Renin–Angiotensin System: An Important Player in the Pathogenesis of Acute Respiratory Distress Syndrome


Abstract
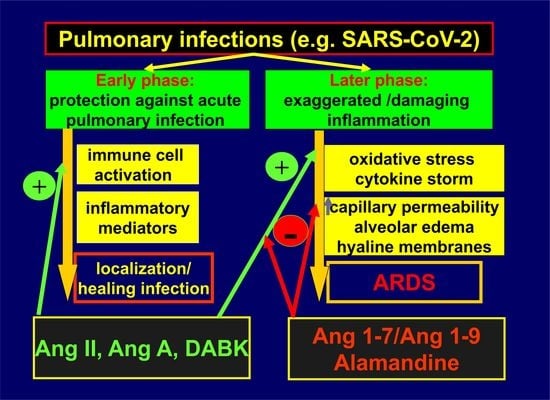
1. Introduction
2. RAS and Pulmonary Diseases
3. Regulatory, Rather than Harmful or Protective
4. ACE2: Conductor of Inflammatory Response in the Lungs
5. Interactions with Infectious Agents
6. Pharmacological Inhibition of ACE and AT1R in Pulmonary Diseases
7. ACEIs and ARBs in COVID-19
8. Other Non-Classical RAS Components
9. Perspectives for Drug Development
10. Conclusions
- —
- First, the roles of membrane-bound and soluble ACE2 in the respiratory tract as well as the biological significance of ACE2-shedding under physiological and pathological conditions should be elucidated.
- —
- The interaction mechanisms of particular infectious agents with the RAS, especially ACE2, are of importance for specific therapeutic approaches.
- —
- Besides ACE2, other non-classical components of the RAS could also play a significant role in the pathogenesis of acute lung injury and ARDS.
- —
- The biological impact of established RAS modulating therapies (ACEIs or ARBs) on RAS molecules should be disclosed in ongoing clinical trials.
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| α-SMA | α-smooth muscle actin |
| ACE | angiotensin converting enzyme |
| ACEIs | angiotensin converting enzyme inhibitors |
| Ac-SDKP | N-acetyl-seryl-aspartyl-lysyl-proline |
| ADAM17 | tumor necrosis factor-α-converting enzyme (TACE) |
| ALI | acute lung injury |
| Ang | angiotensin |
| APA | aminopeptidase A |
| APM | aminopeptidase M |
| ARBs | angiotensin II type 1 receptor blockers |
| ARDS | acute respiratory distress syndrome |
| AT1R | angiotensin II type 1 receptor |
| AT2R | angiotensin II type 2 receptor |
| BAL | bronchoalveolar lavage |
| BLM | bleomycin |
| CATA | cathepsin A |
| CINC-3 | cytokine-induced neutrophil chemoattractant 3 |
| COPD | chronic obstructive pulmonary disease |
| CxA | carboxypeptidase A |
| CXCL5 | C-X-C motif chemokine 5 |
| DABK | des-Arg9-bradykinin |
| DOCA | deoxycorticosterone acetate |
| FiO2 | fraction of inspired oxygen |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| HD | high dose |
| ICU | intensive care unit |
| IL | interleukin |
| i.p. | intraperitoneally |
| i.v. | intravenously |
| KC | C-X-C motif chemokine 1 |
| LD | low dose |
| LPS | lipopolysaccharide |
| mACE2 | membrane-bound angiotensin converting enzyme 2 |
| MasR | Mas receptor |
| MCP-1 | monocyte chemoattractant protein-1 |
| MIP2 | macrophage inflammatory protein-2 |
| MLDAD | mononuclear leukocyte-derived aspartate decarboxylase |
| MrgD | Mas-Related G-Protein Coupled Receptor D |
| NEP | neprilysin |
| PaCO2 | partial pressure of carbon dioxide |
| PaO2 | partial pressure of oxygen |
| POP | propyl oligopeptidase |
| PKC-δ | protein kinase C-δ |
| RAS | renin–angiotensin system |
| rhACE2 | recombinant human angiotensin converting enzyme 2 |
| sACE2 | soluble angiotensin converting enzyme 2 |
| s.c. | subcutaneously |
| T2D | type 2 diabetes mellitus |
| TACE | tumor necrosis factor-α-converting enzyme (ADAM17) |
| TGF-β1 | transforming growth factor β1 |
| TLR | toll like receptor |
| TMPRSS2 | transmembrane protease serine 2 |
| TNF-α | tumor necrosis factor-α |
| WBC | white blood cells |
References
- ARDS Definition Task Force; Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S. Acute respiratory distress syndrome: The Berlin Definition. JAMA 2012, 307, 2526–2533. [Google Scholar] [CrossRef]
- Maca, J.; Jor, O.; Holub, M.; Sklienka, P.; Burša, F.; Burda, M.; Janout, V.; Ševčík, P. Past and Present ARDS Mortality Rates: A Systematic Review. Respir. Care 2017, 62, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Gajic, O. Ventilator settings as a risk factor for acute respiratory distress syndrome in mechanically ventilated patients. Intensive Care Med. 2005, 31, 922–926. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Chai, X.Q.; Magnussen, C.G.; Zosky, G.R.; Shu, S.H.; Wie, X.; Hu, S.S. Renin-angiotensin-system, a potential pharmacological candidate, in acute respiratory distress syndrome during mechanical ventilation. Pulm. Pharmacol. Ther. 2019, 58, 101833. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.; Rubenfeld, G.D. Fifty years of research in ARDS. The epidemiology of acute respiratory distress syndrome. A 50th birthday review. Am. J. Respir. Crit. Care Med. 2017, 195, 860–870. [Google Scholar] [CrossRef]
- Kaku, S.; Nguyen, C.D.; Htet, N.N.; Tutera, D.; Barr, J.; Paintal, H.S.; Kuschner, W.G. Acute respiratory distress syndrome: Etiology, pathogenesis, and summary on management. J. Intensive Care Med. 2019, 35, 723–737. [Google Scholar] [CrossRef]
- Pierrakos, C.; Karanikolas, M.; Scolletta, S.; Karamouzos, V.; Velissaris, D. Acute respiratory distress syndrome: Pathophysiology and therapeutic options. J. Clin. Med. Res. 2012, 4, 7–16. [Google Scholar] [CrossRef]
- Santos, R.A.; Oudit, G.Y.; Verano-Braga, T.; Canta, G.; Steckelings, U.M.; Bader, M. The renin-angiotensin system: Going beyond the classical paradigms. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H958–H970. [Google Scholar] [CrossRef]
- Wu, C.H.; Mohammadmoradi, S.; Chen, J.Z.; Sawada, H.; Daugherty, A.; Lu, H.S. Renin-Angiotensin System and Cardiovascular Functions. Arterioscler. Thromb. Vasc. Biol. 2018, 38, e108–e116. [Google Scholar] [CrossRef]
- Hrenak, J.; Paulis, L.; Simko, F. N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP): Potential target molecule in research of heart, kidney and brain. Curr. Pharm. Des. 2015, 21, 5135–5143. [Google Scholar] [CrossRef]
- Hrenak, J.; Paulis, L.; Simko, F. Angiotensin A/Alamandine/MrgD Axis: Another Clue to Understanding Cardiovascular Pathophysiology. Int. J. Mol. Sci. 2016, 17, 1098. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Simko, J. Heart failure and angiotensin converting enzyme inhibition: Problems and perspectives. Physiol. Res. 1999, 48, 1–8. [Google Scholar] [PubMed]
- Simko, F.; Simko, J.; Fabryova, M. ACE-inhibition and angiotensin II receptor blockers in chronic heart failure: Pathophysiological consideration of the unresolved battle. Cardiovasc. Drugs Ther. 2003, 17, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Pechanova, O.; Repova-Bednarova, K.; Krajcirovicova, K.; Celec, P.; Kamodyova, N.; Zorad, S.; Kucharska, J.; Gvozdjakova, A.; Adamcova, M.; et al. Hypertension and cardiovascular remodelling in rats exposed to continuous light: Protection by ACE-inhibition and melatonin. Mediators Inflamm. 2014, 2014, 703175. [Google Scholar] [CrossRef]
- Simko, F.; Pechanova, O.; Krajcirovicova, K.; Matuskova, J.; Pelouch, V.; Adamcova, M.; Paulis, L. Effects of captopril, spironolactone, and simvastatin on the cardiovascular system of non-diseased Wistar rats. Int. J. Cardiol. 2015, 190, 128–130. [Google Scholar] [CrossRef]
- Patel, S.; Rauf, A.; Khan, H.; Abu-Izneid, T. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed. Pharmacother. 2017, 94, 317–325. [Google Scholar] [CrossRef]
- Mascolo, A.; Sessa, M.; Scavone, C.; De Angelis, A.; Vitale, C.; Berrino, L.; Rossi, F.; Rosano, G.; Capuano, A. New and old roles of the peripheral and brain renin-angiotensin-aldosterone system (RAAS): Focus on cardiovascular and neurological diseases. Int. J. Cardiol. 2017, 227, 734–742. [Google Scholar] [CrossRef]
- Jia, H. Pulmonary Angiotensin-Converting Enzyme 2 (ACE2) and Inflammatory Lung Disease. Shock 2016, 46, 239–248. [Google Scholar] [CrossRef]
- Simko, F.; Baka, T.; Krajcirovicova, K.; Repova, K.; Aziriova, S.; Zorad, S.; Poglitsch, M.; Adamcova, M.; Reiter, R.J.; Paulis, L. Effect of Melatonin on the Renin-Angiotensin-Aldosterone System in l-NAME-Induced Hypertension. Molecules 2018, 23, 265. [Google Scholar] [CrossRef]
- Steckelings, U.M.; Kloet, A.; Sumners, C. Centrally Mediated Cardiovascular Actions of the Angiotensin II Type 2 Receptor. Trends Endocrinol. Metab. 2017, 28, 684–693. [Google Scholar] [CrossRef]
- Paulis, L.; Foulquier, S.; Namsolleck, P.; Recarti, C.; Steckelings, U.M.; Unger, T. Combined Angiotensin Receptor Modulation in the Management of Cardio-Metabolic Disorders. Drugs 2016, 76, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef]
- Santos, R.A. Angiotensin-(1-7). Hypertension 2014, 63, 1138–1147. [Google Scholar] [CrossRef]
- Ocaranza, M.P.; Michea, L.; Chiong, M.; Lagos, C.F.; Lavandero, S.; Jalil, J.E. Recent insights and therapeutic perspectives of angiotensin-(1-9) in the cardiovascular system. Clin. Sci. 2014, 127, 549–557. [Google Scholar] [CrossRef]
- Hrenak, J.; Zorad, S.; Simko, F. Renin-angiotensin system and SARS-CoV-2 interaction: Underlying mechanisms and potential clinical implications. Gen. Physiol. Biophys. 2020, 39, 203–204. [Google Scholar] [CrossRef]
- Van de Veerdonk, F.L.; Netea, M.G.; van Deuren, M.; van der Meer, J.W.; de Mast, Q.; Brüggemann, R.J.; van der Hoeven, H. Kallikrein-kinin blockade in patients with COVID-19 to prevent acute respiratory distress syndrome. Elife 2020, 9, e57555. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, C.P.; Nguyen, J.; Yamaguchi, Y.; Prindle, T.; Fulton, W.B.; Wang, S.; McCray, P.B., Jr.; Chappell, M.; Hackam, D.J.; Jia, H. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg9 bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2018, 314, L17–L31. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Penninger, J.M. Angiotensin-converting enzyme 2 in lung diseases. Curr. Opin. Pharmacol. 2006, 6, 271–276. [Google Scholar] [CrossRef]
- Nie, W.; Zang, Y.; Chen, J.; Liu, T.; Xiao, L.; Xiu, Q. Angiotensin-converting enzyme I/D polymorphism is associated with pneumonia risk: A meta-analysis. J. Renin Angiotensin Aldosterone Syst. 2014, 15, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Liu, Y.; Shang, J.; Yuan, Z.; Ping, F.; Yao, S.; Guo, Y.; Li, Y. Ang-(1-7) treatment attenuates lipopolysaccharide-induced early pulmonary fibrosis. Lab. Investig. 2019, 99, 1770–1783. [Google Scholar] [CrossRef]
- Zou, Z.; Yan, Y.; Shu, Y.; Gao, R.; Sun, Y.; Li, X.; Ju, X.; Liang, Z.; Liu, Q.; Zhao, Y.; et al. Angiotensin-converting enzyme 2 protects from lethal avian influenza A H5N1 infections. Nat. Commun. 2014, 5, 3594. [Google Scholar] [CrossRef]
- Reddy, R.; Asante, I.; Liu, S.; Parikh, P.; Liebler, J.; Borok, Z.; Rodgers, K.; Baydur, A.; Louie, S.G. Circulating angiotensin peptides levels in Acute Respiratory Distress Syndrome correlate with clinical outcomes: A pilot study. PLoS ONE 2019, 14, e0213096. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Guo, J.; Zou, Z.; Liu, J.; Caon, B.; Zhang, S.; Li, H.; Wang, W.; Sheng, M.; Liu, S.; et al. Angiotensin II plasma levels are linked to disease severity and predict fatal outcomes in H7N9-infected patients. Nat. Commun. 2014, 5, 3595. [Google Scholar] [CrossRef]
- Rey-Parra, G.J.; Vadivel, A.; Coltan, L.; Hall, A.; Eaton, F.; Schuster, M.; Loibner, H.; Penninger, J.M.; Kassiri, Z.; Oudit, G.Y.; et al. Angiotensin converting enzyme 2 abrogates bleomycin-induced lung injury. J. Mol. Med. 2012, 90, 637–647. [Google Scholar] [CrossRef]
- Ye, R.; Liu, Z. ACE2 exhibits protective effects against LPS-induced acute lung injury in mice by inhibiting the LPS-TLR4 pathway. Exp. Mol. Pathol. 2020, 113, 104350. [Google Scholar] [CrossRef] [PubMed]
- Zambelli, V.; Bellani, G.; Borsa, R.; Pozzi, F.; Grassi, A.; Scanziani, M.; Castiglioni, V.; Masson, S.; Decio, A.; Laffey, J.G.; et al. Angiotensin-(1-7) improves oxygenation, while reducing cellular infiltrate and fibrosis in experimental Acute Respiratory Distress Syndrome. Intensive Care Med. Exp. 2015, 3, 44. [Google Scholar] [CrossRef] [PubMed]
- Quinton, L.J.; Walkey, A.J.; Mizgerd, J.P. Integrative Physiology of Pneumonia. Physiol. Rev. 2018, 98, 1417–1464. [Google Scholar] [CrossRef]
- Sodhi, C.P.; Nguyen, J.; Yamaguchi, Y.; Werts, A.D.; Lu, P.; Ladd, M.R.; Fulton, W.B.; Kovler, M.L.; Wang, S.; Prindle, T., Jr.; et al. Dynamic variation of pulmonary ACE2 is required to modulate neutrophilic inflammation in response to Pseudomonas aeruginosa lung infection in mice. J. Immunol. 2019, 203, 3000–3012. [Google Scholar] [CrossRef]
- Rice, G.I.; Thomas, D.A.; Grant, P.J.; Turner, A.J.; Hooper, N.M. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem. J. 2004, 383, 45–51. [Google Scholar] [CrossRef]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F.; et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef]
- Wang, W.; McKinnie, S.M.; Farhan, M.; Paul, M.; McDonald, T.; McLean, B.; Llorens-Cortes, C.; Hazra, S.; Murray, A.G.; Vederas, J.C.; et al. Angiotensin-converting enzyme 2 metabolizes and partially inactivates Pyr-Apelin-13 and Apelin-17: Physiological effects in the cardiovascular system. Hypertension 2016, 68, 365–377. [Google Scholar] [CrossRef]
- Campbell, D.J.; Zeitz, C.J.; Esler, M.D.; Horowitz, J.D. Evidence against a major role for angiotensin converting enzyme-related carboxypeptidase (ACE2) in angiotensin peptide metabolism in the human coronary circulation. J. Hypertens. 2004, 22, 1971–1976. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef] [PubMed]
- Doobay, M.F.; Talman, L.S.; Obr, T.D.; Tian, X.; Davisson, R.L.; Lazartigues, E. Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin–angiotensin system. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R373–R381. [Google Scholar] [CrossRef]
- Gembardt, F.; Sterner-Kock, A.; Imboden, H.; Spalteholz, M.; Reibitz, F.; Schultheiss, H.P.; Siems, W.E.; Walther, T. Organ-specific distribution of ACE2 mRNA and correlating peptidase activity in rodents. Peptides 2005, 26, 1270–1277. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhong, L.; Deng, J.; Peng, J.; Dan, H.; Zeng, X.; Li, T.; Chen, Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020, 12, 8. [Google Scholar] [CrossRef]
- Radzikowska, U.; Ding, M.; Tan, G.; Zhakparov, D.; Peng, Y.; Wawrzyniak, P.; Wang, M.; Li, S.; Morita, H.; Altunbulakli, C.; et al. Distribution of ACE2, CD147, CD26 and other SARS-CoV-2 associated molecules in tissues and immune cells in health and in asthma, COPD, obesity, hypertension, and COVID-19 risk factors. Allergy 2020, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.P.; Look, D.C.; Tan, P.; Shi, L.; Hickey, M.; Gakhar, L.; Chappell, M.C.; Wohlford-Lenane, C.; McCray, P.B., Jr. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L84–L96. [Google Scholar] [CrossRef]
- Palau, V.; Pascual, J.; Soler, M.J.; Riera, M. Role of ADAM17 in kidney disease. Am. J. Physiol. Renal Physiol. 2019, 317, F333–F342. [Google Scholar] [CrossRef]
- Iwata, M.; Greenberg, B.H. Ectodomain shedding of ACE and ACE2 as regulators of their protein functions. Curr. Enzym. Inhib. 2011, 7, 42–55. [Google Scholar] [CrossRef]
- Xiao, F.; Zimpelmann, J.; Agaybi, S.; Gurley, S.B.; Puente, L.; Burns, K.D. Characterization of angiotensin-converting enzyme 2 ectodomain shedding from mouse proximal tubular cells. PLoS ONE 2014, 9, e85958. [Google Scholar] [CrossRef]
- Xiao, F.; Zimpelmann, J.; Burger, D.; Kennedy, C.; Hébert, R.L.; Burns, K.D. Protein Kinase C-δ Mediates Shedding of Angiotensin-Converting Enzyme 2 from Proximal Tubular Cells. Front. Pharmacol. 2016, 7, 146. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.B.; Clarke, N.; Wang, Z.; Fan, D.; Parajuli, N.; Basu, R.; Putko, B.; Kassiri, Z.; Turner, A.J.; Oudit, G.Y. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: A positive feedback mechanism in the RAS. J. Mol. Cell. Cardiol. 2014, 66, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yang, J.; Chen, J.; Luo, Q.; Zhang, Q.; Zhang, H. Vitamin D alleviates lipopolysaccharide-induced acute lung injury via regulation of the renin-angiotensin system. Mol. Med. Rep. 2017, 16, 7432–7438. [Google Scholar] [CrossRef] [PubMed]
- Haga, S.; Yamamoto, N.; Nakai-Murakami, C.; Osawa, Y.; Tokunaga, K.; Sata, T.; Yamamoto, N.; Sasazuki, T.; Ishizaka, Y. Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc. Natl. Acad. Sci. USA 2008, 105, 7809–7814. [Google Scholar] [CrossRef]
- Schreiber, B.; Patel, A.; Verma, A. Shedding Light on COVID-19: ADAM17 the Missing Link? Am. J. Ther. 2020, 1–3. [Google Scholar] [CrossRef]
- Kornilov, S.A.; Lucas, I.; Jade, K.; Dai, C.L.; Lovejoy, J.C.; Magis, A.T. Plasma levels of soluble ACE2 are associated with sex, Metabolic Syndrome, and its biomarkers in a large cohort, pointing to a possible mechanism for increased severity in COVID-19. Crit. Care 2020, 24, 452. [Google Scholar] [CrossRef]
- Swärd, P.; Edsfeldt, A.; Reepalu, A.; Jehpsson, L.; Rosengren, B.E.; Karlsson, M.K. Age and sex differences in soluble ACE2 may give insights for COVID-19. Crit. Care 2020, 24, 221. [Google Scholar] [CrossRef]
- Sama, I.E.; Ravera, A.; Santema, B.T.; van Goor, H.; Ter Maaten, J.M.; Cleland, J.G.F.; Rienstra, M.; Friedrich, A.W.; Samani, N.J.; Ng, L.L.; et al. Circulating plasma concentrations of angiotensin-converting enzyme 2 in men and women with heart failure and effects of renin-angiotensin-aldosterone inhibitors. Eur. Heart J. 2020, 41, 1810–1817. [Google Scholar] [CrossRef]
- Xia, H.; Sriramula, S.; Chhabra, K.H.; Lazartigues, E. Brain angiotensin-converting enzyme type 2 shedding contributes to the development of neurogenic hypertension. Circ. Res. 2013, 113, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, N.; Tang, J.; Liu, S.; Luo, D.; Duan, Q.; Wang, X. Downregulation of angiotensin-converting enzyme 2 by the neuraminidase protein of influenza A (H1N1) virus. Virus Res. 2014, 185, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Gu, H.; Zhao, Z.; Wang, W.; Cao, B.; Lai, C.; Yang, X.; Zhang, L.; Duan, Y.; Zhang, S.; et al. Angiotensin-converting enzyme 2 (ACE2) mediates influenza H7N9 virus-induced acute lung injury. Sci. Rep. 2014, 4, 7027. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Pan, J.; Ivanov, Y.P.; Zhou, W.H.; Li, Y.; Greer, A.L. Strain-hardening and suppression of shear-banding in rejuvenated bulk metallic glass. Nature 2020, 578, 559–562. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127–e20. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, Z.; Wang, Y.; Zhou, Y.; Ma, Y.; Zuo, W. Single-cell RNA expression profiling of ACE2, the putative receptor of Wuhan COVID-19. Am. J. Respir. Crit. Care Med. 2020, 202, 756–759. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Saheb Sharif-Askari, N.; Saheb Sharif-Askari, F.; Alabed, M.; Temsah, M.H.; Al Heialy, S.; Hamid, Q.; Halwani, R. Airways expression of SARS-CoV-2 receptor, ACE2, and TMPRSS2 is lower in children than adults and increases with smoking and COPD. Mol. Ther. Methods Clin. Dev. 2020, 18, 1–6. [Google Scholar] [CrossRef]
- Glowacka, I.; Bertram, S.; Herzog, P.; Pfefferle, S.; Steffen, I.; Muench, M.O.; Simmons, G.; Hofmann, H.; Kuri, T.; Weber, F.; et al. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J. Virol. 2010, 84, 1198–1205. [Google Scholar] [CrossRef]
- Hofmann, H.; Geier, M.; Marzi, A.; Krumbiegel, M.; Peipp, M.; Fey, G.H.; Gramberg, T.; Pöhlmann, S. Susceptibility to SARS coronavirus S protein-driven infection correlates with expression of angiotensin converting enzyme 2 and infection can be blocked by soluble receptor. Biochem. Biophys. Res. Commun. 2004, 319, 1216–1221. [Google Scholar] [CrossRef] [PubMed]
- Dijkman, R.; Jebbink, M.F.; Deijs, M.; Milewska, A.; Pyrc, K.; Buelow, E.; van der Bijl, A.; van der Hoek, L. Replication-dependent downregulation of cellular angiotensin-converting enzyme 2 protein expression by human coronavirus NL63. J. Gen. Virol. 2012, 93, 1924–1929. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; McArthur, E.; Farag, A.; Nartey, M.; Fleet, J.L.; Knoll, G.A.; Kim, S.J.; Garg, A.X.; Jain, A.K. Risk of hospitalization for community acquired pneumonia with renin-angiotensin blockade in elderly patients: A population-based study. PLoS ONE 2014, 9, e110165. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, J.K.; Heo, E.Y.; Chung, H.S.; Kim, D.K. The association of renin-angiotensin system blockades and pneumonia requiring admission in patients with COPD. Int. J. Chronic Obstr. Pulm. Dis. 2016, 11, 2159–2166. [Google Scholar] [CrossRef]
- Lai, C.C.; Wang, Y.H.; Wang, C.Y.; Wang, H.C.; Yu, C.J.; Chen, L. Comparative effects of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers on the risk of pneumonia and severe exacerbations in patients with COPD. Int. J. Chronic Obstr. Pulm. Dis. 2018, 13, 867–874. [Google Scholar] [CrossRef]
- Soto, M.; Bang, S.I.; McCombs, J.; Rodgers, K.E. Renin Angiotensin system-modifying therapies are associated with improved pulmonary health. Clin. Diabetes Endocrinol. 2017, 3, 6. [Google Scholar] [CrossRef]
- Kim, J.; Choi, S.M.; Lee, J.; Park, Y.S.; Lee, C.H.; Yim, J.J.; Yoo, C.G.; Kim, Y.W.; Han, S.K.; Lee, S.M. Effect of renin-angiotensin system blockage in patients with acute respiratory distress syndrome: A retrospective case control study. Korean J. Crit. Care Med. 2017, 32, 154–163. [Google Scholar] [CrossRef]
- Davis, T.M.E.; Davis, W.A. Influence of renin-angiotensin system inhibitors on lower-respiratory tract infections in type 2 diabetes: The frmantle diabetes study phase II. Diabetes Care 2020, 43, 2113–2120. [Google Scholar] [CrossRef]
- Fang, L.; Karakiulakis, G.; Roth, M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir. Med. 2020, 8, e21. [Google Scholar] [CrossRef]
- Diaz, J.H. Hypothesis: Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers may increase the risk of severe COVID-19. J. Travel. Med. 2020, 27, taaa041. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Insel, P.A. Risks of ACE inhibitor and ARB usage in COVID-19: Evaluating the evidence. Clin. Pharmacol. Ther. 2020, 108, 236–241. [Google Scholar] [CrossRef]
- Guo, X.; Zhu, Y.; Hong, Y. Decreased mortality of COVID-19 with renin-angiotensin-aldosterone system inhibitors therapy in patients with hypertension: A meta-analysis. Hypertension 2020, 76, e13–e14. [Google Scholar] [CrossRef] [PubMed]
- Barochiner, J.; Martínez, R. Use of inhibitors of the renin-angiotensin system in hypertensive patients and COVID-19 severity: A systematic review and meta-analysis. J. Clin. Pharm. Ther. 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Patoulias, D.; Katsimardou, A.; Stavropoulos, K.; Imprialos, K.; Kalogirou, M.S.; Doumas, M. Renin-angiotensin system inhibitors and COVID-19: A systematic review and meta-analysis. Evidence for significant geographical disparities. Curr. Hypertens. Rep. 2020, 22, 90. [Google Scholar] [CrossRef]
- Vaduganathan, M.; Vardeny, O.; Michel, T.; McMurray, J.J.V.; Pfeffer, M.A.; Solomon, S.D. Renin-angiotensin-aldosterone system inhibitors in patients with Covid-19. N. Engl. J. Med. 2020, 382, 1653–1659. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Pfeffer, M.A. Plasma angiotensin-converting enzyme 2: Novel biomarker in heart failure with implications for COVID-19. Eur. Heart J. 2020, 41, 1818–1820. [Google Scholar] [CrossRef]
- Hamming, I.; van Goor, H.; Turner, A.J.; Rushworth, C.A.; Michaud, A.A.; Corvol, P.; Navis, G. Differential regulation of renal angiotensin-converting enzyme (ACE) and ACE2 during ACE inhibition and dietary sodium restriction in healthy rats. Exp. Physiol. 2008, 93, 631–638. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Jessup, J.; Chappell, M.C.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Diz, D.I.; Gallagher, P.E. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation 2005, 111, 2605–2610. [Google Scholar] [CrossRef]
- Ocaranza, M.P.; Godoy, I.; Jalil, J.E.; Varas, M.; Collantes, P.; Pinto, M.; Roman, M.; Ramirez, C.; Copaja, M.; Diaz-Araya, G.; et al. Enalapril attenuates downregulation of Angiotensin-converting enzyme 2 in the late phase of ventricular dysfunction in myocardial infarcted rat. Hypertension 2006, 48, 572–578. [Google Scholar] [CrossRef]
- Burchill, L.J.; Velkoska, E.; Dean, R.G.; Griggs, K.; Patel, S.K.; Burrell, L.M. Combination renin-angiotensin system blockade and angiotensin-converting enzyme 2 in experimental myocardial infarction: Implications for future therapeutic directions. Clin. Sci. 2012, 123, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Vuille-dit-Bille, R.N.; Camargo, S.M.; Emmenegger, L.; Zahniser, M.S.; Nelson, D.D.; Tuszon, B.; Nakagawa, M.; Toyoda, S.; Yoshida, N.; Emmenegger, L.; et al. Human intestine luminal ACE2 and amino acid transporter expression increased by ACE-inhibitors. Amino Acids 2015, 474, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Furuhashi, M.; Moniwa, N.; Mita, T.; Fuseya, T.; Ishimura, S.; Ohno, K.; Shibata, S.; Tanaka, M.; Watanabe, Y.; Akasaka, H.; et al. Urinary angiotensin-converting enzyme 2 in hypertensive patients may be increased by olmesartan, an angiotensin II receptor blocker. Am. J. Hypertens. 2015, 28, 15–21. [Google Scholar] [CrossRef]
- Arendse, L.B.; Danser, A.H.J.; Poglitsch, M.; Touyz, R.M.; Burnett, J.C., Jr.; Llorens-Cortes, C.; Ehlers, M.R.; Sturrock, E.D. Novel therapeutic approaches targeting the renin-angiotensin system and associated peptides in hypertension and heart failure. Pharmacol. Rev. 2019, 71, 539–570. [Google Scholar] [CrossRef]
- Kobori, H.; Mori, H.; Masaki, T.; Nishiyama, A. Angiotensin II blockade and renal protection. Curr. Pharm. Des. 2013, 19, 3033–3042. [Google Scholar] [CrossRef]
- Mogi, M.; Horiuchi, M. Remote control of brain angiotensin II levels by angiotensin receptor blockers. Hypertens. Res. 2010, 33, 116–117. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xu, X.; Jiang, L.; Dua, K.; Hansbro, P.M.; Liu, G. SARS-CoV-2 induces transcriptional signatures in human lung epithelial cells that promote lung fibrosis. Respir. Res. 2020, 21, 182. [Google Scholar] [CrossRef]
- Cohen, J.B.; Hanff, T.C.; Corrales-Medina, V.; William, P.; Renna, N.; Rosado-Santander, N.R.; Rodriguez-Mori, J.E.; Spaak, J.; Andrade-Villanueva, J. Randomized elimination and prolongation of ACE inhibitors and ARBs in coronavirus 2019 (REPLACE COVID) Trial Protocol. J. Clin. Hypertens. 2020, 1–9. [Google Scholar] [CrossRef]
- Da Silva, A.R.; Lenglet, S.; Carbone, F.; Burger, F.; Roth, A.; Liberale, L.; Bonaventura, A.; Dallegri, F.; Stergiopulos, N.; Santos, R.A.S. Alamandine abrogates neutrophil degranulation in atherosclerotic mice. Eur. J. Clin. Investig. 2017, 47, 117–128. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, S.M.; Dutra, M.F.; Vago, J.P.; Galvão, K.M.I.; Lima, K.M.; Galvão, I.; de Souza-Neto, F.P.; Morais e Silva, M.; Oliveira, A.C.; de Oliveira, F.C.B.; et al. Angiotensin-(1-7) and alamandine promote anti-inflammatory response in macrophages in vitro and in vivo. Mediat. Inflamm. 2019, 2019, 2401081. [Google Scholar] [CrossRef]
- Li, P.; Chen, X.-R.; Xu, F.; Liu, C.; Li, C.; Liu, H.; Wang, H.; Sun, W.; Shen, Y.-H.; Kong, X.-Q. Alamandine attenuates sepsis-associated cardiac dysfunction via inhibiting MAPKs signaling pathways. Life Sci. 2018, 206, 106–116. [Google Scholar] [CrossRef]
- De Souza-Neto, F.P.; Silva, M.M.E.; Santuchi, M.C.; de Alcântara-Leonídio, T.C.; Motta-Santos, D.; Oliveira, A.C.; Melo, M.B.; Canta, G.N.; de Souza, L.E.; Irigoyen, M.C.C.; et al. Alamandine attenuates arterial remodelling induced by transverse aortic constriction in mice. Clin. Sci. 2019, 133, 629–643. [Google Scholar] [CrossRef]
- McKinney, C.A.; Fattah, C.; Loughrey, C.M.; Milligan, G.; Nicklin, S.A. Angiotensin-(1-7) and angiotensin-(1-9): Function in cardiac and vascular remodelling. Clin. Sci. 2014, 126, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Pu, S.Y.; Fan, X.F.; Li, X.S.; Zhang, Y.; Yuan, J.; Zhang, Y.F.; Yang, J.L. Treatment with angiotensin-(1-9) alleviates the cardiomyopathy in streptozotocin-induced diabetic rats. Biochem. Pharmacol. 2015, 95, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, L.; Novoa, U.; Moya, J.; Gabrielli, L.; Jalil, J.E.; García, L.; Chiong, M.; Lavandero, S.; Ocaranza, M.P. Angiotensin-(1-9) reduces cardiovascular and renal inflammation in experimental renin-independent hypertension. Biochem. Pharmacol. 2018, 156, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.A.; Park, B.M.; Kim, S.H. Angiotensin-(1-9) ameliorates pulmonary arterial hypertension via angiotensin type II receptor. Korean J. Physiol. Pharmacol. 2018, 22, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Yang, X.P.; Liu, Y.H.; Xu, J.; Cingolani, O.; Rhaleb, N.E.; Carretero, O.A. Ac-SDKP reverses inflammation and fibrosis in rats with heart failure after myocardial infarction. Hypertension 2004, 43, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, G.E.; Rhaleb, N.E.; Nakagawa, P.; Liao, T.D.; Liu, Y.; Leung, P.; Dai, X.; Yang, X.P.; Carretero, O.A. N-acetyl-seryl-aspartyl-lysyl-proline reduces cardiac collagen cross-linking and inflammation in angiotensin II-induced hypertensive rats. Clin. Sci. 2014, 126, 85–94. [Google Scholar] [CrossRef]
- Lin, C.X.; Rhaleb, N.E.; Yang, X.P.; Liao, T.D.; D’Ambrosio, M.A.; Carretero, O.A. Prevention of aortic fibrosis by N-acetyl-seryl-aspartyl-lysyl-proline in angiotensin II-induced hypertension. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1253–H1261. [Google Scholar] [CrossRef]
- Romero, C.A.; Kumar, N.; Nakagawa, P.; Worou, M.E.; Liao, T.D.; Peterson, E.L.; Carretero, O. A Renal release of N-acetyl-seryl-aspartyl-lysyl-proline is part of an antifibrotic peptidergic system in the kidney. Am. J. Physiol. Ren. Physiol. 2019, 316, F195–F203. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, L.M.; Chen, Y.W.; Ni, Q.W.; Zhou, M.; Qu, C.Y.; Zhang, Y. Antifibrotic effect of N-acetyl-seryl-aspartyl-lysyl-proline on bile duct ligation induced liver fibrosis in rats. World J. Gastroenterol. 2012, 18, 5283–5288. [Google Scholar] [CrossRef] [PubMed]
- Conte, E.; Fagone, E.; Gili, E.; Fruciano, M.; Iemmolo, M.; Pistorio, M.P.; Impellizzeri, D.; Cordaro, M.; Cuzzocrea, S.; Vancheri, C. Preventive and therapeutic effects of thymosin β4 N-terminal fragment Ac-SDKP in the bleomycin model of pulmonary fibrosis. Oncotarget 2016, 7, 33841–33854. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Deng, H.; Yang, F.; Xu, H.; Sun, Y.; Xue, X.; Du, S.; Wang, X.; Li, S.; Liu, Y.; Wang, R. Ac-SDKP suppresses epithelial-mesenchymal transition in A549 cells via HSP27 signaling. Exp. Mol. Pathol. 2014, 97, 176–183. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, D.; Li, Q.; Yang, Y.; Xu, H.; Wie, Z.; Wang, R.; Zhang, W.; Liu, Y.; Geng, Y.; et al. N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) attenuates silicotic fibrosis by suppressing apoptosis of alveolar type II epithelial cells via mediation of endoplasmic reticulum stress. Toxicol. Appl. Pharmacol. 2018, 350, 1–10. [Google Scholar] [CrossRef]
- Wösten-van Asperen, R.M.; Lutter, R.; Specht, P.A.; Moll, G.N.; van Woensel, J.B.; van der Loos, C.M.; van Goor, H.; Kamilic, J.; Florquin, S.; Bos, A.P. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1-7) or an angiotensin II receptor antagonist. J. Pathol. 2011, 225, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef]
- Zhang, H.; Baker, A. Recombinant human ACE2: Acing out angiotensin II in ARDS therapy. Crit. Care 2017, 21, 305. [Google Scholar] [CrossRef]
- Khan, A.; Benthin, C.; Zeno, B.; Albertson, T.E.; Boyd, J.; Christie, J.D.; Hall, R.; Poirier, G.; Ronco, J.J.; Tidswell, M.; et al. A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Crit. Care 2017, 21, 234. [Google Scholar] [CrossRef]
- Chen, Q.; Yang, Y.; Huang, Y.; Pan, C.; Liu, L.; Qiu, H. Angiotensin-(1-7) attenuates lung fibrosis by way of Mas receptor in acute lung injury. J. Surg. Res. 2013, 185, 740–747. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Design | Subjects | Outcome | Ref. |
|---|---|---|---|
| Retrospective cohort study | 254,485 patients > 65 y.o. newly prescribed antihypertensive drugs | ↓ risk of hospitalization with pneumonia within 90 days following treatment initiation with ACEIs/ARBs vs. other antihypertensive drugs | [74] |
| Retrospective nested case–control study | 375 COPD patients | ↓ risk of pneumonia | [75] |
| Retrospective comparative study | 12,452 patients newly prescribed ACEIs/ARBs within 90 days after diagnosis of COPD | ↓ risk of pneumonia, severe pneumonia and ↓ mortality in ARBs group vs. ACEIs | [76] |
| Retrospective cohort study | 215,225 patients | Improved infectious (influenza, pneumonia), inflammatory (COPD) and structural outcomes in ACEIs/ARBs vs. other treatment | [77] |
| Retrospective case–control study | 182 ARDS patients | ↑ duration of mechanical ventilation and ICU stay in ACEIs/ARBs group ↑ survival in ACEIs/ARBs group | [78] |
| Cox regression longitudinal observational study | 1482 T2D patients | ↓ risk of pneumonia/influenza | [79] |
| Treatment | Experimental Model | Effect of Treatment | Ref. |
|---|---|---|---|
| Ang 1–7 LD: 0.27 μg/kg/h i.v. HD: 60 μg/kg/h i.v. Late-ARDS-study: 300 μg/kg/d i.v. | Sprague Dawley rats HCl- and ventilation-induced injury Late-ARDS- study: 2 weeks post-injury | Improved oxygenation (PaO2/FiO2) ↓ WBC in peripheral blood Late-ARDS: ↓ hydroxyproline content in lung HD: ↓ inflammatory cells in BAL | [36] |
| Ang 1–7 2.4 µg/kg/h i.v. | Sprague Dawley rats LPS- and ventilation-induced ARDS | ↑ PaO2 ↑ACE2 and Ang 1–7 in BAL Normalized Ang-(1–7)/Ang II ratio$ ↓ CINC-3, TNF-α, GM-CSF in BAL | [115] |
| Ang-1–7 600 μg/kg/d i.p. | Sprague Dawley rats LPS-induced early pulmonary fibrosis | ↓ lung injury and lung fibrose scores ↓ TGF-β in plasma ↓ Ang II in BAL Normalized E-cadherin and vimentin levels in lung ↓ AT1R mRNA expression in lung ↓ cell-membrane AT1R and ↑ MasR in lung ↓ LPS-induced phosphorylation of Src kinase | [30] |
| Ang 1–7 100 ng/kg/min s.c. | C57BL/6 mice LPS-induced ALI | ↓ edema, bleeding, collagen and septal widening in lung ↓ TGF-β and p-SMAD2/3 in lung | [119] |
| ACE2 2 mg/kg i.p. | C57BL6 mice BLM-induced ALI | ↑ survival, exercise capacity, lung function (dynamic compliance and elastance) ↓ collagen, α-SMA, TGF-β1, TNF- α in lung | [34] |
| ACE2 1.0 mg/kg i.v. | C57BL/6 mice LPS-induced ALI | ↓ lung W/D ↓ PaCO2 and ↑ PaO2 ↓ lung histological score ↓ IL-1β, IL-6 and TNF-α in lung and in serum ↓ protein concentration and neutrophil count in BAL ↓ LPS-activated TLR4-signaling | [35] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hrenak, J.; Simko, F. Renin–Angiotensin System: An Important Player in the Pathogenesis of Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2020, 21, 8038. https://doi.org/10.3390/ijms21218038
Hrenak J, Simko F. Renin–Angiotensin System: An Important Player in the Pathogenesis of Acute Respiratory Distress Syndrome. International Journal of Molecular Sciences. 2020; 21(21):8038. https://doi.org/10.3390/ijms21218038
Chicago/Turabian StyleHrenak, Jaroslav, and Fedor Simko. 2020. "Renin–Angiotensin System: An Important Player in the Pathogenesis of Acute Respiratory Distress Syndrome" International Journal of Molecular Sciences 21, no. 21: 8038. https://doi.org/10.3390/ijms21218038
APA StyleHrenak, J., & Simko, F. (2020). Renin–Angiotensin System: An Important Player in the Pathogenesis of Acute Respiratory Distress Syndrome. International Journal of Molecular Sciences, 21(21), 8038. https://doi.org/10.3390/ijms21218038