Trametinib Induces the Stabilization of a Dual GNAQ p.Gly48Leu- and FGFR4 p.Cys172Gly-Mutated Uveal Melanoma. The Role of Molecular Modelling in Personalized Oncology

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
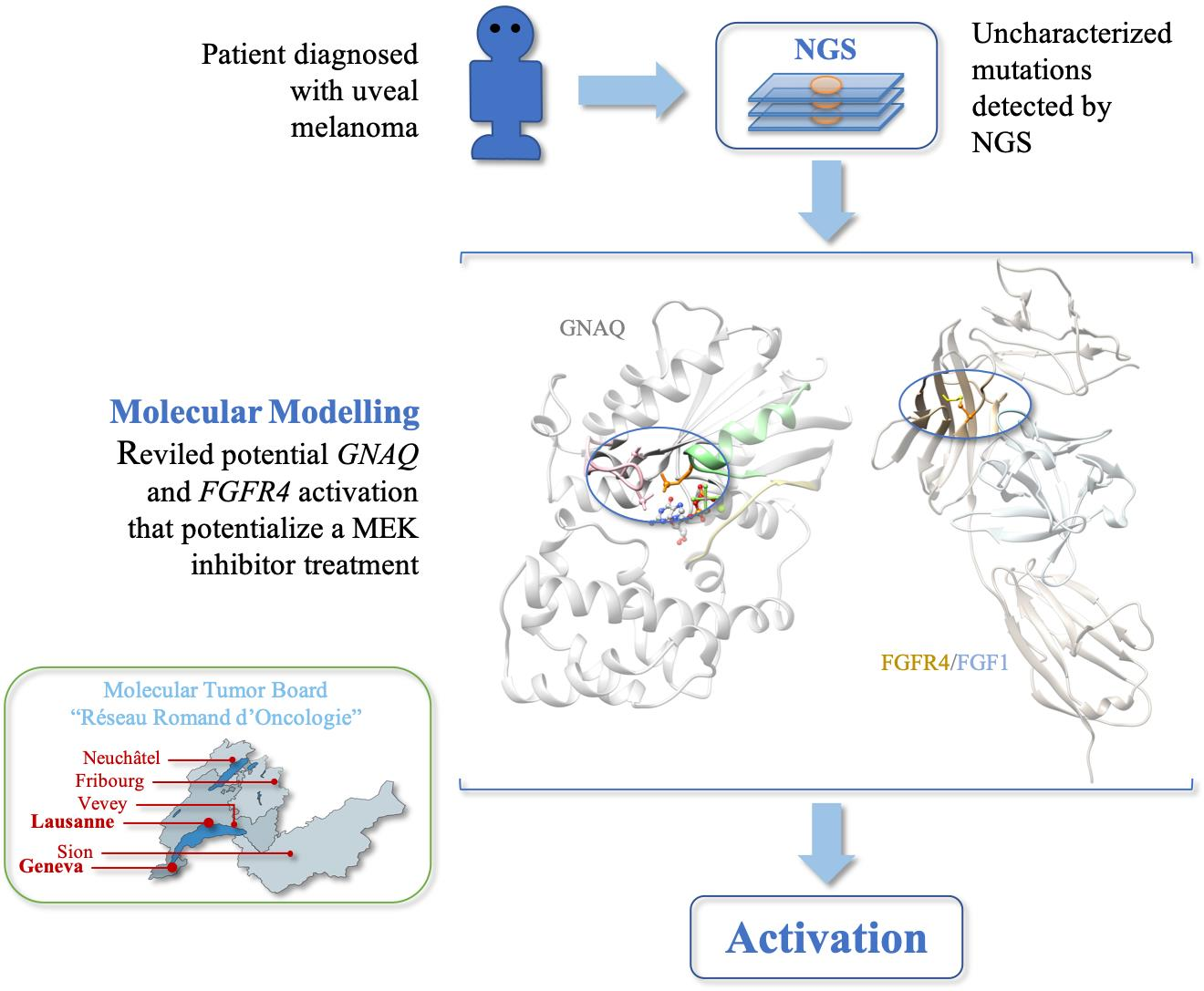
2. Results
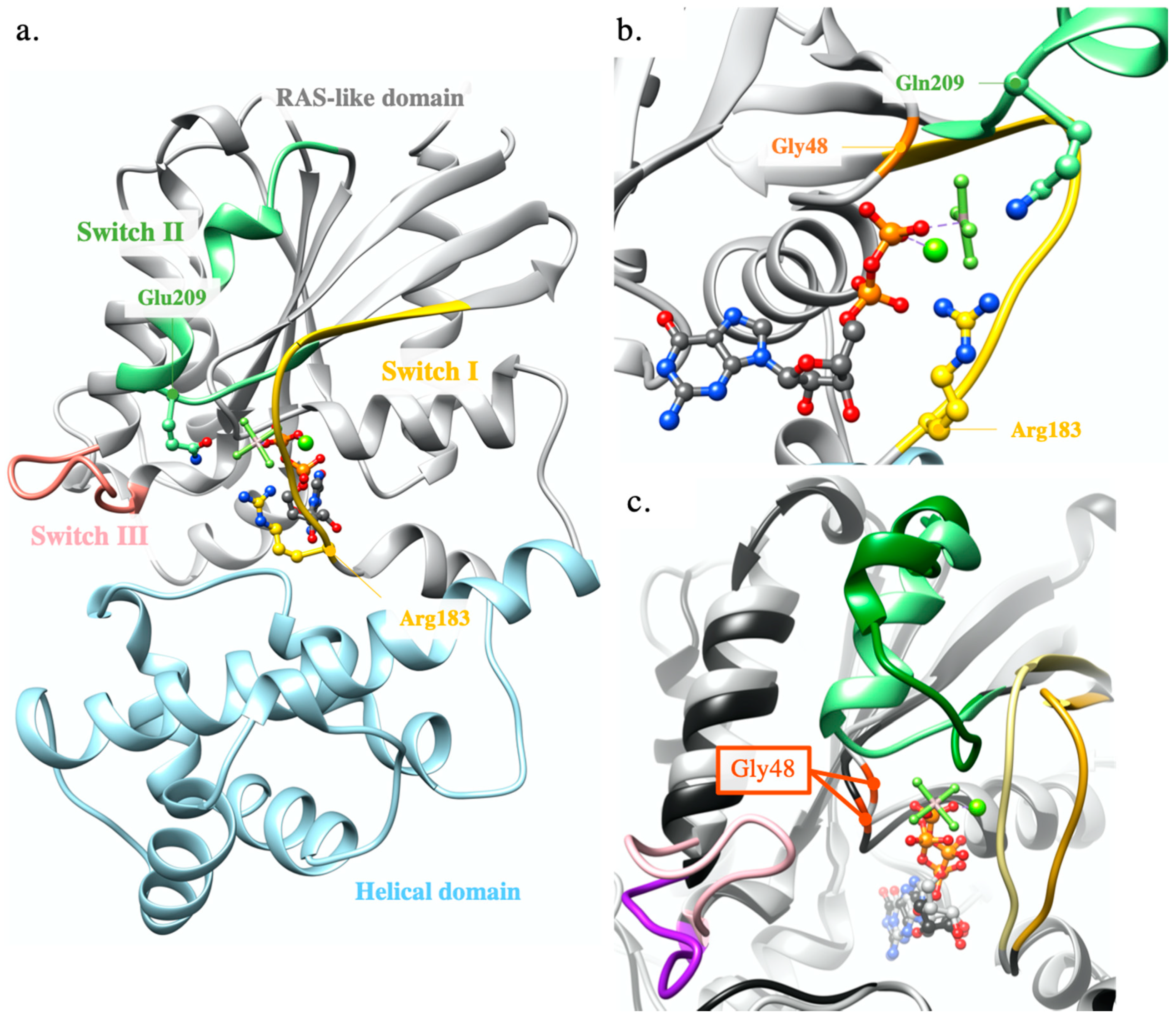
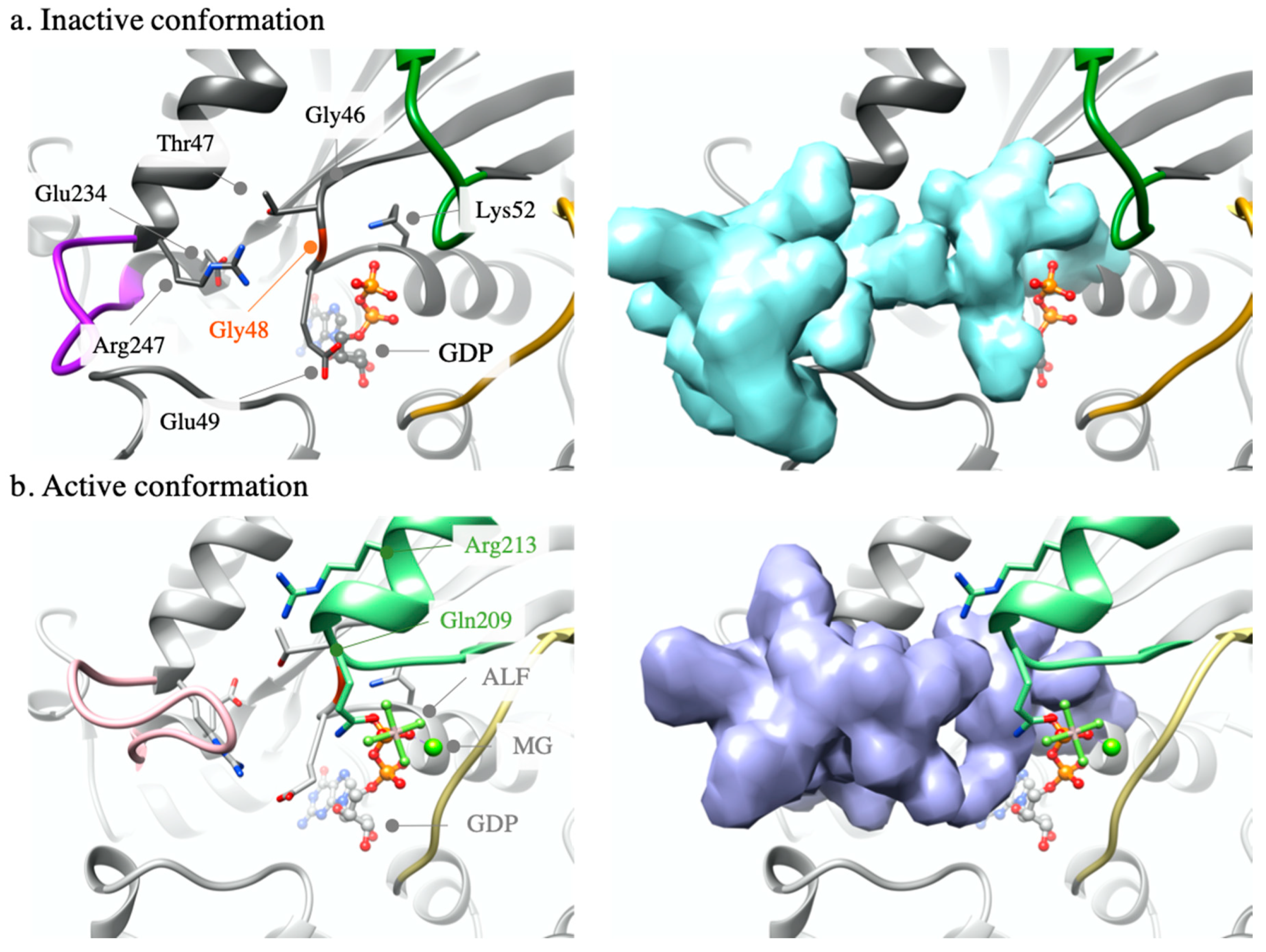
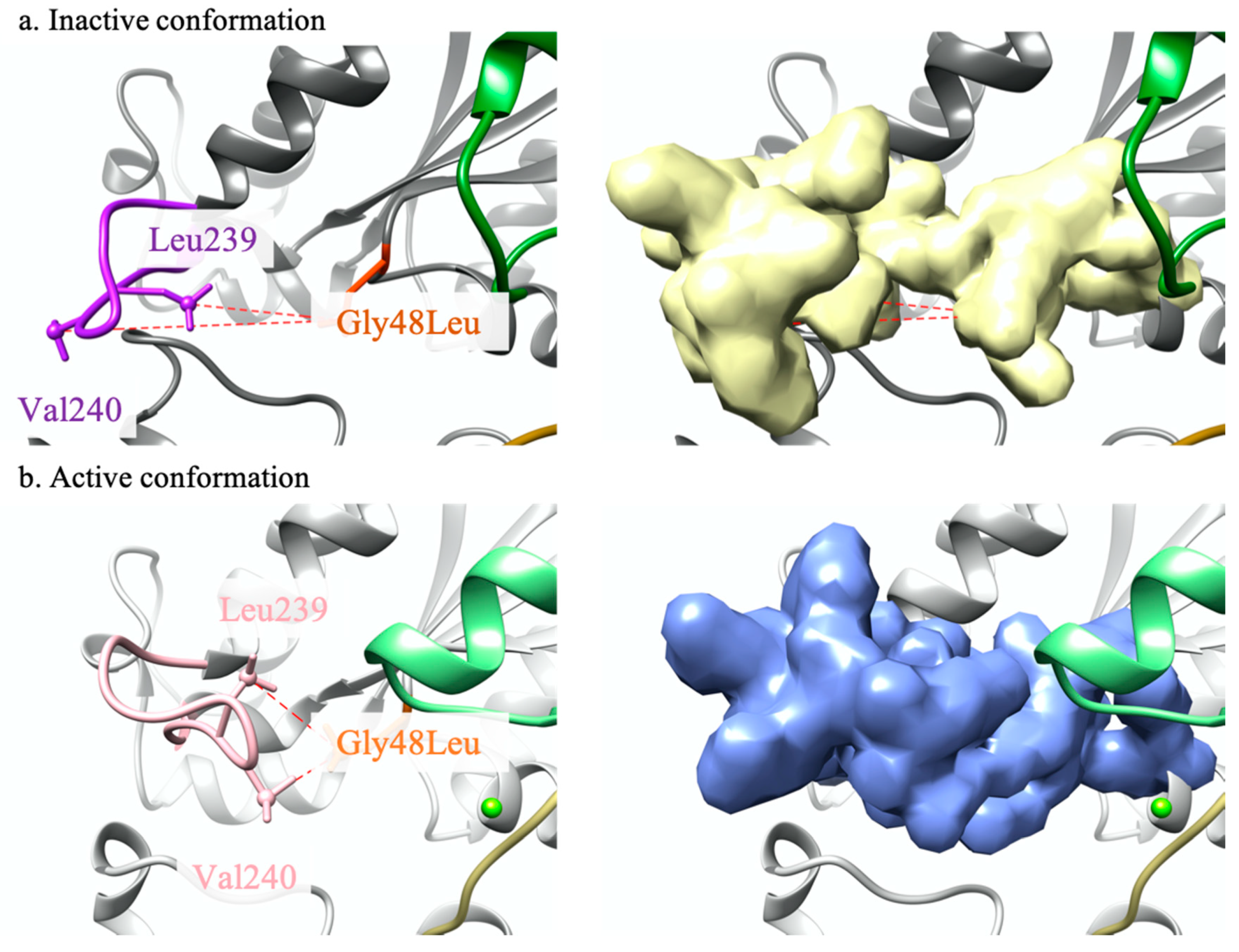
2.1. Modelling Analysis
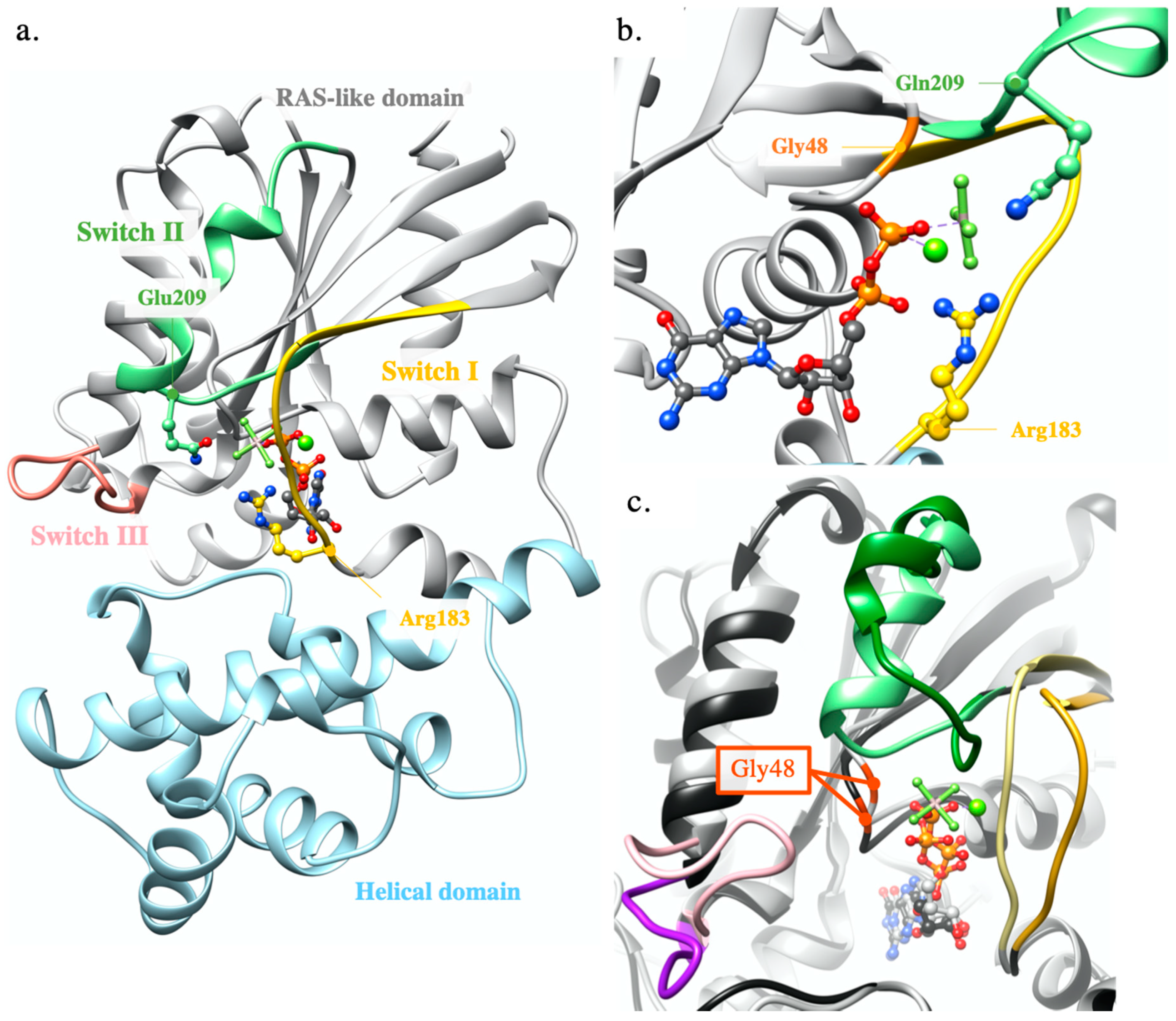
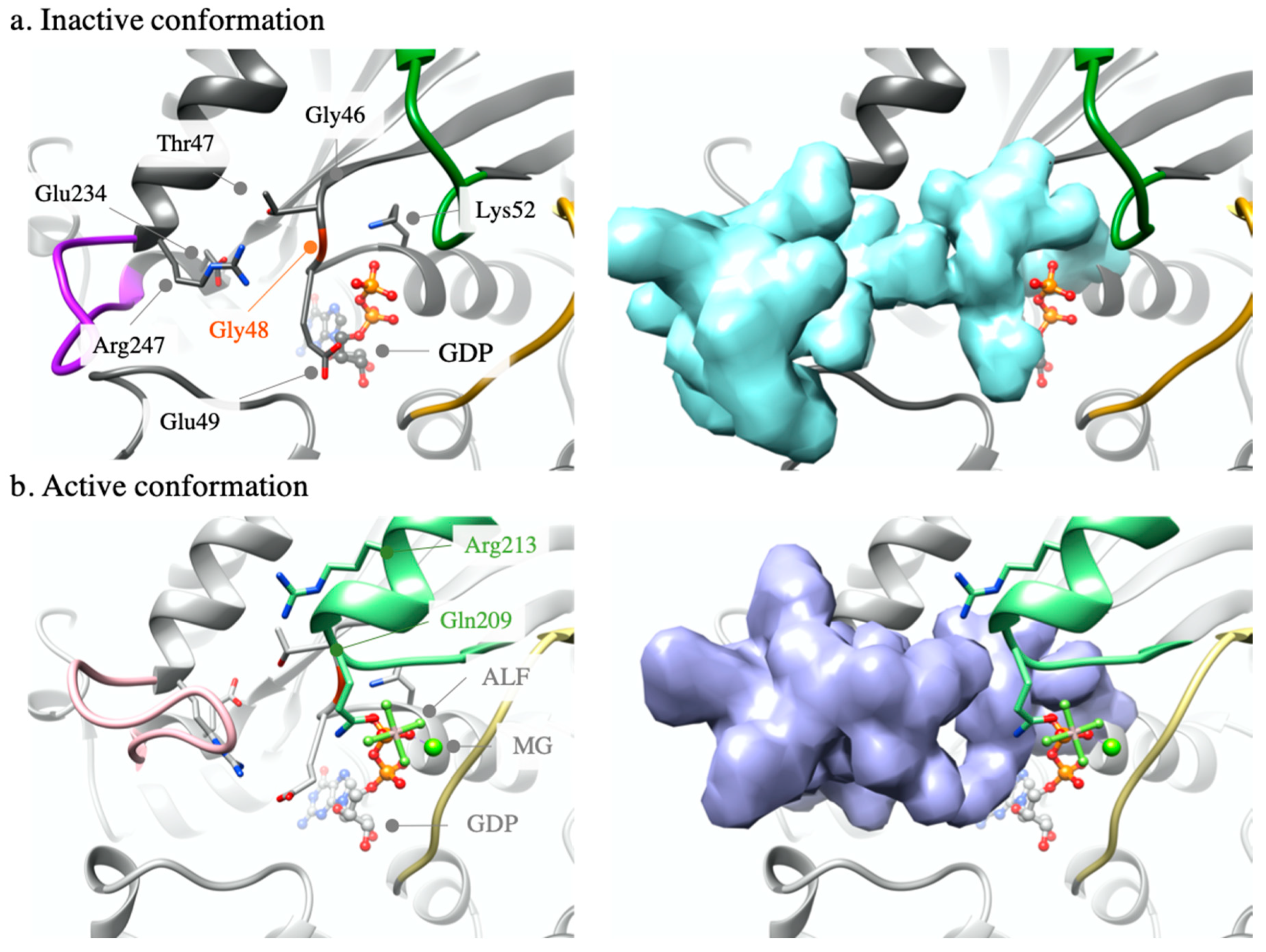
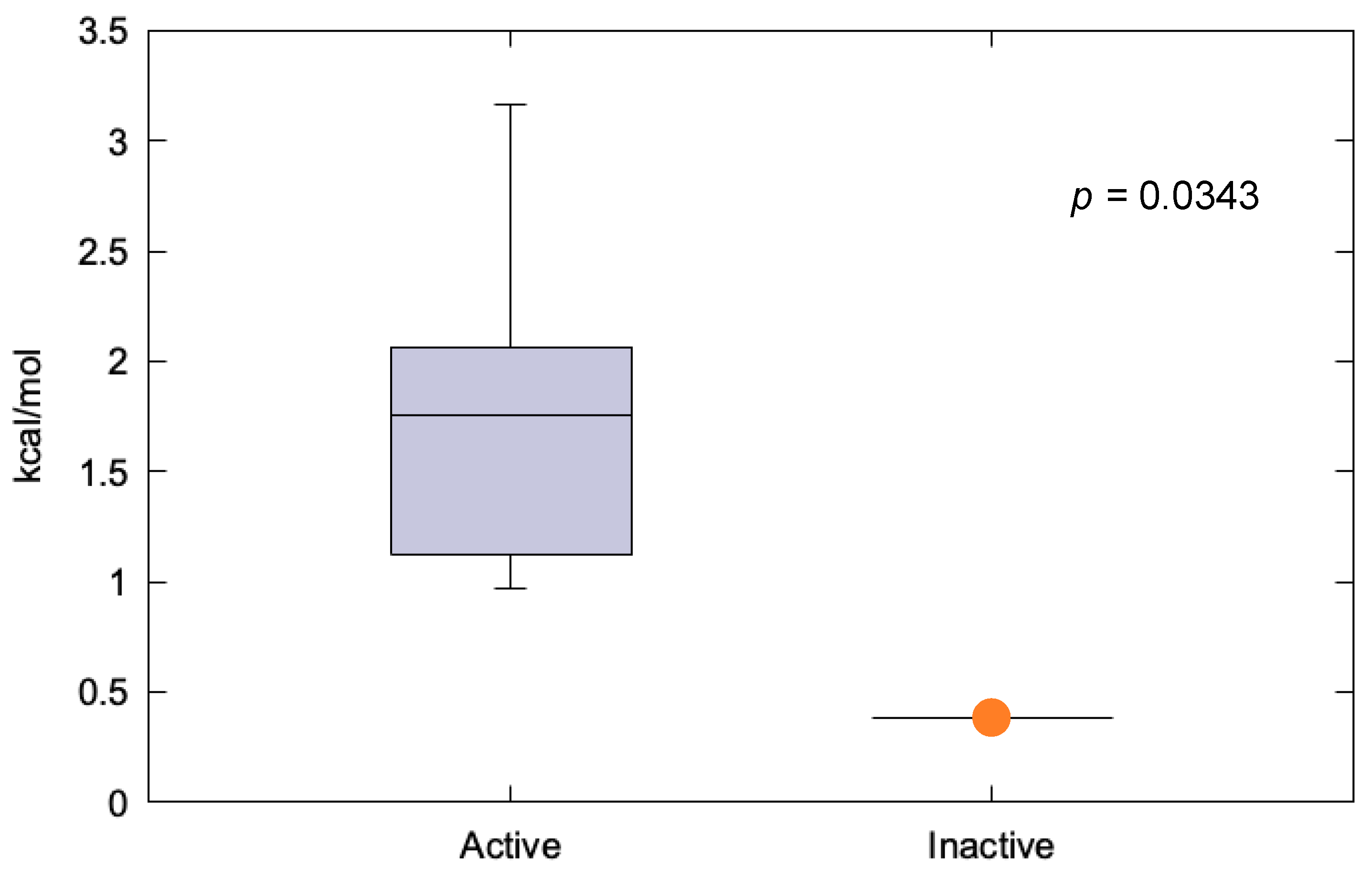
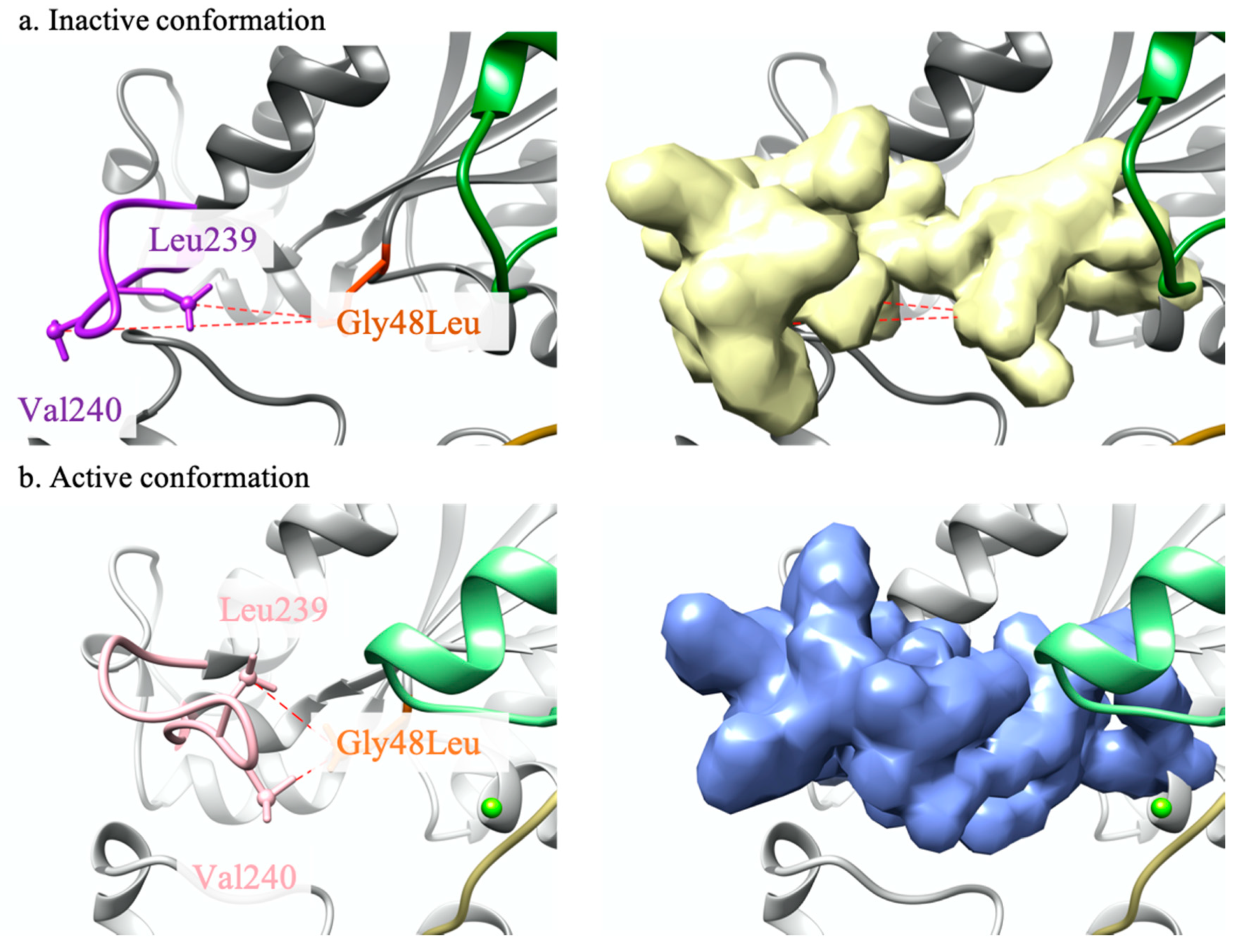
2.1.1. GNAQ p.Gly48Leu
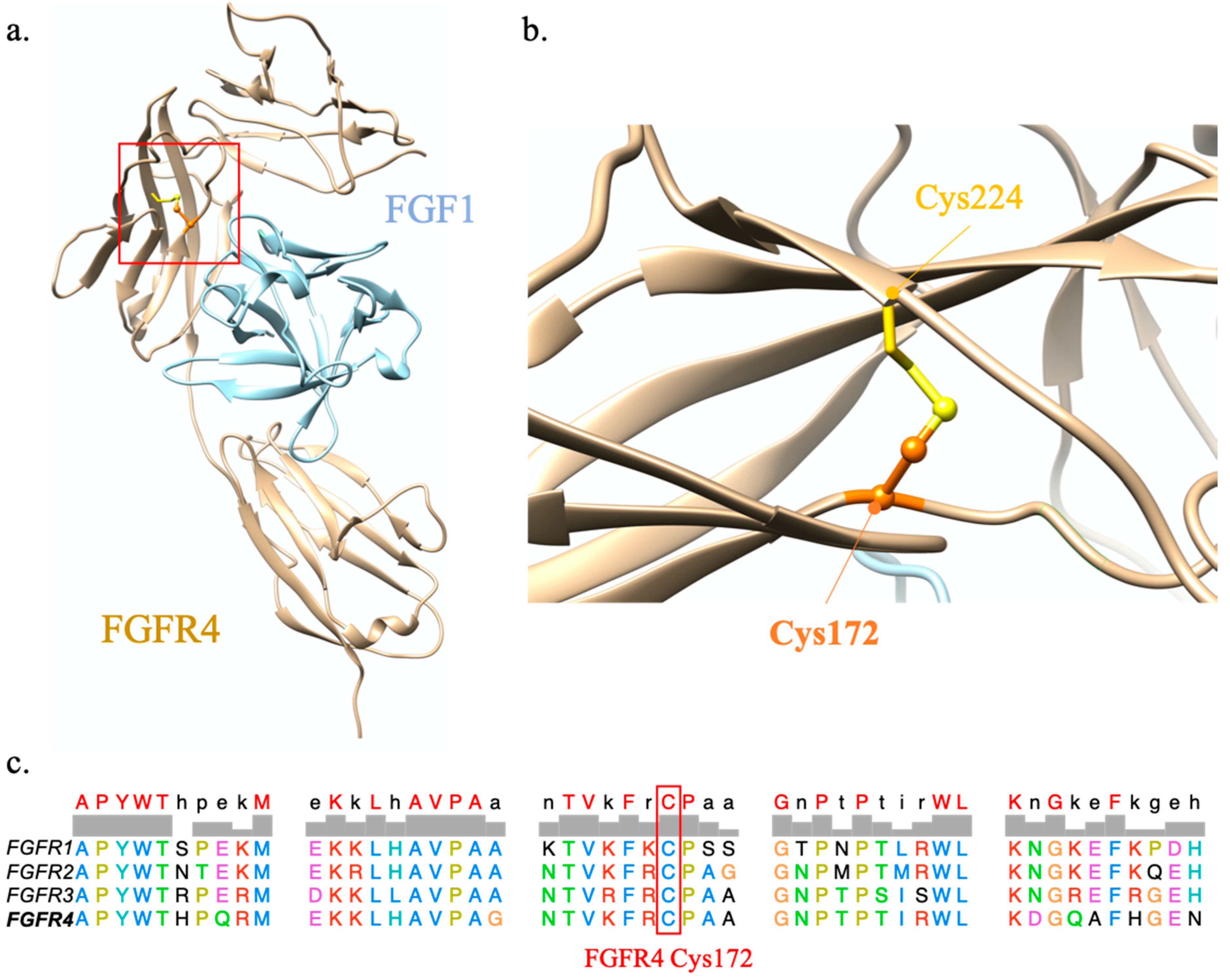
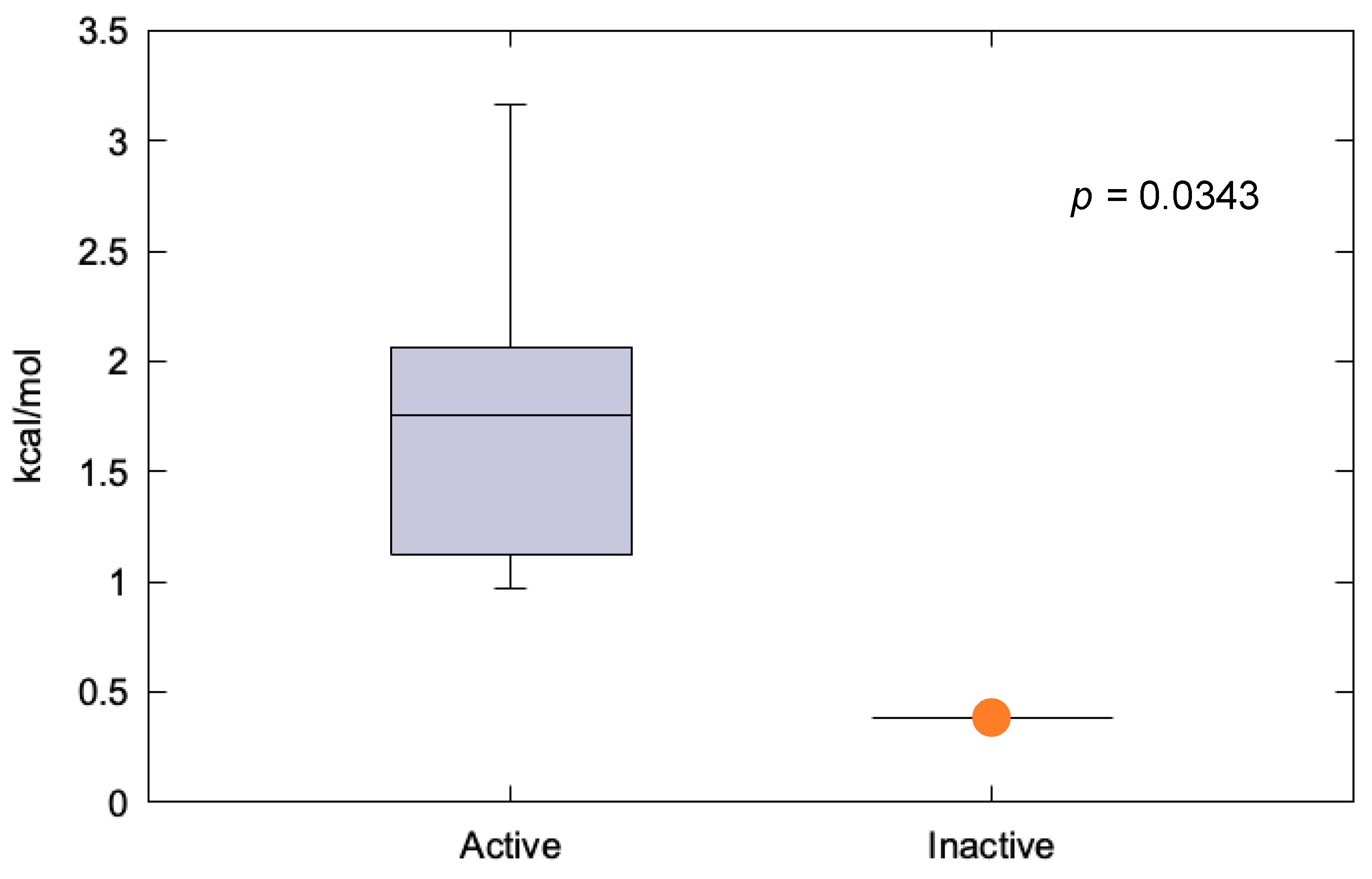
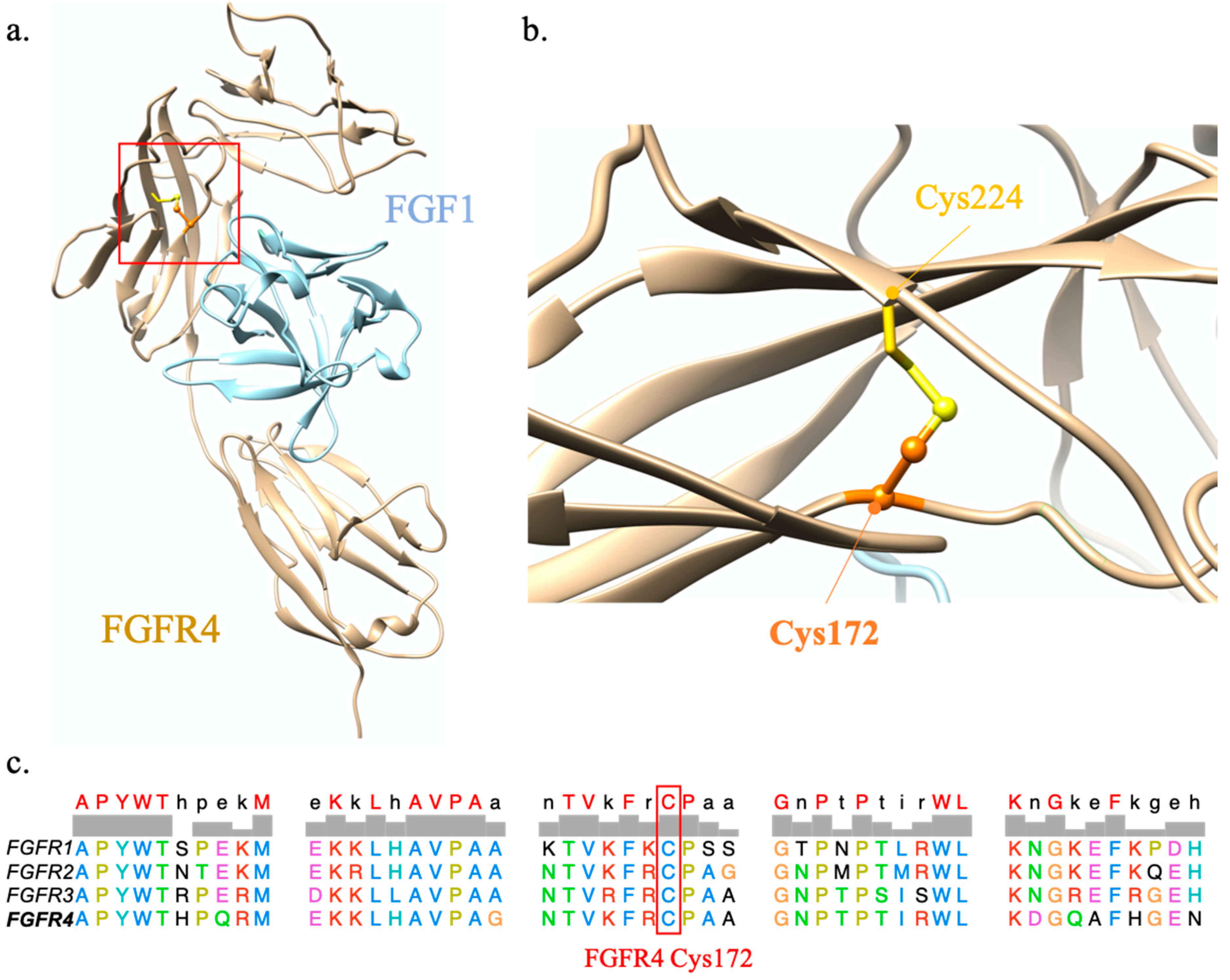
2.1.2. FGFR4 p.Cys172Gly
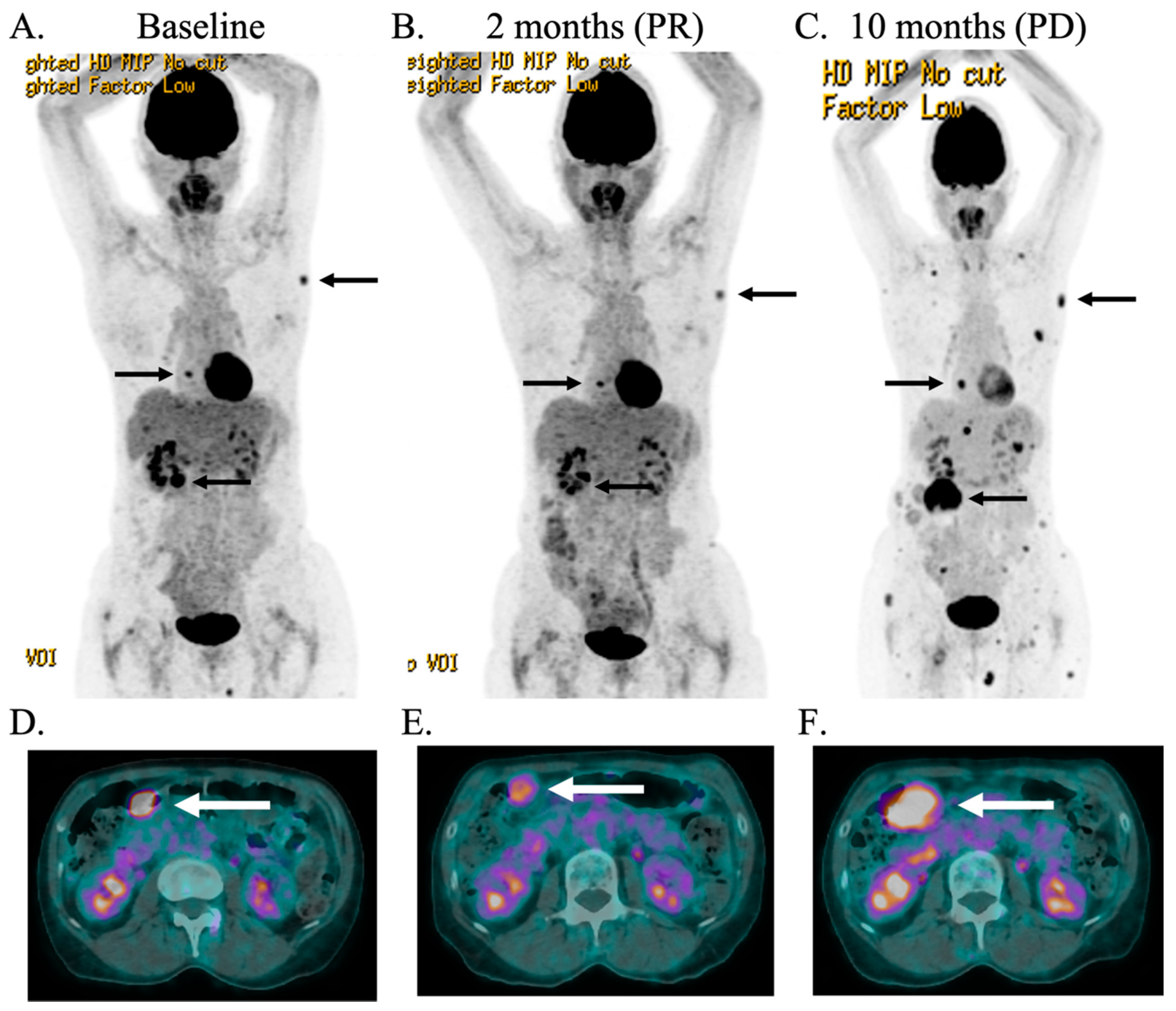
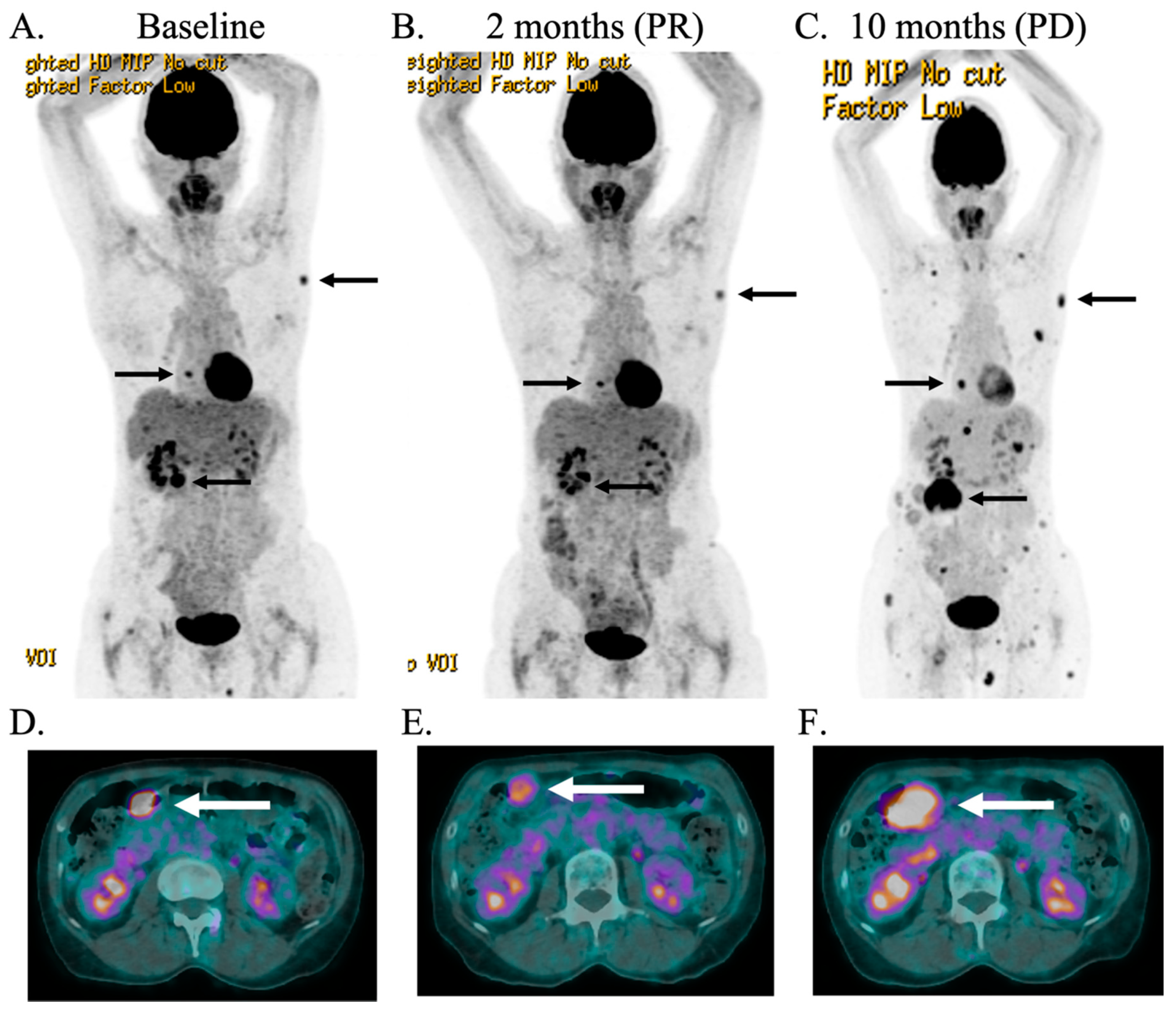
2.2. Case Description
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AJCC | American Joint Committee on Cancer |
| BAM | Binary alignment map |
| FFPE | Formalin-fixed paraffin-embedded |
| FGF | Fibroblast growth factors |
| FGFR | Fibroblast growth factor receptor |
| Gαi | Guanine nucleotide-binding protein G(i) subunit alpha-1 |
| Gαq | Guanine nucleotide-binding protein G(q) subunit alpha chimeric protein |
| GPCR | G protein-coupled receptor family |
| GDP | Guanosine 5′-diphosphate |
| GTP | Guanosine 5′-triphosphate |
| MRI | Magnetic resonance imaging |
| MTB | Molecular tumor board |
| NEMO | NRAS-mutant melanoma |
| NGS | Next generation sequencing |
| PET-CT | Positron emission tomography-computed tomography |
| PDB | Protein Data Bank |
| SRS | Stereotactic radiosurgery |
| SUV | Standardized uptake value |
| SW | Switch region |
| TMB | Tumor mutation burden |
| TPS | Tumor Proportion Score |
References
- Lundstrom, K. An overview on GPCRs and Drug Discovery: Structure-based Drug Design and Structural Biology on GPCRs. In G Protein-Coupled Receptors in Drug Discovery; Leifert, W., Ed.; Humana Press: Totowa, NJ, USA, 2009; pp. 51–66. [Google Scholar] [CrossRef]
- Syrovatkina, V.; Alegre, K.O.; Dey, R.; Huang, X.Y. Regulation, Signaling, and Physiological Functions of G-Proteins. J. Mol. Biol. 2016, 428, 3850–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neves, S.R.; Ram, P.T.; Iyengar, R. G protein pathways. Science 2002, 296, 1636–1639. [Google Scholar] [CrossRef] [PubMed]
- Shoushtari, A.N.; Carvajal, R.D. GNAQ and GNA11 mutations in uveal melanoma. Melanoma Res. 2014, 24, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Decatur, C.L.; Ong, E.; Garg, N.; Anbunathan, H.; Bowcock, A.M.; Field, M.G.; Harbour, J.W. Driver mutations in uveal melanoma associations with gene expression profile and patient outcomes. JAMA Ophthalmol. 2016, 134, 728–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldo, G.L.; Ricks, T.K.; Hicks, S.N.; Cheever, M.L.; Kawano, T.; Tsuboi, K.; Wang, X.; Montell, C.; Kozasa, T.; Sondek, J.; et al. Kinetic scaffolding mediated by a phospholipase C-β and Gq signaling complex. Science 2010, 330, 974–980. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, A.; Kitano, K.; Takasaki, J.; Taniguchi, M.; Mizuno, N.; Tago, K.; Hakoshima, T.; Itoh, H. Structural basis for the specific inhibition of heterotrimeric G q protein by a small molecule. Proc. Natl. Acad. Sci. USA 2010, 107, 13666–13671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, B.; Breeze, A.L. Structure, activation and dysregulation of fibroblast growth factor receptor kinases: Perspectives for clinical targeting. Biochem. Soc. Trans. 2018, 46, 1753–1770. [Google Scholar] [CrossRef]
- Chen, X.; Wu, Q.; Tan, L.; Porter, D.; Jager, M.J.; Emery, C.; Bastian, B.C. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene 2014, 33, 4724–4734. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Tian, H. Current development status of MEK inhibitors. Molecules 2017, 22, 1551. [Google Scholar] [CrossRef] [Green Version]
- Johansson, P.; Aoude, L.G.; Wadt, K.; Glasson, W.J.; Warrier, S.K.; Hewitt, A.W.; Kiilgaard, J.F.; Heegaard, S.; Isaacs, T.; Franchina, M.; et al. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget 2016, 7, 4624–4631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.T.; Asthana, S.; Gao, S.P.; Lee, B.H.; Chapman, J.S.; Kandoth, C.; Gao, J.J.; Socci, N.D.; Solit, D.B.; Olshen, A.B.; et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 2016, 34, 155–163. [Google Scholar] [CrossRef]
- Chang, M.T.; Bhattarai, T.S.; Schram, A.M.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; Chakravarty, D.; Phillips, S.; Kandoth, C.; Penson, A.; et al. Accelerating discovery of functional mutant alleles in cancer. Cancer Discov. 2018, 8, 174–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakhoum, M.F.; Esmaeli, B. Molecular characteristics of uveal melanoma: Insights from the cancer genome atlas (TCGA) project. Cancers 2019, 11, 1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liau, J.-Y.; Lee, J.-C.; Tsai, J.-H.; Chen, C.-C.; Chung, Y.-C.; Wang, Y.-H. High frequency of GNA14, GNAQ, and GNA11 mutations in cherry hemangioma: A histopathological and molecular study of 85 cases indicating GNA14 as the most commonly mutated gene in vascular neoplasms. Mod. Pathol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Joseph, N.M.; Brunt, E.M.; Marginean, C.; Nalbantoglu, I.L.K.; Snover, D.C.; Thung, S.N.; Yeh, M.M.; Umetsu, S.E.; Ferrell, L.D.; Gill, R.M. Frequent GNAQ and GNA14 mutations in hepatic small vessel neoplasm. Am. J. Surg. Pathol. 2018, 42, 1201–1207. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, 382–388. [Google Scholar] [CrossRef] [Green Version]
- Potapov, V.; Cohen, M.; Schreiber, G. Assessing computational methods for predicting protein stability upon mutation: Good on average but not in the details. Protein Eng. Des. Sel. 2009, 22, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Huhtala, M.T.; Pentikainen, O.T.; Johnson, M.S. A dimeric ternary complex of FGFR1, heparin and FGF-1 leads to an “electrostatic sandwich” model for heparin binding. Structure 1999, 7, 699–709. [Google Scholar] [CrossRef] [Green Version]
- Sarabipour, S.; Hristova, K. Pathogenic Cysteine Removal Mutations in FGFR Extracellular Domains Stabilize Receptor Dimers and Perturb the TM Dimer Structure. J. Mol. Biol. 2016, 428, 3903–3910. [Google Scholar] [CrossRef] [Green Version]
- Cleary, J.M.; Raghavan, S.; Li, Y.Y.; Spurr, L.; Wu, Q.; Shi, L.; Brais, L.K.; Odhiambo, Z.; Goyal, L.; Patel, A.K.; et al. Therapeutic targeting of extracellular FGFR2 activating deletions in intrahepatic cholangiocarcinoma. J. Clin. Oncol. 2020, 38, 567. [Google Scholar] [CrossRef]
- Fancello, L.; Gandini, S.; Pelicci, P.G.; Mazzarella, L. Tumor mutational burden quantification from targeted gene panels: Major advancements and challenges. J. Immunother. Cancer 2019, 7, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvajal, R.D.; Piperno-Neumann, S.; Kapiteijn, E.; Chapman, P.B.; Frank, S.; Joshua, A.M.; Piulats, J.M.; Wolter, P.; Cocquyt, V.; Chmielowski, B.; et al. Selumetinib in combination with dacarbazine in patients with metastatic uveal melanoma: A Phase III, Multicenter, Randomized Trial (SUMIT). J. Clin. Oncol. 2018, 36, 1232–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvajal, R.D.; Schwartz, G.K.; Mann, H.; Smith, I.; Nathan, P.D. Study design and rationale for a randomised, placebo-controlled, double-blind study to assess the efficacy of selumetinib (AZD6244; ARRY-142886) in combination with dacarbazine in patients with metastatic uveal melanoma (SUMIT). BMC Cancer 2015, 15, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Shoushtari, A.N.; Kudchadkar, R.R.; Panageas, K.; Murthy, R.K.; Jung, M.; Shah, R.; O’Donnell, B.; Khawaja, T.T.; Shames, Y.; Prempeh-Keteku, N.A.; et al. A randomized phase 2 study of trametinib with or without GSK2141795 in patients with advanced uveal melanoma. J. Clin. Oncol. 2016, 34, 9511. [Google Scholar] [CrossRef]
- Dummer, R.; Schadendorf, D.; Ascierto, P.A.; Arance, A.; Dutriaux, C.; Giacomo, A.M.D.; Rutkowski, P.; Vecchio, M.D.; Gutzmer, R.; Mandala, M.; et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 435–445. [Google Scholar] [CrossRef]
- Kuske, M.; Westphal, D.; Wehner, R.; Schmitz, M.; Beissert, S.; Praetorius, C.; Meier, F. Immunomodulatory effects of BRAF and MEK inhibitors: Implications for Melanoma therapy. Pharmacol. Res. 2018, 136, 151–159. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.C. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Apweiler, R.; Alpi, E.; Antunes, A.; Arganiska, J.; Bely, B.; Bingley, M.; et al. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, 204–212. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesmer, V.M.; Kawano, T.; Shankaranarayanan, A.; Kozasa, T.; Tesmer, J.J.G. Snapshot of Activated G Proteins at the Membrane: The Gαq-GRK2-Gßγ Complex. Science 2005, 310, 1686–1690. [Google Scholar] [CrossRef]
- Lyon, A.M.; Dutta, S.; Boguth, C.A.; Skiniotis, G.; Tesmer, J.J.G. Full-length Gαq-phospholipase C-β3 structure reveals interfaces of the C-terminal coiled-coil domain. Nat. Struct. Mol. Biol. 2013, 20, 355–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyon, A.M.; Tesmer, J.J.G. Structural insights into phospholipase C-β function. Mol. Pharmacol. 2013, 84, 488–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, V.G.; Bommarito, P.A.; Tesmer, J.J.G. Structure of the Regulator of G Protein Signaling 8 (RGS8)-Gα q Complex. J. Biol. Chem. 2016, 291, 5138–5145. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krebs, F.S.; Gérard, C.; Wicky, A.; Aedo-Lopez, V.; Missiaglia, E.; Bisig, B.; Trimech, M.; Michielin, O.; Homicsko, K.; Zoete, V. Trametinib Induces the Stabilization of a Dual GNAQ p.Gly48Leu- and FGFR4 p.Cys172Gly-Mutated Uveal Melanoma. The Role of Molecular Modelling in Personalized Oncology. Int. J. Mol. Sci. 2020, 21, 8021. https://doi.org/10.3390/ijms21218021
Krebs FS, Gérard C, Wicky A, Aedo-Lopez V, Missiaglia E, Bisig B, Trimech M, Michielin O, Homicsko K, Zoete V. Trametinib Induces the Stabilization of a Dual GNAQ p.Gly48Leu- and FGFR4 p.Cys172Gly-Mutated Uveal Melanoma. The Role of Molecular Modelling in Personalized Oncology. International Journal of Molecular Sciences. 2020; 21(21):8021. https://doi.org/10.3390/ijms21218021
Chicago/Turabian StyleKrebs, Fanny S., Camille Gérard, Alexandre Wicky, Veronica Aedo-Lopez, Edoardo Missiaglia, Bettina Bisig, Mounir Trimech, Olivier Michielin, Krisztian Homicsko, and Vincent Zoete. 2020. "Trametinib Induces the Stabilization of a Dual GNAQ p.Gly48Leu- and FGFR4 p.Cys172Gly-Mutated Uveal Melanoma. The Role of Molecular Modelling in Personalized Oncology" International Journal of Molecular Sciences 21, no. 21: 8021. https://doi.org/10.3390/ijms21218021