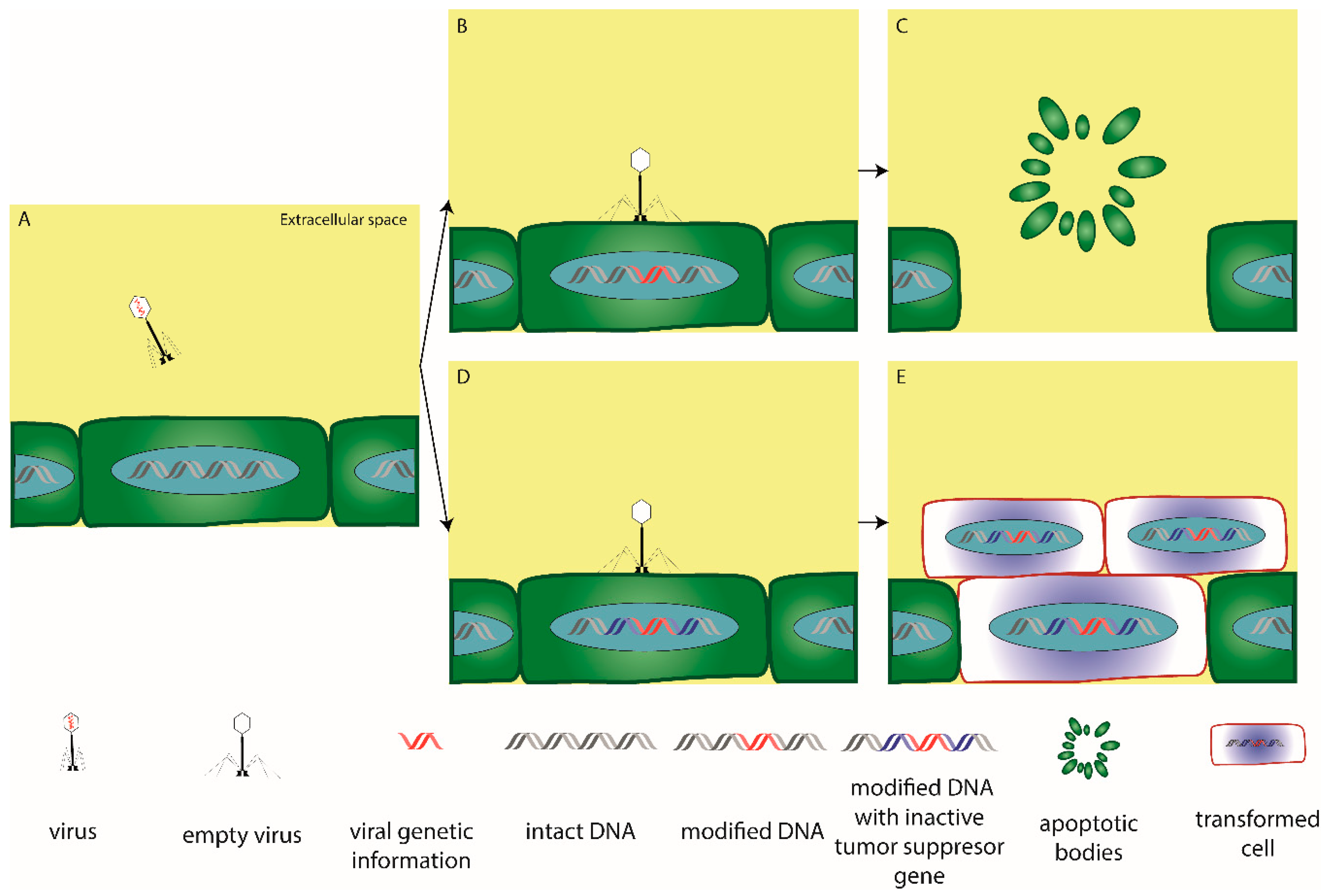

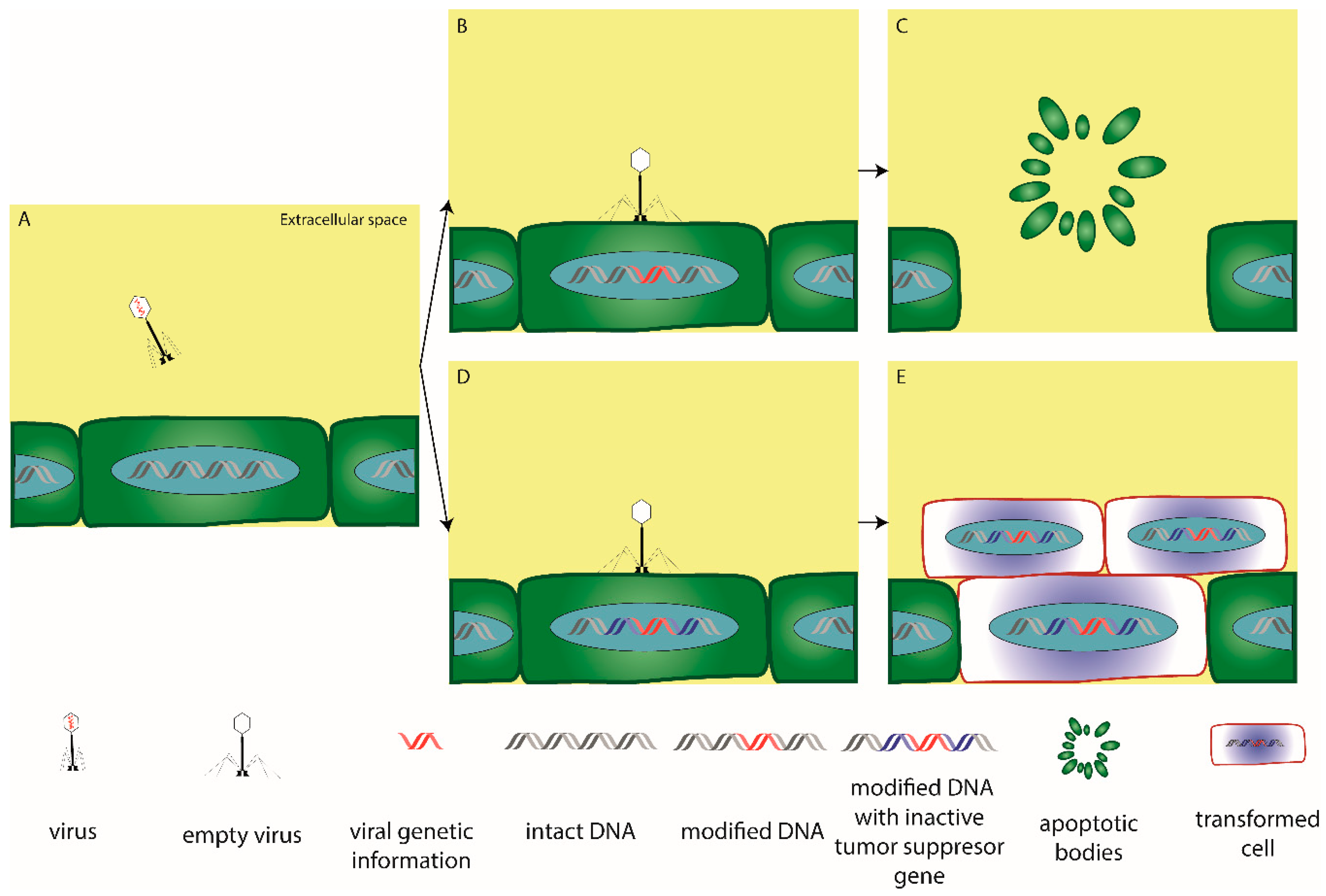
{kind=link}
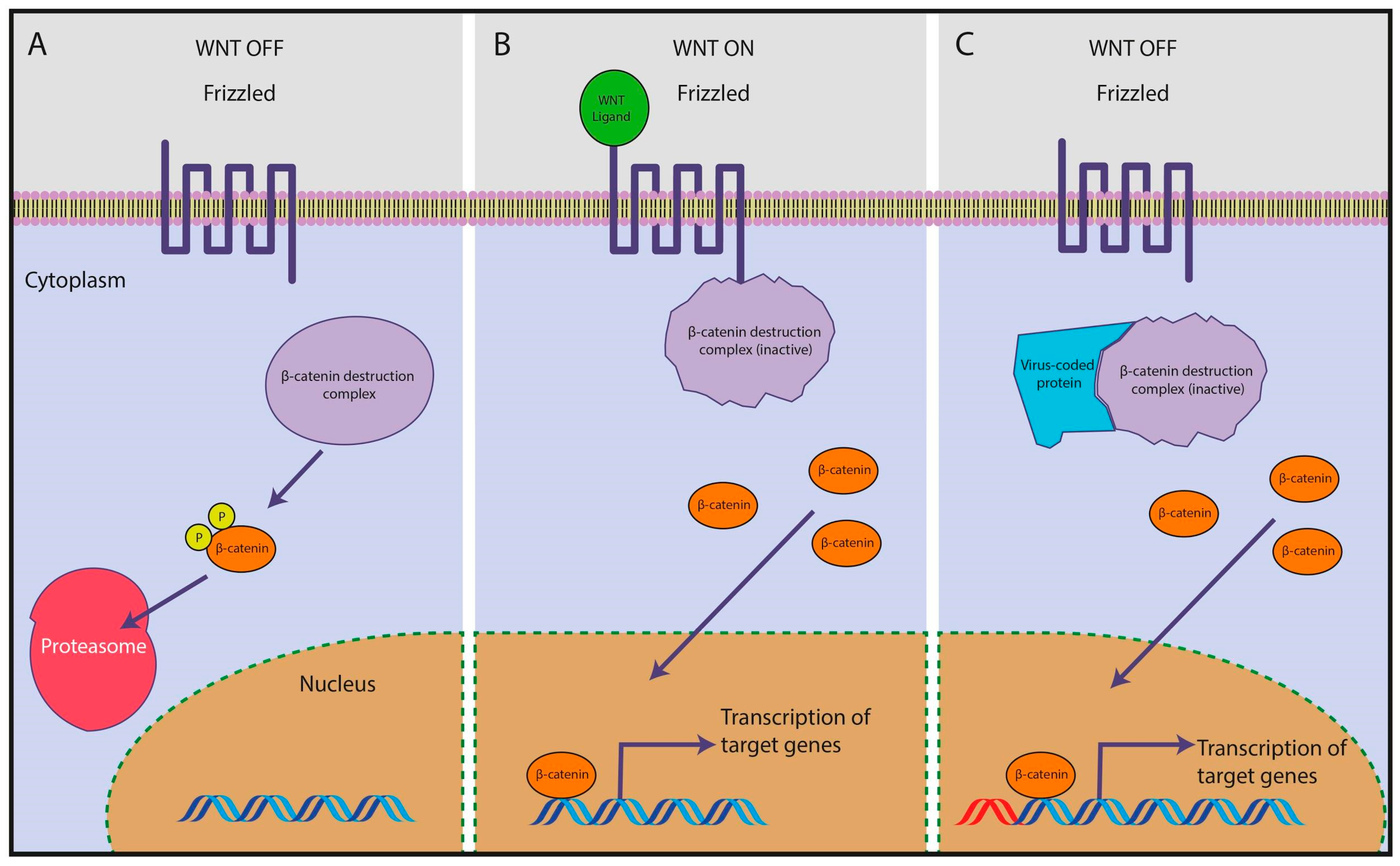
{kind=link}
| First Author | Year | Virus | Cancer Type | Sample Origin | Number of Samples | Observation | Ref. |
|---|---|---|---|---|---|---|---|
| Maden | 1992 | HPV | Oral | USA | 131 | HPV-6 is associated with oral cancer | [36] |
| Jalouli | 2012 | HPV, HSV, EBV | Oral | UK, Sweden, Sudan, Norway, USA, Yemen, India, Sri Lanka | 155 | Higher proportion of samples with HSV and EBV in industrialized countries | [37] |
| Bjørge | 1997 | HPV | Esophageal | Norway | 228 | Increased risk of developing cancer among HPV-16-seropositive subjects | [38] |
| Zhang | 2015 | HPV | Esophageal | China | 3044 | HPV-16 is a possible risk factor | [39] |
| Wang | 2010 | HPV | Esophageal | China, USA | 435 | HPV infection is common in esophageal carcinoma, independent of region and ethnic group of origin | [40] |
| Kirgan | 1990 | HPV | Colorectal | USA | 90 | Association between HPV and colon neoplasia | [41] |
| Lee | 2001 | HPV | Colorectal | Taiwan | 38 | HPV-18 is a possible risk factor | [42] |
| Alexandrou | 2014 | HPV | Anal | Greece | 11 | Lower incidence of HPV in anal cancer compared to other Western countries | [43] |
| Kabarriti | 2019 | HPV | Anal | USA | 5927 | HPV is a significant prognostic marker in anal cancer, especially for locally advanced disease | [44] |
| Muresu | 2020 | HPV | Anal | Italy | 30 | HPV is a possible risk factor | [45] |
| Awerkiew | 2003 | HPV, EBV | Esophageal | Germany | 37 | EBV, but not HPV, was detected in esophageal cancer samples | [46] |
| Martínez-López | 2014 | EBV | Gastric | Mexico | 297 | Possible role for EBV in gastric cancer and early precursor lesions. | [47] |
| Corallo | 2020 | EBV | Gastric | Italy | 175 | Patients with EBV-positive gastric cancer who did not receive ICI had a better response to first-line chemotherapy and better survival compared with EBV-negative patients | [48] |
| Li | 2004 | EBV | Hepatic | China | 141 | Presence of HBV infection in HCC tissues | [49] |
| Song | 2006 | EBV | Colorectal | China | 115 | Possible association of EBV with colorectal carcinoma | [50] |
| Fiorina | 2014 | HPV, EBV, JCV, BKV | Colorectal | Italy | 44 | No or weak association of HPV, EBV, JCV, and BKV with colorectal cancer | [51] |
| Laghi | 1999 | JCV | Colorectal | USA | 23 | JCV DNA may play a role in the chromosomal instability observed in colorectal carcinogenesis | [52] |
| Hori | 2005 | JCV | Colorectal | Japan | 64 | Possible role of JCV in colorectal carcinogenesis | [53] |
| Jung | 2008 | JCV | Colorectal | USA | 74 | JCV T-Antigen is expressed in the early stage of colorectal cancer | [54] |
| Tokita | 2002 | TTV | Hepatic | Japan | 237 | High TTV abundance is an independent risk factor | [55] |
| Iloeje | 2010 | HBV | Pancreatic | Taiwan | 22,471 | Chronic HBV infection may be associated with an increased risk of pancreatic cancer | [56] |
| Zhou | 2012 | HBV, HCV | Bile duct | Meta-analyses (13 case-control studies and 3 cohort studies) | - | HBV and HCV are risk factors in bile duct cancer | [57] |
| Hassan | 2008 | HBV | Pancreatic | USA | 476 | Past exposure to HBV is a possible risk factor in pancreatic cancer | [58] |
| Su | 2020 | HBV | Colorectal | Taiwan | 69,478 | Chronic HBV infection is strongly associated with increased risk of developing colorectal cancer | [59] |
| Dimberg | 2013 | CMV | Colorectal | Sweden, Vietnam | 202 | CMV DNA rate was significantly higher in cancerous tissues compared to normal tissues | [60] |
| Chen | 2016 | CMV | Colorectal | Taiwan | 556 | More favorable disease-free survival rate in non-elderly patients with CMV-positive tumors, specifically in patients with stage III disease | [61] |
| Chen | 2015 | CMV | Colorectal | USA, France, Italy, Japan, China, Taiwan | 92 | Specific genetic CMV polymorphisms are associated with different clinical outcomes | [62] |
| Jin | 2019 | HIV | Anal | Australia | Not applicable | People living with HIV are at markedly higher risk of anal cancer | [63] |
| Colón-López | 2018 | HIV | Anal | USA | Not applicable | Anal cancer incidence is markedly elevated among people with HIV infection | [64] |
| Grew | 2015 | HIV | Anal | USA | Not applicable | HIV-positive patients had significantly worse overall and colostomy-free survival rates than HIV-negative patients | [65] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marônek, M.; Link, R.; Monteleone, G.; Gardlík, R.; Stolfi, C. Viruses in Cancers of the Digestive System: Active Contributors or Idle Bystanders? Int. J. Mol. Sci. 2020, 21, 8133. https://doi.org/10.3390/ijms21218133
Marônek M, Link R, Monteleone G, Gardlík R, Stolfi C. Viruses in Cancers of the Digestive System: Active Contributors or Idle Bystanders? International Journal of Molecular Sciences. 2020; 21(21):8133. https://doi.org/10.3390/ijms21218133
Chicago/Turabian StyleMarônek, Martin, René Link, Giovanni Monteleone, Roman Gardlík, and Carmine Stolfi. 2020. "Viruses in Cancers of the Digestive System: Active Contributors or Idle Bystanders?" International Journal of Molecular Sciences 21, no. 21: 8133. https://doi.org/10.3390/ijms21218133
APA StyleMarônek, M., Link, R., Monteleone, G., Gardlík, R., & Stolfi, C. (2020). Viruses in Cancers of the Digestive System: Active Contributors or Idle Bystanders? International Journal of Molecular Sciences, 21(21), 8133. https://doi.org/10.3390/ijms21218133