
The Role of CaMKII and ERK Signaling in Addiction




Abstract
1. Introduction
2. Mechanisms Involved in Nicotine-Induced Addiction in the Brain
3. CaMKII in Nicotine and Other Psychostimulant-Induced Addiction
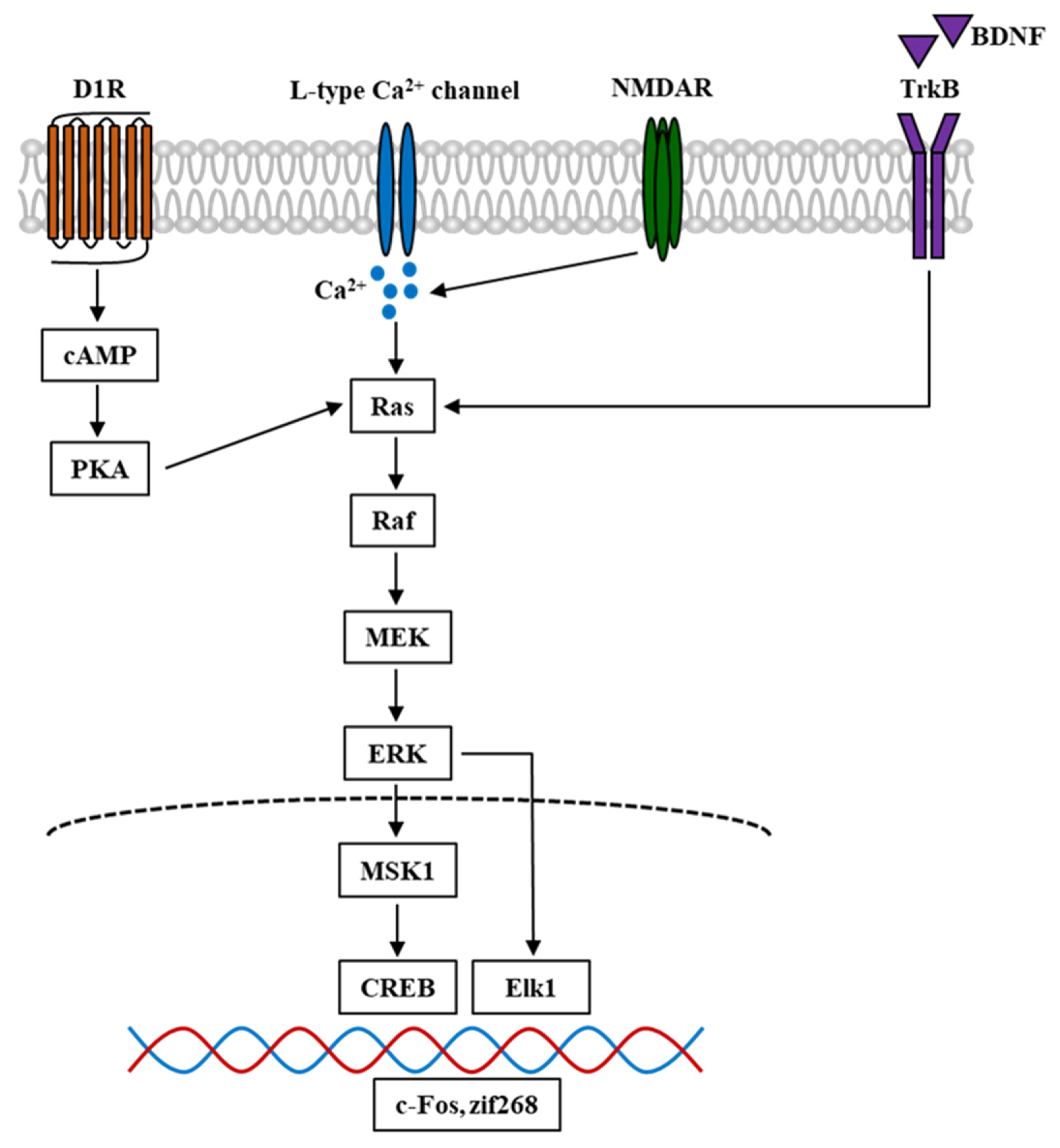
4. ERK in Nicotine and Other Psychostimulant-Induced Addiction
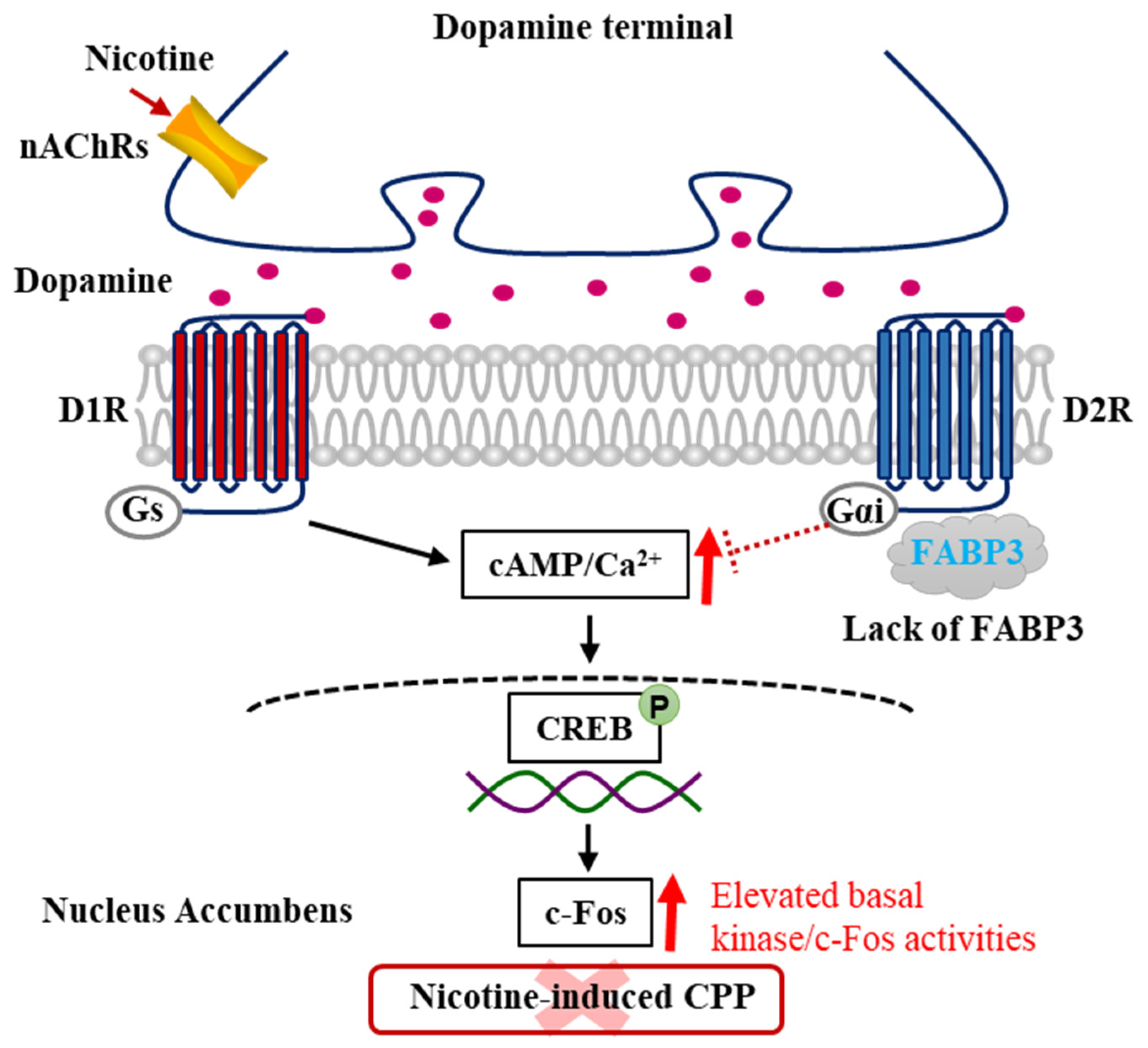
5. Involvement of CaMKII and ERK in the DAergic System in the Nucleus Accumbens
6. Involvement of LCPUFAs and the Novel Target FABP3 in Nicotine and Other Psychostimulant-Induced Addiction
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Forouzanfar, M.H.; Afshin, A.; Alexander, L.T.; Anderson, H.R.; Bhutta, Z.A.; Biryukov, S.; Brauer, M.; Burnett, R.; Cercy, K.; Charlson, F.J.; et al. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1659–1724. [Google Scholar] [CrossRef]
- Reitsma, M.B.; Fullman, N.; Ng, M.; Salama, J.S.; Abajobir, A.; Abate, K.H.; Abbafati, C.; Abera, S.F.; Abraham, B.; Abyu, G.Y.; et al. Smoking prevalence and attributable disease burden in 195 countries and territories, 1990–2015: A systematic analysis from the Global Burden of Disease Study 2015. Lancet 2017, 389, 1885–1906. [Google Scholar] [CrossRef]
- Peto, R.; Boreham, J.; Lopez, A.; Thun, M.; Heath, C. Mortality from tobacco in developed countries: Indirect estimation from national vital statistics. Lancet 1992, 339, 1268–1278. [Google Scholar] [CrossRef]
- Iribarren, C.; Tekawa, I.S.; Sidney, S.; Friedman, G.D. Effect of Cigar Smoking on the Risk of Cardiovascular Disease, Chronic Obstructive Pulmonary Disease, and Cancer in Men. N. Engl. J. Med. 1999, 340, 1773–1780. [Google Scholar] [CrossRef]
- Rose, J.E.; Corrigall, W.A. Nicotine self-administration in animals and humans: Similarities and differences. Psychopharmacology 1997, 130, 28–40. [Google Scholar] [CrossRef]
- Balfour, D.J.; Wright, A.E.; Benwell, M.E.; Birrell, C.E. The putative role of extra-synaptic mesolimbic dopamine in the neurobiology of nicotine dependence. Behav. Brain Res. 2000, 113, 73–83. [Google Scholar] [CrossRef]
- Di Chiara, G. Role of dopamine in the behavioural actions of nicotine related to addiction. Eur. J. Pharmacol. 2000, 393, 295–314. [Google Scholar] [CrossRef]
- Balfour, D.J.; Fagerström, K.O. Pharmacology of nicotine and its therapeutic use in smoking cessation and neurodegenerative disorders. Pharmacol. Ther. 1996, 72, 51–81. [Google Scholar] [CrossRef]
- Shiffman, S.; Scharf, D.M.; Shadel, W.G.; Gwaltney, C.J.; Dang, Q.; Paton, S.M.; Clark, D.B. Analyzing milestones in smoking cessation: Illustration in a nicotine patch trial in adult smokers. J. Consult. Clin. Psychol. 2006, 74, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.M.; Messing, R.O. Protein Kinases and Addiction. Ann. N. Y. Acad. Sci. 2008, 1141, 22–57. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, K.; Stoppini, L.; Miyamoto, E.; Muller, D. Long-term potentiation is associated with an increased activity of Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 1993, 268, 7863–7867. [Google Scholar] [CrossRef]
- Fukunaga, K.; Muller, D.; Miyamoto, E. Increased Phosphorylation of Ca2+/Calmodulin-dependent Protein Kinase II and Its Endogenous Substrates in the Induction of Long Term Potentiation. J. Biol. Chem. 1995, 270, 6119–6124. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, E.X.; Alkondon, M.; Pereira, E.F.; Castro, N.G.; Schrattenholz, A.; Barbosa, C.T.; Bonfante-Cabarcas, R.; Aracava, Y.; Eisenberg, H.M.; Maelicke, A. Properties of neuronal nicotinic acetylcholine receptors: Pharmacological characterization and modulation of synaptic function. J. Pharmacol. Exp. Ther. 1997, 280, 1117–1136. [Google Scholar]
- Unwin, N. Structure and action of the nicotinic acetylcholine receptor explored by electron microscopy. FEBS Lett. 2003, 555, 91–95. [Google Scholar] [CrossRef]
- Dajas-Bailador, F.; Wonnacott, S. Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol. Sci. 2004, 25, 317–324. [Google Scholar] [CrossRef]
- Dani, J.A.; De Biasi, M. Cellular mechanisms of nicotine addiction. Pharmacol. Biochem. Behav. 2001, 70, 439–446. [Google Scholar] [CrossRef]
- Gotti, C.; Clementi, F.; Fornari, A.; Gaimarri, A.; Guiducci, S.; Manfredi, I.; Moretti, M.; Pedrazzi, P.; Pucci, L.; Zoli, M. Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem. Pharmacol. 2009, 78, 703–711. [Google Scholar] [CrossRef]
- Zoli, M.; Pistillo, F.; Gotti, C. Diversity of native nicotinic receptor subtypes in mammalian brain. Neuropharmacology 2015, 96, 302–311. [Google Scholar] [CrossRef]
- Klink, R.; de Kerchove d’Exaerde, A.; Zoli, M.; Changeux, J.P. Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J. Neurosci. 2001, 21, 1452–1463. [Google Scholar] [CrossRef]
- Wonnacott, S.; Sidhpura, N.; Balfour, D.J.K. Nicotine: From molecular mechanisms to behaviour. Curr. Opin. Pharmacol. 2005, 5, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Maskos, U.; Molles, B.E.; Pons, S.; Besson, M.; Guiard, B.P.; Guilloux, J.-P.; Evrard, A.; Cazala, P.; Cormier, A.; Mameli-Engvall, M.; et al. Nicotine reinforcement and cognition restored by targeted expression of nicotinic receptors. Nat. Cell Biol. 2005, 436, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Mineur, Y.S.; Picciotto, M.R. Genetics of nicotinic acetylcholine receptors: Relevance to nicotine addiction. Biochem. Pharmacol. 2008, 75, 323–333. [Google Scholar] [CrossRef]
- Pons, S.; Fattore, L.; Cossu, G.; Tolu, S.; Porcu, E.; McIntosh, J.M.; Changeux, J.; Maskos, U.; Fratta, W. Crucial Role of 4 and 6 Nicotinic Acetylcholine Receptor Subunits from Ventral Tegmental Area in Systemic Nicotine Self-Administration. J. Neurosci. 2008, 28, 12318–12327. [Google Scholar] [CrossRef] [PubMed]
- Gentry, C.L.; Lukas, R.J. Regulation of Nicotinic Acetylcholine Receptor Numbers and Function by Chronic Nicotine Exposure. Curr. Drug Target CNS Neurol. Disord. 2002, 1, 359–385. [Google Scholar] [CrossRef]
- Mineur, Y.S.; Brunzell, D.H.; Grady, S.R.; Lindstrom, J.M.; McIntosh, J.M.; Marks, M.J.; King, S.L.; Picciotto, M.R. Localized low-level re-expression of high-affinity mesolimbic nicotinic acetylcholine receptors restores nicotine-induced locomotion but not place conditioning. Genes Brain Behav. 2009, 8, 257–266. [Google Scholar] [CrossRef]
- Fucile, S. Ca2+ permeability of nicotinic acetylcholine receptors. Cell Calcium 2004, 35, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.D.; Bettegowda, C.; Blosser, J.; Gordon, J. AR-R 17779, an α7 nicotinic agonist, improves learning and memory in rats. Behav. Pharmacol. 1999, 10, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Mansvelder, H.D.; McGehee, D.S. Long-Term Potentiation of Excitatory Inputs to Brain Reward Areas by Nicotine. Neuron 2000, 27, 349–357. [Google Scholar] [CrossRef]
- Mansvelder, H.D.; McGehee, D.S. Cellular and synaptic mechanisms of nicotine addiction. J. Neurobiol. 2002, 53, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Ji, D.; Lape, R.; Dani, J.A. Timing and Location of Nicotinic Activity Enhances or Depresses Hippocampal Synaptic Plasticity. Neuron 2001, 31, 131–141. [Google Scholar] [CrossRef]
- Wonnacott, S. Presynaptic nicotinic ACh receptors. Trends Neurosci. 1997, 20, 92–98. [Google Scholar] [CrossRef]
- Schultz, W.; Dayan, P.; Montague, P.R. A Neural Substrate of Prediction and Reward. Science 1997, 275, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Spanagel, R.; Weiss, F. The dopamine hypothesis of reward: Past and current status. Trends Neurosci. 1999, 22, 521–527. [Google Scholar] [CrossRef]
- Berke, J.D.; Hyman, S.E. Addiction, Dopamine, and the Molecular Mechanisms of Memory. Neuron 2000, 25, 515–532. [Google Scholar] [CrossRef]
- Di Chiara, G.; Imperato, A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc. Natl. Acad. Sci. USA 1988, 85, 5274–5278. [Google Scholar] [CrossRef]
- Wise, R.A.; Bozarth, M.A. A psychomotor stimulant theory of addiction. Psychol. Rev. 1987, 94, 469–492. [Google Scholar] [CrossRef] [PubMed]
- Olds, J.; Milner, P. Positive reinforcement produced by electrical stimulation of Septal area and other regions of rat brain. J. Comp. Physiol. Psychol. 1954, 47, 419–427. [Google Scholar] [CrossRef]
- Wise, R.A.; Rompre, P.P. Brain dopamine and reward. Annu. Rev. Psychol. 1989, 40, 191–225. [Google Scholar] [CrossRef]
- Balfour, D.J.; Benwell, M.E.; E Birrell, C.; Kelly, J.; Al-Aloul, M. Sensitization of the Mesoaccumbens Dopamine Response to Nicotine. Pharmacol. Biochem. Behav. 1998, 59, 1021–1030. [Google Scholar] [CrossRef]
- Pontieri, F.E.; Tanda, G.; Di Chiara, G. Intravenous cocaine, morphine, and amphetamine preferentially increase extracellular dopamine in the “shell” as compared with the “core” of the rat nucleus accumbens. Proc. Natl. Acad. Sci. USA 1995, 92, 12304–12308. [Google Scholar] [CrossRef]
- Benwell, M.E.; Balfour, D.J. The effects of acute and repeated nicotine treatment on nucleus accumbens dopamine and locomotor activity. Br. J. Pharmacol. 1992, 105, 849–856. [Google Scholar] [CrossRef]
- Corrigall, W.A.; Franklin, K.B.J.; Coen, K.M.; Clarke, P.B.S. The mesolimbic dopaminergic system is implicated in the reinforcing effects of nicotine. Psychopharmacology 1992, 107, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Tanda, G.; Di Chiara, G. A dopamine-mu1 opioid link in the rat ventral tegmentum shared by palatable food (Fonzies) and non-psychostimulant drugs of abuse. Eur. J. Neurosci. 1998, 10, 1179–1187. [Google Scholar] [CrossRef]
- Voorn, P.; Vanderschuren, L.J.; Groenewegen, H.J.; Robbins, T.W.; Pennartz, C.M. Putting a spin on the dorsal–ventral divide of the striatum. Trends Neurosci. 2004, 27, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Heimer, L.; Zahm, D.; Churchill, L.; Kalivas, P.; Wohltmann, C. Specificity in the projection patterns of accumbal core and shell in the rat. Neuroscience 1991, 41, 89–125. [Google Scholar] [CrossRef]
- Zahm, D.; Brog, J. On the significance of subterritories in the “accumbens” part of the rat ventral striatum. Neuroscience 1992, 50, 751–767. [Google Scholar] [CrossRef]
- Ito, R.; Robbins, T.W.; Everitt, B.J. Differential control over cocaine-seeking behavior by nucleus accumbens core and shell. Nat. Neurosci. 2004, 7, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Alheid, G.; Heimer, L. New perspectives in basal forebrain organization of special relevance for neuropsychiatric disorders: The striatopallidal, amygdaloid, and corticopetal components of substantia innominata. Neuroscience 1988, 27, 1–39. [Google Scholar] [CrossRef]
- Di Chiara, G. Nucleus accumbens shell and core dopamine: Differential role in behavior and addiction. Behav. Brain Res. 2002, 137, 75–114. [Google Scholar] [CrossRef]
- Cadoni, C.; Di Chiara, G. Differential changes in accumbens shell and core dopamine in behavioral sensitization to nicotine. Eur. J. Pharmacol. 2000, 387, R23–R25. [Google Scholar] [CrossRef]
- Iyaniwura, T.T.; Wright, A.E.; Balfour, D.J. Evidence that mesoaccumbens dopamine and locomotor responses to nicotine in the rat are influenced by pretreatment dose and strain. Psychopharmacology 2001, 158, 73–79. [Google Scholar] [CrossRef]
- Benowitz, N.L. Nicotine addiction. N. Engl. J. Med. 2010, 362, 2295–2303. [Google Scholar] [CrossRef]
- Fenster, C.P.; Hicks, J.H.; Beckman, M.L.; Covernton, P.O.; Quick, M.W.; Lester, R.J. Desensitization of nicotinic receptors in the central nervous system. Ann. N. Y. Acad. Sci. 1999, 868, 620–623. [Google Scholar] [CrossRef]
- Pidoplichko, V.I.; DeBiasi, M.; Williams, J.T.; Dani, J.A. Nicotine activates and desensitizes midbrain dopamine neurons. Nat. Cell Biol. 1997, 390, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-Y.; Mao, X.; Wei, L. Genes and (Common) Pathways Underlying Drug Addiction. PLoS Comput. Biol. 2008, 4, e2. [Google Scholar] [CrossRef]
- Wilar, G.; Shinoda, Y.; Sasaoka, T.; Fukunaga, K. Crucial Role of Dopamine D2 Receptor Signaling in Nicotine-Induced Conditioned Place Preference. Mol. Neurobiol. 2019, 56, 7911–7928. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.J.; Muldoon, P.P.; Walters, C.; Damaj, M.I. Neuronal calcium/calmodulin-dependent protein kinase II mediates nicotine reward in the conditioned place preference test in mice. Behav. Pharmacol. 2016, 27, 50–56. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Risinger, F.; Oakes, R.A. Nicotine-induced conditioned place preference and conditioned place aversion in mice. Pharmacol. Biochem. Behav. 1995, 51, 457–461. [Google Scholar] [CrossRef]
- Grabus, S.D.; Martin, B.R.; Brown, S.E.; Damaj, M.I. Nicotine place preference in the mouse: Influences of prior handling, dose and strain and attenuation by nicotinic receptor antagonists. Psychopharmacology 2006, 184, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Le Foll, B.; Goldberg, S.R. Nicotine induces conditioned place preferences over a large range of doses in rats. Psychopharmacology 2005, 178, 481–492. [Google Scholar] [CrossRef]
- Sadat-Shirazi, M.-S.; Ahmadian-Moghadam, H.; Khalifeh, S.; Zadeh-Tehrani, S.N.; Farahmandfar, M.; Zarrindast, M.-R. The role of calcium-calmodulin-dependent protein kinase II in modulation of spatial memory in morphine sensitized rats. Behav. Brain Res. 2019, 359, 298–303. [Google Scholar] [CrossRef]
- Ma, S.X.; Kim, H.C.; Lee, S.Y.; Jang, C.G. TRPV1 modulates morphine self-administration via activation of the CaMKII-CREB pathway in the nucleus accumbens. Neurochem. Int. 2018, 121, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Matsumura, Y.; Ozaki, S.; Ise, Y.; Yajima, Y.; Suzuki, T. Role of the calcium/calmodulin-dependent protein kinase ii (CaMKII) in the morphine-induced pharmacological effects in the mouse. Neuroscience 2004, 126, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Zhou, X.; Fu, Q.; Peng, Q.; Oh, K.-W.; Hu, Z. A New Insight into the Role of CART in Cocaine Reward: Involvement of CaMKII and Inhibitory G-Protein Coupled Receptor Signaling. Front. Cell. Neurosci. 2017, 11, 244. [Google Scholar] [CrossRef] [PubMed]
- Cates, H.M.; Thibault, M.; Pfau, M.; Heller, E.; Eagle, A.; Gajewski, P.; Bagot, R.; Colangelo, C.; Abbott, T.; Rudenko, G.; et al. Threonine 149 phosphorylation enhances DeltaFosB transcriptional activity to control psychomotor responses to cocaine. J. Neurosci. 2014, 34, 11461–11469. [Google Scholar] [CrossRef]
- Anderson, S.M.; Famous, K.R.; Sadri-Vakili, G.; Kumaresan, V.; Schmidt, H.D.; Bass, C.E.; Terwilliger, E.F.; Cha, J.H.; Pierce, R.C. CaMKII: A biochemical bridge linking accumbens dopamine and glutamate systems in cocaine seeking. Nat. Neurosci. 2008, 11, 344–353. [Google Scholar] [CrossRef]
- Kong, H.; Kuang, W.; Li, S.; Xu, M. Activation of dopamine D3 receptors inhibits reward-related learning induced by cocaine. Neuroscience 2011, 176, 152–161. [Google Scholar] [CrossRef][Green Version]
- Easton, A.C.; Lourdusamy, A.; Havranek, M.; Mizuno, K.; Solati, J.; Golub, Y.; Clarke, T.K.; Vallada, H.; Laranjeira, R.; Desrivières, S.; et al. alphaCaMKII controls the establishment of cocaine’s reinforcing effects in mice and humans. Transl. Psychiatry 2014, 4, e457. [Google Scholar] [CrossRef]
- Caffino, L.; Cassina, C.; Giannotti, G.; Di Clemente, A.; Racagni, G.; Fumagalli, F.; Cervo, L.; Orrù, A.; Moro, F. Short-term abstinence from cocaine self-administration, but not passive cocaine infusion, elevates ?CaMKII autophosphorylation in the rat nucleus accumbens and medial prefrontal cortex. Int. J. Neuropsychopharmacol. 2013, 17, 323–329. [Google Scholar] [CrossRef][Green Version]
- Caffino, L.; Piva, A.; Mottarlini, F.; Di Chio, M.; Giannotti, G.; Chiamulera, C.; Fumagalli, F. Ketamine Self-Administration Elevates alphaCaMKII Autophosphorylation in Mood and Reward-Related Brain Regions in Rats. Mol. Neurobiol. 2018, 55, 5453–5461. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, J.-J.; Liu, X.-D.; Yu, L.-C. Inhibition of CaMKII activity in the nucleus accumbens shell blocks the reinstatement of morphine-seeking behavior in rats. Neurosci. Lett. 2012, 518, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Schopf, I.; Easton, A.C.; Solati, J.; Golub, Y.; Kornhuber, J.; Giese, K.P.; Müller, C.P. alphaCaMKII autophosphorylation mediates neuronal activation in the hippocampal dentate gyrus after alcohol and cocaine in mice. Neurosci. Lett. 2015, 591, 65–68. [Google Scholar] [CrossRef]
- Hu, X.; Huang, F.; Szymusiak, M.; Liu, Y.; Wang, Z.J. Curcumin attenuates opioid tolerance and dependence by inhibiting Ca2+/calmodulin-dependent protein kinase II α activity. J. Pharmacol. Exp. Ther. 2014, 352, 420–428. [Google Scholar] [CrossRef]
- Pierce, R.C.; Kalivas, P.W. Repeated Cocaine Modifies the Mechanism by which Amphetamine Releases Dopamine. J. Neurosci. 1997, 17, 3254–3261. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.-E. Impairing the amphetamine conditioning in rats through the inhibition of hippocampal calcium/calmodulin-dependent protein kinase II activity. Neuropharmacology 2002, 42, 540–547. [Google Scholar] [CrossRef]
- Choe, E.S.; Wang, J.Q. CaMKII regulates amphetamine-induced ERK1/2 phosphorylation in striatal neurons. Neuroreport 2002, 13, 1013–1016. [Google Scholar] [CrossRef] [PubMed]
- Kauer, J.A.; Malenka, R.C. Synaptic plasticity and addiction. Nat. Rev. Neurosci. 2007, 8, 844–858. [Google Scholar] [CrossRef]
- Robison, A.J.; Nestler, E.J. Transcriptional and epigenetic mechanisms of addiction. Nat. Rev. Neurosci. 2011, 12, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Kauer, J.A. Learning Mechanisms in Addiction: Synaptic Plasticity in the Ventral Tegmental Area as a Result of Exposure to Drugs of Abuse. Annu. Rev. Physiol. 2004, 66, 447–475. [Google Scholar] [CrossRef]
- Kalivas, P.W. The glutamate homeostasis hypothesis of addiction. Nat. Rev. Neurosci. 2009, 10, 561–572. [Google Scholar] [CrossRef]
- Vorel, S.R.; Liu, X.; Hayes, R.J.; Spector, J.A.; Gardner, E.L. Relapse to Cocaine-Seeking After Hippocampal Theta Burst Stimulation. Science 2001, 292, 1175–1178. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Zhou, T.; Liao, L.; Yang, Z.; Wong, C.; Henn, F.; Malinow, R.; Yates, J.R.; Hu, H. betaCaMKII in lateral habenula mediates core symptoms of depression. Science 2013, 341, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Shepard, P.D. Lateral habenula stimulation inhibits rat midbrain dopamine neurons through a GABA(A) receptor-mediated mechanism. J. Neurosci. 2007, 27, 6923–6930. [Google Scholar] [CrossRef] [PubMed]
- Kyosseva, S.V. Mitogen-Activated Protein Kinase Signaling. Int. Rev. Neurobiol. 2004, 59, 201–220. [Google Scholar] [CrossRef]
- Hetman, M.; Gozdz, A. Role of extracellular signal regulated kinases 1 and 2 in neuronal survival. JBIC J. Biol. Inorg. Chem. 2004, 271, 2050–2055. [Google Scholar] [CrossRef] [PubMed]
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-wide atlas of gene expression in the adult mouse brain. Nat. Cell Biol. 2006, 445, 168–176. [Google Scholar] [CrossRef]
- Lu, L.; Koya, E.; Zhai, H.; Hope, B.T.; Shaham, Y. Role of ERK in cocaine addiction. Trends Neurosci. 2006, 29, 695–703. [Google Scholar] [CrossRef]
- Valjent, E.; Corvol, J.-C.; Pagès, C.; Besson, M.-J.; Maldonado, R.; Caboche, J. Involvement of the Extracellular Signal-Regulated Kinase Cascade for Cocaine-Rewarding Properties. J. Neurosci. 2000, 20, 8701–8709. [Google Scholar] [CrossRef] [PubMed]
- Mattson, B.J.; Bossert, J.M.; Simmons, D.E.; Nozaki, N.; Nagarkar, D.; Kreuter, J.D.; Hope, B.T. Cocaine-induced CREB phosphorylation in nucleus accumbens of cocaine-sensitized rats is enabled by enhanced activation of extracellular signal-related kinase, but not protein kinase A. J. Neurochem. 2005, 95, 1481–1494. [Google Scholar] [CrossRef] [PubMed]
- Brami-Cherrier, K.; Valjent, E.; Hervé, D.; Darragh, J.; Corvol, J.-C.; Pagès, C.; Simon, A.J.; Girault, J.-A.; Caboche, J. Parsing Molecular and Behavioral Effects of Cocaine in Mitogen- and Stress-Activated Protein Kinase-1-Deficient Mice. J. Neurosci. 2005, 25, 11444–11454. [Google Scholar] [CrossRef] [PubMed]
- Girault, J.A.; Valjent, E.; Caboche, J.; Herve, D. ERK2: A logical AND gate critical for drug-induced plasticity? Curr. Opin. Pharmacol. 2007, 7, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.; Bargas, J.; Hemmings, H.C., Jr.; Nairn, A.C.; Greengard, P. Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron 1995, 14, 385–397. [Google Scholar] [CrossRef]
- Farnsworth, C.L.; Freshney, N.W.; Rosen, L.B.; Ghosh, A.; Greenberg, M.E.; Feig, L.A. Calcium activation of Ras mediated by neuronal exchange factor Ras-GRF. Nat. Cell Biol. 1995, 376, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Steiner, R.C.; Heath, C.J.; Picciotto, M.R. Nicotine-induced phosphorylation of ERK in mouse primary cortical neurons: Evidence for involvement of glutamatergic signaling and CaMKII. J. Neurochem. 2007, 103, 666–678. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Numakawa, T.; Ikeuchi, T.; Hatanaka, H. Nicotine-induced phosphorylation of extracellular signal-regulated protein kinase and CREB in PC12h cells. J. Neurochem. 2008, 79, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Gardoni, F.; Caputi, A.; Cimino, M.; Pastorino, L.; Cattabeni, F.; Di Luca, M. Calcium/calmodulin-dependent protein kinase II is associated with NR2A/B subunits of NMDA receptor in postsynaptic densities. J. Neurochem. 2002, 71, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Leonard, A.S.; Lim, I.A.; Hemsworth, D.E.; Horne, M.C.; Hell, J.W. Calcium/calmodulin-dependent protein kinase II is associated with the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 3239–3244. [Google Scholar] [CrossRef] [PubMed]
- Delint-Ramirez, I.; Segev, A.; Pavuluri, A.; Self, D.W.; Kourrich, S. Cocaine-Induced Synaptic Redistribution of NMDARs in Striatal Neurons Alters NMDAR-Dependent Signal Transduction. Front. Neurosci. 2020, 14, 698. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Sohn, S.; Kim, S.; Kim, J.; Oh, J.H.; Ryu, I.S.; Go, B.S.; Choe, E.S. Repeated nicotine exposure increases the intracellular interaction between ERK-mGluR5 in the nucleus accumbens more in adult than adolescent rats. Addict. Biol. 2021, 26, e12913. [Google Scholar] [CrossRef]
- Hudson, R.; Green, M.; Wright, D.J.; Renard, J.; Jobson, C.E.; Jung, T.; Rushlow, W.; Laviolette, S.R. Adolescent nicotine induces depressive and anxiogenic effects through ERK 1-2 and Akt-GSK-3 pathways and neuronal dysregulation in the nucleus accumbens. Addict. Biol. 2021, 26, e12891. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, Q.; Sun, Q.; Qin, F.; Nie, D.; Li, Q.; Gu, Y.; Jiang, Y.; Lu, S.; Lu, Z. Effects of Compound 511 on BDNF-TrkB Signaling in the Mice Ventral Tegmental Area in Morphine-Induced Conditioned Place Preference. Cell. Mol. Neurobiol. 2020. [Google Scholar] [CrossRef]
- Wen, D.; Hui, R.; Liu, Y.; Luo, Y.; Wang, J.; Shen, X.; Xie, B.; Yu, F.; Cong, B.; Ma, C. Molecular hydrogen attenuates methamphetamine-induced behavioral sensitization and activation of ERK-DeltaFosB signaling in the mouse nucleus accumbens. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 97, 109781. [Google Scholar] [CrossRef]
- Kim, B.; Jha, S.; Seo, J.H.; Jeong, C.-H.; Lee, S.; Lee, S.; Seo, Y.H.; Park, B. MeBib Suppressed Methamphetamine Self-Administration Response via Inhibition of BDNF/ERK/CREB Signal Pathway in the Hippocampus. Biomol. Ther. 2020, 28, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, R.A.; Hoffman, J.L.; Maldonado-Devincci, A.M.; Faccidomo, S.; Hodge, C.W. MGluR5 activity is required for the induction of ethanol behavioral sensitization and associated changes in ERK MAP kinase phosphorylation in the nucleus accumbens shell and lateral habenula. Behav. Brain Res. 2019, 367, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Valjent, E.; Pagès, C.; Hervé, D.; Girault, J.-A.; Caboche, J. Addictive and non-addictive drugs induce distinct and specific patterns of ERK activation in mouse brain. Eur. J. Neurosci. 2004, 19, 1826–1836. [Google Scholar] [CrossRef]
- Brunzell, D.H.; Russell, D.S.; Picciotto, M.R. In vivo nicotine treatment regulates mesocorticolimbic CREB and ERK signaling in C57Bl/6J mice. J. Neurochem. 2003, 84, 1431–1441. [Google Scholar] [CrossRef]
- Berhow, M.T.; Hiroi, N.; Nestler, E.J. Regulation of ERK (Extracellular Signal Regulated Kinase), Part of the Neurotrophin Signal Transduction Cascade, in the Rat Mesolimbic Dopamine System by Chronic Exposure to Morphine or Cocaine. J. Neurosci. 1996, 16, 4707–4715. [Google Scholar] [CrossRef]
- Gerdjikov, T.V.; Ross, G.M.; Beninger, R.J. Place Preference Induced by Nucleus Accumbens Amphetamine Is Impaired by Antagonists of ERK or p38 MAP Kinases in Rats. Behav. Neurosci. 2004, 118, 740–750. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.-M.; Reusch, J.M.; Fibuch, E.E.; Liu, Z.; Wang, J.Q. Amphetamine increases phosphorylation of MAPK/ERK at synaptic sites in the rat striatum and medial prefrontal cortex. Brain Res. 2013, 1494, 101–108. [Google Scholar] [CrossRef]
- Shi, X.; McGinty, J. Extracellular signal-regulated mitogen-activated protein kinase inhibitors decrease amphetamine-induced behavior and neuropeptide gene expression in the striatum. Neuroscience 2006, 138, 1289–1298. [Google Scholar] [CrossRef]
- Payne, T.J.; Smith, P.O.; McCracken, L.M.; McSherry, W.; Antony, M.M. Assessing nicotine dependence: A comparison of the fagerström tolerance questionnaire (FTQ) with the fagerström test for nicotine dependence (FTND) in a clinical sample. Addict. Behav. 1994, 19, 307–317. [Google Scholar] [CrossRef]
- Wesson, D.R.; Ling, W. The Clinical Opiate Withdrawal Scale (COWS). J. Psychoact. Drugs 2003, 35, 253–259. [Google Scholar] [CrossRef]
- Farrell, M. Opiate withdrawal. Addiction 1994, 89, 1471–1475. [Google Scholar] [CrossRef]
- Sofuoglu, M.; Dudish-Poulsen, S.; Poling, J.; Mooney, M.; Hatsukami, D.K. The effect of individual cocaine withdrawal symptoms on outcomes in cocaine users. Addict. Behav. 2005, 30, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Cantwell, B.; McBride, A.J. Self detoxication by amphetamine dependent patients: A pilot study. Drug Alcohol Depend. 1998, 49, 157–163. [Google Scholar] [CrossRef]
- Kebabian, J.W.; Calne, D.B. Multiple receptors for dopamine. Nat. Cell Biol. 1979, 277, 93–96. [Google Scholar] [CrossRef]
- Missale, C.; Nash, S.R.; Robinson, S.W.; Jaber, M.; Caron, M.G. Dopamine Receptors: From Structure to Function. Physiol. Rev. 1998, 78, 189–225. [Google Scholar] [CrossRef]
- Shi, W.; Rayport, S. GABA synapses formed in vitro by local axon collaterals of nucleus accumbens neurons. J. Neurosci. 1994, 14, 4548–4560. [Google Scholar] [CrossRef]
- Smith, R.J.; Lobo, M.K.; Spencer, S.; Kalivas, P.W. Cocaine-induced adaptations in D1 and D2 accumbens projection neurons (a dichotomy not necessarily synonymous with direct and indirect pathways). Curr. Opin. Neurobiol. 2013, 23, 546–552. [Google Scholar] [CrossRef]
- Perreault, M.L.; Hasbi, A.; Alijaniaram, M.; Fan, T.; Varghese, G.; Fletcher, P.J.; Seeman, P.; O’Dowd, B.F.; George, S.R. The dopamine D1-D2 receptor heteromer localizes in dynorphin/enkephalin neurons: Increased high affinity state following amphetamine and in schizophrenia. J. Biol. Chem. 2010, 285, 36625–36634. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Song, W.J.; Yan, Z. Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J. Neurosci. 1996, 16, 6579–6591. [Google Scholar] [CrossRef]
- Bahk, J.Y.; Li, S.; Park, M.S.; Kim, M.O. Dopamine D1 and D2 receptor mRNA up-regulation in the caudate–putamen and nucleus accumbens of rat brains by smoking. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2002, 26, 1095–1104. [Google Scholar] [CrossRef]
- Ahmed, S.H.; Koob, G.F. Changes in response to a dopamine receptor antagonist in rats with escalating cocaine intake. Psychopharmacology 2003, 172, 450–454. [Google Scholar] [CrossRef]
- Yawata, S.; Yamaguchi, T.; Danjo, T.; Hikida, T.; Nakanishi, S. Pathway-specific control of reward learning and its flexibility via selective dopamine receptors in the nucleus accumbens. Proc. Natl. Acad. Sci. USA 2012, 109, 12764–12769. [Google Scholar] [CrossRef]
- Hasbi, A.; Fan, T.; Alijaniaram, M.; Nguyen, T.; Perreault, M.L.; O’Dowd, B.F.; George, S.R. Calcium signaling cascade links dopamine D1–D2 receptor heteromer to striatal BDNF production and neuronal growth. Proc. Natl. Acad. Sci. USA 2009, 106, 21377–21382. [Google Scholar] [CrossRef] [PubMed]
- Self, D.W.; Genova, L.M.; Hope, B.T.; Barnhart, W.J.; Spencer, J.J.; Nestler, E.J. Involvement of cAMP-dependent protein kinase in the nucleus accumbens in cocaine self-administration and relapse of cocaine-seeking behavior. J. Neurosci. 1998, 18, 1848–1859. [Google Scholar] [CrossRef] [PubMed]
- Lynch, W.J.; Taylor, J.R. Persistent changes in motivation to self-administer cocaine following modulation of cyclic AMP-dependent protein kinase A (PKA) activity in the nucleus accumbens. Eur. J. Neurosci. 2005, 22, 1214–1220. [Google Scholar] [CrossRef]
- Oliver, R.J.; Purohit, D.C.; Kharidia, K.M.; Mandyam, C.D. Transient Chemogenetic Inhibition of D1-MSNs in the Dorsal Striatum Enhances Methamphetamine Self-Administration. Brain Sci. 2019, 9, 330. [Google Scholar] [CrossRef]
- Xing, B.; Kong, H.; Meng, X.; Wei, S.; Xu, M.; Li, S. Dopamine D1 but not D3 receptor is critical for spatial learning and related signaling in the hippocampus. Neuroscience 2010, 169, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Ma, J.Z.; Payne, T.J.; Beuten, J.; Dupont, R.T.; Li, M.D. Significant association of DRD1 with nicotine dependence. Qual. Life Res. 2007, 123, 133–140. [Google Scholar] [CrossRef]
- Goutier, W.; Lowry, J.P.; McCreary, A.C.; O’Connor, J.J. Frequency-Dependent Modulation of Dopamine Release by Nicotine and Dopamine D1 Receptor Ligands: An In Vitro Fast Cyclic Voltammetry Study in Rat Striatum. Neurochem. Res. 2016, 41, 945–950. [Google Scholar] [CrossRef]
- Goutier, W.; O’Connor, J.; Lowry, J.; McCreary, A. The effect of nicotine induced behavioral sensitization on dopamine D1 receptor pharmacology: An in vivo and ex vivo study in the rat. Eur. Neuropsychopharmacol. 2015, 25, 933–943. [Google Scholar] [CrossRef]
- Ehlinger, D.G.; Bergstrom, H.C.; Burke, J.C.; Fernandez, G.M.; McDonald, C.G.; Smith, R.F. Adolescent nicotine-induced dendrite remodeling in the nucleus accumbens is rapid, persistent, and D1-dopamine receptor dependent. Brain Struct. Funct. 2016, 221, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Spina, L.; Fenu, S.; Longoni, R.; Rivas, E.; Di Chiara, G. Nicotine-conditioned single-trial place preference: Selective role of nucleus accumbens shell dopamine D1 receptors in acquisition. Psychopharmacology 2005, 184, 447–455. [Google Scholar] [CrossRef]
- Valjent, E.; Pascoli, V.; Svenningsson, P.; Paul, S.; Enslen, H.; Corvol, J.-C.; Stipanovich, A.; Caboche, J.; Lombroso, P.J.; Nairn, A.C.; et al. From The Cover: Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc. Natl. Acad. Sci. USA 2005, 102, 491–496. [Google Scholar] [CrossRef]
- Zhang, L.; Lou, D.; Jiao, H.; Zhang, N.; Wang, X.; Xia, Y.; Zhang, J.; Xu, M. Cocaine-Induced Intracellular Signaling and Gene Expression Are Oppositely Regulated by the Dopamine D1 and D3 Receptors. J. Neurosci. 2004, 24, 3344–3354. [Google Scholar] [CrossRef]
- Valjent, E.; Caboche, J.; Vanhoutte, P. Mitogen-activated protein kinase/extracellular signal-regulated kinase induced gene regulation in brain: A molecular substrate for learning and memory? Mol. Neurobiol. 2001, 23, 83–99. [Google Scholar]
- Dal Toso, R.; Sommer, B.; Ewert, M.; Herb, A.; Pritchett, D.B.; Bach, A.; Shivers, B.D.; Seeburg, P.H. The dopamine D2 receptor: Two molecular forms generated by alternative splicing. EMBO J. 1989, 8, 4025–4034. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, K.; Shioda, N. Novel Dopamine D2 Receptor Signaling through Proteins Interacting with the Third Cytoplasmic Loop. Mol. Neurobiol. 2011, 45, 144–152. [Google Scholar] [CrossRef]
- Zhang, S.; Xie, C.; Wang, Q.; Liu, Z. Interactions of CaMKII with dopamine D2 receptors: Roles in levodopa-induced dyskinesia in 6-hydroxydopamine lesioned Parkinson’s rats. Sci. Rep. 2014, 4, 6811. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, Y.; Benoit-Marand, M.; Gonon, F.; Sulzer, D. Presynaptic regulation of dopaminergic neurotransmission. J. Neurochem. 2003, 87, 273–289. [Google Scholar] [CrossRef] [PubMed]
- Neve, K.A.; Seamans, J.K.; Trantham-Davidson, H. Dopamine receptor signaling. J. Recept. Signal Transduct. Res. 2004, 24, 165–205. [Google Scholar] [CrossRef]
- Beaulieu, J.-M.; Gainetdinov, R.R. The Physiology, Signaling, and Pharmacology of Dopamine Receptors. Pharmacol. Rev. 2011, 63, 182–217. [Google Scholar] [CrossRef] [PubMed]
- Lintas, A.; Chi, N.; Lauzon, N.M.; Bishop, S.F.; Gholizadeh, S.; Sun, N.; Tan, H.; LaViolette, S.R. Identification of a Dopamine Receptor-Mediated Opiate Reward Memory Switch in the Basolateral Amygdala–Nucleus Accumbens Circuit. J. Neurosci. 2011, 31, 11172–11183. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Fukunaga, K.; Miyamoto, E. Activation of nuclear Ca2+/calmodulin-dependent protein kinase II and brain-derived neurotrophic factor gene expression by stimulation of dopamine D2 receptor in transfected NG108-15 cells. J. Neurochem. 2002, 82, 316–328. [Google Scholar] [CrossRef]
- Baik, J.-H.; Picetti, R.; Saiardi, A.; Thiriet, G.; Dierich, A.; Depaulis, A.; Le Meur, M.; Borrelli, E. Parkinsonian-like locomotor impairment in mice lacking dopamine D2 receptors. Nature 1995, 377, 424–428. [Google Scholar] [CrossRef]
- Cunningham, C.L.; Howard, M.A.; Gill, S.J.; Rubinstein, M.; Low, M.J.; Grandy, D.K. Ethanol-conditioned place preference is reduced in dopamine D2 receptor-deficient mice. Pharmacol. Biochem. Behav. 2000, 67, 693–699. [Google Scholar] [CrossRef]
- Chausmer, A.L.; Elmer, G.I.; Rubinstein, M.; Low, M.J.; Grandy, D.K.; Katz, J.L. Cocaine-induced locomotor activity and cocaine discrimination in dopamine D2 receptor mutant mice. Psychopharmacology 2002, 163, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Rosen, L.G.; Zunder, J.; Renard, J.; Fu, J.; Rushlow, W.; Laviolette, S.R. Opiate Exposure State Controls a D2-CaMKIIalpha-Dependent Memory Switch in the Amygdala-Prefrontal Cortical Circuit. Neuropsychopharmacology 2016, 41, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Gordon, N. Nutrition and cognitive function. Brain Dev. 1997, 19, 165–170. [Google Scholar] [CrossRef]
- Sakayori, N.; Kikkawa, T.; Tokuda, H.; Kiryu, E.; Yoshizaki, K.; Kawashima, H.; Yamada, T.; Arai, H.; Kang, J.X.; Katagiri, H.; et al. Maternal dietary imbalance between omega-6 and omega-3 polyunsaturated fatty acids impairs neocortical development via epoxy metabolites. Stem Cells 2016, 34, 470–482. [Google Scholar] [CrossRef]
- Horrocks, L.A.; Yeo, Y.K. Health benefits of docosahexaenoic acid (DHA). Pharmacol. Res. 1999, 40, 211–225. [Google Scholar] [CrossRef]
- Kotani, S.; Sakaguchi, E.; Warashina, S.; Matsukawa, N.; Ishikura, Y.; Kiso, Y.; Sakakibara, M.; Yoshimoto, T.; Guo, J.; Yamashima, T. Dietary supplementation of arachidonic and docosahexaenoic acids improves cognitive dysfunction. Neurosci. Res. 2006, 56, 159–164. [Google Scholar] [CrossRef]
- Arvindakshan, M.; Ghate, M.; Ranjekar, P.K.; Evans, D.R.; Mahadik, S.P. Supplementation with a combination of omega-3 fatty acids and antioxidants (vitamins E and C) improves the outcome of schizophrenia. Schizophr. Res. 2003, 62, 195–204. [Google Scholar] [CrossRef]
- Kuhn, F.T.; Trevizol, F.; Dias, V.T.; Barcelos, R.C.S.; Pase, C.S.; Roversi, K.; Antoniazzi, C.T.D.D.; Roversi, K.; Boufleur, N.; Benvegnú, D.M.; et al. Toxicological aspects of trans fat consumption over two sequential generations of rats: Oxidative damage and preference for amphetamine. Toxicol. Lett. 2015, 232, 58–67. [Google Scholar] [CrossRef]
- Anggadiredja, K.; Nakamichi, M.; Hiranita, T.; Tanaka, H.; Shoyama, Y.; Watanabe, S.; Yamamoto, T. Endocannabinoid System Modulates Relapse to Methamphetamine Seeking: Possible Mediation by the Arachidonic Acid Cascade. Neuropsychopharmacology 2004, 29, 1470–1478. [Google Scholar] [CrossRef]
- Ramadan, E.; Basselin, M.; Taha, A.Y.; Cheon, Y.; Chang, L.; Chen, M.; Rapoport, S.I. Chronic valproate treatment blocks D2-like receptor-mediated brain signaling via arachidonic acid in rats. Neuropharmacology 2011, 61, 1256–1264. [Google Scholar] [CrossRef]
- Bhattacharjee, A.K.; Chang, L.; White, L.; Bazinet, R.P.; I Rapoport, S. D-Amphetamine Stimulates D2Dopamine Receptor-Mediated Brain Signaling Involving Arachidonic Acid in Unanesthetized Rats. Br. J. Pharmacol. 2006, 26, 1378–1388. [Google Scholar] [CrossRef]
- Metz, V.G.; Segat, H.J.; Dias, V.T.; Barcelos, R.C.S.; Maurer, L.H.; Stiebe, J.; Emanuelli, T.; Burger, M.E.; Pase, C.S. Omega-3 decreases D1 and D2 receptors expression in the prefrontal cortex and prevents amphetamine-induced conditioned place preference in rats. J. Nutr. Biochem. 2019, 67, 182–189. [Google Scholar] [CrossRef]
- Buydens-Branchey, L.; Branchey, M.; McMakin, D.L.; Hibbeln, J.R. Polyunsaturated fatty acid status and aggression in cocaine addicts. Drug Alcohol Depend. 2003, 71, 319–323. [Google Scholar] [CrossRef]
- Coe, N.R.; Bernlohr, D.A. Physiological properties and functions of intracellular fatty acid-binding proteins. Biochim. Biophys. Acta (BBA) Lipids Lipid Metab. 1998, 1391, 287–306. [Google Scholar] [CrossRef]
- Elmes, M.W.; Kaczocha, M.; Berger, W.T.; Leung, K.; Ralph, B.P.; Wang, L.; Sweeney, J.M.; Miyauchi, J.T.; Tsirka, S.E.; Ojima, I.; et al. Fatty acid-binding proteins (FABPs) are intracellular carriers for Delta9-tetrahydrocannabinol (THC) and cannabidiol (CBD). J. Biol. Chem. 2015, 290, 8711–8721. [Google Scholar] [CrossRef]
- Figueiredo, A.; Hamilton, J.; Marion, M.; Blum, K.; Kaczocha, M.; Haj-Dahmane, S.; Deutsch, D.; Thanos, P.K. Pharmacological Inhibition of Brain Fatty Acid Binding Protein Reduces Ethanol Consumption in Mice. J. Reward. Defic. Syndr. Addict. Sci. 2017, 3, 21–27. [Google Scholar] [CrossRef]
- Hamilton, J.; Marion, M.; Figueiredo, A.; Clavin, B.H.; Deutsch, D.; Kaczocha, M.; Haj-Dahmane, S.; Thanos, P.K. Fatty acid binding protein deletion prevents stress-induced preference for cocaine and dampens stress-induced corticosterone levels. Synapse 2018, 72, e22031. [Google Scholar] [CrossRef]
- Crofton, E.J.; Nenov, M.N.; Zhang, Y.; Tapia, C.M.; Donnelly, J.; Koshy, S.; Laezza, F.; Green, T.A. Topographic transcriptomics of the nucleus accumbens shell: Identification and validation of fatty acid binding protein 5 as target for cocaine addiction. Neuropharmacology 2021, 183, 108398. [Google Scholar] [CrossRef] [PubMed]
- Owada, Y.; Yoshimoto, T.; Kondo, H. Spatio-temporally differential expression of genes for three members of fatty acid binding proteins in developing and mature rat brains. J. Chem. Neuroanat. 1996, 12, 113–122. [Google Scholar] [CrossRef]
- Shioda, N.; Yamamoto, Y.; Watanabe, M.; Binas, B.; Owada, Y.; Fukunaga, K. Heart-Type Fatty Acid Binding Protein Regulates Dopamine D2 Receptor Function in Mouse Brain. J. Neurosci. 2010, 30, 3146–3155. [Google Scholar] [CrossRef] [PubMed]
- Yabuki, Y.; Takahata, I.; Matsuo, K.; Owada, Y.; Fukunaga, K. Ramelteon Improves Post-traumatic Stress Disorder-Like Behaviors Exhibited by Fatty Acid-Binding Protein 3 Null Mice. Mol. Neurobiol. 2017, 55, 3577–3591. [Google Scholar] [CrossRef]
- Islam, A.; Kagawa, Y.; Sharifi, K.; Ebrahimi, M.; Miyazaki, H.; Yasumoto, Y.; Kawamura, S.; Yamamoto, Y.; Sakaguti, S.; Sawada, T.; et al. Fatty Acid Binding Protein 3 Is Involved in n–3 and n–6 PUFA Transport in Mouse Trophoblasts. J. Nutr. 2014, 144, 1509–1516. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Nishi, D.; Nakaya, N.; Sone, T.; Hamazaki, K.; Hamazaki, T.; Koido, Y. Attenuating posttraumatic distress with omega-3 polyunsaturated fatty acids among disaster medical assistance team members after the Great East Japan Earthquake: The APOP randomized controlled trial. BMC Psychiatry 2011, 11, 132. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Drug | Region | CaMKII Activity |
|---|---|---|
| Nicotine | Nucleus accumbens | Phosphorylation level increased [56] |
| Hippocampal CA1 | Phosphorylation level increased [56] | |
| Ventral tegmental area | Activity increased [57] | |
| Morphine | Nucleus accumbens shell | Activity is crucial for morphine-seeking [71] |
| Hippocampus | CaMKIIα level increased in synaptosomes [72] | |
| Prefrontal cortex | Phosphorylation level increased [73] | |
| Cocaine | Nucleus accumbens shell | Activity is crucial for cocaine-seeking [65,66] |
| Hippocampal dentate gyrus | CaMKIIα phosphorylation mediates neuronal activation [72] | |
| Medial prefrontal cortex | CaMKIIα phosphorylation level increased after cocaine withdrawal [69] | |
| Amphetamine | Nucleus accumbens shell | Activity is crucial for amphetamine-induced DA release [74] |
| Hippocampus | Activity is crucial for amphetamine-induced CPP [75] | |
| Striatum | Phosphorylation level increased [76] |
| Drug | Region | ERK Activity |
|---|---|---|
| Nicotine | Nucleus accumbens | Phosphorylation level increased [56] |
| Prefrontal cortex | Phosphorylation level increased [105,106] | |
| Hippocampal CA1 | Phosphorylation level increased [56] | |
| Morphine | Nucleus accumbens | Phosphorylation level increased [105] |
| Prefrontal cortex | Phosphorylation level increased [105] | |
| Ventral tegmental area | Phosphorylation level increased [107] | |
| Cocaine | Nucleus accumbens | Phosphorylation level increased [105] |
| Prefrontal cortex | Phosphorylation level increased [10,105] | |
| Striatum | Activity increased [88] | |
| Amphetamine | Nucleus accumbens | Activity is crucial for amphetamine-induced CPP [108] |
| Medial prefrontal cortex | Phosphorylation level increased at synaptic sites [109] | |
| Striatum | Phosphorylation level increased [76,110] |
| Drug | Clinical Signs of Drug Withdrawal |
|---|---|
| Nicotine | Anxiety, Depression, Craving, Restlessness and impatience, Increased appetite and body weight [111] |
| Morphine | Anxiety, Nausea, Emesis, Diarrhea, Body aches, Restlessness, Agitation and dysphoria [112,113] |
| Cocaine | Anxiety, Depression, Craving, Restlessness, Vivid, unpleasant dreams, Fatigue and exhaustion, Increased appetite [114] |
| Amphetamine | Anxiety, Depression, Irritability, Body aches, Impaired social functioning Vivid, unpleasant dreams, Fatigue, Increased appetite [115] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, W.; Kawahata, I.; Cheng, A.; Fukunaga, K. The Role of CaMKII and ERK Signaling in Addiction. Int. J. Mol. Sci. 2021, 22, 3189. https://doi.org/10.3390/ijms22063189
Jia W, Kawahata I, Cheng A, Fukunaga K. The Role of CaMKII and ERK Signaling in Addiction. International Journal of Molecular Sciences. 2021; 22(6):3189. https://doi.org/10.3390/ijms22063189
Chicago/Turabian StyleJia, Wenbin, Ichiro Kawahata, An Cheng, and Kohji Fukunaga. 2021. "The Role of CaMKII and ERK Signaling in Addiction" International Journal of Molecular Sciences 22, no. 6: 3189. https://doi.org/10.3390/ijms22063189
APA StyleJia, W., Kawahata, I., Cheng, A., & Fukunaga, K. (2021). The Role of CaMKII and ERK Signaling in Addiction. International Journal of Molecular Sciences, 22(6), 3189. https://doi.org/10.3390/ijms22063189