The Emerging Role of LHb CaMKII in the Comorbidity of Depressive and Alcohol Use Disorders

Abstract
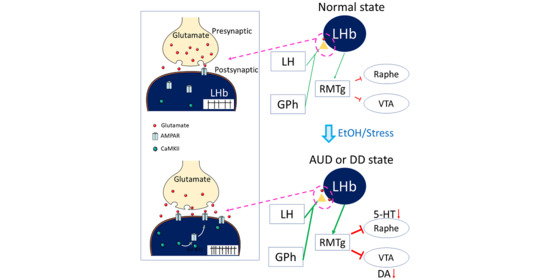
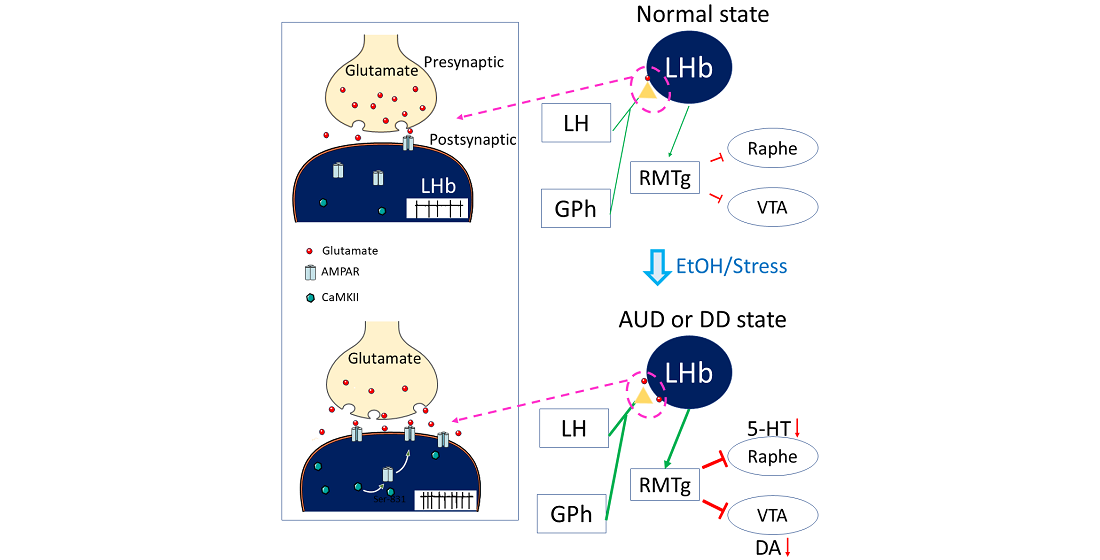
:
1. Introduction
2. The Role of the Lateral Habenula in Depressive Disorders and Alcohol Use Disorders
3. CaMKII Structure and Regulation
4. The Role of CaMKII in Alcohol Use Disorders
5. The Role of CaMKII in Depressive Disorders
6. The Comorbidity of Depressive Disorders and Alcohol Use Disorders
7. Conclusions
Funding
Conflicts of Interest
References
- Sacks, J.J.; Gonzales, K.R.; Bouchery, E.E.; Tomedi, L.E.; Brewer, R.D. 2010 national and state costs of excessive alcohol consumption. Am. J. Prev. Med. 2015, 49, e73–e79. [Google Scholar] [CrossRef] [PubMed]
- Ripke, S.; Wray, N.R.; Lewis, C.M.; Hamilton, S.P.; Weissman, M.M.; Breen, G.; Byrne, E.M.; Blackwood, D.H.R.; Boomsma, D.I.; Cichon, S.; et al. Major Depressive Disorder Working Group of the Psychiatric, G.C. A mega-analysis of genome-wide association studies for major depressive disorder. Mol. Psychiatry 2013, 18, 497–511. [Google Scholar] [PubMed] [Green Version]
- Cassano, P.; Fava, M. Depression and public health, and overview. J.Psychosom. Res. 2002, 53, 849–857. [Google Scholar] [CrossRef]
- Burns, L.; Teesson, M. Alcohol use disorders comorbid with anxiety, depression and drug use disorders. Findings from the Australian National Survey of Mental Health and Well Being. Drug Alcohol Depend. 2002, 68, 299–307. [Google Scholar] [CrossRef]
- Petrakis, I.L.; Gonzalez, G.; Rosenheck, R.; Krystal, J.H. Comorbidity of alcoholism and psychiatric disorders: An overview. Alcohol Res. Health 2002, 26, 81–89. [Google Scholar]
- Akbar, M.; Egli, M.; Cho, Y.E.; Song, B.J.; Noronha, A. Medications for alcohol use disorders: An overview. Pharmacy 2018, 185, 64–85. [Google Scholar] [CrossRef]
- Li, J.; Kang, S.; Fu, R.; Wu, L.; Wu, W.; Liu, H.; Gregor, D.; Zuo, W.; Bekker, A.; Ye, J.H. Inhibition of AMPA receptor and CaMKII activity in the lateral habenula reduces depressive-like behavior and alcohol intake in rats. Neuropharmacology 2017, 126, 108–120. [Google Scholar] [CrossRef]
- Hikosaka, O. The habenula: From stress evasion to value-based decision-making. Nat. Rev. Neurosci 2010, 11, 503–513. [Google Scholar] [CrossRef]
- Aizawa, H.; Amo, R.; Okamoto, H. Phylogeny and ontogeny of the habenular structure. Front. Neurosci. 2011, 5, 138. [Google Scholar] [CrossRef] [Green Version]
- Namboodiri, V.M.; Rodriguez-Romaguera, J.; Stuber, G.D. The habenula. Curr. Biol. 2016, 26, R873–R877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisler, S.; Trimble, M. The lateral habenula: No longer neglected. CNS Spectr. 2008, 13, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Sartorius, A.; Kiening, K.L.; Kirsch, P.; von Gall, C.C.; Haberkorn, U.; Unterberg, A.W.; Henn, F.A.; Meyer-Lindenberg, A. Remission of major depression under deep brain stimulation of the lateral habenula in a therapy-refractory patient. Biol. Psychiatry 2010, 67, e9–e11. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Piriz, J.; Mirrione, M.; Chung, C.; Proulx, C.D.; Schulz, D.; Henn, F.; Malinow, R. Synaptic potentiation onto habenula neurons in the learned helplessness model of depression. Nature 2011, 470, 535–539. [Google Scholar] [CrossRef]
- Shabel, S.J.; Proulx, C.D.; Piriz, J.; Malinow, R. Mood regulation. GABA/glutamate co-release controls habenula output and is modified by antidepressant treatment. Science 2014, 345, 1494–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecca, S.; Pelosi, A.; Tchenio, A.; Moutkine, I.; Lujan, R.; Hervé, D.; Mameli, M. Rescue of GABAB and GIRK function in the lateral habenula by protein phosphatase 2A inhibition ameliorates depression-like phenotypes in mice. Nat. Med. 2016, 22, 254–261. [Google Scholar] [CrossRef]
- Tchenio, A.; Lecca, S.; Valentinova, K.; Mameli, M. Limiting habenular hyperactivity ameliorates maternal separation-driven depressive-like symptoms. Nat. Commun. 2017, 8, 1135. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Yang, Y.; Ni, Z.; Dong, Y.; Cai, G.; Foncelle, A.; Ma, S.; Sang, K.; Tang, S.; Li, Y.; et al. Astroglial Kir4.1 in the lateral habenula drives neuronal bursts in depression. Nature 2018, 554, 323–327. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Cui, Y.; Sang, K.; Dong, Y.; Ni, Z.; Ma, S.; Hu, H. Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature 2018, 554, 317–322. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Hu, J.; Hu, H. Lateral habenula in the pathophysiology of depression. Curr. Opin. Neurobiol. 2018, 48, 90–96. [Google Scholar] [CrossRef]
- Wang, R.Y.; Aghajanian, G.K. Physiological evidence for habenula as major link between forebrain and midbrain raphe. Science 1977, 197, 89–91. [Google Scholar] [CrossRef]
- Christoph, G.R.; Leonzio, R.J.; Wilcox, K.S. Stimulation of the lateral habenula inhibits dopamine-containing neurons in the substantia nigra and ventral tegmental area of the rat. J. Neurosci. 1986, 6, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Varga, V.; Kocsis, B.; Sharp, T. Electrophysiological evidence for convergence of inputs from the medial prefrontal cortex and lateral habenula on single neurons in the dorsal raphe nucleus. Eur. J. Neurosci. 2003, 17, 280–286. [Google Scholar] [CrossRef]
- Velasquez, K.; Molfese, D.; Salas, R. The role of the habenula in drug addiction. Front. Hum. Neurosci. 2014, 8, 174. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Zhang, B.-L.; Yang, S.-J.; Rusak, B. The role of lateral habenula–dorsal raphe nucleus circuits in higher brain functions and psychiatric illness. Behav. Brain Res. 2015, 277, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Keiflin, R.; Janak, P.H. Dopamine prediction errors in reward learning and addiction: From theory to neural circuitry. Neuron 2015, 88, 247–263. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Cui, Y.; Yang, Y. Circuits and functions of the lateral habenula in health and in disease. Nat. Rev. Neurosci 2020, 21, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Jhou, T.C.; Fields, H.L.; Baxter, M.G.; Saper, C.B.; Holland, P.C. The rostromedial tegmental nucleus (RMTg), a GABAergic afferent to midbrain dopamine neurons, encodes aversive stimuli and inhibits motor responses. Neuron 2009, 61, 786–800. [Google Scholar] [CrossRef] [Green Version]
- Kaufling, J.; Veinante, P.; Pawlowski, S.A.; Freund-Mercier, M.J.; Barrot, M. Afferents to the GABAergic tail of the ventral tegmental area in the rat. J. Comp. Neurol. 2009, 513, 597–621. [Google Scholar] [CrossRef]
- Lecourtier, L.; Defrancesco, A.; Moghaddam, B. Differential tonic influence of lateral habenula on prefrontal cortex and nucleus accumbens dopamine release. Eur. J. Neurosci. 2008, 27, 1755–1762. [Google Scholar] [CrossRef] [Green Version]
- Lammel, S.; Lim, B.K.; Ran, C.; Huang, K.W.; Betley, M.J.; Tye, K.M.; Deisseroth, K.; Malenka, R.C. Input-specific control of reward and aversion in the ventral tegmental area. Nature 2012, 491, 212–217. [Google Scholar] [CrossRef] [Green Version]
- Browne, C.A.; Hammack, R.; Lucki, I. Dysregulation of the lateral habenula in major depressive disorder. Front. Synaptic Neurosci. 2018, 10, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omelchenko, N.; Bell, R.; Sesack, S.R. Lateral habenula projections to dopamine and GABA neurons in the rat ventral tegmental area. Eur. J. Neurosci. 2009, 30, 1239–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proulx, C.D.; Aronson, S.; Milivojevic, D.; Molina, C.; Loi, A.; Monk, B.; Shabel, S.J.; Malinow, R. A neural pathway controlling motivation to exert effort. Proc. Natl. Acad. Sci. USA 2018, 115, 5792–5797. [Google Scholar] [CrossRef] [Green Version]
- Proulx, C.D.; Hikosaka, O.; Malinow, R. Reward processing by the lateral habenula in normal and depressive behaviors. Nat. Neurosci. 2014, 17, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Root, D.H.; Melendez, R.I.; Zaborszky, L.; Napier, T.C. The ventral pallidum: Subregion-specific functional anatomy and roles in motivated behaviors. Prog. Neurobiol. 2015, 130, 29–70. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A.M.; Stuber, G.D. Activation of lateral habenula inputs to the ventral midbrain promotes behavioral avoidance. Nat. Neurosci. 2012, 15, 1105–1107. [Google Scholar] [CrossRef] [PubMed]
- Lecca, S.; Meye, F.J.; Mameli, M. The lateral habenula in addiction and depression: An anatomical, synaptic and behavioral overview. Eur. J. Neurosci. 2014, 39, 1170–1178. [Google Scholar] [CrossRef]
- Matsumoto, M.; Hikosaka, O. Lateral habenula as a source of negative reward signals in dopamine neurons. Nature 2007, 447, 1111–1115. [Google Scholar] [CrossRef] [Green Version]
- Volkow, N.D.; Fowler, J.S.; Wang, G.-J.; Swanson, J.M.; Telang, F. Dopamine in drug abuse and addiction: Results of imaging studies and treatment implications. Arch. Neurol. 2007, 64, 1575–1579. [Google Scholar] [CrossRef]
- Diana, M. The dopamine hypothesis of drug addiction and its potential therapeutic value. Front. Psychiatry 2011, 2, 64. [Google Scholar] [CrossRef] [Green Version]
- Wise, R.A.; Robble, M.A. Dopamine and addiction. Annu. Rev. Psychol. 2020, 71, 79–106. [Google Scholar] [CrossRef]
- You, C.; Vandegrift, B.; Brodie, M.S. Ethanol actions on the ventral tegmental area: Novel potential targets on reward pathway neurons. Psychopharmacol. 2018, 235, 1711–1726. [Google Scholar] [CrossRef] [Green Version]
- Wise, R.A.; Rompre, P.-P. Brain dopamine and reward. Annu. Rev. Psychol. 1989, 40, 191–225. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, M.; Bonci, A. Role of dopamine neurons in reward and aversion: A synaptic plasticity perspective. Neuron 2015, 86, 1145–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graziane, N.M.; Neumann, P.A.; Dong, Y. A Focus on reward prediction and the lateral habenula: Functional alterations and the behavioral outcomes induced by drugs of abuse. Front. Synaptic Neurosci. 2018, 10, 12. [Google Scholar] [CrossRef] [Green Version]
- Faulkner, P.; Deakin, J.F. The role of serotonin in reward, punishment and behavioural inhibition in humans: Insights from studies with acute tryptophan depletion. Neurosci. Biobehav. Rev. 2014, 46, 365–378. [Google Scholar] [CrossRef]
- Müller, C.P.; Homberg, J.R. The role of serotonin in drug use and addiction. Behav. Brain Res. 2015, 277, 146–192. [Google Scholar] [CrossRef]
- Cannon, D.M.; Ichise, M.; Rollis, D.; Klaver, J.M.; Gandhi, S.K.; Charney, D.S.; Manji, H.K.; Drevets, W.C. Elevated serotonin transporter binding in major depressive disorder assessed using positron emission tomography and [11C] DASB.; Comparison with bipolar disorder. Biol. Psychiatry 2007, 62, 870–877. [Google Scholar] [CrossRef]
- Bernard, R.; Veh, R.W. Individual neurons in the rat lateral habenular complex project mostly to the dopaminergic ventral tegmental area or to the serotonergic raphe nuclei. J. Comp. Neurol. 2012, 520, 2545–2558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quina, L.A.; Tempest, L.; Ng, L.; Harris, J.A.; Ferguson, S.; Jhou, T.C.; Turner, E.E. Efferent pathways of the mouse lateral habenula. J. Comp. Neurol. 2015, 523, 32–60. [Google Scholar] [CrossRef] [Green Version]
- Pollak Dorocic, I.; Fürth, D.; Xuan, Y.; Johansson, Y.; Pozzi, L.; Silberberg, G.; Carlén, M.; Meletis, K. A whole-brain atlas of inputs to serotonergic neurons of the dorsal and median raphe nuclei. Neuron 2014, 83, 663–678. [Google Scholar] [CrossRef] [Green Version]
- Weissbourd, B.; Ren, J.; DeLoach, K.E.; Guenthner, C.J.; Miyamichi, K.; Luo, L. Presynaptic partners of dorsal raphe serotonergic and GABAergic neurons. Neuron 2014, 83, 645–662. [Google Scholar] [CrossRef] [Green Version]
- Vertes, R.P.; Fortin, W.J.; Crane, A.M. Projections of the median raphe nucleus in the rat. J. Comp. Neurol. 1999, 407, 555–582. [Google Scholar] [CrossRef]
- Vertes, R.P. A PHA-L analysis of ascending projections of the dorsal raphe nucleus in the rat. J. Comp. Neurol. 1991, 313, 643–668. [Google Scholar] [CrossRef]
- Morin, L.P.; Meyer-Bernstein, E.L. The ascending serotonergic system in the hamster: Comparison with projections of the dorsal and median raphe nuclei. Neuroscience 1999, 91, 81–105. [Google Scholar] [CrossRef]
- Muzerelle, A.; Scotto-Lomassese, S.; Bernard, J.F.; Soiza-Reilly, M.; Gaspar, P. Conditional anterograde tracing reveals distinct targeting of individual serotonin cell groups (B5–B9) to the forebrain and brainstem. Brain Struct. Funct. 2016, 221, 535–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiyasova, V.; Fernandez, S.P.; Laine, J.; Stankovski, L.; Muzerelle, A.; Doly, S.; Gaspar, P. A genetically defined morphologically and functionally unique subset of 5-HT neurons in the mouse raphe nuclei. J. Neurosci. 2011, 31, 2756–2768. [Google Scholar] [CrossRef] [Green Version]
- Fu, R.; Zuo, W.; Shiwalkar, N.; Mei, Q.; Fan, Q.; Chen, X.; Li, J.; Bekker, A.; Ye, J.H. Alcohol withdrawal drives depressive behaviors by activating neurons in the rostromedial tegmental nucleus. Neuropsychopharmacology 2019, 44, 1464–1475. [Google Scholar] [CrossRef]
- Geisler, S.; Andres, K.H.; Veh, R.W. Morphologic and cytochemical criteria for the identification and delineation of individual subnuclei within the lateral habenular complex of the rat. J. Comp. Neurol. 2003, 458, 78–97. [Google Scholar] [CrossRef]
- Zhang, L.; Hernández, V.S.; Vázquez-Juárez, E.; Chay, F.K.; Barrio, R.A. Thirst is associated with suppression of habenula output and active stress coping: Is there a role for a non-canonical vasopressin-glutamate pathway? Front. Neural Circuits 2016, 10, 13. [Google Scholar] [CrossRef]
- Tchenio, A.; Valentinova, K.; Mameli, M. Can the lateral habenula crack the serotonin code? Front. Synaptic Neurosci. 2016, 8, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aizawa, H.; Kobayashi, M.; Tanaka, S.; Fukai, T.; Okamoto, H. Molecular characterization of the subnuclei in rat habenula. J. Comp. Neurol. 2012, 520, 4051–4066. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, H.; Bianco, I.H.; Hamaoka, T.; Miyashita, T.; Uemura, O.; Concha, M.L.; Russell, C.; Wilson, S.W.; Okamoto, H. Laterotopic representation of left-right information onto the dorso-ventral axis of a zebrafish midbrain target nucleus. Curr. Biol. 2005, 15, 238–243. [Google Scholar] [CrossRef] [Green Version]
- Ji, H.; Shepard, P.D. Lateral habenula stimulation inhibits rat midbrain dopamine neurons through a GABA(A) receptor-mediated mechanism. J. Neurosci. 2007, 27, 6923–6930. [Google Scholar] [CrossRef]
- Tian, J.; Uchida, N. Habenula lesions reveal that multiple mechanisms underlie dopamine prediction errors. Neuron 2015, 87, 1304–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Liu, M.-Z.; Li, Q.; Deng, J.; Mu, D.; Sun, Y.-G. Organization of functional long-range circuits controlling the activity of serotonergic neurons in the dorsal raphe nucleus. Cell Rep. 2017, 18, 3018–3032. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Li, J.; Zuo, W.; Fu, R.; Gregor, D.; Krnjevic, K.; Bekker, A.; Ye, J.-H. Ethanol withdrawal drives anxiety-related behaviors by reducing m-type potassium channel activity in the lateral habenula. Neuropsychopharmacology 2017, 42, 1813–1824. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, L.; Sego, C.; Metzger, M. Differential projections from the lateral habenula to the rostromedial tegmental nucleus and ventral tegmental area in the rat. J. Comp. Neurol. 2012, 520, 1278–1300. [Google Scholar] [CrossRef]
- Sego, C.; Gonçalves, L.; Lima, L.; Furigo, I.C.; Donato, J., Jr.; Metzger, M. Lateral habenula and the rostromedial tegmental nucleus innervate neurochemically distinct subdivisions of the dorsal raphe nucleus in the rat. J. Comp. Neurol. 2014, 522, 1454–1484. [Google Scholar] [CrossRef]
- Sun, Y.; Cao, J.; Xu, C.; Liu, X.; Wang, Z.; Zhao, H. Rostromedial tegmental nucleus-substantia nigra pars compacta circuit mediates aversive and despair behavior in mice. Exp. Neurol. 2020, 333, 113433. [Google Scholar] [CrossRef]
- Kang, S.; Li, J.; Bekker, A.; Ye, J.H. Rescue of glutamate transport in the lateral habenula alleviates depression- and anxiety-like behaviors in ethanol-withdrawn rats. Neuropharmacology 2018, 129, 47–56. [Google Scholar] [CrossRef]
- Zuo, W.; Fu, R.; Hopf, F.W.; Xie, G.; Krnjević, K.; Li, J.; Ye, J.H. Ethanol drives aversive conditioning through dopamine 1 receptor and glutamate receptor-mediated activation of lateral habenula neurons. Addict. Biol. 2017, 22, 103–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, J.; Smith, K.; Cowen, P.; Friston, K.; Dolan, R.J. Covariation of activity in habenula and dorsal raphe nuclei following tryptophan depletion. Neuroimage 1999, 10, 163–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shumake, J.; Edwards, E.; Gonzalez-Lima, F. Opposite metabolic changes in the habenula and ventral tegmental area of a genetic model of helpless behavior. Brain Res. 2003, 963, 274–281. [Google Scholar] [CrossRef]
- Li, K.; Zhou, T.; Liao, L.; Yang, Z.; Wong, C.; Henn, F.; Malinow, R.; Yates, J.R., III; Hu, H. βCaMKII in lateral habenula mediates core symptoms of depression. Science 2013, 341, 1016–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, R.P.; Drevets, W.C.; Roiser, J.P. Defining the habenula in human neuroimaging studies. Neuroimage 2013, 64, 722–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.; Li, J.; Zuo, W.; Chen, P.; Gregor, D.; Fu, R.; Han, X.; Bekker, A.; Ye, J.H. Downregulation of M-channels in lateral habenula mediates hyperalgesia during alcohol withdrawal in rats. Sci. Rep. 2019, 9, 2714. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.-M.; Hu, B.; Xia, Y.-H.; Zhang, B.-L.; Zhao, H. Lateral habenula lesions improve the behavioral response in depressed rats via increasing the serotonin level in dorsal raphe nucleus. Behav. Brain Res. 2008, 188, 84–90. [Google Scholar] [CrossRef]
- Meng, H.; Wang, Y.; Huang, M.; Lin, W.; Wang, S.; Zhang, B. Chronic deep brain stimulation of the lateral habenula nucleus in a rat model of depression. Brain Res. 2011, 1422, 32–38. [Google Scholar] [CrossRef]
- Winter, C.; Vollmayr, B.; Djodari-Irani, A.; Klein, J.; Sartorius, A. Pharmacological inhibition of the lateral habenula improves depressive-like behavior in an animal model of treatment resistant depression. Behav. Brain Res. 2011, 216, 463–465. [Google Scholar] [CrossRef]
- Shabel, S.J.; Wang, C.; Monk, B.; Aronson, S.; Malinow, R. Stress transforms lateral habenula reward responses into punishment signals. Proc. Natl. Acad. Sci. USA 2019, 116, 12488–12493. [Google Scholar] [CrossRef] [Green Version]
- Wirtshafter, D.; Asin, K.E.; Pitzer, M.R. Dopamine agonists and stress produce different patterns of Fos-like immunoreactivity in the lateral habenula. Brain Res. 1994, 633, 21–26. [Google Scholar] [CrossRef]
- Park, H.; Rhee, J.; Park, K.; Han, J.-S.; Malinow, R.; Chung, C. Exposure to stressors facilitates long-term synaptic potentiation in the lateral habenula. J. Neurosci. 2017, 37, 6021–6030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregor, D.M.; Zuo, W.; Fu, R.; Bekker, A.; Ye, J.H. Elevation of transient receptor potential vanilloid 1 function in the lateral habenula mediates aversive behaviors in alcohol-withdrawn rats. Anesthesiology 2019, 130, 592–608. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Fowler, J.S.; Wang, G.J.; Swanson, J.M. Dopamine in drug abuse and addiction: Results from imaging studies and treatment implications. Mol. Psychiatry 2004, 9, 557–569. [Google Scholar] [CrossRef] [Green Version]
- Volkow, N.D.; Fowler, J.S.; Wang, G.J.; Baler, R.; Telang, F. Imaging dopamine’s role in drug abuse and addiction. Neuropharmacology 2009, 56, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Spanagel, R. Alcoholism: A systems approach from molecular physiology to addictive behavior. Physiol. Rev. 2009, 89, 649–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhou, T.C.; Good, C.H.; Rowley, C.S.; Xu, S.P.; Wang, H.; Burnham, N.W.; Hoffman, A.F.; Lupica, C.R.; Ikemoto, S. Cocaine drives aversive conditioning via delayed activation of dopamine-responsive habenular and midbrain pathways. J. Neurosci. 2013, 33, 7501–7512. [Google Scholar] [CrossRef]
- Nowak, K.L.; McBride, W.J.; Lumeng, L.; Li, T.K.; Murphy, J.M. Involvement of dopamine D2 autoreceptors in the ventral tegmental area on alcohol and saccharin intake of the alcohol-preferring P rat. Alcohol Clin. Exp. Res. 2000, 24, 476–483. [Google Scholar] [CrossRef]
- Gonzales, R.A.; Job, M.O.; Doyon, W.M. The role of mesolimbic dopamine in the development and maintenance of ethanol reinforcement. Pharmacol. Ther. 2004, 103, 121–146. [Google Scholar] [CrossRef]
- Koob, G.F.; Volkow, N.D. Neurocircuitry of addiction. Neuropsychopharmacology 2010, 35, 217–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diana, M.; Pistis, M.; Muntoni, A.; Rossetti, Z.L.; Gessa, G. Marked decrease of A10 dopamine neuronal firing during ethanol withdrawal syndrome in rats. Eur. J. Pharmacol. 1992, 221, 403–404. [Google Scholar] [CrossRef]
- Diana, M.; Pistis, M.; Carboni, S.; Gessa, G.L.; Rossetti, Z.L. Profound decrement of mesolimbic dopaminergic neuronal activity during ethanol withdrawal syndrome in rats: Electrophysiological and biochemical evidence. Proc. Natl. Acad. Sci. USA 1993, 90, 7966–7969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, R.Y. Ethanol withdrawal reduces the number of spontaneously active ventral tegmental area dopamine neurons in conscious animals. J. Pharm. Exp. 2003, 307, 566–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, F.; Parsons, L.H.; Schulteis, G.; Hyytiä, P.; Lorang, M.T.; Bloom, F.E.; Koob, G.F. Ethanol self-administration restores withdrawal-associated deficiencies in accumbal dopamine and 5-hydroxytryptamine release in dependent rats. J. Neurosci. 1996, 16, 3474–3485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barak, S.; Carnicella, S.; Yowell, Q.V.; Ron, D. Glial cell line-derived neurotrophic factor reverses alcohol-induced allostasis of the mesolimbic dopaminergic system: Implications for alcohol reward and seeking. J. Neurosci. 2011, 31, 9885–9894. [Google Scholar] [CrossRef] [Green Version]
- Szczypiński, J.J.; Gola, M. Dopamine dysregulation hypothesis: The common basis for motivational anhedonia in major depressive disorder and schizophrenia? Rev. Neurosci. 2018, 29, 727–744. [Google Scholar] [CrossRef]
- Belujon, P.; Grace, A.A. Dopamine system dysregulation in major depressive disorders. Int. J. Neuropsychopharmacol. 2017, 20, 1036–1046. [Google Scholar] [CrossRef] [Green Version]
- Hudmon, A.; Schulman, H. Structure–function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem. J. 2002, 364, 593–611. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, T. Neuronal Ca2+/calmodulin-dependent protein kinase II—Discovery, progress in a quarter of a century, and perspective: Implication for learning and memory. Biol. Pharm. Bull. 2005, 28, 1342–1354. [Google Scholar] [CrossRef] [Green Version]
- Swulius, M.; Waxham, M. Ca2+/Calmodulin-dependent Protein Kinases. Cell. Mol. Life Sci. 2008, 65, 2637–2657. [Google Scholar] [CrossRef] [Green Version]
- Zalcman, G.; Federman, N.; Romano, A. CaMKII isoforms in learning and memory: Localization and function. Front. Mol. Neurosci. 2018, 11, 445. [Google Scholar] [CrossRef]
- Griffith, L.C. Calcium/calmodulin-dependent protein kinase II: An unforgettable kinase. J. Neurosci. 2004, 24, 8391–8393. [Google Scholar] [CrossRef] [Green Version]
- Soderling, T.R.; Stull, J.T. Structure and regulation of calcium/calmodulin-dependent protein kinases. Chem. Rev. 2001, 101, 2341–2352. [Google Scholar] [CrossRef] [PubMed]
- Colbran, R.J. Protein phosphatases and calcium/calmodulin-dependent protein kinase ii-dependent synaptic plasticity. J. Neurosci. 2004, 24, 8404–8409. [Google Scholar] [CrossRef]
- Braun, A.P.; Schulman, H. The multifunctional calcium/calmodulin-dependent protein kinase: From form to function. Annu. Rev. Physiol. 1995, 57, 417–445. [Google Scholar] [CrossRef]
- Colbran, R.J.; Brown, A.M. Calcium/calmodulin-dependent protein kinase II and synaptic plasticity. Curr. Opin. Neurobiol. 2004, 14, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Coultrap, S.J.; Buard, I.; Kulbe, J.R.; Dell’Acqua, M.L.; Bayer, K.U. CaMKII autonomy is substrate-dependent and further stimulated by Ca2+/calmodulin. J. Biol. Chem. 2010, 285, 17930–17937. [Google Scholar] [CrossRef] [Green Version]
- Cannady, R.; Fisher, K.R.; Graham, C.; Crayle, J.; Besheer, J.; Hodge, C.W. Potentiation of amygdala AMPA receptor activity selectively promotes escalated alcohol self-administration in a CaMKII-dependent manner. Addict. Biol. 2017, 22, 652–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easton, A.C.; Lucchesi, W.; Lourdusamy, A.; Lenz, B.; Solati, J.; Golub, Y.; Lewczuk, P.; Fernandes, C.; Desrivieres, S.; Dawirs, R.R.; et al. αCaMKII autophosphorylation controls the establishment of alcohol drinking behavior. Neuropsychopharmacology 2013, 38, 1636–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salling, M.C.; Hodge, C.J.; Psilos, K.E.; Eastman, V.R.; Faccidomo, S.P.; Hodge, C.W. Cue-induced reinstatement of alcohol-seeking behavior is associated with increased CaMKII T286 phosphorylation in the reward pathway of mice. Pharm. Biochem. Behav. 2017, 163, 20–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zuo, W.; Fu, R.; Xie, G.; Kaur, A.; Bekker, A.; Ye, J.H. High Frequency electrical stimulation of lateral habenula reduces voluntary ethanol consumption in rats. Int. J. Neuropsychopharmacol. 2016, 19, 10. [Google Scholar] [CrossRef] [Green Version]
- Easton, A.C.; Lucchesi, W.; Mizuno, K.; Fernandes, C.; Schumann, G.; Giese, K.P.; Muller, C.P. alphaCaMKII autophosphorylation controls the establishment of alcohol-induced conditioned place preference in mice. Behav. Brain. Res. 2013, 252, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Schopf, I.; Easton, A.C.; Solati, J.; Golub, Y.; Kornhuber, J.; Giese, K.P.; Muller, C.P. alphaCaMKII autophosphorylation mediates neuronal activation in the hippocampal dentate gyrus after alcohol and cocaine in mice. Neurosci. Lett. 2015, 591, 65–68. [Google Scholar] [CrossRef]
- Faccidomo, S.; Reid, G.T.; Agoglia, A.E.; Ademola, S.A.; Hodge, C.W. CaMKII inhibition in the prefrontal cortex specifically increases the positive reinforcing effects of sweetened alcohol in C57BL/6J mice. Behav. Brain Res. 2016, 298, 286–290. [Google Scholar] [CrossRef] [Green Version]
- Natividad, L.A.; Steinman, M.Q.; Laredo, S.A.; Irimia, C.; Polis, I.Y.; Lintz, R.; Buczynski, M.W.; Martin-Fardon, R.; Roberto, M.; Parsons, L.H. Phosphorylation of calcium/calmodulin-dependent protein kinase II in the rat dorsal medial prefrontal cortex is associated with alcohol-induced cognitive inflexibility. Addict. Biol. 2018, 23, 1117–1129. [Google Scholar] [CrossRef]
- de Paiva Lima, C.; da Silva, E.S.D.A.; Damasceno, S.; Ribeiro, A.F.; Rocha, C.S.; Berenguer de Matos, A.H.; Correia, D.; Boerngen-Lacerda, R.; Brunialti Godard, A.L. Loss of control over the ethanol consumption: Differential transcriptional regulation in prefrontal cortex. J. Neurogenet. 2017, 31, 170–177. [Google Scholar] [CrossRef]
- Yuanyuan, J.; Junyan, Z.; Cuola, D.; Jingjing, C.; Yuhui, S.; Dan, X.; Wei, D.; Yongsheng, Z. Memantine attenuated alcohol withdrawal-induced anxiety-like behaviors through down-regulating NR1-CaMKII-ERK signaling pathway. Neurosci. Lett. 2018, 686, 133–139. [Google Scholar] [CrossRef]
- Somkuwar, S.S.; Mandyam, C.D. Individual differences in ethanol drinking and seeking behaviors in rats exposed to chronic intermittent ethanol vapor exposure is associated with altered CaMKII autophosphorylation in the nucleus accumbens shell. Brain Sci 2019, 9, 367. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Wang, Y.; Li, Y.; Qiao, X.; Yan, P.; Zhu, Y.; Lai, J. Differential phosphorylation of NMDAR1-CaMKII-MAPKs in the rat nucleus accumbens following chronic ethanol exposure. Neurosci. Lett. 2015, 597, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Coelho, M.A.; McGregor, H.A.; Solton, N.R.; Cohen, M.; Szumlinski, K.K. adolescent mice are resilient to alcohol withdrawal-induced anxiety and changes in indices of glutamate function within the nucleus accumbens. Front. Cell Neurosci 2016, 10, 265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kash, T.L.; Baucum, A.J.; Conrad, K.L.; Colbran, R.J.; Winder, D.G. Alcohol exposure alters NMDAR function in the bed nucleus of the stria terminalis. Neuropsychopharmacology 2009, 34, 2420–2429. [Google Scholar] [CrossRef]
- Zuo, W.; Wu, L.; Mei, Q.; Zuo, Q.; Zhou, Z.; Fu, R.; Li, W.; Wu, W.; Matthew, L.; Ye, J.H. Adaptation in 5-HT(2) receptors-CaMKII signaling in lateral habenula underlies increased nociceptive-sensitivity in ethanol-withdrawn rats. Neuropharmacology 2019, 158, 107747. [Google Scholar] [CrossRef] [PubMed]
- Salling, M.C.; Faccidomo, S.P.; Li, C.; Psilos, K.; Galunas, C.; Spanos, M.; Agoglia, A.E.; Kash, T.L.; Hodge, C.W. Moderate alcohol drinking and the amygdala proteome: Identification and validation of calcium/calmodulin dependent kinase II and AMPA receptor activity as novel molecular mechanisms of the positive reinforcing effects of alcohol. Biol. Psychiatry 2016, 79, 430–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agoglia, A.E.; Holstein, S.E.; Reid, G.; Hodge, C.W. CaMKIIalpha-GluA1 activity underlies vulnerability to adolescent binge alcohol drinking. Alcohol Clin. Exp. Res. 2015, 39, 1680–1690. [Google Scholar] [CrossRef] [Green Version]
- Christian, D.T.; Alexander, N.J.; Diaz, M.R.; Robinson, S.; McCool, B.A. Chronic intermittent ethanol and withdrawal differentially modulate basolateral amygdala AMPA-type glutamate receptor function and trafficking. Neuropharmacology 2012, 62, 2430–2439. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Cui, H.; Wang, W.; Zhao, B.; Lai, J. The region-specific activation of Ca2+/calmodulin dependent protein kinase II and extracellular signal-regulated kinases in hippocampus following chronic alcohol exposure. Brain Res. Bull. 2012, 89, 191–196. [Google Scholar] [CrossRef]
- Mahadev, K.; Chetty, C.S.; Vemuri, M.C. Effect of prenatal and postnatal ethanol exposure on Ca2+ /calmodulin-dependent protein kinase II in rat cerebral cortex. Alcohol 2001, 23, 183–188. [Google Scholar] [CrossRef]
- Fu, R.; Gregor, D.; Peng, Z.; Li, J.; Bekker, A.; Ye, J. Chronic intermittent voluntary alcohol drinking induces hyperalgesia in Sprague-Dawley rats. Int. J. Physiol. Pathophysiol. Pharmacol. 2015, 7, 136–144. [Google Scholar]
- Li, J.; Li, Y.; Zhang, B.; Shen, X.; Zhao, H. Why depression and pain often coexist and mutually reinforce: Role of the lateral habenula. Exp. Neurol. 2016, 284, 106–113. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Xuan, C.; Li, Y.; Piao, L.; Li, J.; Zhao, H. Role of the lateral habenula in pain-associated depression. Front. Behav. Neurosci. 2017, 11, 31. [Google Scholar] [CrossRef] [Green Version]
- Sheng, J.; Liu, S.; Wang, Y.; Cui, R.; Zhang, X. The link between depression and chronic pain: Neural mechanisms in the brain. Neural Plast. 2017, 2017, 9724371. [Google Scholar] [CrossRef] [PubMed]
- Delicata, F.; Bombardi, C.; Pierucci, M.; Di Maio, R.; De Deurwaerdère, P.; Di Giovanni, G. Preferential modulation of the lateral habenula activity by serotonin-2A rather than -2C receptors: Electrophysiological and neuroanatomical evidence. CNS Neurosci. Ther. 2018, 24, 721–733. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Zhang, L.; Zhang, J.; Du, C.-X.; Lv, S.-X.; Wang, T.; Wang, H.-S.; Xie, W.; Liu, J. Activation and blockade of serotonin4 receptors in the lateral habenula improve working memory in unilateral 6-hydroxydopamine-lesioned Parkinson’s rats. Neurol. Res. 2019, 41, 585–593. [Google Scholar] [PubMed]
- Shabel, S.J.; Proulx, C.D.; Trias, A.; Murphy, R.T.; Malinow, R. Input to the lateral habenula from the basal ganglia is excitatory, aversive, and suppressed by serotonin. Neuron 2012, 74, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Xie, G.; Zuo, W.; Wu, L.; Li, W.; Wu, W.; Bekker, A.; Ye, J.-H. Serotonin modulates glutamatergic transmission to neurons in the lateral habenula. Sci. Rep. 2016, 6, 23798. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Carnicella, S.; Phamluong, K.; Jeanblanc, J.; Ronesi, J.A.; Chaudhri, N.; Janak, P.H.; Lovinger, D.M.; Ron, D. Ethanol induces long-term facilitation of NR2B-NMDA receptor activity in the dorsal striatum: Implications for alcohol drinking behavior. J. Neurosci. 2007, 27, 3593–3602. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, P.L.; Tabakoff, B. The role of the NMDA receptor in ethanol withdrawal. Exs 1994, 71, 61–70. [Google Scholar]
- Mirshahi, T.; Woodward, J.J. Ethanol sensitivity of heteromeric NMDA receptors: Effects of subunit assembly, glycine and NMDAR1 Mg2+-insensitive mutants. Neuropharmacology 1995, 34, 347–355. [Google Scholar] [CrossRef]
- Chu, B.; Anantharam, V.; Treistman, S.N. Ethanol inhibition of recombinant heteromeric NMDA channels in the presence and absence of modulators. J. Neurochem. 1995, 65, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Lovinger, D.M.; White, G.; Weight, F.F. Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science 1989, 243, 1721–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovinger, D.M.; White, G.; Weight, F.F. NMDA receptor-mediated synaptic excitation selectively inhibited by ethanol in hippocampal slice from adult rat. J. Neurosci. 1990, 10, 1372–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, H.S.; Ticku, M.K. Effect of ethanol on phosphorylation of the NMDAR2B subunit in mouse cortical neurons. Mol. Brain Res. 1999, 68, 159–168. [Google Scholar] [CrossRef]
- Kash, T.L.; Matthews, R.T.; Winder, D.G. Alcohol inhibits NR2B-containing NMDA receptors in the ventral bed nucleus of the stria terminalis. Neuropsychopharmacology 2008, 33, 1379–1390. [Google Scholar] [CrossRef]
- Kalluri, H.S.; Mehta, A.K.; Ticku, M.K. Up-regulation of NMDA receptor subunits in rat brain following chronic ethanol treatment. Brain Res. Mol. Brain Res. 1998, 58, 221–224. [Google Scholar] [CrossRef]
- Carpenter-Hyland, E.P.; Chandler, L.J. Homeostatic plasticity during alcohol exposure promotes enlargement of dendritic spines. Eur. J. Neurosci. 2006, 24, 3496–3506. [Google Scholar] [CrossRef]
- Chandrasekar, R. Alcohol and NMDA receptor: Current research and future direction. Front. Mol. Neurosci. 2013, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- Hell, J.W. CaMKII: Claiming center stage in postsynaptic function and organization. Neuron 2014, 81, 249–265. [Google Scholar] [CrossRef] [Green Version]
- Fukunaga, K.; Stoppini, L.; Miyamoto, E.; Muller, D. Long-term potentiation is associated with an increased activity of Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 1993, 268, 7863–7867. [Google Scholar]
- Kasahara, J.; Fukunaga, K.; Miyamoto, E. Activation of calcium/calmodulin-dependent protein kinase IV in long term potentiation in the rat hippocampal CA1 region. J. Biol. Chem. 2001, 276, 24044–24050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukunaga, K.; Muller, D.; Miyamoto, E. Increased phosphorylation of Ca2+/calmodulin-dependent protein kinase II and its endogenous substrates in the induction of long-term potentiation. J. Biol. Chem. 1995, 270, 6119–6124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukunaga, K.; Muller, D.; Miyamoto, E. CaM kinase II in long-term potentiation. Neurochem. Int. 1996, 28, 343–358. [Google Scholar] [CrossRef]
- Bredt, D.S.; Nicoll, R.A. AMPA receptor trafficking at excitatory synapses. Neuron 2003, 40, 361–379. [Google Scholar] [CrossRef] [Green Version]
- Kryger, R.; Wilce, P. The effects of alcoholism on the human basolateral amygdala. Neuroscience 2010, 167, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hamida, S.B.; Darcq, E.; Zhu, W.; Gibb, S.L.; Lanfranco, M.F.; Carnicella, S.; Ron, D. Ethanol-mediated facilitation of AMPA receptor function in the dorsomedial striatum: Implications for alcohol drinking behavior. J. Neurosci. 2012, 32, 15124–15132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meye, F.J.; Lecca, S.; Valentinova, K.; Mameli, M. Synaptic and cellular profile of neurons in the lateral habenula. Front. Hum. Neurosci. 2013, 7, 860. [Google Scholar] [CrossRef] [Green Version]
- Rao, V.R.; Finkbeiner, S. NMDA and AMPA receptors: Old channels, new tricks. Trends Neurosci. 2007, 30, 284–291. [Google Scholar] [CrossRef]
- Cannady, R.; Fisher, K.R.; Durant, B.; Besheer, J.; Hodge, C.W. Enhanced AMPA receptor activity increases operant alcohol self-administration and cue-induced reinstatement. Addict. Biol 2013, 18, 54–65. [Google Scholar] [CrossRef]
- Mao, L.M.; Guo, M.L.; Jin, D.Z.; Fibuch, E.E.; Choe, E.S.; Wang, J.Q. Post-translational modification biology of glutamate receptors and drug addiction. Front. Neuroanat. 2011, 5, 19. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Bian, E.; Li, J.; Zuo, W.; Mei, Q.; Fu, R.; Shiwalkar, N.; Fan, Q.; Gajewski, M.; Ye, J.-H. Pain, anxiety- and depression-like behaviors in alcohol-preferring and -non-preferring rats. Neurol. Neurobiol. 2020, 3, 1–8. [Google Scholar]
- Wang, J.; Li, M.; Wang, P.; Zha, Y.; He, Z.; Li, Z. Inhibition of the lateral habenular CaMKII abolishes naloxone-precipitated conditioned place aversion in morphine-dependent mice. Neurosci. Lett. 2017, 653, 64–70. [Google Scholar] [CrossRef]
- Shah, A.; Zuo, W.; Kang, S.; Li, J.; Fu, R.; Zhang, H.; Bekker, A.; Ye, J.H. The lateral habenula and alcohol: Role of glutamate and M-type potassium channels. Pharm. Biochem. Behav. 2017, 162, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Berman, R.M.; Cappiello, A.; Anand, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S.; Krystal, J.H. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 2000, 47, 351–354. [Google Scholar] [CrossRef]
- Regier, D.A.; Farmer, M.E.; Rae, D.S.; Locke, B.Z.; Keith, S.J.; Judd, L.L.; Goodwin, F.K. Comorbidity of mental disorders with alcohol and other drug abuse: Results from the epidemiologic catchment area (ECA) study. JAMA 1990, 264, 2511–2518. [Google Scholar] [CrossRef]
- Kessler, R.C.; Nelson, C.B.; McGonagle, K.A.; Edlund, M.J.; Frank, R.G.; Leaf, P.J. The epidemiology of co-occurring addictive and mental disorders: Implications for prevention and service utilization. Am. J. Orthopsychiatry 1996, 66, 17–31. [Google Scholar] [CrossRef] [Green Version]
- Grant, B.F.; Harford, T.C. Comorbidity between DSM-IV alcohol use disorders and major depression: Results of a national survey. Drug Alcohol Depend. 1995, 39, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Vaillant, G.E. Is alcoholism more often the cause or the result of depression? Harv Rev. Psychiatry 1993, 1, 94–99. [Google Scholar] [CrossRef]
- Pettinati, H.M. Antidepressant treatment of co-occurring depression and alcohol dependence. Biol. Psychiatry 2004, 56, 785–792. [Google Scholar] [CrossRef]
- Davis, L.; Uezato, A.; Newell, J.M.; Frazier, E. Major depression and comorbid substance use disorders. Curr. Opin. Psychiatry 2008, 21, 14–18. [Google Scholar] [CrossRef]
- Hasin, D.S.; Stinson, F.S.; Ogburn, E.; Grant, B.F. Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: Results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch. Gen. Psychiatry 2007, 64, 830–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R.K.; Gale, G.; Walsh, K.; Hennessy, V.E.; Iskandar, G.; Mordecai, L.A.; Brandner, B.; Kindt, M.; Curran, H.V.; Kamboj, S.K. Ketamine can reduce harmful drinking by pharmacologically rewriting drinking memories. Nat. Commun. 2019, 10, 5187. [Google Scholar] [CrossRef]
- Getachew, B.; Tizabi, Y. Both ketamine and NBQX attenuate alcohol-withdrawal induced depression in male rats. J. Drug Alcohol Res. 2019, 8, 236069. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Brain Region | Alcohol Treatment | Tested In | CaMKII Levels | Findings | Reference |
|---|---|---|---|---|---|
| Global | 2 g/kg, i.p. | pCaMKIIα-deficient, heterozygous, and WT mice | N/A | ↑ EtOH’s negative reinforcing action in pCaMKIIα-deficient mice | [114] |
| Three-bottle free choice | Onset of EtOH consumption delayed in pCaMKIIα-deficient mice | [111] | |||
| 2 g/kg, i.p. | pCaMKII is crucial to hippocampal DG neuron activation after EtOH exposure | [115] | |||
| PFC | Operant self-admin | C57BL/6J mice | CaMKIIα (↑) | ↓ mPFC CaMKII ↑ the positive reinforcing effects of sweetened EtOH | [116] |
| CIE | L-E rats (M) | CaMKIIα (↑) | pCaMKII in dorsal mPFC is associated with EtOH-induced cognitive inflexibility | [117] | |
| Three-bottle free choice | Mice | CaMKIIα (↓) | PFC CaMkIIα gene is linked to EtOH consumption | [118] | |
| mPFC and NAc Sh | Self-admin (20%) (28 d) | S-D rats (M) | pCaMKII (↑) at 6 h deprivation | CaMKII mediates EtOH-WD-induced anxiety and neurochemical adaptations in mPFC and NAc | [119] |
| NAc Sh | CIE | Adult Wistar rats (M) | pCaMKII (↓) in high responders | ↓ pCaMKII is involved in EtOH-seeking behaviors | [120] |
| Self-admin (6%) (28 d) | S-D rats (M) | pCaMKII CIE (↓) 24 h WD (↑) | Activation of NMDAR1–CaMKII contributes to EtOH drinking and negative emotional states | [121] | |
| Drink in the dark (14 d) | Adult and adolescent C57BL/6J mice (M) | CaMKII at 24 h WD Adult (↑) Adolescent (↔) | CaMKII is positively linked to negative affective symptoms in EtOH-WD adults | [122] | |
| BNST | 0.8 g/kg, i.p. + CIE | C57BL/6J mice (M) | CaMKIIα (↓) | ↓ CaMKIIα in EtOH-exposed vBNST contributes to changes in synaptic NMDAR kinetics | [123] |
| Amygdala, NAc, septum, thalamus, piriform cortex | Self-admin | C57BL/6J mice (M) | pCaMKII-T286 (↑) | Cue-induced reinstatement of EtOH-seeking is associated with pCaMKII-T286 in reward- and memory-related brain regions | [112] |
| LHb | Two-bottle free choice (8–12 w) | S-D and L-E rats (M) | CaMKII (↑) at 24 h WD | ↓ CaMKII in the LHb ↓ EtOH intake and depressive symptoms | [7,124] |
| Amygdala | Self-admin | Adult P rats (M) | pCaMKII-T286 and CaMKIIα (↑) | ↓ Acb CaMKII ↓ EtOH self-administration | [110] |
| Operant self-admin (15%) | Adult C57BL/6J mice (M) | pCaMKII (↑) | EtOH drinking ↑ CaMKII in the amygdala that regulates EtOH’s positive reinforcing effects | [125] | |
| Home-cage drink and operant self-admin | Adolescent and adult C57BL/6J mice (M) | pCaMKIIα-T286 Adolescent (↓) Adults (↔) | Differential CaMKIIα-dependent AMPAR activation underlies age-related escalation of binge drinking | [126] | |
| Basolateral amygdala | CIE | S-D rats (M) | pCaMKII-T286 CIE (↑), WD (↔) | CIE- and WD-induced changes in CaMKII activity contribute to ↑ GluA1R phosphorylation/trafficking | [127] |
| pCaMKII-T305 CIE (↔), WD (↑) | |||||
| Hippocampal CA1 and DG | Self-admin (20%) (28 d) | S-D rats (M) | pCaMKII-T286 CIE (↓) WD (↑) | CaMKII activation in hippocampal subregions contributes to EtOH dependence | [128] |
| Cerebral cortex | Self-admin (10%) | Wistar rats | CaMKIIα (↑) | Pre- and post-natal EtOH exposure ↑ CaMKII levels in membrane and cytosolic fractions | [129] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shor, C.; Zuo, W.; Eloy, J.D.; Ye, J.-H. The Emerging Role of LHb CaMKII in the Comorbidity of Depressive and Alcohol Use Disorders. Int. J. Mol. Sci. 2020, 21, 8123. https://doi.org/10.3390/ijms21218123
Shor C, Zuo W, Eloy JD, Ye J-H. The Emerging Role of LHb CaMKII in the Comorbidity of Depressive and Alcohol Use Disorders. International Journal of Molecular Sciences. 2020; 21(21):8123. https://doi.org/10.3390/ijms21218123
Chicago/Turabian StyleShor, Chaya, Wanhong Zuo, Jean D. Eloy, and Jiang-Hong Ye. 2020. "The Emerging Role of LHb CaMKII in the Comorbidity of Depressive and Alcohol Use Disorders" International Journal of Molecular Sciences 21, no. 21: 8123. https://doi.org/10.3390/ijms21218123
APA StyleShor, C., Zuo, W., Eloy, J. D., & Ye, J.-H. (2020). The Emerging Role of LHb CaMKII in the Comorbidity of Depressive and Alcohol Use Disorders. International Journal of Molecular Sciences, 21(21), 8123. https://doi.org/10.3390/ijms21218123