Interaction between Trinuclear Regium Complexes of Pyrazolate and Anions, a Computational Study





Abstract
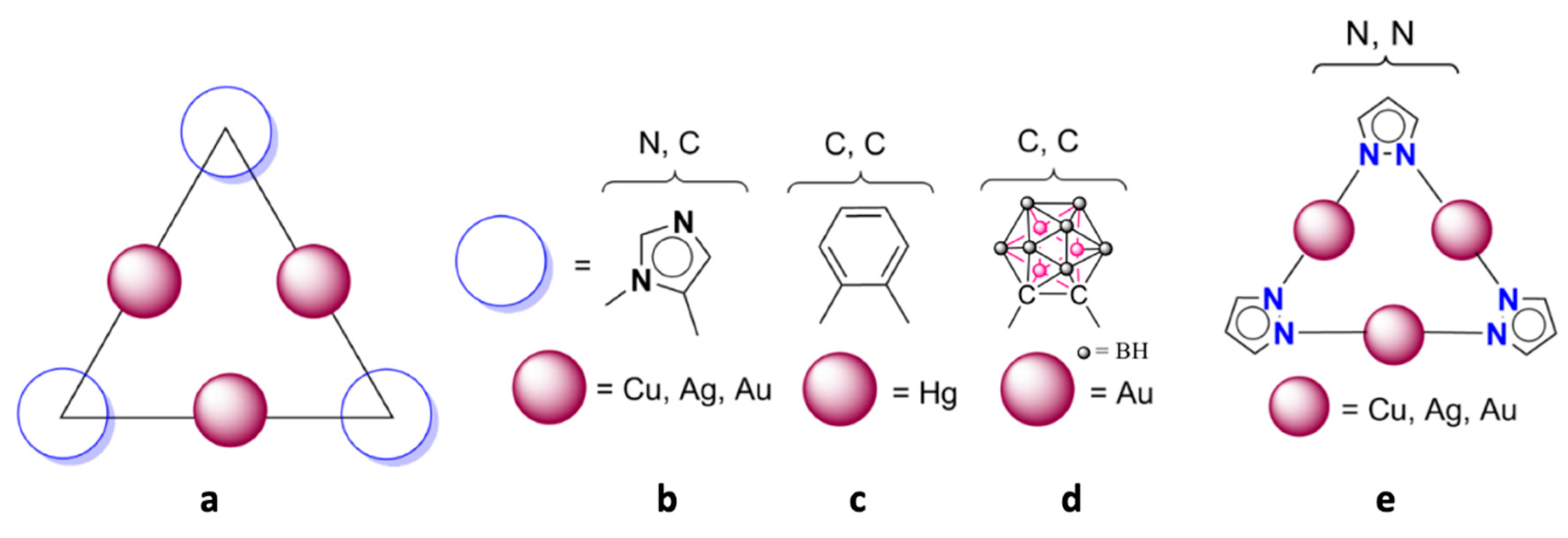
1. Introduction
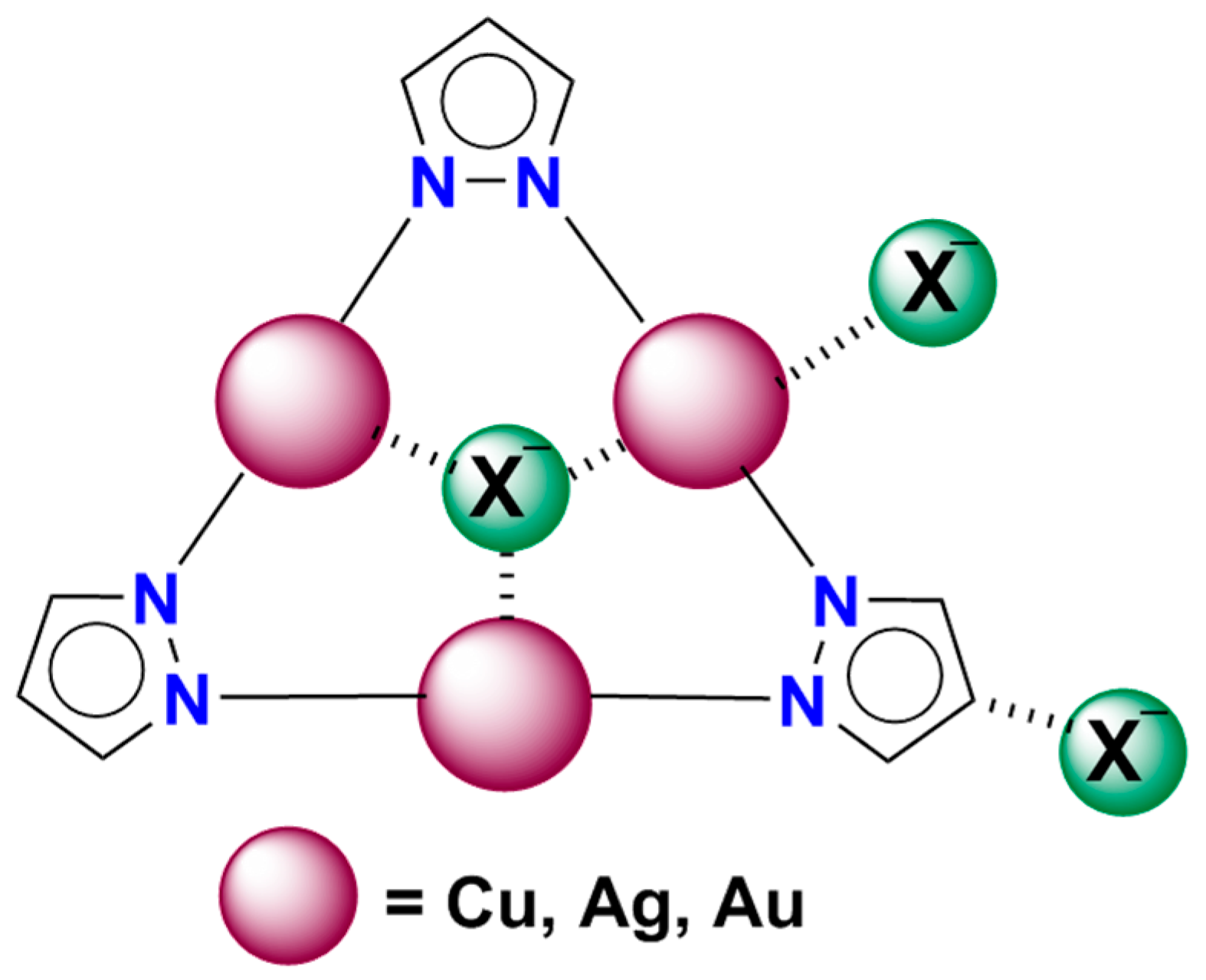
2. Results and Discussion
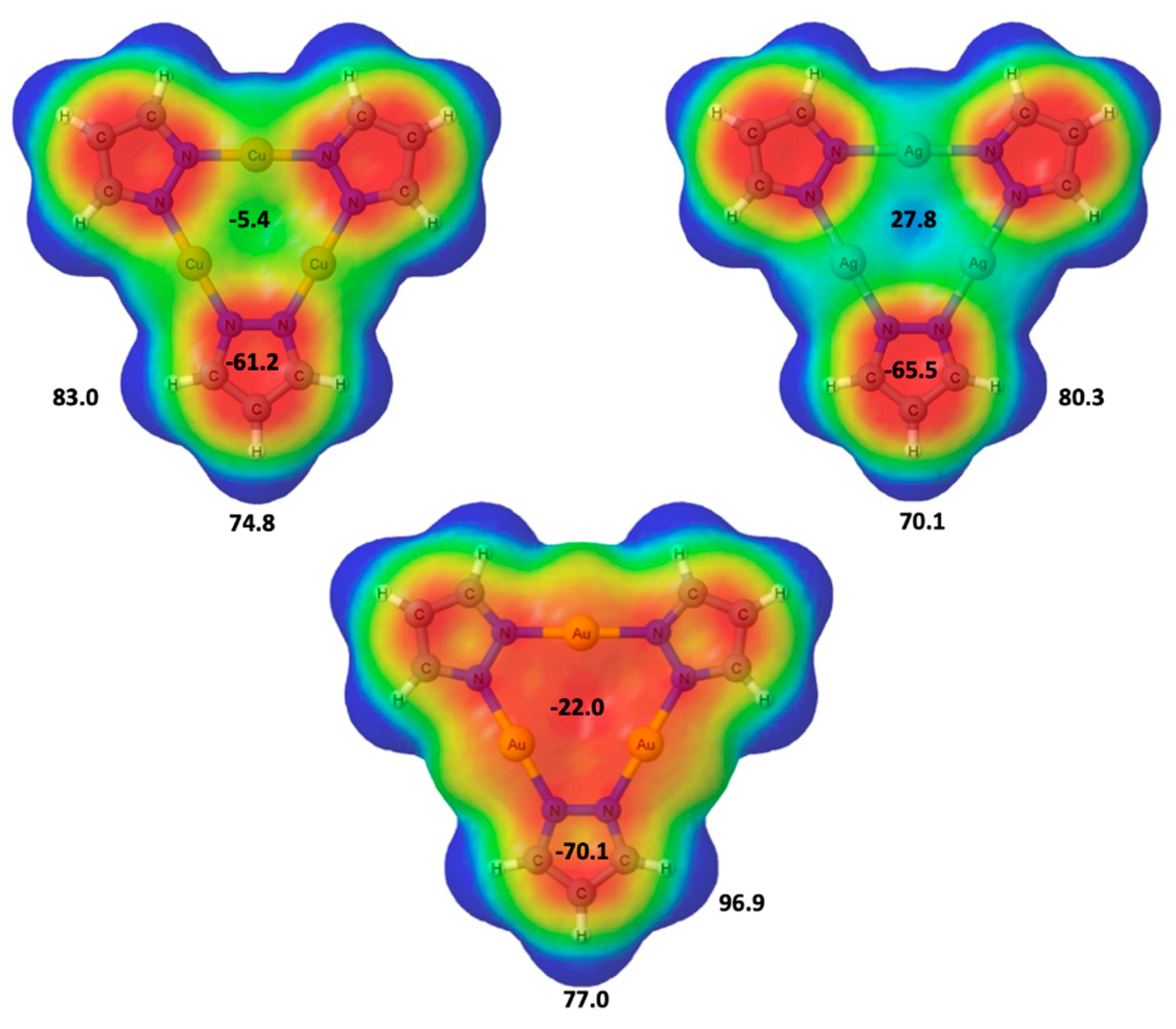
2.1. Isolated (Pz-M)3 Monomers
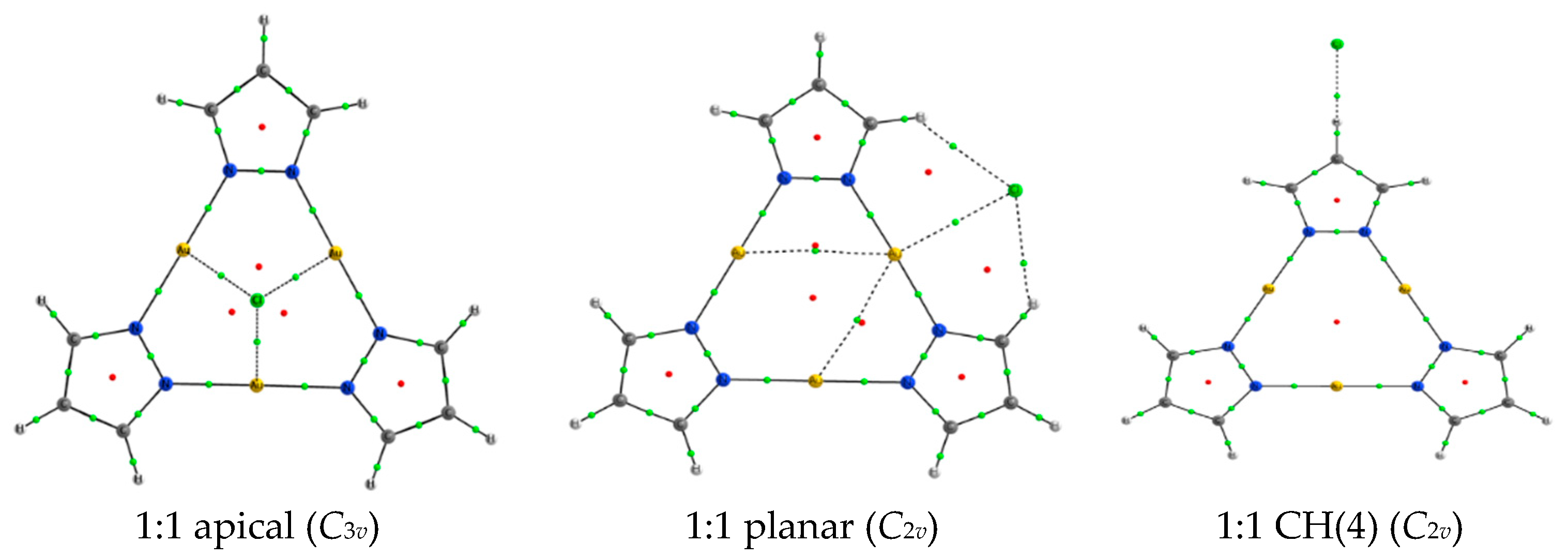
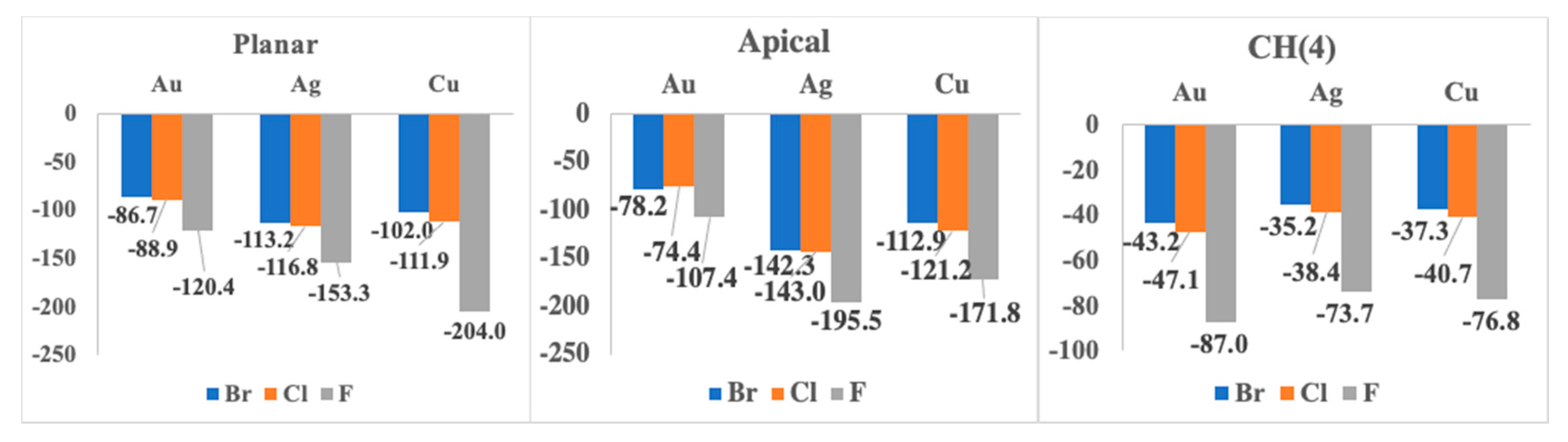
2.2. 1:1 Complexes
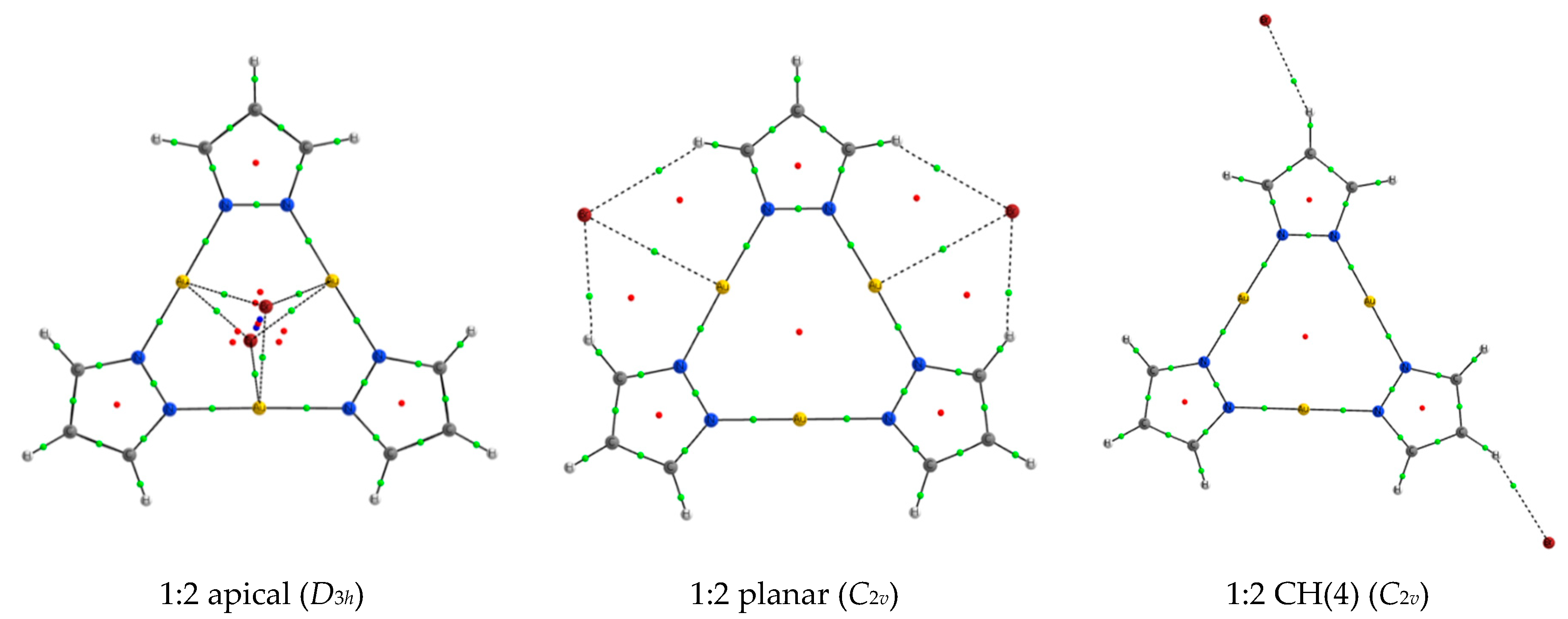
2.3. 1:2 Complexes
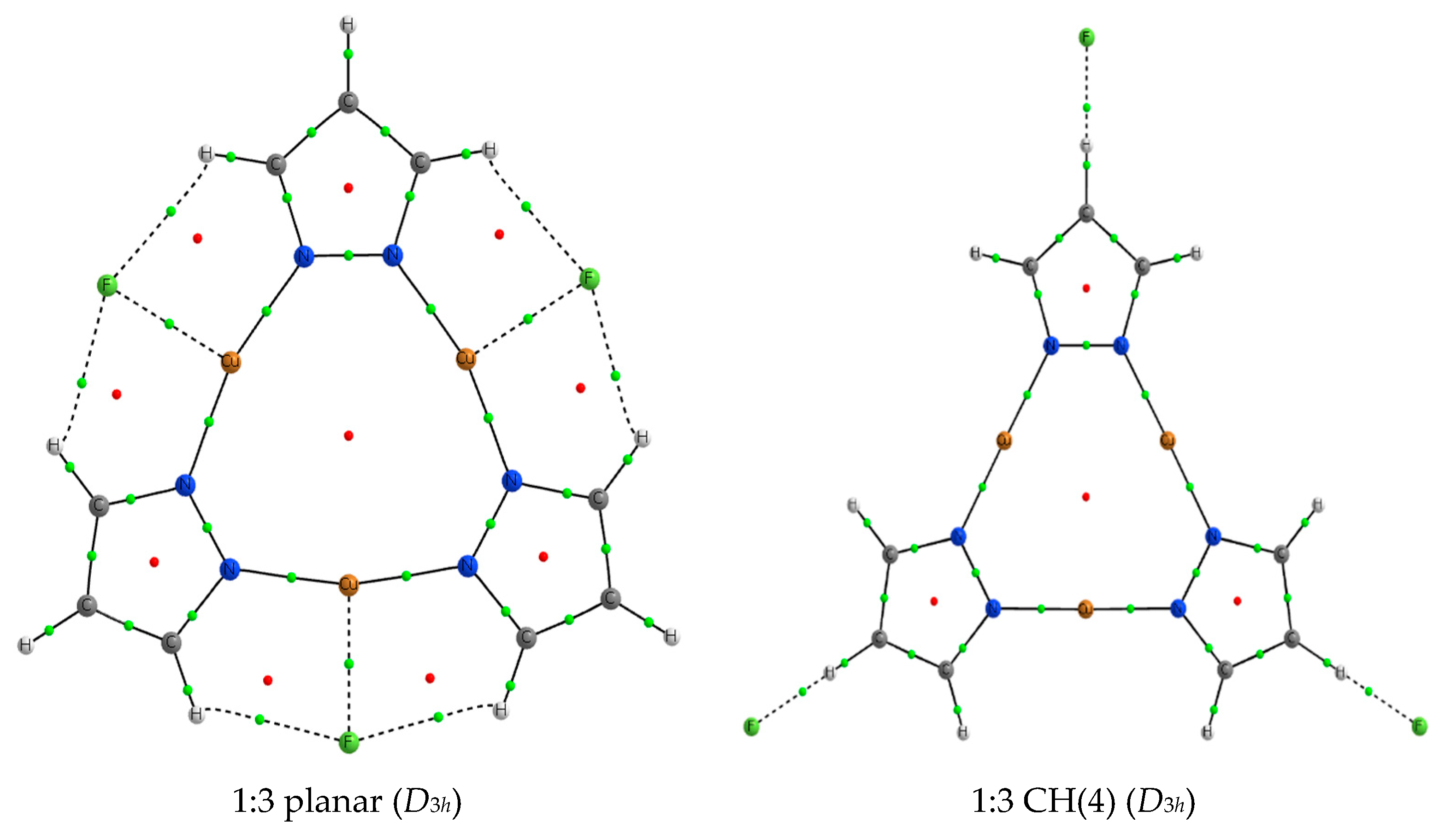
2.4. 1:3 Complexes
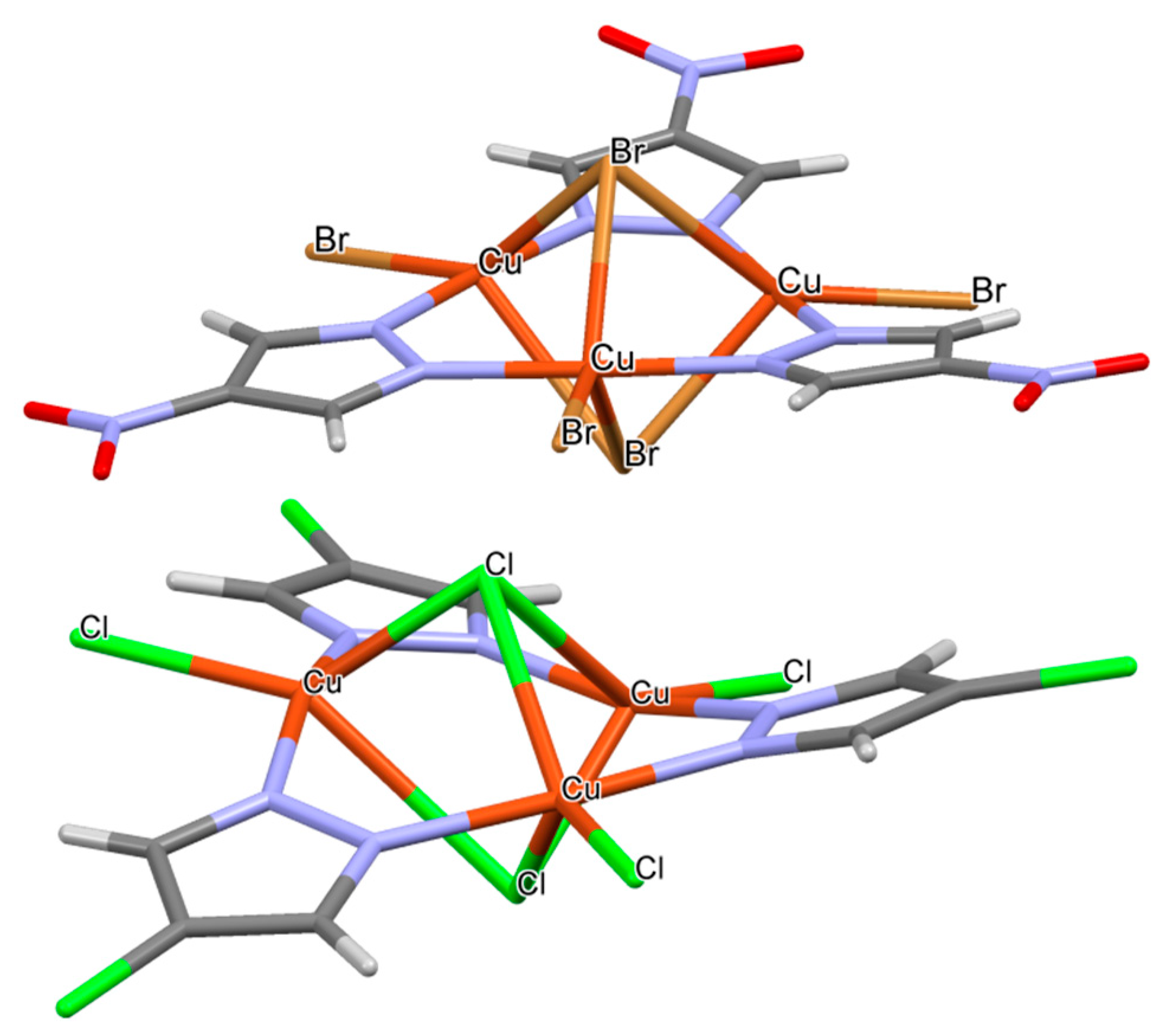
2.5. CSD Search
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Frieden, E. Non-covalent interactions: Key to biological flexibility and specificity. J. Chem. Educ. 1975, 52, 754. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Sommer, I.; Lengauer, T.; Domingues, F.S. Alignment of Non-Covalent Interactions at Protein-Protein Interfaces. PLoS ONE 2008, 3, e1926. [Google Scholar] [CrossRef] [PubMed]
- Busschaert, N.; Caltagirone, C.; Van Rossom, W.; Gale, P.A. Applications of Supramolecular Anion Recognition. Chem. Rev. 2015, 115, 8038–8155. [Google Scholar] [CrossRef] [PubMed]
- Molina, P.; Zapata, F.; Caballero, A. Anion Recognition Strategies Based on Combined Noncovalent Interactions. Chem. Rev. 2017, 117, 9907–9972. [Google Scholar] [CrossRef]
- Cockroft, S.L.; Hunter, C.A. Chemical double-mutant cycles: Dissecting non-covalent interactions. Chem. Soc. Rev. 2007, 36, 172–188. [Google Scholar] [CrossRef] [PubMed]
- Maharramov, A.M.; Mahmudov, K.T.; Kopylovich, M.N.; Pombeiro, A.J.L. Non-Covalent Interactions in the Synthesis and Design of New Compounds; Wiley: Hoboken, NJ, USA, 2016. [Google Scholar]
- Mahadevi, A.S.; Sastry, G.N. Cation−π Interaction: Its Role and Relevance in Chemistry, Biology, and Material Science. Chem. Rev. 2013, 113, 2100–2138. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, G.; McClellan, A. The Hydrogen Bond; W.H. Freeman and Company: San Francisco, CA, USA, 1960. [Google Scholar]
- Hadzi, D. Hydrogen Bonding. In Proceedings of the Symposium on Hydrogen Bonding, Ljubljana, Slovenia, 29 July–3 August 1957; Pergamon Press: London, UK, 1957. [Google Scholar]
- Speakman, J.C. The Hydrogen Bond and other Inter-Molecular Forces; The Chemical Society: London, UK, 1975. [Google Scholar]
- Jeffrey, G.A.; Saenger, W. Hydrogen Bonding in Biological Structures; Springer: Berlin, Germany, 1994. [Google Scholar]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Alkorta, I.; Elguero, J.; Frontera, A. Not Only Hydrogen Bonds: Other Noncovalent Interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef]
- Wang, W.; Ji, B.; Zhang, Y. Chalcogen Bond: A Sister Noncovalent Bond to Halogen Bond. J. Phys. Chem. A 2009, 113, 8132–8135. [Google Scholar] [CrossRef]
- Minyaev, R.M.; Minkin, V.I. Theoretical study of O - > X (S, Se, Te) coordination in organic compounds. Can. J. Chem. 1998, 76, 776–788. [Google Scholar] [CrossRef]
- Zahn, S.; Frank, R.; Hey-Hawkins, E.; Kirchner, B. Pnicogen Bonds: A New Molecular Linker? Chem. Eur. J. 2011, 17, 6034–6038. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. The Pnicogen Bond in Review. In Structures, Binding Energies, Bonding Properties, and Spin-Spin Coupling Constants of Complexes Stabilized by Pnicogen Bonds; Scheiner, S., Ed.; Noncovalent Forces; Springer International Publishing: Cham, Germany, 2015; pp. 191–263. [Google Scholar]
- Alkorta, I.; Rozas, I.; Elguero, J. Molecular Complexes between Silicon Derivatives and Electron-Rich Groups. J. Phys. Chem. A 2001, 105, 743–749. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel-Bonding Interaction: Rediscovered Supramolecular Force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other [sigma]-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Stenlid, J.H.; Brinck, T. Extending the σ-Hole Concept to Metals: An Electrostatic Interpretation of the Effects of Nanostructure in Gold and Platinum Catalysis. J. Am. Chem. Soc. 2017, 139, 11012–11015. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Frontera, A. Regium-π vs Cation-π Interactions in M2 and MCl (M = Cu, Ag and Au) Complexes with Small Aromatic Systems: An ab Initio Study. Inorganics 2018, 6, 64. [Google Scholar] [CrossRef]
- Frontera, A.; Bauzá, A. Regium–π bonds: An Unexplored Link between Noble Metal Nanoparticles and Aromatic Surfaces. Chem. Eur. J. 2018, 24, 7228–7234. [Google Scholar] [CrossRef]
- Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Regium bonds between Mn clusters (M = Cu, Ag, Au and n = 2–6) and nucleophiles NH3 and HCN. Phys. Chem. Chem. Phys. 2018, 20, 22498–22509. [Google Scholar] [CrossRef]
- Legon, A.C.; Walker, N.R. What’s in a name? ‘Coinage-metal’ non-covalent bonds and their definition. Phys. Chem. Chem. Phys. 2018, 20, 19332–19338. [Google Scholar] [CrossRef]
- Halldin Stenlid, J.; Johansson, A.J.; Brinck, T. σ-Holes and σ-lumps direct the Lewis basic and acidic interactions of noble metal nanoparticles: Introducing regium bonds. Phys. Chem. Chem. Phys. 2018, 20, 2676–2692. [Google Scholar] [CrossRef]
- Assadollahzadeh, B.; Schwerdtfeger, P. A systematic search for minimum structures of small gold clusters Aun (n = 2–20) and their electronic properties. J. Chem. Phys. 2009, 131, 064306. [Google Scholar] [CrossRef] [PubMed]
- Slaughter, L.M. Homogeneous Gold Catalysis; Springer International Publishing: Cham, Switzerland, 2015. [Google Scholar]
- Evans, C.J.; Reynard, L.M.; Gerry, M.C.L. Pure Rotational Spectra, Structures, and Hyperfine Constants of OC−AuX (X = F, Cl, Br). Inorg. Chem. 2001, 40, 6123–6131. [Google Scholar] [CrossRef] [PubMed]
- Medcraft, C.; Bittner, D.M.; Tew, D.P.; Walker, N.R.; Legon, A.C. Geometries of H2S⋯MI (M = Cu, Ag, Au) complexes studied by rotational spectroscopy: The effect of the metal atom. J. Chem. Phys. 2016, 145, 194306. [Google Scholar] [CrossRef] [PubMed]
- Obenchain, D.A.; Frank, D.S.; Grubbs, G.S.; Pickett, H.M.; Novick, S.E. The covalent interaction between dihydrogen and gold: A rotational spectroscopic study of H2–AuCl. J. Chem. Phys. 2017, 146, 204302. [Google Scholar] [CrossRef]
- Blakey, I.; Merican, Z.; Rintoul, L.; Chuang, Y.-M.; Jack, K.S.; Micallef, A.S. Interactions of iodoperfluorobenzene compounds with gold nanoparticles. Phys. Chem. Chem. Phys. 2012, 14, 3604–3611. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Dias, H.V.R.; Parasar, D.; Martín-Pastor, M. An experimental and computational NMR study of organometallic nine-membered rings: Trinuclear silver(I) complexes of pyrazolate ligands. Mag. Res. Chem. 2020, 58, 319–328. [Google Scholar] [CrossRef]
- Zhang, G.; Yue, H.; Weinhold, F.; Wang, H.; Li, H.; Chen, D. Resonance Character of Copper/Silver/Gold Bonding in Small Molecule⋅⋅⋅M-X (X=F, Cl, Br, CH3, CF3) Complexes. ChemPhysChem 2015, 16, 2424–2431. [Google Scholar] [CrossRef]
- Li, H.; Li, Q.; Li, R.; Li, W.; Cheng, J. Prediction and characterization of HCCH⋅⋅⋅AuX (X = OH, F, Cl, Br, CH3, CCH, CN, and NC) complexes: A π Au-bond. J. Chem. Phys. 2011, 135, 074304. [Google Scholar] [CrossRef]
- Gao, M.; Li, Q.; Li, H.-B.; Li, W.; Cheng, J. How do organic gold compounds and organic halogen molecules interact? Comparison with hydrogen bonds. RSC Adv. 2015, 5, 12488–12497. [Google Scholar] [CrossRef]
- Zhang, G.; Zhao, X.; Chen, D. Dual Bonding between H2O/H2S and AgCl/CuCl: Cu/Ag Bond, Sister Bond to Au Bond. J. Phys. Chem. A 2013, 117, 10944–10950. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Y.; Zheng, B.; Zhou, F.; Jiao, Y.; Liu, Y.; Ding, X.; Lu, T. A theoretical investigation on Cu/Ag/Au bonding in XH2P⋯MY(X = H, CH3, F, CN, NO2; M = Cu, Ag, Au; Y = F, Cl, Br, I) complexes. J. Chem. Phys. 2018, 148, 194106. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Yang, X.; Cheng, J.; Li, Q.; Li, W.; Loffredo, R.E. Interplay between Metal⋯π Interactions and Hydrogen Bonds: Some Unusual Synergetic Effects of Coinage Metals and Substituents. ChemPhysChem 2013, 14, 3341–3347. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Li, Q.; Li, W.; Cheng, J. Interplay between Cation–π and Coinage-Metal–Oxygen Interactions: An Ab Initio Study and Cambridge Structural Database Survey. ChemPhysChem 2015, 16, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Cheng, J.; Li, W.; Li, Q. Regulation of coin metal substituents and cooperativity on the strength and nature of tetrel bonds. RSC Adv. 2017, 7, 46321–46328. [Google Scholar] [CrossRef]
- Zheng, B.; Liu, Y.; Huang, L.; Wang, Z.; Liu, H.; Liu, Y. Cooperative effects between F … Ag bonded and X … Br (Cl) halogen-bonded interaction in BrF(ClF) … AgX … BrF(ClF) (X = F, Cl, Br) complexes: A theoretical study. Mol. Phys. 2018, 116, 1834–1843. [Google Scholar] [CrossRef]
- Trujillo, C.; Sánchez-Sanz, G.; Elguero, J.; Alkorta, I. The Lewis acidities of gold(I) and gold(III) derivatives: A theoretical study of complexes of AuCl and AuCl3. Struct. Chem. 2020, 31, 1909–1918. [Google Scholar] [CrossRef]
- Samanta, D.; Wu, M.M.; Jena, P. Unique Spectroscopic Signature of Nearly Degenerate Isomers of Au(CN)3 Anion. J. Phys. Chem. Lett. 2011, 2, 3027–3031. [Google Scholar] [CrossRef]
- Wang, X.; Wan, X.; Zhou, H.; Takami, S.; Kubo, M.; Miyamoto, A. Electronic structures and spectroscopic properties of dimers Cu2, Ag2, and Au2 calculated by density functional theory. J. Mol. Struct. THEOCHEM 2002, 579, 221–227. [Google Scholar] [CrossRef]
- Wesendrup, R.; Laerdahl, J.K.; Schwerdtfeger, P. Relativistic effects in gold chemistry. VI. Coupled cluster calculations for the isoelectronic series AuPt−, Au2, and AuHg+. J. Chem. Phys. 1999, 110, 9457–9462. [Google Scholar] [CrossRef]
- Jena, P. Beyond the Periodic Table of Elements: The Role of Superatoms. J. Phys. Chem. Lett. 2013, 4, 1432–1442. [Google Scholar] [CrossRef]
- Schwerdtfeger, P.; Lein, M.; Krawczyk, R.P.; Jacob, C.R. The adsorption of CO on charged and neutral Au and Au2: A comparison between wave-function based and density functional theory. J. Chem. Phys. 2008, 128, 124302. [Google Scholar] [CrossRef]
- Kryachko, E.S.; Remacle, F. Complexes of DNA Bases and Gold Clusters Au3 and Au4 Involving Nonconventional N−H⋯Au Hydrogen Bonding. Nano Lett. 2005, 5, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Kryachko, E.S.; Remacle, F. Three-gold clusters form nonconventional hydrogen bonds O–H⋯Au and N–H⋯Au with formamide and formic acid. Chem. Phys. Lett. 2005, 404, 142–149. [Google Scholar] [CrossRef]
- Kryachko, E.S.; Karpfen, A.; Remacle, F. Nonconventional Hydrogen Bonding between Clusters of Gold and Hydrogen Fluoride. J. Phys. Chem. A 2005, 109, 7309–7318. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q. The X⋯Au interactions in the CF3X (X = Cl, Br) ⋯Aun (n = 2, 3, and 4) complexes. J. Mol. Model. 2014, 20, 2133. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Understanding Regium Bonds and their Competition with Hydrogen Bonds in Au2:HX Complexes. ChemPhysChem 2019, 20, 1572–1580. [Google Scholar] [CrossRef]
- Sanchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Rivalry between regium and hydrogen bonds established within diatomic coinage molecules and Lewis acids/bases. ChemPhysChem 2020, in press. [Google Scholar] [CrossRef]
- Tekarli, S.M.; Cundari, T.R.; Omary, M.A. Rational Design of Macrometallocyclic Trinuclear Complexes with Superior π-Acidity and π-Basicity. JACS 2008, 130, 1669–1675. [Google Scholar] [CrossRef]
- Awad, S.B.; Brown, D.S.; Cohen, S.C.; Humphries, R.E.; Massey, A.G. A reinvestigation of phenylene- and polyphenylene-mercurials. J. Organomet. Chem. 1977, 127, 127–138. [Google Scholar] [CrossRef]
- Tsunoda, M.; Gabbaï, F.P. μ6-η2:η2:η2:η2:η2:η2 As a New Bonding Mode for Benzene. J. Am. Chem. Soc. 2000, 122, 8335–8336. [Google Scholar] [CrossRef]
- Aullón, G.; Laguna, A.; Oliva, J.M. Electronic structure and geometries of o-carborane derived cyclic structures [{μ-1,2-(C2B10H10)nMn}Agm]z−, M = {Au, Hg}, n = {3, 4}, m = {0, 1, 2}, z = {n − m, −m}. Dalton Trans. 2012, 41, 14146–14150. [Google Scholar] [CrossRef] [PubMed]
- Aullón, G.; Laguna, A.; Filippov, O.A.; Oliva-Enrich, J.M. Trinuclear Gold–Carborane Cluster as a Host Structure. Eur. J. Inorg. Chem. 2019, 2019, 18–22. [Google Scholar] [CrossRef]
- Yang, G.; Raptis, R.G. Supramolecular Assembly of Trimeric Gold(I) Pyrazolates through Aurophilic Attractions. Inorg. Chem. 2003, 42, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Martínez, J.R.; Raptis, R.G. Dinuclear gold(III) pyrazolato complexes—Synthesis, structural characterization and transformation to their trinuclear gold(I) and gold(I/III) analogues. Inorg. Chim. Acta 2009, 362, 1546–1552. [Google Scholar] [CrossRef]
- Osuga, T.; Murase, T.; Hoshino, M.; Fujita, M. A Tray-Shaped, PdII-Clipped Au3 Complex as a Scaffold for the Modular Assembly of [3×n] Au Ion Clusters. Angew. Chem. Int. Ed. 2014, 53, 11186–11189. [Google Scholar] [CrossRef]
- Rasika Dias, H.V.; Palehepitiya Gamage, C.S. Arene-Sandwiched Silver(I) Pyrazolates. Angew. Chem. Int. Ed. 2007, 46, 2192–2194. [Google Scholar] [CrossRef]
- Saotome, M.; Shimizu, D.; Itagaki, A.; Young, D.J.; Fujisawa, K. Structures and Photoluminescence of Silver(I) and Gold(I) Cyclic Trinuclear Complexes with Aryl Substituted Pyrazolates. Chem. Lett. 2019, 48, 533–536. [Google Scholar] [CrossRef]
- Zheng, J.; Lu, Z.; Wu, K.; Ning, G.-H.; Li, D. Coinage-Metal-Based Cyclic Trinuclear Complexes with Metal–Metal Interactions: Theories to Experiments and Structures to Functions. Chem. Rev. 2020, 120, 9675–9742. [Google Scholar] [CrossRef]
- Caramori, G.F.; Piccoli, R.M.; Segala, M.; Muñoz-Castro, A.; Guajardo-Maturana, R.; Andrada, D.M.; Frenking, G. Cyclic trinuclear copper(i), silver(i), and gold(i) complexes: A theoretical insight. Dalton Trans. 2015, 44, 377–385. [Google Scholar] [CrossRef]
- Pandolfo, L.; Pettinari, C. Trinuclear copper(ii) pyrazolate compounds: A long story of serendipitous discoveries and rational design. CrystEngComm 2017, 19, 1701–1720. [Google Scholar] [CrossRef]
- Mata, I.; Alkorta, I.; Molins, E.; Espinosa, E. Electrostatics at the Origin of the Stability of Phosphate-Phosphate Complexes Locked by Hydrogen Bonds. ChemPhysChem 2012, 13, 1421–1424. [Google Scholar] [CrossRef] [PubMed]
- Mata, I.; Alkorta, I.; Molins, E.; Espinosa, E. Tracing environment effects that influence the stability of anion–anion complexes: The case of phosphate–phosphate interactions. Chem. Phys. Lett. 2013, 555, 106–109. [Google Scholar] [CrossRef]
- Weinhold, F.; Klein, R.A. Anti-Electrostatic Hydrogen Bonds. Angew. Chem. Int. Ed. 2014, 126, 11396–11399. [Google Scholar] [CrossRef]
- Frenking, G.; Caramori, G.F. No Need for a Re-examination of the Electrostatic Notation of the Hydrogen Bonding: A Comment. Angew. Chem. Int. Ed. 2015, 54, 2596–2599. [Google Scholar] [CrossRef]
- Mata, I.; Molins, E.; Alkorta, I.; Espinosa, E. The paradox of hydrogen-bonded anion-anion aggregates in oxoanions: A fundamental electrostatic problem explained in terms of electrophilic...nucleophilic interactions. J. Phys. Chem. A 2015, 119, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Chen, Z.; Xu, Z.; Wang, J.; Yang, Y.; Cai, T.; Shi, J.; Zhu, W. Stability and Characteristics of the Halogen Bonding Interaction in an Anion-Anion Complex: A Computational Chemistry Study. J. Phys. Chem. B 2016, 120, 610–620. [Google Scholar] [CrossRef]
- Chalanchi, S.M.; Alkorta, I.; Elguero, J.; Quinonero, D. Hydrogen Bond versus Halogen Bond in Cation-Cation Complexes: Effect of the Solvent. ChemPhysChem 2017, 18, 3462–3468. [Google Scholar] [CrossRef]
- Prohens, R.; Portell, A.; Font-Bardia, M.; Bauzá, A.; Frontera, A. H-Bonded anion–anion complex trapped in a squaramido-based receptor. Chem. Comm. 2018, 54, 1841–1844. [Google Scholar] [CrossRef]
- Iribarren, Í.; Montero-Campillo, M.M.; Alkorta, I.; Elguero, J.; Quiñonero, D. Cations brought together by hydrogen bonds: The protonated pyridine–boronic acid dimer explained. Phys. Chem. Chem. Phys. 2019, 21, 5796–5802. [Google Scholar] [CrossRef]
- Azofra, L.M.; Elguero, J.; Alkorta, I. A Conceptual DFT Study of Phosphonate Dimers: Dianions Supported by H-Bonds. J. Phys. Chem. A 2020, 124, 2207–2214. [Google Scholar] [CrossRef]
- Azofra, L.M.; Elguero, J.; Alkorta, I. Stabilisation of dianion dimers trapped inside cyanostar macrocycles. Phys. Chem. Chem. Phys 2020, 22, 11348–11353. [Google Scholar] [CrossRef] [PubMed]
- Chesman, A.S.R.; Hodgson, J.L.; Izgorodina, E.I.; Urbatsch, A.; Turner, D.R.; Deacon, G.B.; Batten, S.R. Anion-Anion Interactions in the Crystal Packing of Functionalized Methanide Anions: An Experimental and Computational Study. Crys.Growth Des. 2014, 14, 1922–1932. [Google Scholar] [CrossRef]
- Martínez-Camarena, Á.; Savastano, M.; Bazzicalupi, C.; Bianchi, A.; García-España, E. Stabilisation of Exotic Tribromide (Br3−) Anions via Supramolecular Interaction with a Tosylated Macrocyclic Pyridinophane. A Serendipitous Case. Molecules 2020, 25, 3155. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Mata, I.; Molins, E.; Espinosa, E. Charged versus Neutral Hydrogen-Bonded Complexes: Is There a Difference in the Nature of the Hydrogen Bonds? Chem. Eur. J. 2016, 22, 9226–9234. [Google Scholar] [CrossRef] [PubMed]
- Quinonero, D.; Alkorta, I.; Elguero, J. Cation-cation and anion-anion complexes stabilized by halogen bonds. Phys. Chem. Chem. Phys 2016, 18, 27939–27950. [Google Scholar] [CrossRef]
- Varadwaj, A.; Varadwaj, P.R.; Yamashita, K. Do surfaces of positive electrostatic potential on different halogen derivatives in molecules attract? like attracting like! J. Comput. Chem. 2018, 39, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; Yamashita, K. Can Combined Electrostatic and Polarization Effects Alone Explain the F⋯F Negative-Negative Bonding in Simple Fluoro-Substituted Benzene Derivatives? A First-Principles Perspective. Computation 2018, 6, 51. [Google Scholar] [CrossRef]
- Varadwaj, A.; Marques, H.M.; Varadwaj, P.R. Is the Fluorine in Molecules Dispersive? Is Molecular Electrostatic Potential a Valid Property to Explore Fluorine-Centered Non-Covalent Interactions? Molecules 2019, 24, 379. [Google Scholar] [CrossRef]
- Dau, T.M.; Asamoah, B.D.; Belyaev, A.; Chakkaradhari, G.; Hirva, P.; Jänis, J.; Grachova, E.V.; Tunik, S.P.; Koshevoy, I.O. Adjustable coordination of a hybrid phosphine–phosphine oxide ligand in luminescent Cu, Ag and Au complexes. Dalton Trans. 2016, 45, 14160–14173. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Kinjo, R. Coordination of Asymmetric Diborenes towards Cationic Coinage Metals (Au, Ag, Cu). Chem. Eur. J. 2018, 24, 15656–15662. [Google Scholar] [CrossRef]
- Rozas, I.; Alkorta, I.; Elguero, J. Behavior of Ylides Containing N, O, and C Atoms as Hydrogen Bond Acceptors. J. Am. Chem. Soc. 2000, 122, 11154–11161. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. A description of the chemical bond in terms of local properties of electron density and energy. Croat. Chem. Acta 1984, 57, 1259–1281. [Google Scholar]
- Mata, I.; Alkorta, I.; Molins, E.; Espinosa, E. Universal Features of the Electron Density Distribution in Hydrogen-Bonding Regions: A Comprehensive Study Involving H⋅⋅⋅X (X=H, C, N, O, F, S, Cl, π) Interactions. Chem. Eur. J. 2010, 16, 2442–2452. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Sanz, G.; Alkorta, I.; Elguero, J. Theoretical study of the HXYH dimers (X, Y = O, S, Se). Hydrogen bonding and chalcogen-chalcogen interactions. Mol. Phys. 2011, 109, 2543–2552. [Google Scholar] [CrossRef]
- Alkorta, I.; Solimannejad, M.; Provasi, P.; Elguero, J. Theoretical Study of Complexes and Fluoride Cation Transfer between N2F+ and Electron Donors. J. Phys. Chem. A 2007, 111, 7154–7161. [Google Scholar] [CrossRef] [PubMed]
- Mathivathanan, L.; Al-Ameed, K.; Lazarou, K.; Trávníček, Z.; Sanakis, Y.; Herchel, R.; McGrady, J.E.; Raptis, R.G. A trigonal prismatic Cu6-pyrazolato complex containing a μ6-F ligand. Dalton Trans. 2015, 44, 20685–20691. [Google Scholar] [CrossRef]
- Shi, K.; Mathivathanan, L.; Raptis, R.G. Crystal structure of [mu]6-chlorido-nonakis([mu]-4-chloropyrazolato)bis-[mu]3-methoxo-hexacopper(II). Acta Crystallogr. E 2017, 73, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Mezei, G.; Raptis, R.G. Effect of pyrazole-substitution on the structure and nuclearity of Cu(II)-pyrazolato complexes. Inorg. Chim. Acta 2004, 357, 3279–3288. [Google Scholar] [CrossRef]
- Mezei, G.; Raptis, R.G.; Telser, J. Trinuclear, Antiferromagnetically Coupled CuII Complex with an EPR Spectrum of Mononuclear CuII: Effect of Alcoholic Solvents. Inorg. Chem. 2006, 45, 8841–8843. [Google Scholar] [CrossRef]
- Angaridis, P.A.; Baran, P.; Boča, R.; Cervantes-Lee, F.; Haase, W.; Mezei, G.; Raptis, R.G.; Werner, R. Synthesis and Structural Characterization of Trinuclear CuII−Pyrazolato Complexes Containing μ3-OH, μ3-O, and μ3-Cl Ligands. Magnetic Susceptibility Study of [PPN]2[(μ3-O)Cu3(μ-pz)3Cl3]. Inorg. Chem. 2002, 41, 2219–2228. [Google Scholar] [CrossRef]
- Surmann, S.A.; Mezei, G. Halogen-bonded network of trinuclear copper(II) 4-iodopyrazolate complexes formed by mutual breakdown of chloroform and nanojars. Acta Crystallogr. E 2016, 72, 1517–1520. [Google Scholar] [CrossRef]
- Kamiyama, A.; Kajiwara, T.; Ito, T. Cage-Type Hexacopper(II) Complex Formed by Chloride Template. Chem. Lett. 2002, 31, 980–981. [Google Scholar]
- Boudalis, A.K.; Rogez, G.; Heinrich, B.; Raptis, R.G.; Turek, P. Towards ionic liquids with tailored magnetic properties: Bmim+ salts of ferro- and antiferromagnetic CuII3 triangles. Dalton Trans. 2017, 46, 12263–12273. [Google Scholar] [CrossRef]
- Boča, R.; Dlháň, L.; Mezei, G.; Ortiz-Pérez, T.; Raptis, R.G.; Telser, J. Triangular, Ferromagnetically-Coupled CuII3−Pyrazolato Complexes as Possible Models of Particulate Methane Monooxygenase (pMMO). Inorg. Chem. 2003, 42, 5801–5803. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian-Basis Sets for Use in Correlated Molecular Calculations .1. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Peterson, K.A.; Puzzarini, C. Systematically convergent basis sets for transition metals. II. Pseudopotential-based correlation consistent basis sets for the group 11 (Cu, Ag, Au) and 12 (Zn, Cd, Hg) elements. Theor. Chem. Acc. 2005, 114, 283–296. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 c01; Gaussian. Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Popelier, P.L.A. Atoms In Molecules; An introduction; Prentice Hall: Harlow, UK, 2000. [Google Scholar]
- Keith, T.A. In TK Gristmill Software, Overland Park, KS, USA. Version 17.11.14 B. Available online: aim.tkgristmill.com (accessed on 1 June 2020).
- Jmol: An open-Source Java Viewer for Chemical Structures in 3D. Available online: http://www.jmol.org/ (accessed on 1 June 2020).
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B 2016, 72, 171–179. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| F− | 1:1 Apical | 1:1 Planar | 1:1 CH (4) |
|---|---|---|---|
| Au | −107.4 | −120.4 | −87.0 |
| Ag | −195.5 | −153.3 | −73.7 |
| Cu | −171.8 | −204.0 | −76.8 |
| Cl− | |||
| Au | −74.4 | −88.9 | −47.1 |
| Ag | −143.0 | −116.8 | −38.4 |
| Cu | −121.2 | −111.9 | −40.7 |
| Br− | |||
| Au | −78.2 | −86.7 | −43.2 |
| Ag | −142.3 | −113.2 | −35.2 |
| Cu | −112.9 | −102.0 | −37.3 |
| F− | Cl− | Br− | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Complex | Dist. | Au | Ag | Cu | Au | Ag | Cu | Au | Ag | Cu |
| Apical | M⋯X | 2.558 | 2.379 | 2.135 | 3.017 | 2.758 | 2.556 | 3.127 | 2.856 | 2.692 |
| Planar | M⋯X | 2.434 | 2.256 | 2.016 | 2.956 | 2.623 | 2.430 | 3.088 | 2.722 | 2.575 |
| H(3)⋯X | 2.336 | 2.483 | 2.279 | 2.614 | 2.790 | 2.575 | 2.706 | 2.889 | 2.665 | |
| CH(4) | H(4)⋯X | 1.508 | 1.554 | 1.551 | 2.317 | 2.366 | 2.357 | 2.507 | 2.556 | 2.547 |
| 1:2 Apical | 1:2 Planar | 1:2 CH(4) | |
|---|---|---|---|
| F(-) | |||
| Au | 113.1 | −35.7 | −39.6 |
| Ag | −46.4 | −79.0 | −18.6 |
| Cu | −34.7 | −82.3 | −20.3 |
| Cl(-) | |||
| Au | 139.5 | 23.9 | 15.8 |
| Ag | 23.3 | −25.8 | 29.2 |
| Cu | 93.5 | 2.1 | 28.1 |
| Br(-) | |||
| Au | 120.4 | 20.2 | 18.8 |
| Ag | 15.3 | −24.0 | 31.2 |
| Cu | 94.1 | 10.6 | 30.3 |
| F− | Cl− | Br− | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Complex | Dist. | Au | Ag | Cu | Au | Ag | Cu | Au | Ag | Cu |
| Apical | M⋯X | 2.613 | 2.437 | 2.211 | 3.226 | 2.845 | 2.707 | 3.339 | 2.949 | 2.892 |
| Planar | M⋯X | 4.424 a | 2.327 | 2.090 | 3.325 | 2.734 | 2.639 | 3.455 | 2.849 | 2.962 |
| H(3)⋯·X b | 1.592 | 2.179 | 2.260 | 2.591 | 2.720 | 2.550 | 2.727 | 2.827 | 2.640 | |
| CH(4) | H(4)⋯·X | 1.619 | 1.660 | 1.659 | 2.478 | 2.538 | 2.532 | 2.696 | 2.775 | 2.756 |
| F− | Cl− | Br− | ||||
|---|---|---|---|---|---|---|
| 1:3 Planar | 1:3 CH(4) | 1:3 Planar | 1:3 CH(4) | 1:3 Planar | 1:3 CH(4) | |
| Au | Dissociation | 136.2 | Dissociation | 188.1 | Dissociation | 188.2 |
| Ag | 224.6 | 160.3 | 260.2 | 203.1 | 252.3 | 201.9 |
| Cu | 252.1 | 163.9 | Dissociation | 206.3 | Dissociation | 205.0 |
| F− | Cl− | Br− | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Complex | Dist. | Au | Ag | Cu | Au | Ag | Cu | Au | Ag | Cu |
| Planar | M⋯X | 2.411 | 2.211 | 3.025 | 3.238 | |||||
| H(3)⋯·X | 2.461 | 2.387 | 2.863 | 2.942 | ||||||
| CH(4) | H(4)⋯·X | 1.735 | 1.777 | 1.781 | 2.651 | 2.740 | 2.743 | 2.864 | 2.969 | 2.970 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkorta, I.; Elguero, J.; Trujillo, C.; Sánchez-Sanz, G. Interaction between Trinuclear Regium Complexes of Pyrazolate and Anions, a Computational Study. Int. J. Mol. Sci. 2020, 21, 8036. https://doi.org/10.3390/ijms21218036
Alkorta I, Elguero J, Trujillo C, Sánchez-Sanz G. Interaction between Trinuclear Regium Complexes of Pyrazolate and Anions, a Computational Study. International Journal of Molecular Sciences. 2020; 21(21):8036. https://doi.org/10.3390/ijms21218036
Chicago/Turabian StyleAlkorta, Ibon, José Elguero, Cristina Trujillo, and Goar Sánchez-Sanz. 2020. "Interaction between Trinuclear Regium Complexes of Pyrazolate and Anions, a Computational Study" International Journal of Molecular Sciences 21, no. 21: 8036. https://doi.org/10.3390/ijms21218036
APA StyleAlkorta, I., Elguero, J., Trujillo, C., & Sánchez-Sanz, G. (2020). Interaction between Trinuclear Regium Complexes of Pyrazolate and Anions, a Computational Study. International Journal of Molecular Sciences, 21(21), 8036. https://doi.org/10.3390/ijms21218036