Stem Cell-Based Therapies for Parkinson Disease


Abstract
1. Introduction
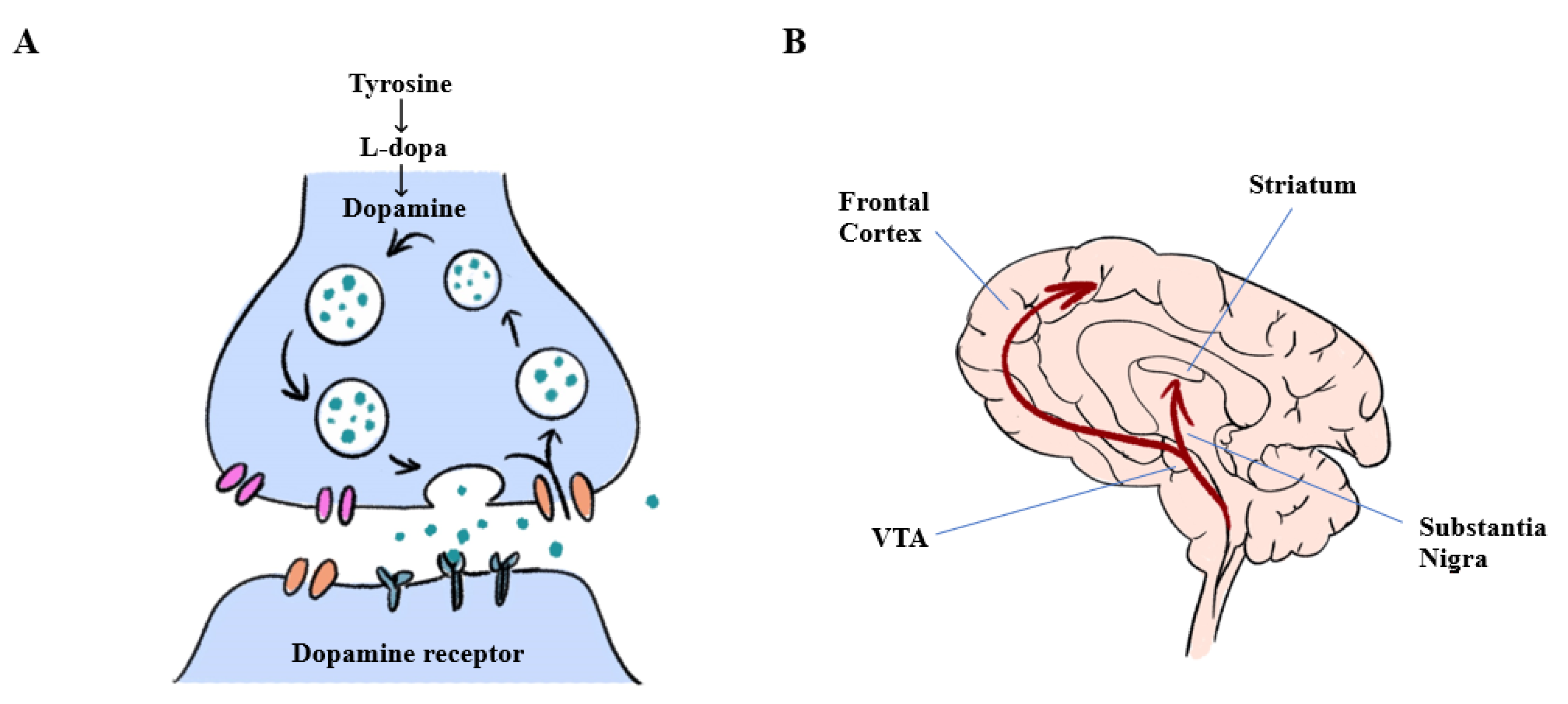
2. Pathophysiology
3. Animal Models of PD
3.1. Neurotoxin-Induced PD Models
3.2. Transgenic PD Models
4. Treatments
5. Stem Cell-Based Treatments
5.1. Fetal Ventral Mesencephalon Tissue
5.2. MSCs
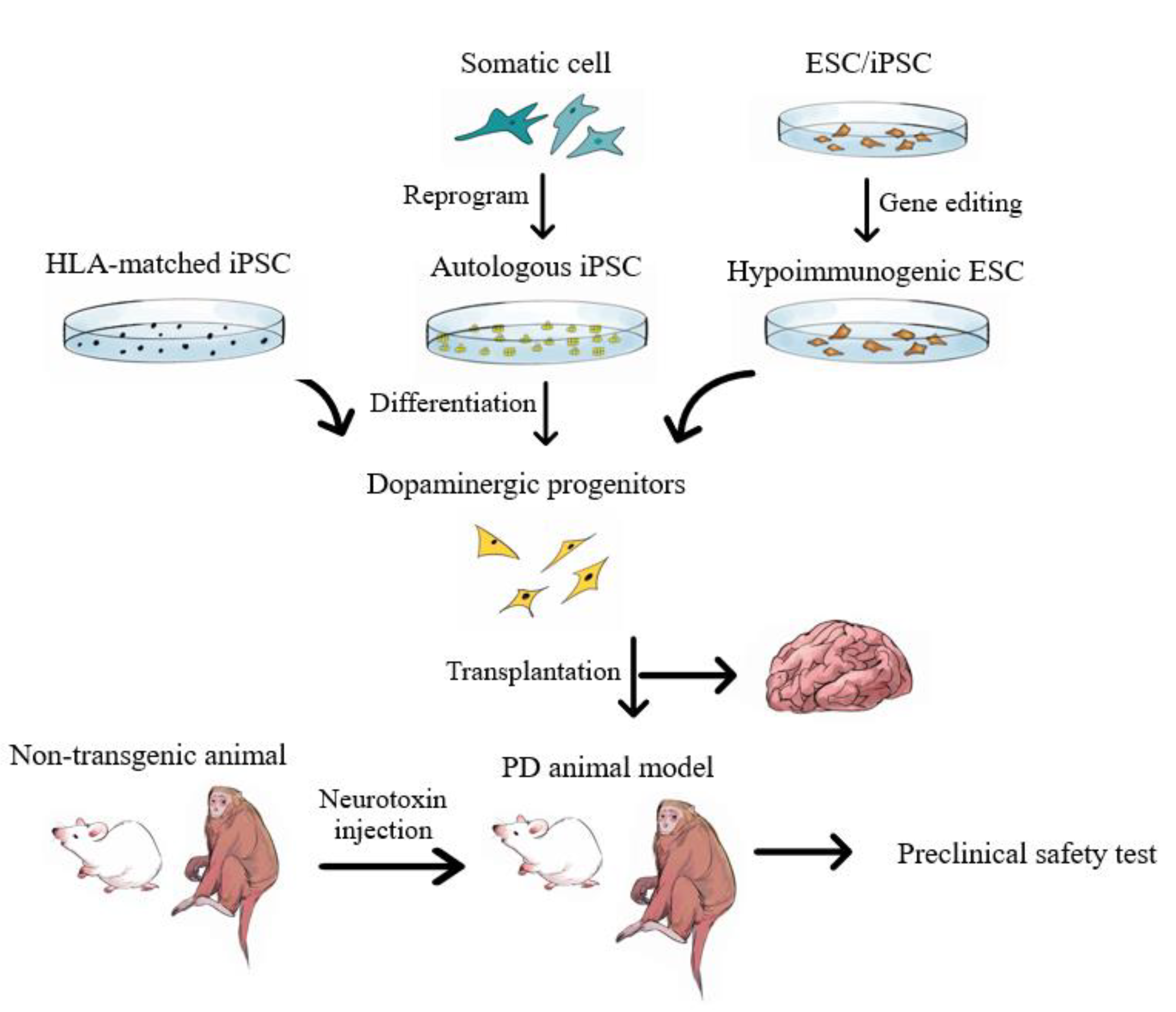
5.3. Pluripotent Stem Cell-Derived Neuron Progenitor Cells
5.4. Advantages and Disadvantages of iPSC-Based Therapy
6. Future Prospects
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Marchetti, B.; Tirolo, C.; L’Episcopo, F.; Caniglia, S.; Testa, N.; Smith, J.A.; Pluchino, S.; Serapide, M.F. Parkinson’s disease, aging and adult neurogenesis: Wnt/β-catenin signalling as the key to unlock the mystery of endogenous brain repair. Aging Cell 2020, 19, e13101. [Google Scholar] [CrossRef] [PubMed]
- Schrag, A.; Schott, J.M. Epidemiological, clinical, and genetic characteristics of early-onset parkinsonism. Lancet Neurol. 2006, 5, 355–363. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Pagano, G.; Ferrara, N.; Brooks, D.J.; Pavese, N. Age at onset and Parkinson disease phenotype. Neurology 2016, 86, 1400–1407. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- De Lau, L.M.; Breteler, M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Samii, A.; Nutt, J.G.; Ransom, B.R. Parkinson’s disease. Lancet 2004, 363, 1783–1793. [Google Scholar] [CrossRef]
- Aarsland, D.; Beyer, M.K.; Kurz, M.W. Dementia in Parkinson’s disease. Curr. Opin. Neurol. 2008, 21, 676–682. [Google Scholar] [CrossRef]
- Aarsland, D.; Zaccai, J.; Brayne, C. A systematic review of prevalence studies of dementia in Parkinson’s disease. Mov. Disord. 2005, 20, 1255–1263. [Google Scholar] [CrossRef]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef]
- Levy, G. The relationship of Parkinson disease with aging. Arch. Neurol. 2007, 64, 1242–1246. [Google Scholar] [CrossRef] [PubMed]
- Trinh, J.; Farrer, M. Advances in the genetics of Parkinson disease. Nat. Rev. Neurol. 2013, 9, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Kieburtz, K.; Wunderle, K.B. Parkinson’s disease: Evidence for environmental risk factors. Mov. Disord. 2013, 28, 8–13. [Google Scholar] [CrossRef]
- Blesa, J.; Przedborski, S. Parkinson’s disease: Animal models and dopaminergic cell vulnerability. Front. Neuroanat. 2014, 8, 155. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W.; Braak, H.; Duda, J.E.; Duyckaerts, C.; Gasser, T.; Halliday, G.M.; Hardy, J.; Leverenz, J.B.; Del Tredici, K.; Wszolek, Z.K.; et al. Neuropathological assessment of Parkinson’s disease: Refining the diagnostic criteria. Lancet Neurol. 2009, 8, 1150–1157. [Google Scholar] [CrossRef]
- Eriksen, J.L.; Dawson, T.M.; Dickson, D.W.; Petrucelli, L. Caught in the act: Alpha-synuclein is the culprit in Parkinson’s disease. Neuron 2003, 40, 453–456. [Google Scholar] [CrossRef]
- Brundin, P.; Dave, K.D.; Kordower, J.H. Therapeutic approaches to target alpha-synuclein pathology. Exp. Neurol. 2017, 298, 225–235. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Lee, V.M.; Trojanowski, J.Q. Mechanisms of Parkinson’s disease linked to pathological alpha-synuclein: New targets for drug discovery. Neuron 2006, 52, 33–38. [Google Scholar] [CrossRef]
- Cheng, J.; Lu, Q.; Song, L.; Ho, M.S. α-Synuclein trafficking in Parkinson’s Disease: Insights from fly and mouse models. ASN Neuro 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Noyce, A.J.; Bestwick, J.P.; Silveira-Moriyama, L.; Hawkes, C.H.; Giovannoni, G.; Lees, A.J.; Schrag, A. Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Ann. Neurol. 2012, 72, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Thakur, P.; Breger, L.S.; Lundblad, M.; Wan, O.W.; Mattsson, B.; Luk, K.C.; Lee, V.M.Y.; Trojanowski, J.Q.; Björklund, A. Modeling Parkinson’s disease pathology by combination of fibril seeds and α-synuclein overexpression in the rat brain. Proc. Natl. Acad. Sci. USA 2017, 114, E8284–E8293. [Google Scholar] [CrossRef] [PubMed]
- Koprich, J.B.; Kalia, L.V.; Brotchie, J.M. Animal models of α-synucleinopathy for Parkinson disease drug development. Nat. Rev. Neurosci. 2017, 18, 515–529. [Google Scholar] [CrossRef] [PubMed]
- Carvey, P.M.; Zhao, C.H.; Hendey, B.; Lum, H.; Trachtenberg, J.; Desai, B.S.; Snyder, J.; Zhu, Y.G.; Ling, Z.D. 6-Hydroxydopamine-induced alterations in blood-brain barrier permeability. Eur. J. Neurosci. 2005, 22, 1158–1168. [Google Scholar] [CrossRef]
- Blandini, F.; Armentero, M.T.; Martignoni, E. The 6-hydroxydopamine model: News from the past. Park. Relat. Disord. 2008, 14 (Suppl. 2), S124–S129. [Google Scholar] [CrossRef]
- Kopin, I.J. MPTP: An industrial chemical and contaminant of illicit narcotics stimulates a new era in research on Parkinson’s disease. Environ. Health Perspect. 1987, 75, 45–51. [Google Scholar] [CrossRef]
- Blesa, J.; Pifl, C.; Sánchez-González, M.A.; Juri, C.; García-Cabezas, M.A.; Adánez, R.; Iglesias, E.; Collantes, M.; Peñuelas, I.; Sánchez-Hernández, J.J.; et al. The nigrostriatal system in the presymptomatic and symptomatic stages in the MPTP monkey model: A PET, histological and biochemical study. Neurobiol. Dis. 2012, 48, 79–91. [Google Scholar] [CrossRef]
- Fornai, F.; Schlüter, O.M.; Lenzi, P.; Gesi, M.; Ruffoli, R.; Ferrucci, M.; Lazzeri, G.; Busceti, C.L.; Pontarelli, F.; Battaglia, G.; et al. Parkinson-like syndrome induced by continuous MPTP infusion: Convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 3413–3418. [Google Scholar] [CrossRef]
- Duty, S.; Jenner, P. Animal models of Parkinson’s disease: A source of novel treatments and clues to the cause of the disease. Br. J. Pharmacol. 2011, 164, 1357–1391. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.M.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J. Exp. Med. 2012, 209, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.F.; Collier, T.J.; Patterson, J.R.; Kemp, C.J.; Fischer, D.L.; Stoll, A.C.; Sortwell, C.E. Quality over quantity: Advantages of using alpha-synuclein preformed fibril triggered synucleinopathy to model idiopathic Parkinson’s Disease. Front. Neurosci. 2018, 12, 621. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.J.; Bonifati, V. The genetics of Parkinson’s disease: Progress and therapeutic implications. Mov. Disord. 2013, 28, 14–23. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Lee, Y.; Dawson, V.L.; Dawson, T.M. Animal models of Parkinson’s disease: Vertebrate genetics. Cold Spring Harb. Perspect. Med. 2012, 2, a009324. [Google Scholar] [CrossRef]
- Fishbein, I.; Kuo, Y.M.; Giasson, B.I.; Nussbaum, R.L. Augmentation of phenotype in a transgenic Parkinson mouse heterozygous for a Gaucher mutation. Brain 2014, 137, 3235–3247. [Google Scholar] [CrossRef]
- Toft, M.; Mata, I.F.; Kachergus, J.M.; Ross, O.A.; Farrer, M.J. LRRK2 mutations and Parkinsonism. Lancet 2005, 365, 1229–1230. [Google Scholar] [CrossRef]
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 2019, 14, 36. [Google Scholar] [CrossRef]
- Bardien, S.; Lesage, S.; Brice, A.; Carr, J. Genetic characteristics of leucine-rich repeat kinase 2 (LRRK2) associated Parkinson’s disease. Park. Relat. Disord. 2011, 17, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Farrer, M.; Kachergus, J.; Forno, L.; Lincoln, S.; Wang, D.S.; Hulihan, M.; Maraganore, D.; Gwinn-Hardy, K.; Wszolek, Z.; Dickson, D.; et al. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann. Neurol. 2004, 55, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef]
- Houlden, H.; Singleton, A.B. The genetics and neuropathology of Parkinson’s disease. Acta Neuropathol. 2012, 124, 325–338. [Google Scholar] [CrossRef]
- Neumann, J.; Bras, J.; Deas, E.; O’Sullivan, S.S.; Parkkinen, L.; Lachmann, R.H.; Li, A.; Holton, J.; Guerreiro, R.; Paudel, R.; et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 2009, 132, 1783–1794. [Google Scholar] [CrossRef]
- Zhao, Y.; Dzamko, N. Recent developments in LRRK2-targeted therapy for Parkinson’s Disease. Drugs 2019, 79, 1037–1051. [Google Scholar] [CrossRef]
- Lees, A.J.; Tolosa, E.; Olanow, C.W. Four pioneers of L-dopa treatment: Arvid Carlsson, Oleh Hornykiewicz, George Cotzias, and Melvin Yahr. Mov. Disord. 2015, 30, 19–36. [Google Scholar] [CrossRef]
- Tabar, V.; Studer, L. Pluripotent stem cells in regenerative medicine: Challenges and recent progress. Nat. Rev. Genet. 2014, 15, 82–92. [Google Scholar] [CrossRef]
- Maiti, P.; Manna, J.; Dunbar, G.L. Current understanding of the molecular mechanisms in Parkinson’s disease: Targets for potential treatments. Transl. Neurodegener. 2017, 6, 28. [Google Scholar] [CrossRef]
- Stoker, T.B.; Torsney, K.M.; Barker, R.A. Emerging treatment approaches for Parkinson’s Disease. Front. Neurosci. 2018, 12, 693. [Google Scholar] [CrossRef]
- Salat, D.; Tolosa, E. Levodopa in the treatment of Parkinson’s disease: Current status and new developments. J. Park. Dis. 2013, 3, 255–269. [Google Scholar] [CrossRef]
- Tambasco, N.; Romoli, M.; Calabresi, P. Levodopa in Parkinson’s Disease: Current Status and Future Developments. Curr. Neuropharmacol. 2018, 16, 1239–1252. [Google Scholar] [CrossRef]
- Miocinovic, S.; Somayajula, S.; Chitnis, S.; Vitek, J.L. History, applications, and mechanisms of deep brain stimulation. JAMA Neurol. 2013, 70, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Bruno, V.; Arena, J.; Cammarota, Á.; Merello, M. Challenges in PD patient management after DBS: A pragmatic review. Mov. Disord. Clin. Pract. 2018, 5, 246–254. [Google Scholar] [CrossRef]
- Martínez-Fernández, R.; Rodríguez-Rojas, R.; Del Álamo, M.; Hernández-Fernández, F.; Pineda-Pardo, J.A.; Dileone, M.; Alonso-Frech, F.; Foffani, G.; Obeso, I.; Gasca-Salas, C.; et al. Focused ultrasound subthalamotomy in patients with asymmetric Parkinson’s disease: A pilot study. Lancet Neurol. 2018, 17, 54–63. [Google Scholar] [CrossRef]
- Fan, Y.; Winanto; Ng, S.Y. Replacing what’s lost: A new era of stem cell therapy for Parkinson’s disease. Transl. Neurodegener. 2020, 9, 2. [Google Scholar] [CrossRef]
- Olanow, C.W.; Goetz, C.G.; Kordower, J.H.; Stoessl, A.J.; Sossi, V.; Brin, M.F.; Shannon, K.M.; Nauert, G.M.; Perl, D.P.; Godbold, J.; et al. A double-blind controlled trial of bilateral fetal nigral transplantation in Parkinson’s disease. Ann. Neurol. 2003, 54, 403–414. [Google Scholar] [CrossRef]
- Kefalopoulou, Z.; Politis, M.; Piccini, P.; Mencacci, N.; Bhatia, K.; Jahanshahi, M.; Widner, H.; Rehncrona, S.; Brundin, P.; Björklund, A.; et al. Long-term clinical outcome of fetal cell transplantation for Parkinson disease: Two case reports. JAMA Neurol. 2014, 71, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Obeso, J.A.; Rodriguez-Oroz, M.C.; Goetz, C.G.; Marin, C.; Kordower, J.H.; Rodriguez, M.; Hirsch, E.C.; Farrer, M.; Schapira, A.H.; Halliday, G. Missing pieces in the Parkinson’s disease puzzle. Nat. Med. 2010, 16, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Puelles, E. Genetic control of basal midbrain development. J. Neurosci. Res. 2007, 85, 3530–3534. [Google Scholar] [CrossRef]
- Lindvall, O.; Rehncrona, S.; Brundin, P.; Gustavii, B.; Astedt, B.; Widner, H.; Lindholm, T.; Björklund, A.; Leenders, K.L.; Rothwell, J.C.; et al. Human fetal dopamine neurons grafted into the striatum in two patients with severe Parkinson’s disease. A detailed account of methodology and a 6-month follow-up. Arch. Neurol. 1989, 46, 615–631. [Google Scholar] [CrossRef]
- Lindvall, O.; Brundin, P.; Widner, H.; Rehncrona, S.; Gustavii, B.; Frackowiak, R.; Leenders, K.L.; Sawle, G.; Rothwell, J.C.; Marsden, C.D.; et al. Grafts of fetal dopamine neurons survive and improve motor function in Parkinson’s disease. Science 1990, 247, 574–577. [Google Scholar] [CrossRef] [PubMed]
- Piccini, P.; Lindvall, O.; Björklund, A.; Brundin, P.; Hagell, P.; Ceravolo, R.; Oertel, W.; Quinn, N.; Samuel, M.; Rehncrona, S.; et al. Delayed recovery of movement-related cortical function in Parkinson’s disease after striatal dopaminergic grafts. Ann. Neurol. 2000, 48, 689–695. [Google Scholar] [CrossRef]
- Farag, E.S.; Vinters, H.V.; Bronstein, J. Pathologic findings in retinal pigment epithelial cell implantation for Parkinson disease. Neurology 2009, 73, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506. [Google Scholar] [CrossRef] [PubMed]
- Brundin, P.; Kordower, J.H. Neuropathology in transplants in Parkinson’s disease: Implications for disease pathogenesis and the future of cell therapy. Prog. Brain Res. 2012, 200, 221–241. [Google Scholar]
- Kelly, C.M.; Precious, S.V.; Torres, E.M.; Harrison, A.W.; Williams, D.; Scherf, C.; Weyrauch, U.M.; Lane, E.L.; Allen, N.D.; Penketh, R.; et al. Medical terminations of pregnancy: A viable source of tissue for cell replacement therapy for neurodegenerative disorders. Cell Transplant. 2011, 20, 503–513. [Google Scholar] [CrossRef]
- Barker, R.A.; Barrett, J.; Mason, S.L.; Björklund, A. Fetal dopaminergic transplantation trials and the future of neural grafting in Parkinson’s disease. Lancet Neurol. 2013, 12, 84–91. [Google Scholar] [CrossRef]
- Tronci, E.; Fidalgo, C.; Carta, M. Foetal cell transplantation for Parkinson’s Disease: Focus on graft-induced dyskinesia. Park. Dis. 2015, 2015, 563820. [Google Scholar] [CrossRef][Green Version]
- Barker, R.A. Designing stem-cell-based dopamine cell replacement trials for Parkinson’s disease. Nat. Med. 2019, 25, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Baremberg, D.; Gao, J.; Duan, J.; Lu, X.; Zhang, N.; Chen, Q. Development of stem cell-based therapy for Parkinson’s disease. Transl. Neurodegener. 2015, 4, 16. [Google Scholar] [CrossRef]
- Liu, J.; Huang, H.Y. How to improve the survival of the fetal ventral mesencephalic cell transplanted in Parkinson’s disease? Neurosci. Bull. 2007, 23, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Gross, R.E.; Watts, R.L.; Hauser, R.A.; Bakay, R.A.; Reichmann, H.; von Kummer, R.; Ondo, W.G.; Reissig, E.; Eisner, W.; Steiner-Schulze, H.; et al. Intrastriatal transplantation of microcarrier-bound human retinal pigment epithelial cells versus sham surgery in patients with advanced Parkinson’s disease: A double-blind, randomised, controlled trial. Lancet Neurol. 2011, 10, 509–519. [Google Scholar] [CrossRef]
- Gugliandolo, A.; Bramanti, P.; Mazzon, E. Mesenchymal stem cell therapy in Parkinson’s disease animal models. Curr. Res. Transl. Med. 2017, 65, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Xiong, N.; Cao, X.; Zhang, Z.; Huang, J.; Chen, C.; Zhang, Z.; Jia, M.; Xiong, J.; Liang, Z.; Sun, S.; et al. Long-term efficacy and safety of human umbilical cord mesenchymal stromal cells in rotenone-induced hemiparkinsonian rats. Biol. Blood Marrow Transplant. 2010, 16, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Venkataramana, N.K.; Kumar, S.K.; Balaraju, S.; Radhakrishnan, R.C.; Bansal, A.; Dixit, A.; Rao, D.K.; Das, M.; Jan, M.; Gupta, P.K.; et al. Open-labeled study of unilateral autologous bone-marrow-derived mesenchymal stem cell transplantation in Parkinson’s disease. Transl. Res. 2010, 155, 62–70. [Google Scholar] [CrossRef]
- Arutyunyan, I.; Elchaninov, A.; Makarov, A.; Fatkhudinov, T. Umbilical cord as prospective source for mesenchymal stem cell-based therapy. Stem Cells Int. 2016, 2016, 6901286. [Google Scholar] [CrossRef]
- Da Silva Meirelles, L.; Chagastelles, P.C.; Nardi, N.B. Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J. Cell Sci. 2006, 119, 2204–2213. [Google Scholar] [CrossRef]
- Minteer, D.M.; Marra, K.G.; Rubin, J.P. Adipose stem cells: Biology, safety, regulation, and regenerative potential. Clin. Plast. Surg. 2015, 42, 169–179. [Google Scholar] [CrossRef]
- Chung, T.H.; Hsu, S.C.; Wu, S.H.; Hsiao, J.K.; Lin, C.P.; Yao, M.; Huang, D.M. Dextran-coated iron oxide nanoparticle-improved therapeutic effects of human mesenchymal stem cells in a mouse model of Parkinson’s disease. Nanoscale 2018, 10, 2998–3007. [Google Scholar] [CrossRef] [PubMed]
- Phinney, D.G.; Prockop, D.J. Concise review: Mesenchymal stem/multipotent stromal cells: The state of transdifferentiation and modes of tissue repair—Current views. Stem Cells 2007, 25, 2896–2902. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, Y.; Deng, Z. Pre-conditioned mesenchymal stem cells: A better way for cell-based therapy. Stem Cell Res. Ther. 2013, 4, 63. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, M.; Cimini, A.; Castelli, V. Insights into the effects of mesenchymal stem cell-derived secretome in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 5241. [Google Scholar] [CrossRef] [PubMed]
- Hofer, H.R.; Tuan, R.S. Secreted trophic factors of mesenchymal stem cells support neurovascular and musculoskeletal therapies. Stem Cell Res. Ther. 2016, 7, 131. [Google Scholar] [CrossRef] [PubMed]
- Gasparotto, J.; Ribeiro, C.T.; Bortolin, R.C.; Somensi, N.; Rabelo, T.K.; Kunzler, A.; Souza, N.C.; Pasquali, M.A.B.; Moreira, J.C.F.; Gelain, D.P. Targeted inhibition of RAGE in substantia nigra of rats blocks 6-OHDA-induced dopaminergic denervation. Sci. Rep. 2017, 7, 8795. [Google Scholar] [CrossRef]
- Saeedi, P.; Halabian, R.; Imani Fooladi, A.A. A revealing review of mesenchymal stem cells therapy, clinical perspectives and Modification strategies. Stem Cell Investig. 2019, 6, 34. [Google Scholar] [CrossRef]
- Han, Y.; Li, X.; Zhang, Y.; Han, Y.; Chang, F.; Ding, J. Mesenchymal stem cells for regenerative medicine. Cells 2019, 8, 886. [Google Scholar] [CrossRef]
- Gao, F.; Chiu, S.M.; Motan, D.A.; Zhang, Z.; Chen, L.; Ji, H.L.; Tse, H.F.; Fu, Q.L.; Lian, Q. Mesenchymal stem cells and immunomodulation: Current status and future prospects. Cell Death Dis. 2016, 7, e2062. [Google Scholar] [CrossRef]
- Oh, S.H.; Lee, S.C.; Kim, D.Y.; Kim, H.N.; Shin, J.Y.; Ye, B.S.; Lee, P.H. Mesenchymal stem cells stabilize axonal transports for autophagic clearance of α-synuclein in parkinsonian models. Stem Cells 2017, 35, 1934–1947. [Google Scholar] [CrossRef]
- Denu, R.A.; Nemcek, S.; Bloom, D.D.; Goodrich, A.D.; Kim, J.; Mosher, D.F.; Hematti, P. Fibroblasts and mesenchymal stromal/stem cells are phenotypically indistinguishable. Acta Haematol. 2016, 136, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.; Andrade, P.Z.; Abecasis, M.M.; Gimble, J.M.; Chase, L.G.; Campbell, A.M.; Boucher, S.; Vemuri, M.C.; Silva, C.L.; Cabral, J.M. Toward a clinical-grade expansion of mesenchymal stem cells from human sources: A microcarrier-based culture system under xeno-free conditions. Tissue Eng. Part C Methods 2011, 17, 1201–1210. [Google Scholar] [CrossRef]
- Sonntag, K.C.; Song, B.; Lee, N.; Jung, J.H.; Cha, Y.; Leblanc, P.; Neff, C.; Kong, S.W.; Carter, B.S.; Schweitzer, J.; et al. Pluripotent stem cell-based therapy for Parkinson’s disease: Current status and future prospects. Prog. Neurobiol. 2018, 168, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Kriks, S.; Shim, J.W.; Piao, J.; Ganat, Y.M.; Wakeman, D.R.; Xie, Z.; Carrillo-Reid, L.; Auyeung, G.; Antonacci, C.; Buch, A.; et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 2011, 480, 547–551. [Google Scholar] [CrossRef]
- Tao, Y.; Zhang, S.C. Neural subtype specification from human pluripotent stem cells. Cell Stem Cell 2016, 19, 573–586. [Google Scholar] [CrossRef]
- Jo, J.; Xiao, Y.; Sun, A.X.; Cukuroglu, E.; Tran, H.D.; Goke, J.; Tan, Z.Y.; Saw, T.Y.; Tan, C.P.; Lokman, H.; et al. Midbrain-like organoids from human pluripotent stem cells contain functional dopaminergic and neuromelanin-producing neurons. Cell Stem Cell 2016, 19, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Reubinoff, B.E.; Itsykson, P.; Turetsky, T.; Pera, M.F.; Reinhartz, E.; Itzik, A.; Ben-Hur, T. Neural progenitors from human embryonic stem cells. Nat. Biotechnol. 2001, 19, 1134–1140. [Google Scholar] [CrossRef]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef]
- Kirkeby, A.; Grealish, S.; Wolf, D.A.; Nelander, J.; Wood, J.; Lundblad, M.; Lindvall, O.; Parmar, M. Generation of regionally specified neural progenitors and functional neurons from human embryonic stem cells under defined conditions. Cell Rep. 2012, 1, 703–714. [Google Scholar] [CrossRef]
- Hargus, G.; Cooper, O.; Deleidi, M.; Levy, A.; Lee, K.; Marlow, E.; Yow, A.; Soldner, F.; Hockemeyer, D.; Hallett, P.J.; et al. Differentiated Parkinson patient-derived induced pluripotent stem cells grow in the adult rodent brain and reduce motor asymmetry in Parkinsonian rats. Proc. Natl. Acad. Sci. USA 2010, 107, 15921–15926. [Google Scholar] [CrossRef]
- Martínez-Cerdeño, V.; Noctor, S.C.; Espinosa, A.; Ariza, J.; Parker, P.; Orasji, S.; Daadi, M.M.; Bankiewicz, K.; Alvarez-Buylla, A.; Kriegstein, A.R. Embryonic MGE precursor cells grafted into adult rat striatum integrate and ameliorate motor symptoms in 6-OHDA-lesioned rats. Cell Stem Cell 2010, 6, 238–250. [Google Scholar] [CrossRef]
- Grealish, S.; Diguet, E.; Kirkeby, A.; Mattsson, B.; Heuer, A.; Bramoulle, Y.; Van Camp, N.; Perrier, A.L.; Hantraye, P.; Björklund, A.; et al. Human ESC-derived dopamine neurons show similar preclinical efficacy and potency to fetal neurons when grafted in a rat model of Parkinson’s disease. Cell Stem Cell 2014, 15, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Steinbeck, J.A.; Choi, S.J.; Mrejeru, A.; Ganat, Y.; Deisseroth, K.; Sulzer, D.; Mosharov, E.V.; Studer, L. Optogenetics enables functional analysis of human embryonic stem cell-derived grafts in a Parkinson’s disease model. Nat. Biotechnol. 2015, 33, 204–209. [Google Scholar] [CrossRef]
- Chen, Y.; Xiong, M.; Dong, Y.; Haberman, A.; Cao, J.; Liu, H.; Zhou, W.; Zhang, S.C. Chemical control of grafted human PSC-derived neurons in a mouse model of Parkinson’s Disease. Cell Stem Cell 2016, 18, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Laperle, A.H.; Sances, S.; Yucer, N.; Dardov, V.J.; Garcia, V.J.; Ho, R.; Fulton, A.N.; Jones, M.R.; Roxas, K.M.; Avalos, P.; et al. iPSC modeling of young-onset Parkinson’s disease reveals a molecular signature of disease and novel therapeutic candidates. Nat. Med. 2020, 26, 289–299. [Google Scholar] [CrossRef]
- Hallett, P.J.; Deleidi, M.; Astradsson, A.; Smith, G.A.; Cooper, O.; Osborn, T.M.; Sundberg, M.; Moore, M.A.; Perez-Torres, E.; Brownell, A.L.; et al. Successful function of autologous iPSC-derived dopamine neurons following transplantation in a non-human primate model of Parkinson’s disease. Cell Stem Cell 2015, 16, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Morizane, A.; Doi, D.; Magotani, H.; Onoe, H.; Hayashi, T.; Mizuma, H.; Takara, S.; Takahashi, R.; Inoue, H.; et al. Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nature 2017, 548, 592–596. [Google Scholar] [CrossRef]
- Zhou, T.; Kim, T.W.; Chong, C.N.; Tan, L.; Amin, S.; Sadat Badieyan, Z.; Mukherjee, S.; Ghazizadeh, Z.; Zeng, H.; Guo, M.; et al. A hPSC-based platform to discover gene-environment interactions that impact human β-cell and dopamine neuron survival. Nat. Commun. 2018, 9, 4815. [Google Scholar] [CrossRef]
- Doi, D.; Magotani, H.; Kikuchi, T.; Ikeda, M.; Hiramatsu, S.; Yoshida, K.; Amano, N.; Nomura, M.; Umekage, M.; Morizane, A.; et al. Pre-clinical study of induced pluripotent stem cell-derived dopaminergic progenitor cells for Parkinson’s disease. Nat. Commun. 2020, 11, 3369. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, J.; Tsang, K.S.; Yang, H.; Gao, W.Q. Therapeutic potential of human amniotic epithelial cells on injuries and disorders in the central nervous system. Stem Cells Int. 2019, 2019, 5432301. [Google Scholar] [CrossRef]
- Li, H.; Niederkorn, J.Y.; Neelam, S.; Mayhew, E.; Word, R.A.; McCulley, J.P.; Alizadeh, H. Immunosuppressive factors secreted by human amniotic epithelial cells. Investig. Ophthalmol. Vis. Sci. 2005, 46, 900–907. [Google Scholar] [CrossRef]
- Lukomska, B.; Stanaszek, L.; Zuba-Surma, E.; Legosz, P.; Sarzynska, S.; Drela, K. Challenges and controversies in human mesenchymal stem cell therapy. Stem Cells Int. 2019, 2019, 9628536. [Google Scholar] [CrossRef] [PubMed]
- Hoban, D.B.; Shrigley, S.; Mattsson, B.; Breger, L.S.; Jarl, U.; Cardoso, T.; Nelander Wahlestedt, J.; Luk, K.C.; Björklund, A.; Parmar, M. Impact of α-synuclein pathology on transplanted hESC-derived dopaminergic neurons in a humanized α-synuclein rat model of PD. Proc. Natl. Acad. Sci. USA 2020, 117, 15209–15220. [Google Scholar] [CrossRef]
- Hansen, C.; Angot, E.; Bergström, A.L.; Steiner, J.A.; Pieri, L.; Paul, G.; Outeiro, T.F.; Melki, R.; Kallunki, P.; Fog, K.; et al. α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Investig. 2011, 121, 715–725. [Google Scholar] [CrossRef]
- Deuse, T.; Hu, X.; Gravina, A.; Wang, D.; Tediashvili, G.; De, C.; Thayer, W.O.; Wahl, A.; Garcia, J.V.; Reichenspurner, H.; et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol. 2019, 37, 252–258. [Google Scholar] [CrossRef]
- Taylor, C.J.; Peacock, S.; Chaudhry, A.N.; Bradley, J.A.; Bolton, E.M. Generating an iPSC bank for HLA-matched tissue transplantation based on known donor and recipient HLA types. Cell Stem Cell 2012, 11, 147–152. [Google Scholar] [CrossRef]
- Nakatsuji, N.; Nakajima, F.; Tokunaga, K. HLA-haplotype banking and iPS cells. Nat. Biotechnol. 2008, 26, 739–740. [Google Scholar] [CrossRef]
- Lee, S.; Huh, J.Y.; Turner, D.M.; Lee, S.; Robinson, J.; Stein, J.E.; Shim, S.H.; Hong, C.P.; Kang, M.S.; Nakagawa, M.; et al. Repurposing the cord blood bank for haplobanking of HLA-Homozygous iPSCs and their usefulness to multiple populations. Stem Cells 2018, 36, 1552–1566. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Choi, J.; Park, N.; Kang, J.; Kim, M.; Kim, Y.; Ju, J.H. Development of immunocompatible pluripotent stem cells via CRISPR-based human leukocyte antigen engineering. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Aron Badin, R.; Bugi, A.; Williams, S.; Vadori, M.; Michael, M.; Jan, C.; Nassi, A.; Lecourtois, S.; Blancher, A.; Cozzi, E.; et al. MHC matching fails to prevent long-term rejection of iPSC-derived neurons in non-human primates. Nat. Commun. 2019, 10, 4357. [Google Scholar] [CrossRef]
- Rivetti di Val Cervo, P.; Romanov, R.A.; Spigolon, G.; Masini, D.; Martín-Montañez, E.; Toledo, E.M.; La Manno, G.; Feyder, M.; Pifl, C.; Ng, Y.H.; et al. Induction of functional dopamine neurons from human astrocytes in vitro and mouse astrocytes in a Parkinson’s disease model. Nat. Biotechnol. 2017, 35, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Kang, X.; Hu, J.; Zhang, D.; Liang, Z.; Meng, F.; Zhang, X.; Xue, Y.; Maimon, R.; Dowdy, S.F.; et al. Reversing a model of Parkinson’s disease with in situ converted nigral neurons. Nature 2020, 582, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Su, J.; Hu, X.; Zhou, C.; Li, H.; Chen, Z.; Xiao, Q.; Wang, B.; Wu, W.; Sun, Y.; et al. Glia-to-neuron conversion by CRISPR-CasRx alleviates symptoms of neurological disease in mice. Cell 2020, 181, 590–603. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Target | Action | Agent (Country) | Trial Phase | Clinical Trial Identifier | Year for Starting | Status |
|---|---|---|---|---|---|---|
| Human neural stem cells | Human fetal neural stem cell injection delivered nasally | Second Affiliated Hospital of Soochow University (China) | Phase 2/3 | NCT03128450 | 2017 | Unknown |
| Human parthenogenetic neural stem cells injected intracerebrally to the striatum and substantia nigra | Cyto Therapeutics Pty Limited (Australia) | Phase 1 | NCT02452723 | 2015 | Active, not recruiting | |
| Differentiated neurons derived from adult central nervous system progenitor cells transplanted in patients. Stereotactic delivery of cell suspension into basal ganglia structures | NeuroGeneration | Phase 1/2 | NCT03309514 | 2017 | Not yet recruiting | |
| Mesenchymal stem cells | Umbilical cord-derived MSC from mesoderm possesses strong proliferation ability and multiple differentiation potentials, delivered by intravenous infusion to patients with PD | Guangzhou General Hospital of Guangzhou Military Command (China) | Phase 1 | NCT03550183 | 2018 | Enrolling by invitation |
| Allogenic umbilical cord-derived stem cells injected intravenously to enrolled patients with PD | University of Jordan (Jordan) | Phase 1/2 | NCT03684122 | 2018 | Recruiting | |
| Phase IIa double-blind randomized placebo-controlled trial | The University of Texas Health Science Center (USA) | Phase 2 | NCT04506073 | 2020 | Not yet recruiting | |
| Human stem cells (OK99) | Implantation of Celavie human stem cells (OK99) to address the underlying pathology of the disease by replacing damaged/destroyed cells of the brain and stimulating the patient’s brain to repair itself | Celavie Bioscences, LLC (Mexico) | Phase 1 | NCT02780895 | 2016 | Unknown |
| Human amniotic epithelial stem Cells (hAESCs) | Stereotactic transplantation of hAESCs into the lateral ventricle | Shanghai East Hospital (China) | Early Phase 1 | NCT04414813 | 2020 | Not yet recruiting |
| Human embryonic stem cells | Transplantation of human embryonic stem cell-derived neural precursor cells into the striatum. | Chinese Academy of Sciences (China) | Phase 1/2 | NCT03119636 | 2017 | Recruiting |
| Induced pluripotent stem cells | Develop human-induced pluripotent stem cells from cell cultures taken from skin biopsies or patients’ hair | Hadassah Medical Organization (Israel) | Not Applicable | NCT00874783 | 2009 | Recruiting |
| Total dose of iPSC-derived neural stem cells administered on day 0 | Allife Medical Science and Technology Co. Ltd. (China) | Early Phase 1 | NCT03815071 | 2019 | Not yet recruiting | |
| Bone marrow–derived stem cells | Isolation of autologous bone marrow-derived stem cells and transfer to the vascular system and interior third of the nasal passages | MD Stem Cells (USA) | Not Applicable | NCT02795052 | 2016 | Terminated |
| Autologous bone marrow-derived stem cells stereotactically transplanted into the patient’s striatum | Jaslok Hospital and Research Centre (India) | Not Applicable | NCT00976430 | 2009 | Recruiting | |
| Allogeneic bone marrow-derived MSCs delivered intravenously | The University of Texas Health Science Center (USA) | Phase 1/2 | NCT02611167 | 2015 | Completed |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Cheung, H.-H. Stem Cell-Based Therapies for Parkinson Disease. Int. J. Mol. Sci. 2020, 21, 8060. https://doi.org/10.3390/ijms21218060
Liu Z, Cheung H-H. Stem Cell-Based Therapies for Parkinson Disease. International Journal of Molecular Sciences. 2020; 21(21):8060. https://doi.org/10.3390/ijms21218060
Chicago/Turabian StyleLiu, Zhaohui, and Hoi-Hung Cheung. 2020. "Stem Cell-Based Therapies for Parkinson Disease" International Journal of Molecular Sciences 21, no. 21: 8060. https://doi.org/10.3390/ijms21218060
APA StyleLiu, Z., & Cheung, H.-H. (2020). Stem Cell-Based Therapies for Parkinson Disease. International Journal of Molecular Sciences, 21(21), 8060. https://doi.org/10.3390/ijms21218060