Role of Polycomb Complexes in Normal and Malignant Plasma Cells

 , ,
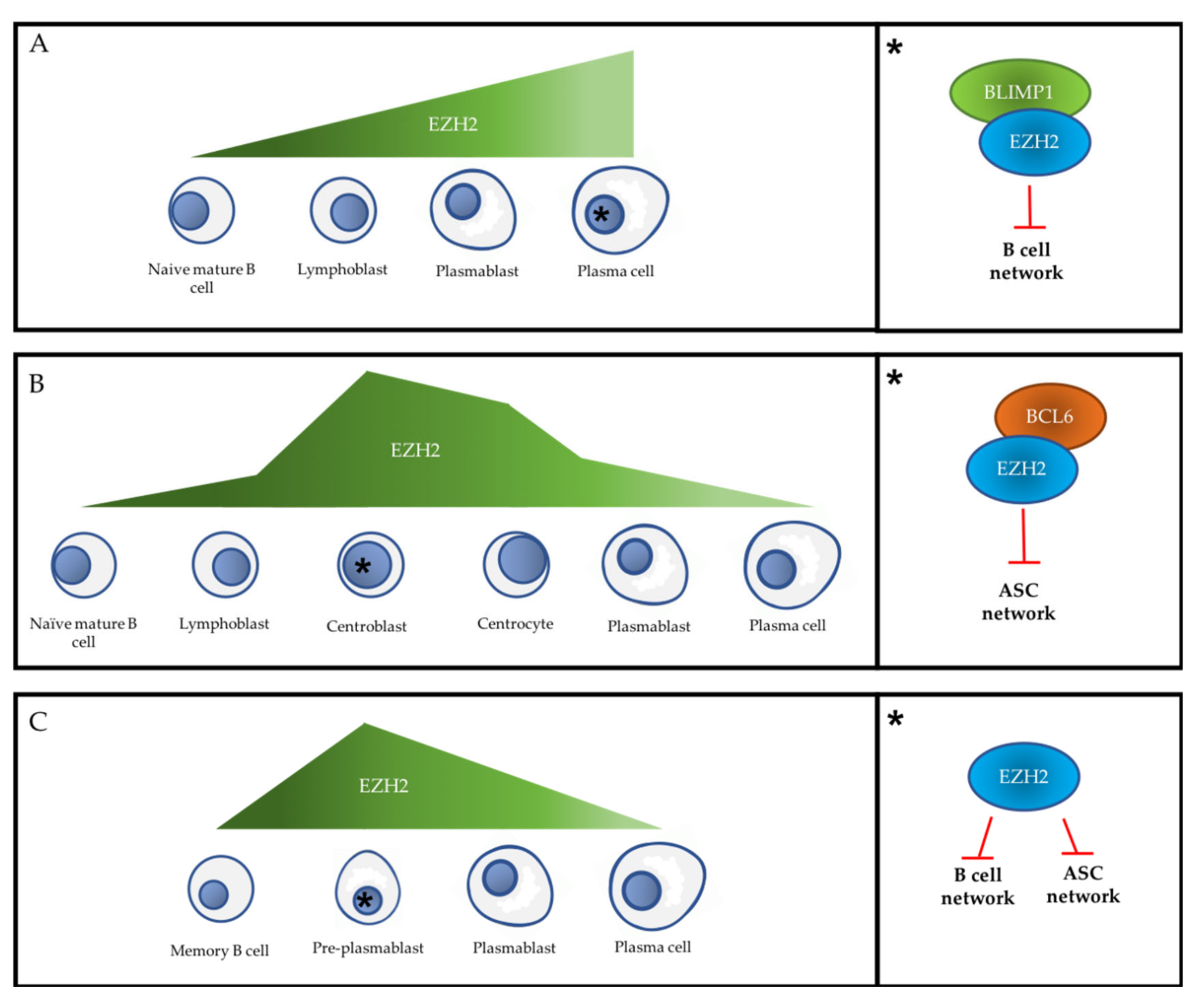
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
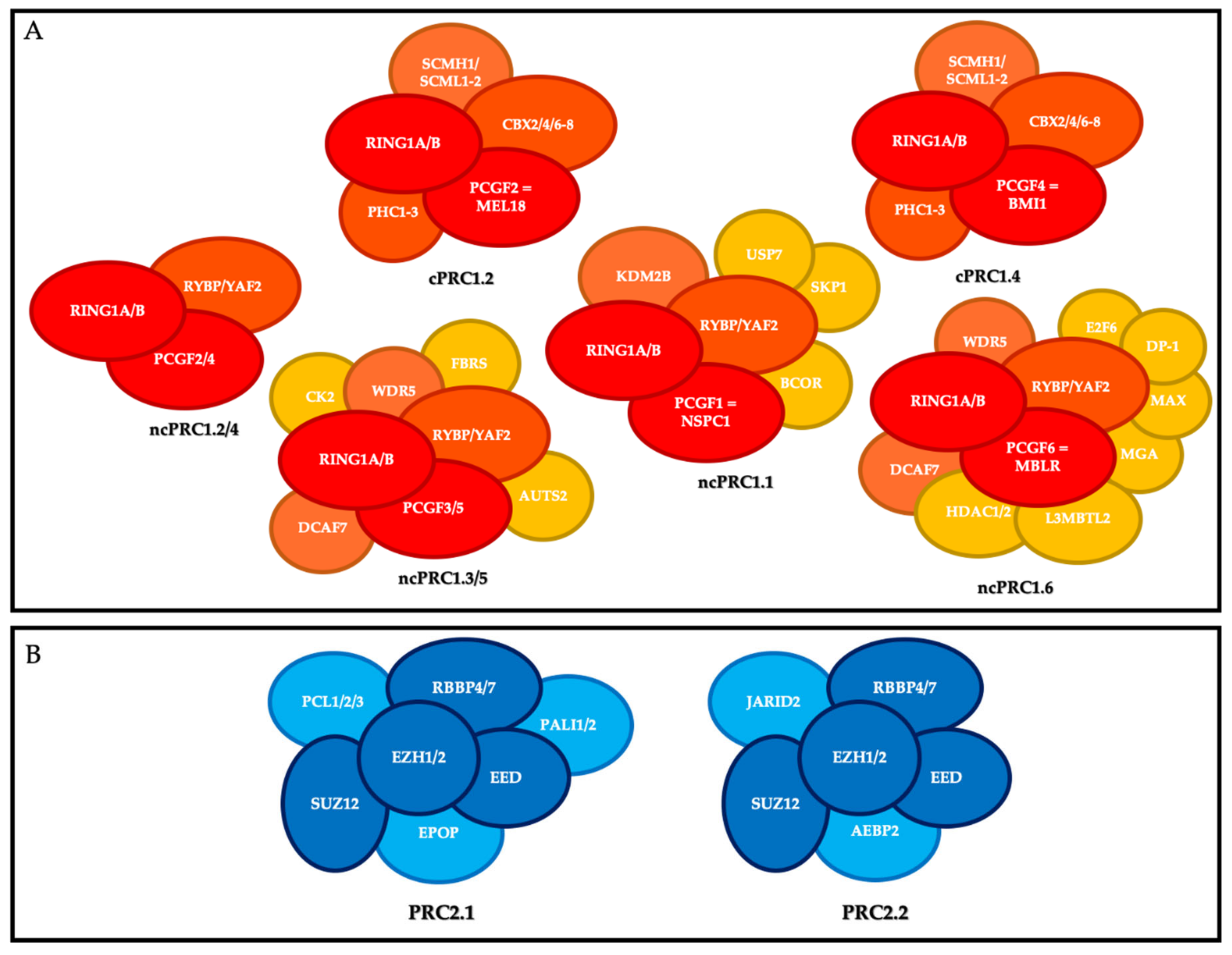
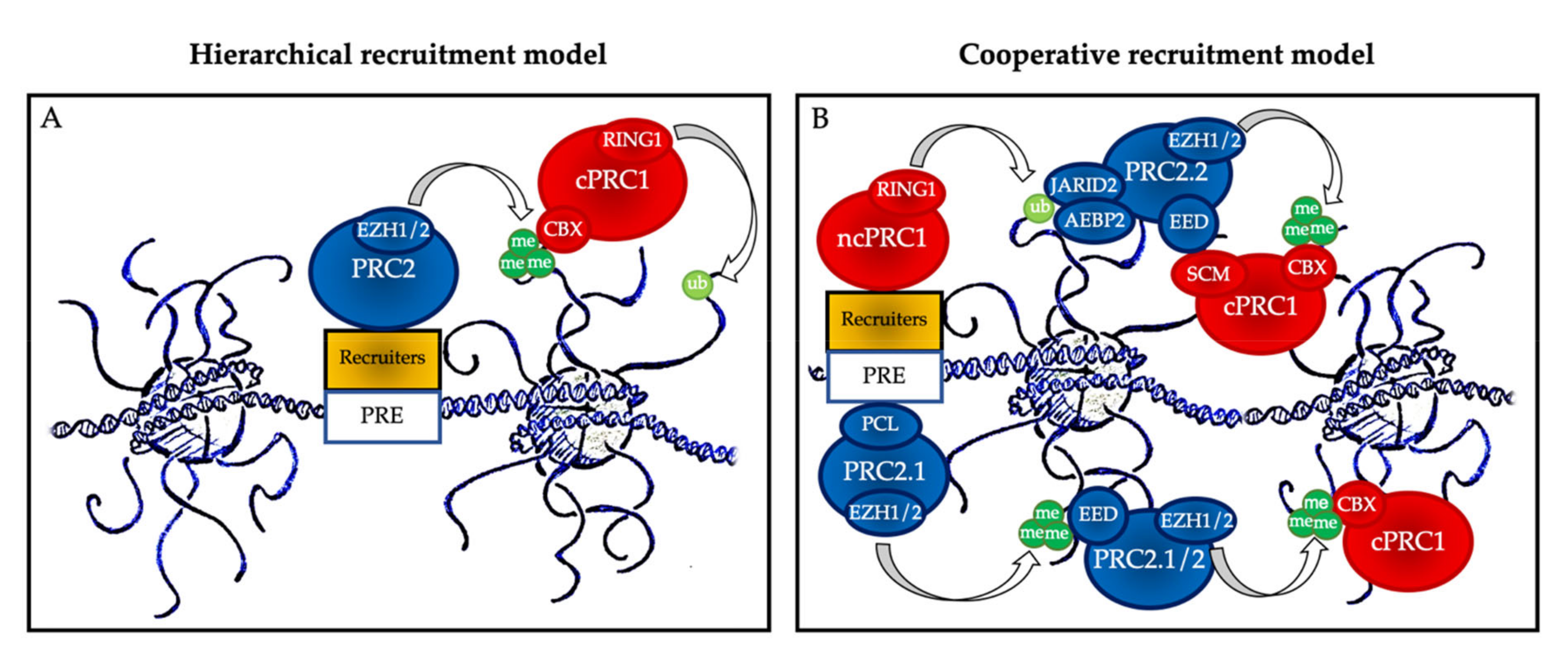
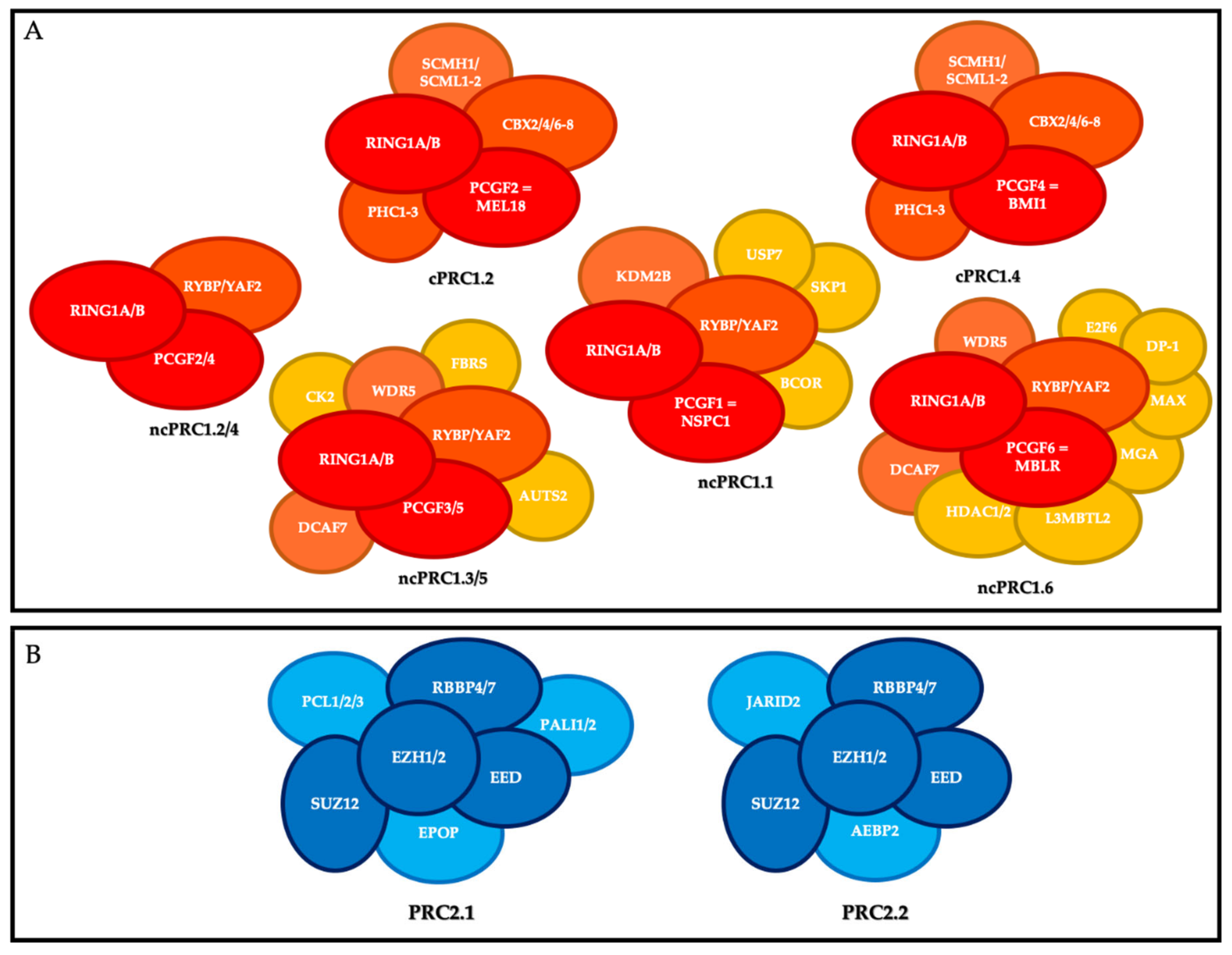
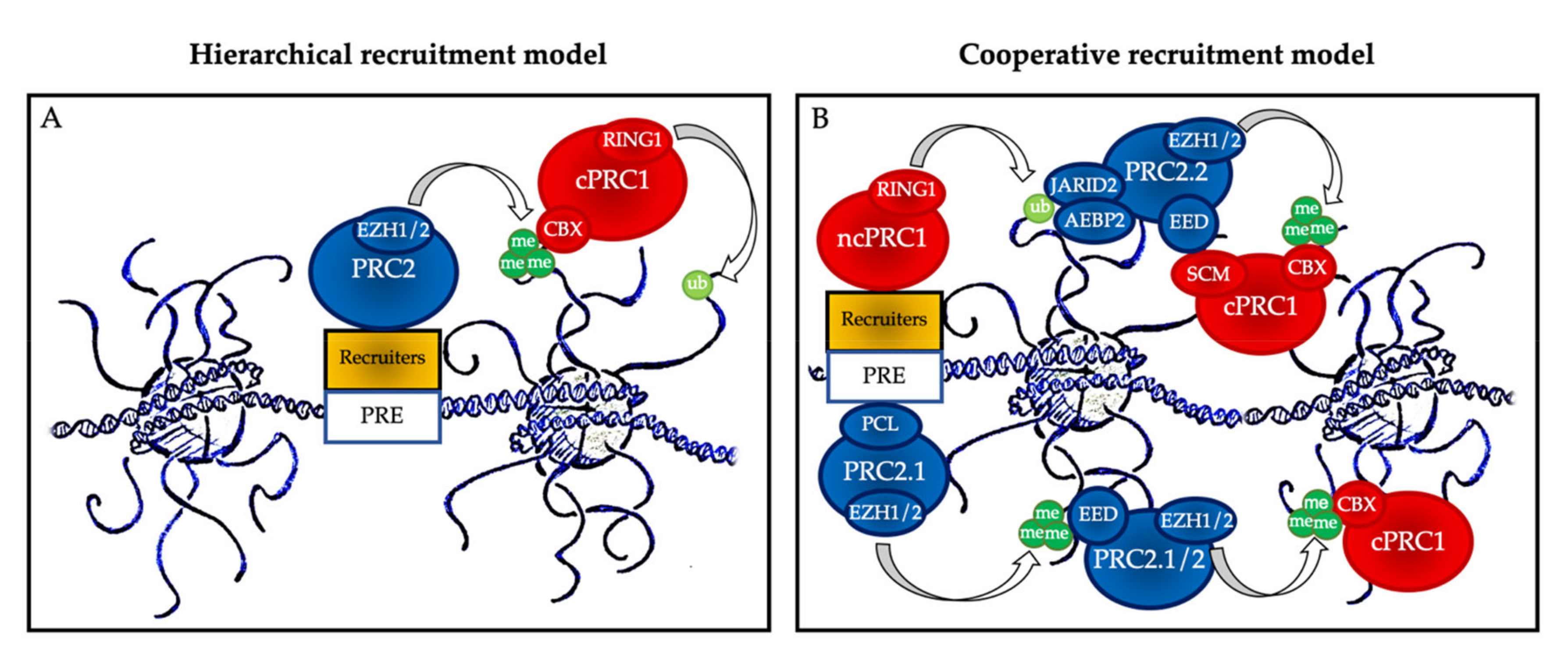
2. PcG Complexes
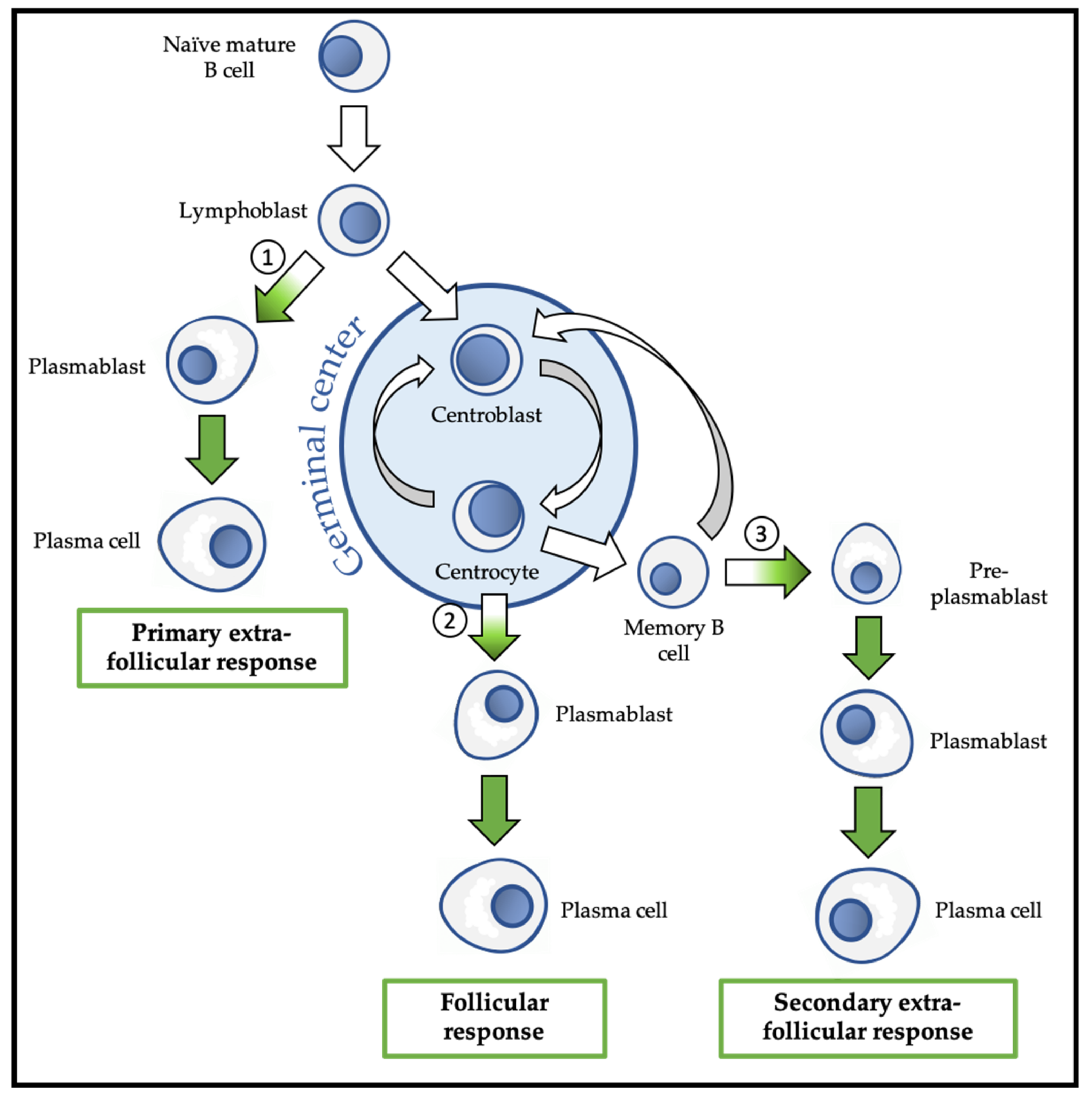
3. Lymphopoiesis and Plasma Cell Differentiation
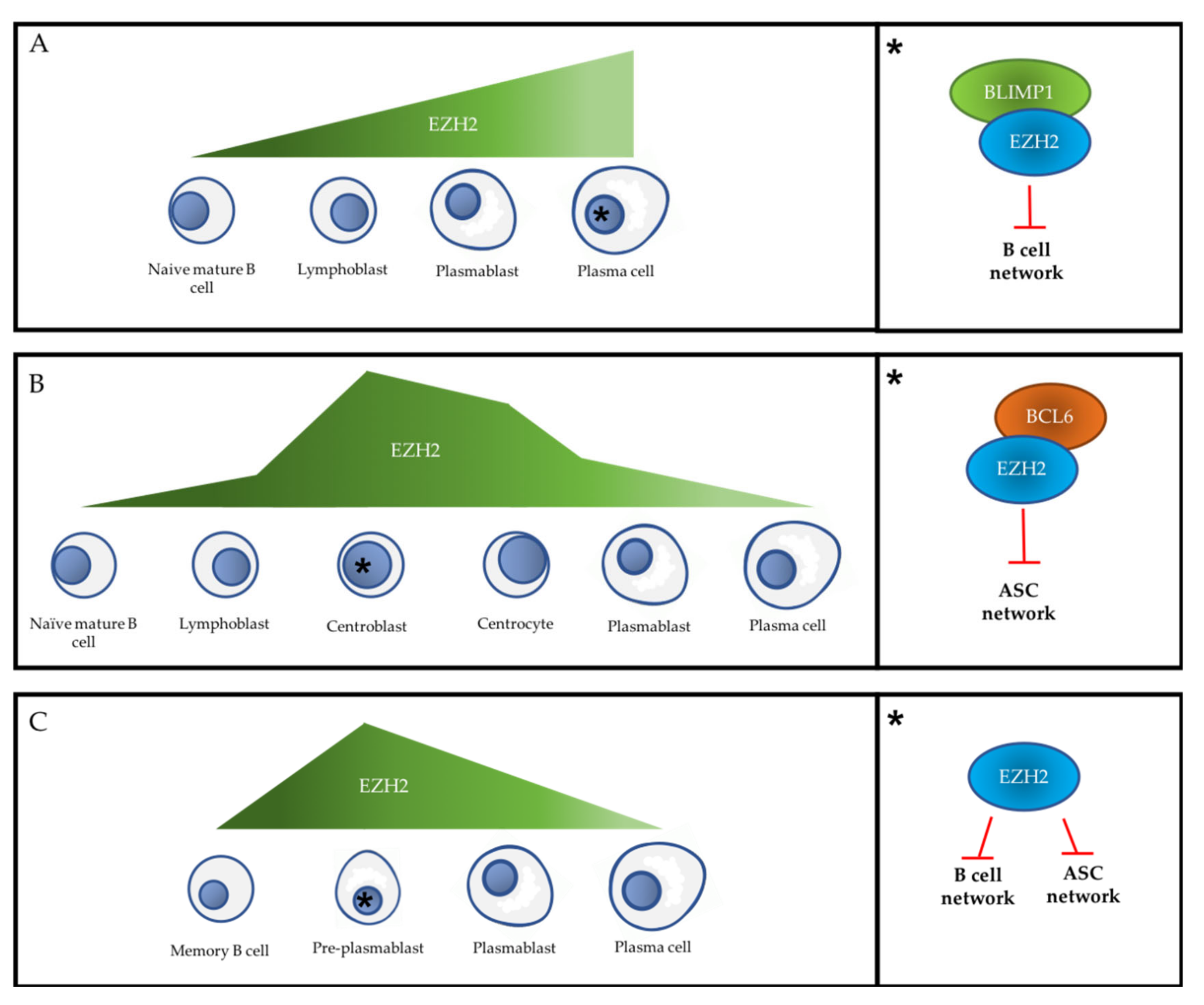
3.1. PcG Proteins and Early B Cell Differentiation
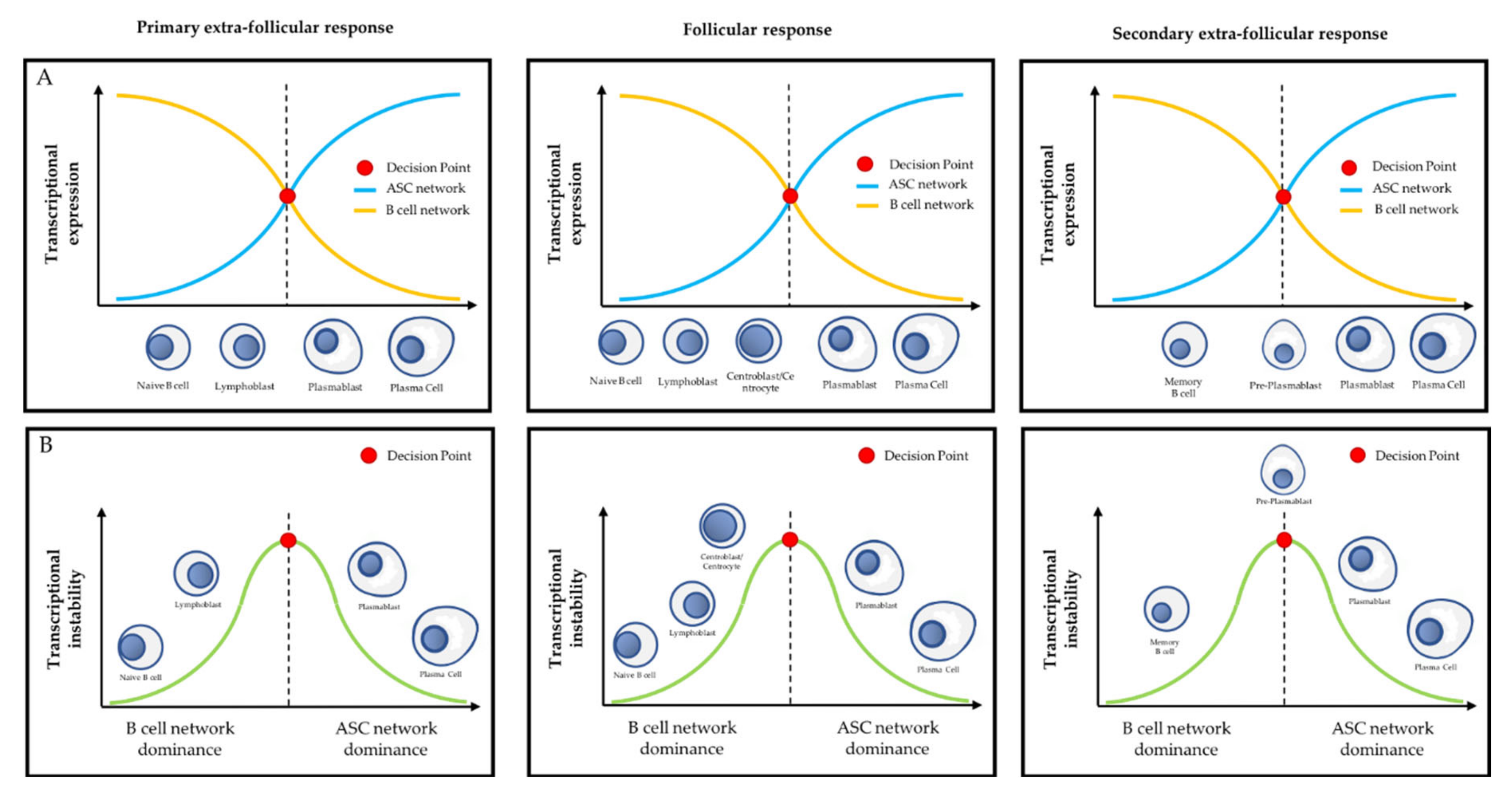
3.2. PcG Proteins and the Primary Extra-Follicular Response
3.3. PcG Proteins and the Follicular Response
3.4. PcG Proteins and the Secondary Extra-Follicular Response
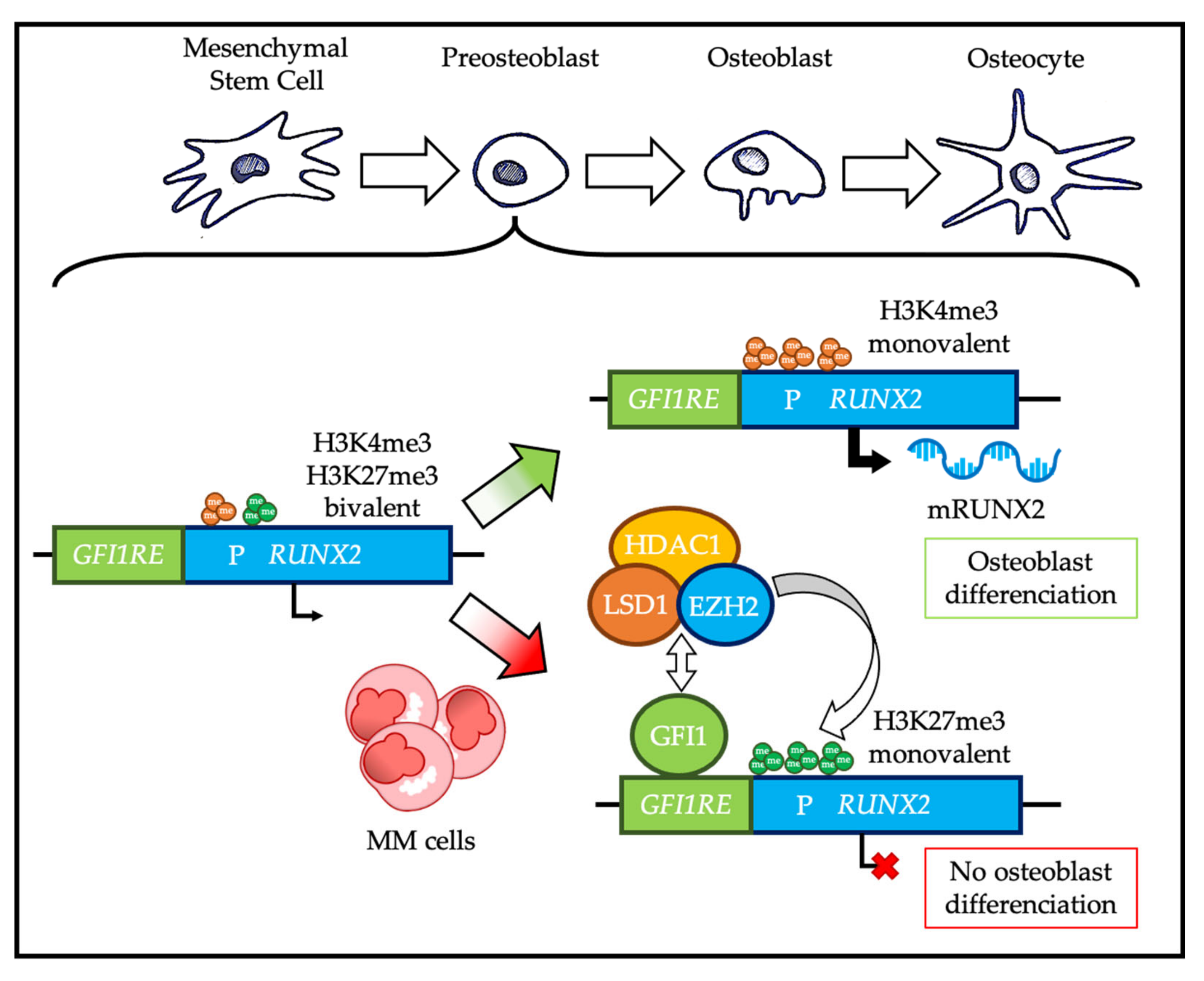
4. Multiple Myeloma
4.1. PRC2 in Multiple Myeloma Pathophysiology
4.2. PRC1 in Multiple Myeloma Pathophysiology
4.3. PcG Proteins and Multiple Myeloma Tumor Microenvironment
4.4. PcG Proteins and Drug Resistance in Multiple Myeloma
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Ribatti, D. The discovery of plasma cells: An historical note. Immunol. Lett. 2017, 188, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Maximow, A.A. Chapter III-C. In Experimentelle Untersuchungen über Entzündliche Neubildung von Bindegewebe; Fischer: Leipzig, Germany, 1902. [Google Scholar]
- Fagraeus, A. Plasma Cellular Reaction and its Relation to the Formation of Antibodies in vitro. Nature 1947, 159, 499. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.D.; Peterson, R.D.A.; Good, R.A. Delineation of the thymic and bursal lymphoid systems in chicken. Nature 1965, 205, 103–146. [Google Scholar] [CrossRef] [PubMed]
- Cyster, J.G.; Allen, C.D.C. B cell responses—Cell interaction dynamics and decisions. Cell 2019, 177, 524–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, P.H. Melanogaster-New mutants: Report of Pamela H. Lewis. Dros. Inform. Serv. 1947, 21, 69. [Google Scholar]
- Chittock, E.C.; Latwiel, S.; Miller, T.C.R.; Müller, C.W. Molecular architecture of polycomb repressive complexes. Biochem. Soc. Trans. 2017, 45, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Blackledge, N.P.; Rose, N.R.; Klose, R.J. Targeting polycomb systems to regulate gene expression: Modifications to a complex story. Nat. Rev. Mol. Cell Biol. 2015, 16, 643–649. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.; Zhang, Y.; Sun, T.; Cheng, B. Epigenetic regulation by polycomb group complexes: Focus on roles of CBX proteins. J. Zhejiang Univ. Sci. B 2014, 15, 412–428. [Google Scholar] [CrossRef] [Green Version]
- Schuettengruber, B.; Bourbon, H.-M.; Di Croce, L.; Cavalli, G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 2017, 171, 34–57. [Google Scholar] [CrossRef] [Green Version]
- Healy, E.; Mucha, M.; Glancy, E.; Fitzpatrick, D.J.; Conway, E.; Neikes, H.K.; Monger, C.; Van Mierlo, G.; Baltissen, M.P.; Koseki, Y.; et al. PRC2.1 and PRC2.2 Synergize to Coordinate H3K27 Trimethylation. Mol. Cell 2019, 76, 437–452.e6. [Google Scholar] [CrossRef]
- Blackledge, N.P.; Farcas, A.M.; Kondo, T.; King, H.W.; McGouran, J.F.; Hanssen, L.L.P.; Ito, S.; Cooper, S.; Kondo, K.; Koseki, Y.; et al. Variant PRC1 Complex-Dependent H2A Ubiquitylation Drives PRC2 Recruitment and Polycomb Domain Formation. Cell 2014, 157, 1445–1459. [Google Scholar] [CrossRef] [Green Version]
- Simon, J.A.; Kingston, R.E. Occupying chromatin: Polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol. Cell 2013, 49, 808–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mårtensson, I.-L.; Almqvist, N.; Grimsholm, O.; Bernardi, A.I. The pre-B cell receptor checkpoint. FEBS Lett. 2010, 584, 2572–2579. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Mazor, T.; Huang, H.; Huang, H.-T.; Kathrein, K.L.; Woo, A.J.; Chouinard, C.R.; Labadorf, A.; Akie, T.E.; Moran, T.B.; et al. Direct Recruitment of Polycomb Repressive Complex 1 (PRC1) to Chromatin by Core Binding Transcription Factors. Mol. Cell 2012, 45, 330–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalb, R.; Latwiel, S.; Baymaz, H.I.; Jansen, P.W.T.C.; Müller, C.W.; Vermeulen, M.; Müller, J. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat. Struct. Mol. Biol. 2014, 21, 569–571. [Google Scholar] [CrossRef]
- Cooper, S.; Grijzenhout, A.; Underwood, E.; Ancelin, K.; Zhang, T.; Nesterova, T.B.; Anil-Kirmizitas, B.; Bassett, A.; Kooistra, S.M.; Agger, K.; et al. Jarid2 binds mono-ubiquitylated H2A lysine 119 to mediate crosstalk between Polycomb complexes PRC1 and PRC2. Nat. Commun. 2016, 7, 13661. [Google Scholar] [CrossRef] [Green Version]
- Tamburri, S.; Lavarone, E.; Fernández-Pérez, D.; Conway, E.; Zanotti, M.; Manganaro, D.; Pasini, D. Histone H2AK119 Mono-Ubiquitination Is Essential for Polycomb-Mediated Transcriptional Repression. Mol. Cell 2020, 77, 840–856.e5. [Google Scholar] [CrossRef] [Green Version]
- Perino, M.; van Mierlo, G.; Loh, C.; Wardle, S.M.T.; Zijlmans, D.W.; Marks, H.; Veenstra, G.J.C. Two Functional Axes of Feedback-Enforced PRC2 Recruitment in Mouse Embryonic Stem Cells. Stem Cell Rep. 2020. [Google Scholar] [CrossRef]
- Shapiro-Shelef, M.; Calame, K. Regulation of plasma-cell development. Nat. Rev. Immunol. 2005, 5, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Tellier, J.; Nutt, S.L. Plasma cells: The programming of an antibody-secreting machine. Eur. J. Immunol. 2019, 49, 30–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBien, T.W.; Tedder, T.F. B lymphocytes: How they develop and function. Blood 2008, 112, 1570–1580. [Google Scholar] [CrossRef]
- Loder, B.F.; Mutschler, B.; Ray, R.J.; Paige, C.J.; Sideras, P.; Torres, R.; Lamers, M.C.; Carsetti, R. B Cell Development in the Spleen Takes Place in Discrete Steps and Is Determined by the Quality of B Cell Receptor–Derived Signals. J. Exp. Med. 1999, 190, 75–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, J.B.; Silverman, M.; Monroe, J.G. Transitional B cells: Step by step towards immune competence. Trends Immunol. 2003, 24, 342–348. [Google Scholar] [CrossRef]
- Liu, Y.-J.; Zhang, J.; Lane, P.J.L.; Chan, E.Y.-T.; Maclennan, I.C.M. Sites of specific B cell activation in primary and secondary responses to T cell-dependent and T cell-independent antigens. Eur. J. Immunol. 1991, 21, 2951–2962. [Google Scholar] [CrossRef]
- Kurosaki, T.; Shinohara, H.; Baba, Y. B Cell Signaling and Fate Decision. Annu. Rev. Immunol. 2010, 28, 21–55. [Google Scholar] [CrossRef]
- Murphy, K.; Weaver, C. Chapter 1-18: Lymphocytes activated by antigen proliferate in the peripheral lymphoid organs, generating effector cells and immunological memory. In Janeway’s Immunobiology; Garland Science: New York, NY, USA, 2016. [Google Scholar]
- Kupfer, A.; Singer, S.J. The specific interaction of helper T cells and antigen-presenting B cells. IV. Membrane and cytoskeletal reorganizations in the bound T cell as a function of antigen dose. J. Exp. Med. 1989, 170, 1697–1713. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L. The immunological synapse. Cancer Immunol. Res. 2014, 2, 1023–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nutt, S.L.; Hodgkin, P.D.; Tarlinton, D.M.; Corcoran, L.M. The generation of antibody-secreting plasma cells. Nat. Rev. Immunol. 2015, 15, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.C.; Joyner, C.J.; Sanz, I.; Lee, F.E.-H. Factors Affecting Early Antibody Secreting Cell Maturation Into Long-Lived Plasma Cells. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Shlomchik, M.J.; Weisel, F. Germinal center selection and the development of memory B and plasma cells. Immunol. Rev. 2012, 247, 52–63. [Google Scholar] [CrossRef]
- Klein, U.; Dalla-Favera, R. Germinal centres: Role in B-cell physiology and malignancy. Nat. Rev. Immunol 2008, 8, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, M.; Sankaranand, V.S.; Anant, S.; Sugai, M.; Kinoshita, K.; Davidson, N.O.; Honjo, T. Specific Expression of Activation-induced Cytidine Deaminase (AID), a Novel Member of the RNA-editing Deaminase Family in Germinal Center B Cells. J. Biol. Chem. 1999, 274, 18470–18476. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Pone, E.J.; Al-Qahtani, A.; Park, S.-R.; Zan, H.; Casali, P. Regulation of aicda expression and AID activity: Relevance to somatic hypermutation and class switch DNA recombination. Crit. Rev. Immunol. 2007, 27, 367–397. [Google Scholar] [CrossRef] [Green Version]
- Caganova, M.; Carrisi, C.; Varano, G.; Mainoldi, F.; Zanardi, F.; Germain, P.-L.; George, L.; Alberghini, F.; Ferrarini, L.; Talukder, A.K.; et al. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J. Clin. Investig. 2013, 123, 5009–5022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, R.-H.; Yu, Y.-L.; Hung, M.-C. The roles of EZH2 in cell lineage commitment. Am. J. Transl. Res. 2011, 3, 243–250. [Google Scholar] [PubMed]
- Herviou, L.; Cavalli, G.; Cartron, G.; Klein, B.; Moreaux, J. EZH2 in normal hematopoiesis and hematological malignancies. Oncotarget 2015, 7, 2284–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mochizuki-Kashio, M.; Mishima, Y.; Miyagi, S.; Negishi, M.; Saraya, A.; Konuma, T.; Shinga, J.; Koseki, H.; Iwama, A. Dependency on the polycomb gene Ezh2 distinguishes fetal from adult hematopoietic stem cells. Blood 2011, 118, 6553–6561. [Google Scholar] [CrossRef]
- Xie, H.; Xu, J.; Hsu, J.H.; Nguyen, M.; Fujiwara, Y.; Peng, C.; Orkin, S.H. Polycomb repressive complex 2 regulates hematopoietic stem cell maintenance and differentiation in a developmental stage-specific manner. Cell Stem Cell 2014, 14, 68–80. [Google Scholar] [CrossRef] [Green Version]
- Su, I.; Basavaraj, A.; Krutchinsky, A.N.; Hobert, O.; Ullrich, A.; Chait, B.T.; Tarakhovsky, A. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat. Immunol. 2003, 4, 124–131. [Google Scholar] [CrossRef]
- Vidal, M.; Starowicz, K. Polycomb complexes PRC1 and their function in hematopoiesis. Exp. Hematol. 2017, 48, 12–31. [Google Scholar] [CrossRef] [Green Version]
- Isshiki, Y.; Nakajima-Takagi, Y.; Oshima, M.; Aoyama, K.; Rizk, M.; Kurosawa, S.; Saraya, A.; Kondo, T.; Sakaida, E.; Nakaseko, C.; et al. KDM2B in polycomb repressive complex 1.1 functions as a tumor suppressor in the initiation of T-cell leukemogenesis. Blood Adv. 2019, 3, 2537–2549. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.; Price, M.J.; Patterson, D.G.; Barwick, B.G.; Haines, R.R.; Kania, A.K.; Bradley, J.E.; Randall, T.D.; Boss, J.M.; Scharer, C.D. EZH2 Represses the B Cell Transcriptional Program and Regulates Antibody-Secreting Cell Metabolism and Antibody Production. J. Immunol. 2018, 200, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Béguelin, W.; Rivas, M.A.; Calvo Fernández, M.T.; Teater, M.; Purwada, A.; Redmond, D.; Shen, H.; Challman, M.F.; Elemento, O.; Singh, A.; et al. EZH2 enables germinal centre formation through epigenetic silencing of CDKN1A and an Rb-E2F1 feedback loop. Nat. Commun. 2017, 8, 877. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Roberts, C.W.M. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Scharer, C.D.; Barwick, B.G.; Guo, M.; Bally, A.P.R.; Boss, J.M. Plasma cell differentiation is controlled by multiple cell division-coupled epigenetic programs. Nat. Commun. 2018, 9, 1698. [Google Scholar] [CrossRef] [Green Version]
- Scharer, C.D.; Patterson, D.G.; Mi, T.; Price, M.J.; Hicks, S.L.; Boss, J.M. Antibody-secreting cell destiny emerges during the initial stages of B-cell activation. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Stebegg, M.; Kumar, S.D.; Silva-Cayetano, A.; Fonseca, V.R.; Linterman, M.A.; Graca, L. Regulation of the Germinal Center Response. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- De Silva, N.S.; Klein, U. Dynamics of B cells in germinal centres. Nat. Rev. Immunol. 2015, 15, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Béguelin, W.; Popovic, R.; Teater, M.; Jiang, Y.; Bunting, K.L.; Rosen, M.; Shen, H.; Yang, S.N.; Wang, L.; Ezponda, T.; et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 2013, 23, 677–692. [Google Scholar] [CrossRef] [Green Version]
- Velichutina, I.; Shaknovich, R.; Geng, H.; Johnson, N.A.; Gascoyne, R.D.; Melnick, A.M.; Elemento, O. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood 2010, 116, 5247–5255. [Google Scholar] [CrossRef] [Green Version]
- Van Galen, J.C.; Dukers, D.F.; Giroth, C.; Sewalt, R.G.A.B.; Otte, A.P.; Meijer, C.J.L.M.; Raaphorst, F.M. Distinct expression patterns of polycomb oncoproteins and their binding partners during the germinal center reaction. Eur. J. Immunol. 2004, 34, 1870–1881. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Kleine-Kohlbrecher, D.; Dietrich, N.; Pasini, D.; Gargiulo, G.; Beekman, C.; Theilgaard-Mönch, K.; Minucci, S.; Porse, B.T.; Marine, J.-C.; et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007, 21, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.-K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef]
- Yu, K.; Lieber, M.R. Current insights into the mechanism of mammalian immunoglobulin class switch recombination. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Bransteitter, R.; Pham, P.; Scharff, M.D.; Goodman, M.F. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc. Natl. Acad. Sci. USA 2003, 100, 4102–4107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhuri, J.; Tian, M.; Khuong, C.; Chua, K.; Pinaud, E.; Alt, F.W. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature 2003, 422, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Sohail, A.; Klapacz, J.; Samaranayake, M.; Ullah, A.; Bhagwat, A.S. Human activation-induced cytidine deaminase causes transcription-dependent, strand-biased C to U deaminations. Nucleic Acids Res. 2003, 31, 2990–2994. [Google Scholar] [CrossRef] [Green Version]
- Ramiro, A.R.; Stavropoulos, P.; Jankovic, M.; Nussenzweig, M.C. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat. Immunol. 2003, 4, 452–456. [Google Scholar] [CrossRef]
- Raaphorst, F.M.; van Kemenade, F.J.; Fieret, E.; Hamer, K.M.; Satijn, D.P.E.; Otte, A.P.; Meijer, C.J.L.M. Cutting Edge: Polycomb Gene Expression Patterns Reflect Distinct B Cell Differentiation Stages in Human Germinal Centers. J. Immunol. 2000, 164, 1–4. [Google Scholar] [CrossRef]
- Kurosaki, T.; Kometani, K.; Ise, W. Memory B cells. Nat. Rev. Immunol. 2015, 15, 149–159. [Google Scholar] [CrossRef]
- Weisel, F.; Shlomchik, M. Memory B Cells of Mice and Humans. Annu. Rev. Immunol. 2017, 35, 255–284. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, M.; Caraux, A.; De Vos, J.; Fiol, G.; Larroque, M.; Cognot, C.; Bret, C.; Duperray, C.; Hose, D.; Klein, B. An in vitro model of differentiation of memory B cells into plasmablasts and plasma cells including detailed phenotypic and molecular characterization. Blood 2009, 114, 5173–5181. [Google Scholar] [CrossRef] [Green Version]
- Jourdan, M.; Caraux, A.; Caron, G.; Robert, N.; Fiol, G.; Rème, T.; Bolloré, K.; Vendrell, J.-P.; Gallou, S.L.; Mourcin, F.; et al. Characterization of a Transitional Preplasmablast Population in the Process of Human B Cell to Plasma Cell Differentiation. J. Immunol. 2011, 187, 3931–3941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung-Hagesteijn, C.; Erdmann, N.; Cheung, G.; Keats, J.J.; Stewart, A.K.; Reece, D.; Chung, K.C.; Tiedemann, R.E. Xbp1s-Negative Tumor B Cells and Pre-Plasmablasts Mediate Therapeutic Proteasome Inhibitor Resistance in Multiple Myeloma. Cancer Cell 2013, 24, 289–304. [Google Scholar] [CrossRef] [Green Version]
- Herviou, L.; Jourdan, M.; Martinez, A.-M.; Cavalli, G.; Moreaux, J. EZH2 is overexpressed in transitional preplasmablasts and is involved in human plasma cell differentiation. Leukemia 2019, 33, 2047–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowan, A.J.; Allen, C.; Barac, A.; Basaleem, H.; Bensenor, I.; Curado, M.P.; Foreman, K.; Gupta, R.; Harvey, J.; Hosgood, H.D.; et al. Global Burden of Multiple Myeloma. JAMA Oncol. 2018, 4, 1221–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuehl, W.M.; Bergsagel, P.L. Multiple myeloma: Evolving genetic events and host interactions. Nat. Rev. Cancer 2002, 2, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Barwick, B.G.; Gupta, V.A.; Vertino, P.M.; Boise, L.H. Cell of Origin and Genetic Alterations in the Pathogenesis of Multiple Myeloma. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.A.; Leone, P.E.; Chiecchio, L.; Dickens, N.J.; Jenner, M.W.; Boyd, K.D.; Johnson, D.C.; Gonzalez, D.; Dagrada, G.P.; Protheroe, R.K.M.; et al. A compendium of myeloma-associated chromosomal copy number abnormalities and their prognostic value. Blood 2010, 116, e56–e65. [Google Scholar] [CrossRef]
- Zhan, F.; Huang, Y.; Colla, S.; Stewart, J.P.; Hanamura, I.; Gupta, S.; Epstein, J.; Yaccoby, S.; Sawyer, J.; Burington, B.; et al. The molecular classification of multiple myeloma. Blood 2006, 108, 2020–2028. [Google Scholar] [CrossRef] [Green Version]
- Vikova, V.; Jourdan, M.; Robert, N.; Requirand, G.; Boireau, S.; Bruyer, A.; Vincent, L.; Cartron, G.; Klein, B.; Elemento, O.; et al. Comprehensive characterization of the mutational landscape in multiple myeloma cell lines reveals potential drivers and pathways associated with tumor progression and drug resistance. Theranostics 2019, 9, 540–553. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.A.; Wardell, C.P.; Chiecchio, L.; Smith, E.M.; Boyd, K.D.; Neri, A.; Davies, F.E.; Ross, F.M.; Morgan, G.J. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood 2011, 117, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.F.; Johnson, D.C.; Wu, P.; Walker, B.A.; Brioli, A.; Mirabella, F.; Wardell, C.P.; Melchor, L.; Davies, F.E.; Morgan, G.J. Global methylation analysis identifies prognostically important epigenetically inactivated tumor suppressor genes in multiple myeloma. Blood 2013, 122, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Ordoñez, R.; Kulis, M.; Russiñol, N.; Chapaprieta, V.; Carrasco-Leon, A.; García-Torre, B.; Charalampopoulou, S.; Clot, G.; Beekman, R.; Meydan, C.; et al. Chromatin activation as a unifying principle underlying pathogenic mechanisms in multiple myeloma. Genome Res. 2020, 30, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Agirre, X.; Castellano, G.; Pascual, M.; Heath, S.; Segura, V.; Bergmann, A.; Esteve, A.; Merkel, A.; Raineri, E.; Agueda, L.; et al. Whole-epigenome analysis in multiple myeloma reveals DNA hypermethylation of B cell-specific enhancers. Genome Res. 2015, 34, 478–487. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Chen, K.; De Paepe, A.; Hellqvist, E.; Krstic, A.D.; Metang, L.; Gustafsson, C.; Davis, R.E.; Levy, Y.M.; Surapaneni, R.; et al. Active enhancer and chromatin accessibility landscapes chart the regulatory network of primary multiple myeloma. Blood 2018, 131, 2138–2150. [Google Scholar] [CrossRef] [Green Version]
- Alzrigat, M.; Párraga, A.A.; Jernberg-Wiklund, H. Epigenetics in multiple myeloma: From mechanisms to therapy. Semin. Cancer Biol. 2018, 51, 101–115. [Google Scholar] [CrossRef]
- De Smedt, E.; Lui, H.; Maes, K.; De Veirman, K.; Menu, E.; Vanderkerken, K.; De Bruyne, E. The Epigenome in Multiple Myeloma: Impact on Tumor Cell Plasticity and Drug Response. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef]
- Dupéré-Richer, D.; Licht, J.D. Epigenetic Regulatory Mutations and Epigenetic Therapy for Multiple Myeloma. Curr. Opin. Hematol. 2017, 24, 336–344. [Google Scholar] [CrossRef]
- Kalushkova, A.; Fryknäs, M.; Lemaire, M.; Fristedt, C.; Agarwal, P.; Eriksson, M.; Deleu, S.; Atadja, P.; Österborg, A.; Nilsson, K.; et al. Polycomb Target Genes Are Silenced in Multiple Myeloma. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Alzrigat, M.; Párraga, A.A.; Enroth, S.; Singh, U.; Ungerstedt, J.; Österborg, A.; Brown, P.J.; Ma, A.; Jin, J.; et al. Genome-wide profiling of histone H3 lysine 27 and lysine 4 trimethylation in multiple myeloma reveals the importance of Polycomb gene targeting and highlights EZH2 as a potential therapeutic target. Oncotarget 2016, 7, 6809–6823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzrigat, M.; Párraga, A.A.; Agarwal, P.; Zureigat, H.; Österborg, A.; Nahi, H.; Ma, A.; Jin, J.; Nilsson, K.; Öberg, F.; et al. EZH2 inhibition in multiple myeloma downregulates myeloma associated oncogenes and upregulates microRNAs with potential tumor suppressor functions. Oncotarget 2016, 8, 10213–10224. [Google Scholar] [CrossRef] [Green Version]
- Rizq, O.; Mimura, N.; Oshima, M.; Saraya, A.; Koide, S.; Kato, Y.; Aoyama, K.; Nakajima-Takagi, Y.; Wang, C.; Chiba, T.; et al. Dual Inhibition of EZH2 and EZH1 Sensitizes PRC2-Dependent Tumors to Proteasome Inhibition. Clin. Cancer Res. 2017, 23, 4817–4830. [Google Scholar] [CrossRef] [Green Version]
- Popovic, R.; Martinez-Garcia, E.; Giannopoulou, E.G.; Zhang, Q.; Zhang, Q.; Ezponda, T.; Shah, M.Y.; Zheng, Y.; Will, C.M.; Small, E.C.; et al. Histone Methyltransferase MMSET/NSD2 Alters EZH2 Binding and Reprograms the Myeloma Epigenome through Global and Focal Changes in H3K36 and H3K27 Methylation. PLoS Genet. 2014, 10. [Google Scholar] [CrossRef]
- Ezponda, T.; Dupéré-Richer, D.; Will, C.M.; Small, E.C.; Varghese, N.; Patel, T.; Nabet, B.; Popovic, R.; Oyer, J.; Bulic, M.; et al. UTX/KDM6A Loss Enhances the Malignant Phenotype of Multiple Myeloma and Sensitizes Cells to EZH2 inhibition. Cell Rep. 2017, 21, 628–640. [Google Scholar] [CrossRef] [Green Version]
- Zeng, D.; Liu, M.; Pan, J. Blocking EZH2 methylation transferase activity by GSK126 decreases stem cell-like myeloma cells. Oncotarget 2016, 8, 3396–3411. [Google Scholar] [CrossRef] [Green Version]
- Harding, T.; Swanson, J.; Van Ness, B. EZH2 inhibitors sensitize myeloma cell lines to panobinostat resulting in unique combinatorial transcriptomic changes. Oncotarget 2018, 9, 21930–21942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, T.A.; Winter, J.N.; Giulino-Roth, L.; Longley, J.; Lopez, J.; Michot, J.-M.; Leonard, J.P.; Ribrag, V.; McCabe, M.T.; Creasy, C.L.; et al. Phase I Study of the Novel Enhancer of Zeste Homolog 2 (EZH2) Inhibitor GSK2816126 in Patients with Advanced Hematologic and Solid Tumors. Clin. Cancer Res. 2019, 25, 7331–7339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernando, H.; Gelato, K.A.; Lesche, R.; Beckmann, G.; Koehr, S.; Otto, S.; Steigemann, P.; Stresemann, C. EZH2 Inhibition Blocks Multiple Myeloma Cell Growth through Upregulation of Epithelial Tumor Suppressor Genes. Mol. Cancer 2016, 15, 287–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herviou, L.; Kassambara, A.; Boireau, S.; Robert, N.; Requirand, G.; Müller-Tidow, C.; Vincent, L.; Seckinger, A.; Goldschmidt, H.; Cartron, G.; et al. PRC2 targeting is a therapeutic strategy for EZ score defined high-risk multiple myeloma patients and overcome resistance to IMiDs. Clin. Epigenetics 2018, 10. [Google Scholar] [CrossRef]
- Kurmasheva, R.T.; Sammons, M.; Favours, E.; Wu, J.; Kurmashev, D.; Cosmopoulos, K.; Keilhack, H.; Klaus, C.R.; Houghton, P.J.; Smith, M.A. Initial Testing (Stage 1) of Tazemetostat (EPZ-6438), a Novel EZH2 Inhibitor, by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2017, 64. [Google Scholar] [CrossRef] [PubMed]
- Pawlyn, C.; Bright, M.D.; Buros, A.F.; Stein, C.K.; Walters, Z.; Aronson, L.I.; Mirabella, F.; Jones, J.R.; Kaiser, M.F.; Walker, B.A.; et al. Overexpression of EZH2 in multiple myeloma is associated with poor prognosis and dysregulation of cell cycle control. Blood Cancer J. 2017, 7, e549. [Google Scholar] [CrossRef]
- Honma, D.; Kanno, O.; Watanabe, J.; Kinoshita, J.; Hirasawa, M.; Nosaka, E.; Shiroishi, M.; Takizawa, T.; Yasumatsu, I.; Horiuchi, T.; et al. Novel orally bioavailable EZH1/2 dual inhibitors with greater antitumor efficacy than an EZH2 selective inhibitor. Cancer Sci. 2017, 108, 2069–2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzrigat, M.; Jernberg-Wiklund, H.; Licht, J.D. Targeting EZH2 in Multiple Myeloma—Multifaceted Anti-Tumor Activity. Epigenomes 2018, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croonquist, P.A.; Linden, M.A.; Zhao, F.; Van Ness, B.G. Gene profiling of a myeloma cell line reveals similarities and unique signatures among IL-6 response, N-ras-activating mutations, and coculture with bone marrow stromal cells. Blood 2003, 102, 2581–2592. [Google Scholar] [CrossRef] [Green Version]
- Croonquist, P.A.; Van Ness, B. The polycomb group protein enhancer of zeste homolog 2 (EZH2) is an oncogene that influences myeloma cell growth and the mutant ras phenotype. Oncogene 2005, 24, 6269–6280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neo, W.H.; Lim, J.F.; Grumont, R.; Gerondakis, S.; Su, I. c-Rel Regulates Ezh2 Expression in Activated Lymphocytes and Malignant Lymphoid Cells. J. Biol. Chem. 2014, 289, 31693–31707. [Google Scholar] [CrossRef] [Green Version]
- Rizk, M.; Rizq, O.; Oshima, M.; Nakajima-Takagi, Y.; Koide, S.; Saraya, A.; Isshiki, Y.; Chiba, T.; Yamazaki, S.; Ma, A.; et al. Akt inhibition synergizes with polycomb repressive complex 2 inhibition in the treatment of multiple myeloma. Cancer Sci. 2019, 110, 3695–3707. [Google Scholar] [CrossRef] [Green Version]
- Pichiorri, F.; Suh, S.-S.; Ladetto, M.; Kuehl, M.; Palumbo, T.; Drandi, D.; Taccioli, C.; Zanesi, N.; Alder, H.; Hagan, J.P.; et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 12885–12890. [Google Scholar] [CrossRef] [Green Version]
- Seckinger, A.; MeiΔner, T.; Moreaux, J.; Benes, V.; Hillengass, J.; Castoldi, M.; Zimmermann, J.; Ho, A.D.; Jauch, A.; Goldschmidt, H.; et al. miRNAs in multiple myeloma—A survival relevant complex regulator of gene expression. Oncotarget 2015, 6, 39165–39183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rastgoo, N.; Pourabdollah, M.; Abdi, J.; Reece, D.; Chang, H. Dysregulation of EZH2/miR-138 axis contributes to drug resistance in multiple myeloma by downregulating RBPMS. Leukemia 2018, 32, 2471–2482. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Kikuchi, J. Epigenetic mechanisms of cell adhesion-mediated drug resistance in multiple myeloma. Int. J. Hematol. 2016, 104, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Zhan, F.; Hardin, J.; Kordsmeier, B.; Bumm, K.; Zheng, M.; Tian, E.; Sanderson, R.; Yang, Y.; Wilson, C.; Zangari, M.; et al. Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells. Blood 2002, 99, 1745–1757. [Google Scholar] [CrossRef]
- Zhan, F.; Tian, E.; Bumm, K.; Smith, R.; Barlogie, B.; Shaughnessy, J. Gene expression profiling of human plasma cell differentiation and classification of multiple myeloma based on similarities to distinct stages of late-stage B-cell development. Blood 2003, 101, 1128–1140. [Google Scholar] [CrossRef]
- Nakagawa, M.; Fujita, S.; Katsumoto, T.; Yamagata, K.; Ogawara, Y.; Hattori, A.; Kagiyama, Y.; Honma, D.; Araki, K.; Inoue, T.; et al. Dual inhibition of enhancer of zeste homolog 1/2 overactivates WNT signaling to deplete cancer stem cells in multiple myeloma. Cancer Sci. 2019, 110, 194–208. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Chen, Y.; Li, R.; Liu, Y.; Wen, L.; Zhang, C. Triptolide alters histone H3K9 and H3K27 methylation state and induces G0/G1 arrest and caspase-dependent apoptosis in multiple myeloma in vitro. Toxicology 2010, 267, 70–79. [Google Scholar] [CrossRef]
- Martinez-Garcia, E.; Popovic, R.; Min, D.-J.; Sweet, S.M.M.; Thomas, P.M.; Zamdborg, L.; Heffner, A.; Will, C.; Lamy, L.; Staudt, L.M.; et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood 2011, 117, 211–220. [Google Scholar] [CrossRef]
- Kuo, A.J.; Cheung, P.; Chen, K.; Zee, B.M.; Kioi, M.; Lauring, J.; Xi, Y.; Park, B.H.; Shi, X.; Garcia, B.A.; et al. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol. Cell 2011, 44, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Alzrigat, M.; Jernberg-Wiklund, H. The miR-125a and miR-320c are potential tumor suppressor microRNAs epigenetically silenced by the polycomb repressive complex 2 in multiple myeloma. RNA Dis. 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Viré, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.-M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef]
- Fujita, S.; Honma, D.; Adachi, N.; Araki, K.; Takamatsu, E.; Katsumoto, T.; Yamagata, K.; Akashi, K.; Aoyama, K.; Iwama, A.; et al. Dual inhibition of EZH1/2 breaks the quiescence of leukemia stem cells in acute myeloid leukemia. Leukemia 2018, 32, 855–864. [Google Scholar] [CrossRef]
- Ren, Z.; Ahn, J.H.; Liu, H.; Tsai, Y.-H.; Bhanu, N.V.; Koss, B.; Allison, D.F.; Ma, A.; Storey, A.J.; Wang, P.; et al. PHF19 promotes multiple myeloma tumorigenicity through PRC2 activation and broad H3K27me3 domain formation. Blood 2019, 134, 1176–1189. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Du, C.; Ma, X.; Sui, W.; Yu, Z.; Liu, L.; Zhao, L.; Li, Z.; Xu, J.; Wei, X.; et al. Polycomb-like Protein 3 Induces Proliferation and Drug Resistance in Multiple Myeloma and Is Regulated by miRNA-15a. Mol Cancer Res. 2020, 18, 1063–1073. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, J.S.; Li, N.; Weinhold, N.; Försti, A.; Ali, M.; van Duin, M.; Thorleifsson, G.; Johnson, D.C.; Chen, B.; Halvarsson, B.-M.; et al. Genome-wide association study identifies multiple susceptibility loci for multiple myeloma. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Bolomsky, A.; Schlangen, K.; Schreiner, W.; Zojer, N.; Ludwig, H. Targeting of BMI-1 with PTC-209 shows potent anti-myeloma activity and impairs the tumour microenvironment. J. Hematol. Oncol. 2016, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzrigat, M.; Párraga, A.A.; Majumder, M.M.; Ma, A.; Jin, J.; Österborg, A.; Nahi, H.; Nilsson, K.; Heckman, C.A.; Öberg, F.; et al. The polycomb group protein BMI-1 inhibitor PTC-209 is a potent anti-myeloma agent alone or in combination with epigenetic inhibitors targeting EZH2 and the BET bromodomains. Oncotarget 2017, 8, 103731–103743. [Google Scholar] [CrossRef] [Green Version]
- Bolomsky, A.; Muller, J.; Stangelberger, K.; Lejeune, M.; Duray, E.; Breid, H.; Vrancken, L.; Pfeiffer, C.; Hübl, W.; Willheim, M.; et al. The anti-mitotic agents PTC-028 and PTC596 display potent activity in pre-clinical models of multiple myeloma but challenge the role of BMI-1 as an essential tumour gene. Br. J. Haematol. 2020. [Google Scholar] [CrossRef]
- Bartucci, M.; Hussein, M.S.; Huselid, E.; Flaherty, K.; Patrizii, M.; Laddha, S.V.; Kui, C.; Bigos, R.A.; Gilleran, J.A.; El Ansary, M.M.S.; et al. Synthesis and Characterization of Novel BMI1 Inhibitors Targeting Cellular Self-Renewal in Hepatocellular Carcinoma. Target. Oncol. 2017, 12, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.J.L.; Scheijen, B.; Voncken, J.-W.; Kieboom, K.; Berns, A.; van Lohuizen, M. Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes Dev. 1999, 13, 2678–2690. [Google Scholar] [CrossRef] [PubMed]
- De Vos, J.; Thykjær, T.; Tarte, K.; Ensslen, M.; Raynaud, P.; Requirand, G.; Pellet, F.; Pantesco, V.; Rème, T.; Jourdan, M.; et al. Comparison of gene expression profiling between malignant and normal plasma cells with oligonucleotide arrays. Oncogene 2002, 21, 6848–6857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chng, W.J.; Kumar, S.; VanWier, S.; Ahmann, G.; Price-Troska, T.; Henderson, K.; Chung, T.-H.; Kim, S.; Mulligan, G.; Bryant, B.; et al. Molecular Dissection of Hyperdiploid Multiple Myeloma by Gene Expression Profiling. Cancer Res. 2007, 67, 2982–2989. [Google Scholar] [CrossRef] [Green Version]
- Zhan, F.; Barlogie, B.; Arzoumanian, V.; Huang, Y.; Williams, D.R.; Hollmig, K.; Pineda-Roman, M.; Tricot, G.; van Rhee, F.; Zangari, M.; et al. Gene-expression signature of benign monoclonal gammopathy evident in multiple myeloma is linked to good prognosis. Blood 2007, 109, 1692–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.-J.; Datta, S.; Band, V.; Dimri, G.P. Mel-18, a Polycomb Group Protein, Regulates Cell Proliferation and Senescence via Transcriptional Repression of Bmi-1 and c-Myc Oncoproteins. MBoC 2006, 18, 536–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.-H.; Dimri, M.; Dimri, G.P. A Positive Feedback Loop Regulates the Expression of Polycomb Group Protein BMI1 via WNT Signaling Pathway. J. Biol. Chem. 2013, 288, 3406–3418. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.-Q.; Niu, W.-Y.; Li, Y.-P.; Huang, H.-B.; Zhan, R. miR-203 inhibits cell growth and regulates G1/S transition by targeting Bmi-1 in myeloma cells. Mol. Med. Rep. 2016, 14, 4795–4801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, K.-Y.; Liang, R.; So, C.-C.; Jin, D.-Y.; Costello, J.F.; Chim, C.-S. Epigenetic silencing of MIR203 in multiple myeloma. Br. J. Haematol. 2011, 154, 569–578. [Google Scholar] [CrossRef]
- Jagani, Z.; Wiederschain, D.; Loo, A.; He, D.; Mosher, R.; Fordjour, P.; Monahan, J.; Morrissey, M.; Yao, Y.-M.; Lengauer, C.; et al. The Polycomb Group Protein Bmi-1 Is Essential for the Growth of Multiple Myeloma Cells. Cancer Res. 2010, 70, 5528–5538. [Google Scholar] [CrossRef] [Green Version]
- Tagde, A.; Markert, T.; Rajabi, H.; Hiraki, M.; Alam, M.; Bouillez, A.; Avigan, D.; Anderson, K.; Kufe, D. Targeting MUC1-C suppresses polycomb repressive complex 1 in multiple myeloma. Oncotarget 2017, 8, 69237–69249. [Google Scholar] [CrossRef] [Green Version]
- Treon, S.P.; Mollick, J.A.; Urashima, M.; Teoh, G.; Chauhan, D.; Ogata, A.; Raje, N.; Hilgers, J.H.; Nadler, L.; Belch, A.R.; et al. Muc-1 core protein is expressed on multiple myeloma cells and is induced by dexamethasone. Blood 1999, 93, 1287–1298. [Google Scholar] [CrossRef]
- Kawano, T.; Ahmad, R.; Nogi, H.; Agata, N.; Anderson, K.; Kufe, D. MUC1 oncoprotein promotes growth and survival of human multiple myeloma cells. Int. J. Oncol. 2008, 33, 153–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MUC1 (EMA) expressing plasma cells in bone marrow infiltrated by plasma cell myeloma. Histol. Histopathol. 2007, 889–893. [CrossRef]
- Huang, L.; Ren, J. MUC1 Cytoplasmic Domain Coactivates Wnt Target Gene Transcription and Confers Transformation. Cancer Biol. Ther. 2003, 2, 700–704. [Google Scholar] [CrossRef]
- Rajabi, H.; Ahmad, R.; Jin, C.; Kosugi, M.; Alam, M.; Joshi, M.D.; Kufe, D. MUC1-C Oncoprotein Induces TCF7L2 Transcription Factor Activation and Promotes Cyclin D1 Expression in Human Breast Cancer Cells. J. Biol. Chem. 2012, 287, 10703–10713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagde, A.; Rajabi, H.; Bouillez, A.; Alam, M.; Gali, R.; Bailey, S.; Tai, Y.-T.; Hideshima, T.; Anderson, K.; Avigan, D.; et al. MUC1-C drives MYC in multiple myeloma. Blood 2016, 127, 2587–2597. [Google Scholar] [CrossRef] [Green Version]
- Hiraki, M.; Maeda, T.; Bouillez, A.; Alam, M.; Tagde, A.; Hinohara, K.; Suzuki, Y.; Markert, T.; Miyo, M.; Komura, K.; et al. MUC1-C Activates BMI1 in human cancer cells. Oncogene 2017, 36, 2791–2801. [Google Scholar] [CrossRef] [Green Version]
- Polakis, P. Wnt signaling and cancer. Genes Dev. 2000, 14, 1837–1851. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, A. Tumor formation by genetic mutations in the components of the Wnt signaling pathway. Cancer Sci. 2003, 94, 225–229. [Google Scholar] [CrossRef]
- Le, P.; McDermott, J.D.; Jimeno, A. Targeting the Wnt pathway in human cancers: Therapeutic targeting with a focus on OMP-54F28. Pharmacol. Ther. 2015, 146, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Gavriatopoulou, M.; Dimopoulos, M.A. Pathogenesis of bone disease in multiple myeloma: From bench to bedside. Blood Cancer J. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Abe, M.; Hiasa, M.; Oda, A.; Amou, H.; Nakano, A.; Takeuchi, K.; Kitazoe, K.; Kido, S.; Inoue, D.; et al. Myeloma Cell-Osteoclast Interaction Enhances Angiogenesis Together with Bone Resorption: A Role for Vascular Endothelial Cell Growth Factor and Osteopontin. Clin. Cancer Res. 2007, 13, 816–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, J.; Takeshita, S.; Barker, J.E.; Kanagawa, O.; Ross, F.P.; Teitelbaum, S.L. TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J. Clin. Invest. 2000, 106, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Politou, M.; Viniou, N.; Rahemtulla, A. Significance of macrophage inflammatory protein-1 alpha (MIP-1α) in multiple myeloma. Leuk. Lymphoma 2005, 46, 1699–1707. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Shimizu, N.; Dallas, M.; Niewolna, M.; Story, B.; Williams, P.J.; Mundy, G.R.; Yoneda, T. Anti-α4 integrin antibody suppresses the development of multiple myeloma and associated osteoclastic osteolysis. Blood 2004, 104, 2149–2154. [Google Scholar] [CrossRef]
- Vanderkerken, K.; Medicherla, S.; Coulton, L.; Raeve, H.D.; Willems, A.; Lawson, M.; Camp, B.V.; Protter, A.A.; Higgins, L.S.; Menu, E.; et al. Inhibition of p38α Mitogen-Activated Protein Kinase Prevents the Development of Osteolytic Bone Disease, Reduces Tumor Burden, and Increases Survival in Murine Models of Multiple Myeloma. Cancer Res. 2007, 67, 4572–4577. [Google Scholar] [CrossRef] [Green Version]
- Giuliani, N.; Colla, S.; Sala, R.; Moroni, M.; Lazzaretti, M.; La Monica, S.; Bonomini, S.; Hojden, M.; Sammarelli, G.; Barillè, S.; et al. Human myeloma cells stimulate the receptor activator of nuclear factor-κB ligand (RANKL) in T lymphocytes: A potential role in multiple myeloma bone disease. Blood 2002, 100, 4615–4621. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Calle, J.; Anderson, J.; Cregor, M.D.; Hiasa, M.; Chirgwin, J.M.; Carlesso, N.; Yoneda, T.; Mohammad, K.S.; Plotkin, L.I.; Roodman, G.D.; et al. Bidirectional Notch signaling and osteocyte-derived factors in the bone marrow microenvironment promote tumor cell proliferation and bone destruction in multiple myeloma. Cancer Res. 2016, 76, 1089–1100. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.E.; Ward, C.; Rathour, L.; Galasko, C.B. Myeloma affects both the growth and function of human osteoblast-like cells. Clin. Exp. Metast. 1992, 10, 33–38. [Google Scholar] [CrossRef]
- Terpos, E.; Berenson, J.; Raje, N.; Roodman, G.D. Management of bone disease in multiple myeloma. Expert Rev. Hematol. 2014, 7, 113–125. [Google Scholar] [CrossRef]
- Terpos, E.; Christoulas, D.; Gavriatopoulou, M.; Dimopoulos, M.A. Mechanisms of bone destruction in multiple myeloma. Eur. J. Cancer Care 2017, 26, e12761. [Google Scholar] [CrossRef]
- Adamik, J.; Jin, S.; Sun, Q.; Zhang, P.; Weiss, K.R.; Anderson, J.L.; Silbermann, R.; Roodman, G.D.; Galson, D.L. EZH2 or HDAC1 Inhibition Reverses Multiple Myeloma-Induced Epigenetic Suppression of Osteoblast Differentiation. Mol. Cancer Res. 2017, 15, 405–417. [Google Scholar] [CrossRef] [Green Version]
- Hemming, S.; Cakouros, D.; Vandyke, K.; Davis, M.J.; Zannettino, A.C.W.; Gronthos, S. Identification of Novel EZH2 Targets Regulating Osteogenic Differentiation in Mesenchymal Stem Cells. Stem Cells Dev. 2016, 25, 909–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemming, S.; Cakouros, D.; Codrington, J.; Vandyke, K.; Arthur, A.; Zannettino, A.; Gronthos, S. EZH2 deletion in early mesenchyme compromises postnatal bone microarchitecture and structural integrity and accelerates remodeling. FASEB J. 2017, 31, 1011–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakhri, B.; Vij, R. Clonal Evolution in Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2016, 16, S130–S134. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Chesi, M.; Egan, J.B.; Garbitt, V.M.; Palmer, S.E.; Braggio, E.; Van Wier, S.; Blackburn, P.R.; Baker, A.S.; Dispenzieri, A.; et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012, 120, 1067–1076. [Google Scholar] [CrossRef]
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.-V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Primers 2017, 3, 17046. [Google Scholar] [CrossRef]
- Wu, S.-Q.; Xu, Z.-Z.; Niu, W.-Y.; Huang, H.-B.; Zhan, R. shRNA-mediated Bmi-1 silencing sensitizes multiple myeloma cells to bortezomib. Int. J. Mol. Med. 2014, 34, 616–623. [Google Scholar] [CrossRef] [Green Version]
- Bjorklund, C.C.; Lu, L.; Kang, J.; Hagner, P.R.; Havens, C.G.; Amatangelo, M.; Wang, M.; Ren, Y.; Couto, S.; Breider, M.; et al. Rate of CRL4CRBN substrate Ikaros and Aiolos degradation underlies differential activity of lenalidomide and pomalidomide in multiple myeloma cells by regulation of c-Myc and IRF4. Blood Cancer J. 2015, 5, e354. [Google Scholar] [CrossRef]
- Guirguis, A.A.; Ebert, B.L. Lenalidomide: Deciphering mechanisms of action in myeloma, myelodysplastic syndrome and beyond. Curr. Opin. Cell Biol. 2015, 37, 61–67. [Google Scholar] [CrossRef]
- Dimopoulos, K.; Søgaard Helbo, A.; Fibiger Munch-Petersen, H.; Sjö, L.; Christensen, J.; Sommer Kristensen, L.; Asmar, F.; Hermansen, N.E.U.; O’Connel, C.; Gimsing, P.; et al. Dual inhibition of DNMTs and EZH2 can overcome both intrinsic and acquired resistance of myeloma cells to IMiDs in a cereblon-independent manner. Mol. Oncol. 2018, 12, 180–195. [Google Scholar] [CrossRef]
- Shaffer, A.L.; Lin, K.-I.; Kuo, T.C.; Yu, X.; Hurt, E.M.; Rosenwald, A.; Giltnane, J.M.; Yang, L.; Zhao, H.; Calame, K.; et al. Blimp-1 Orchestrates Plasma Cell Differentiation by Extinguishing the Mature B Cell Gene Expression Program. Immunity 2002, 17, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Chan, H.L.; Morey, L. Emerging Roles for Polycomb-Group Proteins in Stem Cells and Cancer. Trends Biochem. Sci. 2019, 44, 688–700. [Google Scholar] [CrossRef]
- Loubière, V.; Delest, A.; Thomas, A.; Bonev, B.; Schuettengruber, B.; Sati, S.; Martinez, A.-M.; Cavalli, G. Coordinate redeployment of PRC1 proteins suppresses tumor formation during Drosophila development. Nat. Genet. 2016, 48, 1436–1442. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varlet, E.; Ovejero, S.; Martinez, A.-M.; Cavalli, G.; Moreaux, J. Role of Polycomb Complexes in Normal and Malignant Plasma Cells. Int. J. Mol. Sci. 2020, 21, 8047. https://doi.org/10.3390/ijms21218047
Varlet E, Ovejero S, Martinez A-M, Cavalli G, Moreaux J. Role of Polycomb Complexes in Normal and Malignant Plasma Cells. International Journal of Molecular Sciences. 2020; 21(21):8047. https://doi.org/10.3390/ijms21218047
Chicago/Turabian StyleVarlet, Emmanuel, Sara Ovejero, Anne-Marie Martinez, Giacomo Cavalli, and Jerome Moreaux. 2020. "Role of Polycomb Complexes in Normal and Malignant Plasma Cells" International Journal of Molecular Sciences 21, no. 21: 8047. https://doi.org/10.3390/ijms21218047
APA StyleVarlet, E., Ovejero, S., Martinez, A.-M., Cavalli, G., & Moreaux, J. (2020). Role of Polycomb Complexes in Normal and Malignant Plasma Cells. International Journal of Molecular Sciences, 21(21), 8047. https://doi.org/10.3390/ijms21218047