The P72R Polymorphism in R248Q/W p53 Mutants Modifies the Mutant Effect on Epithelial to Mesenchymal Transition Phenotype and Cell Invasion via CXCL1 Expression


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
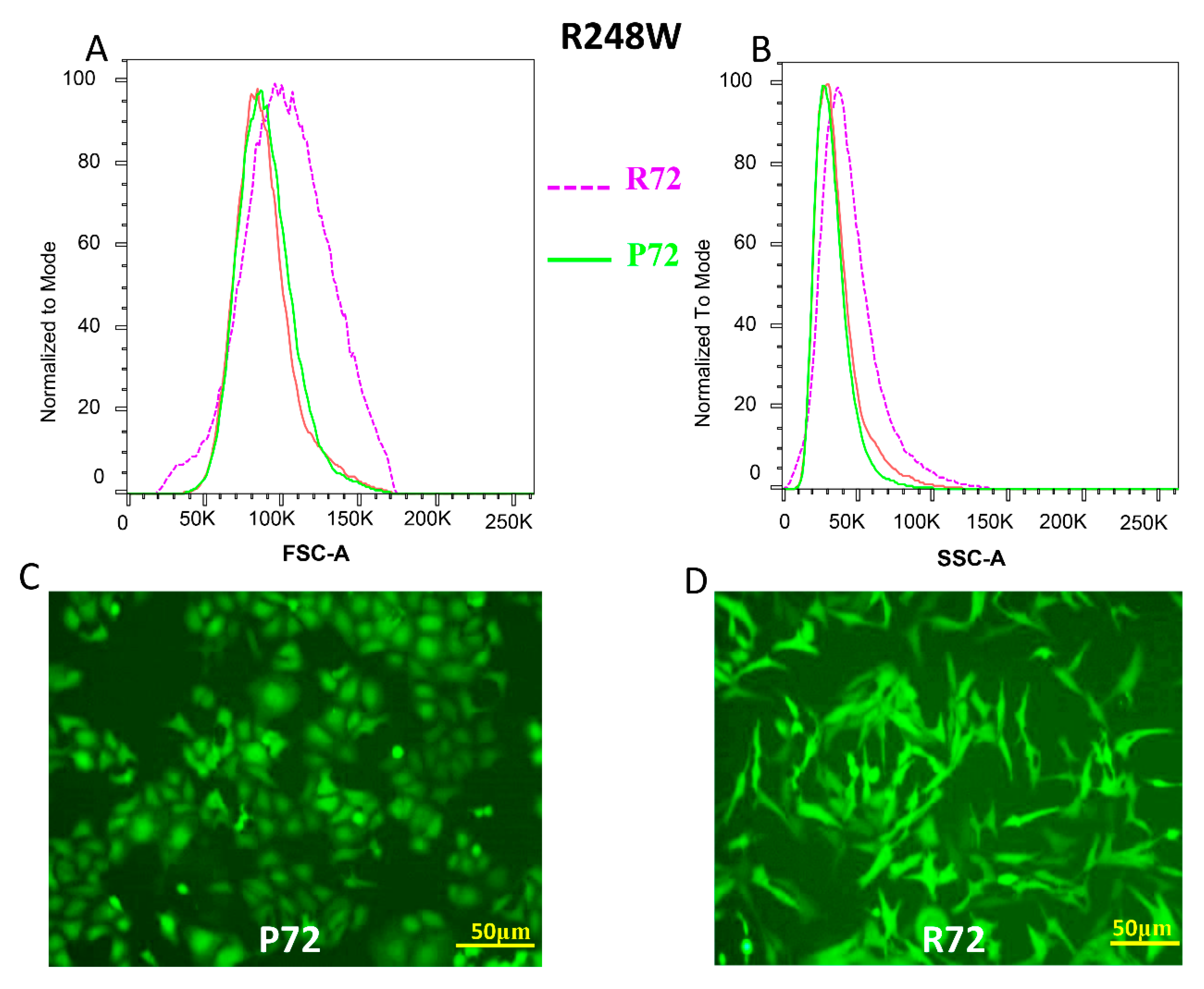
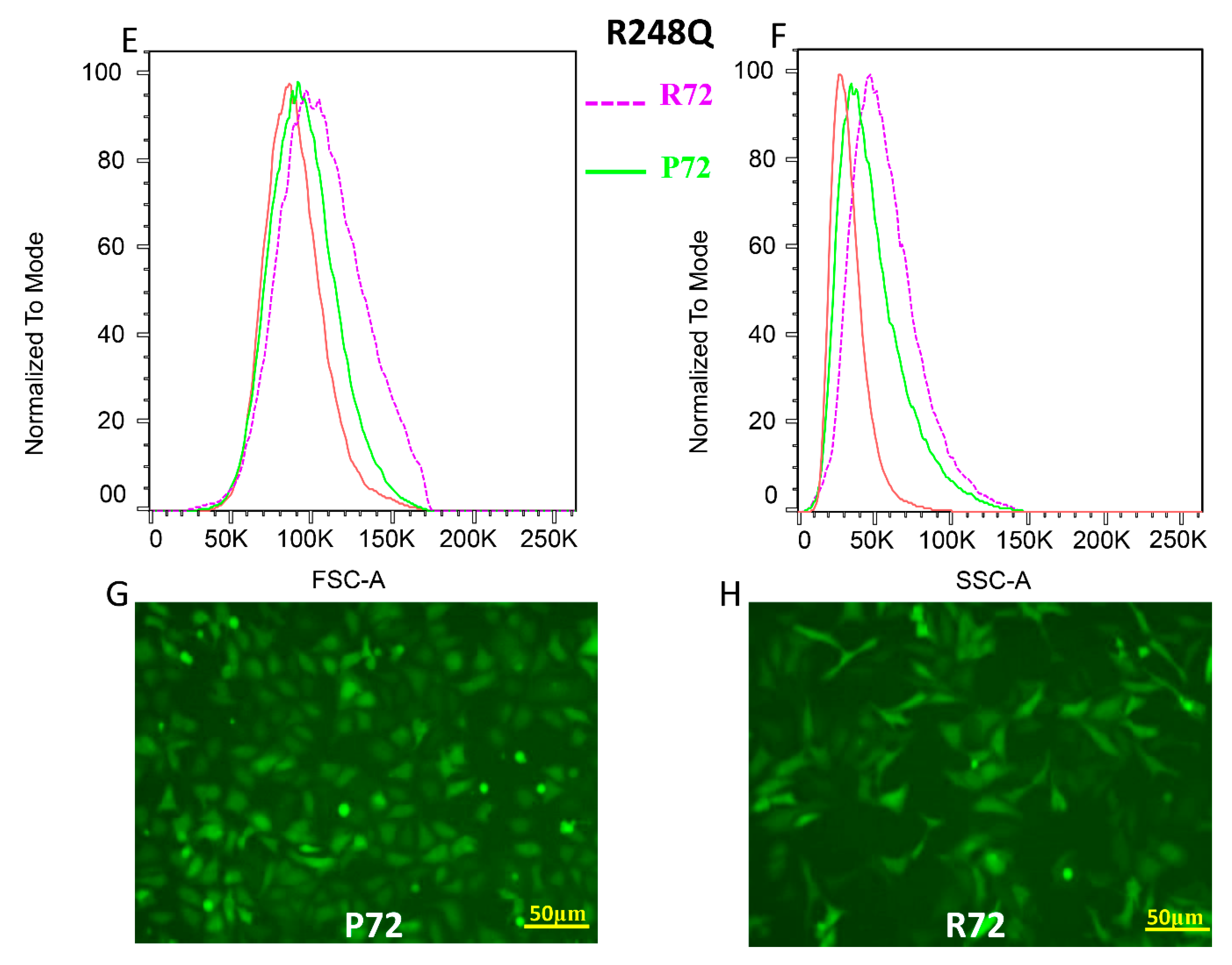
2.1. The P72R SNP alters the Morphology of R248Q/W p53 Mutants
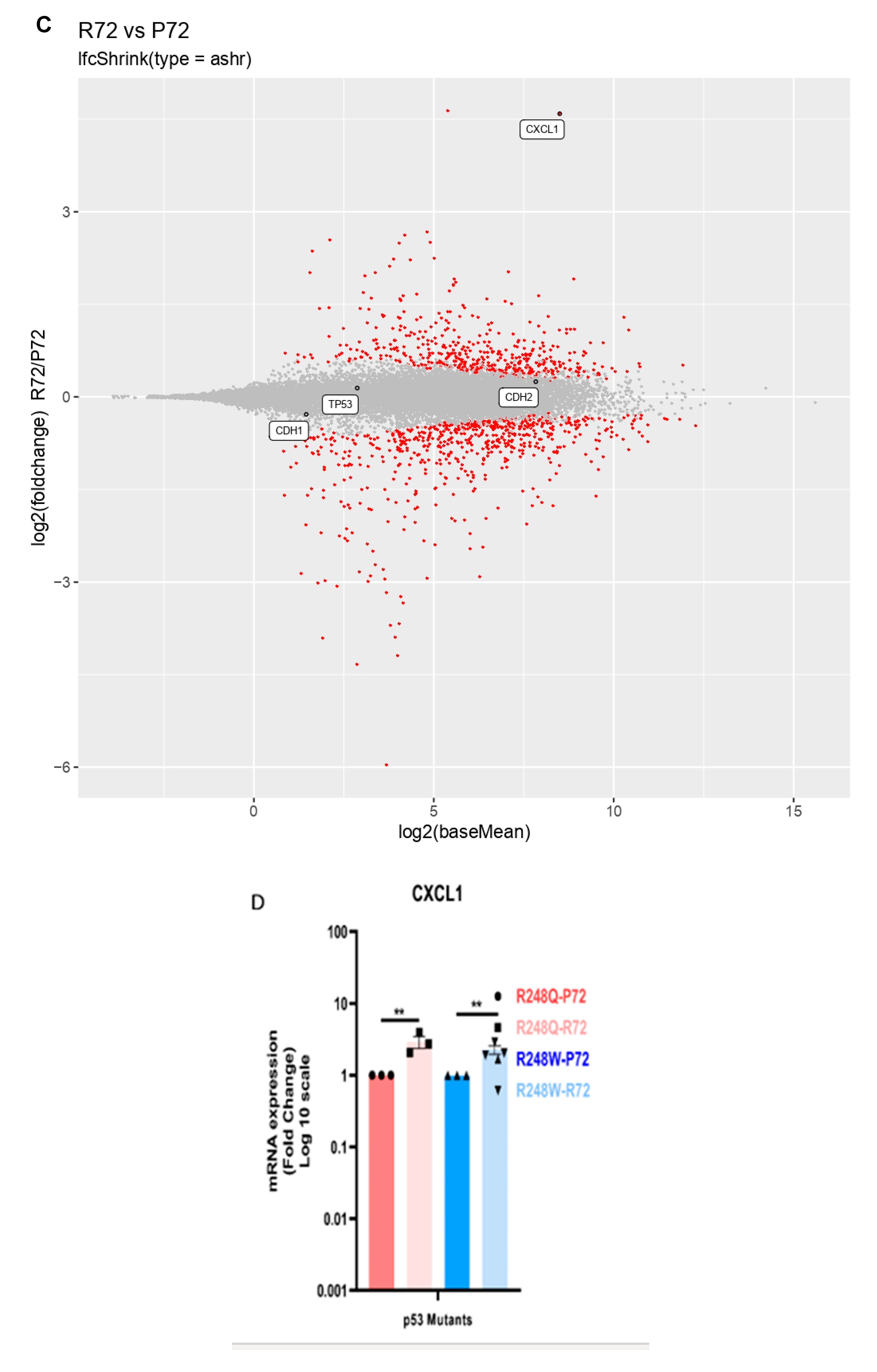
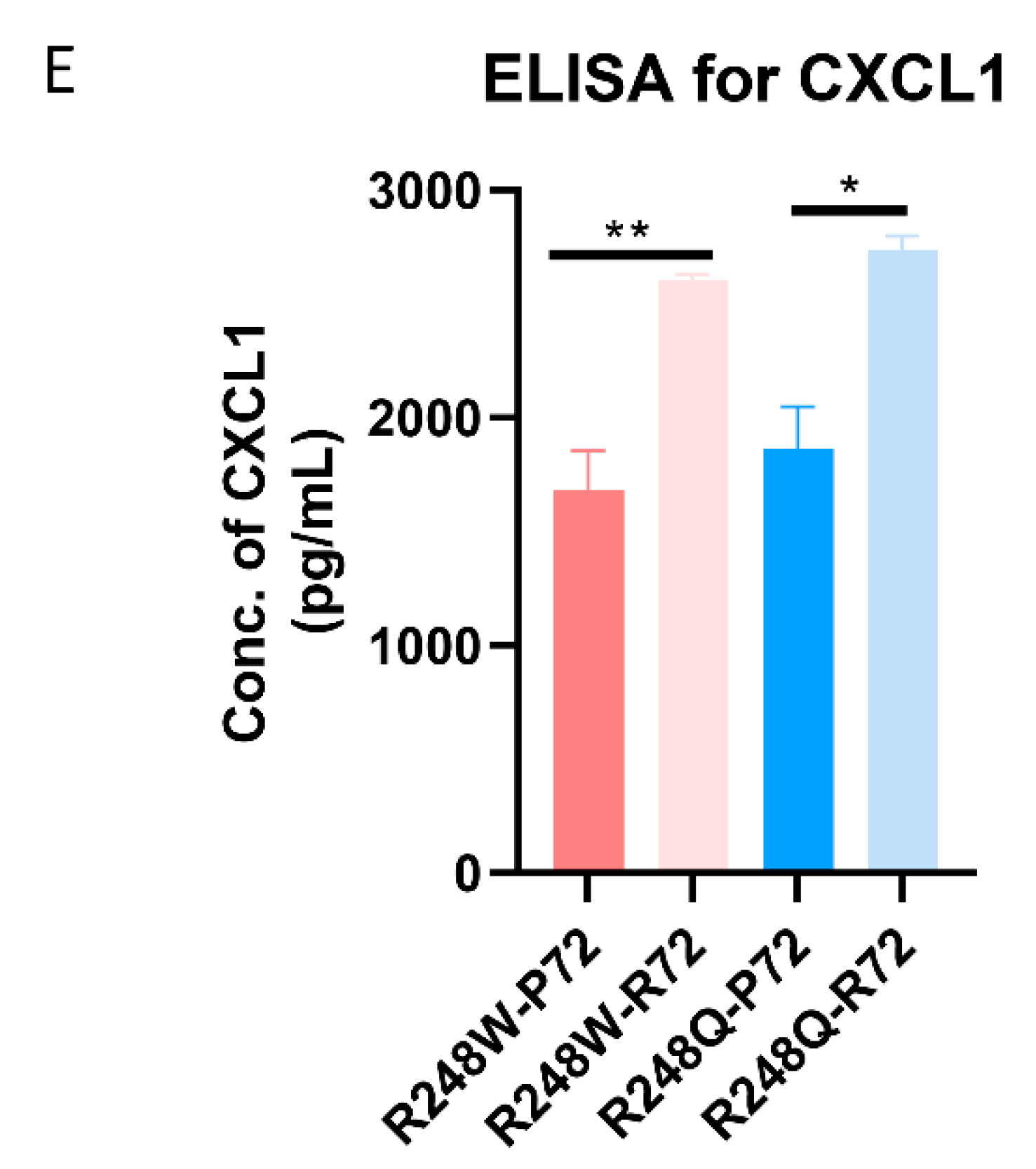
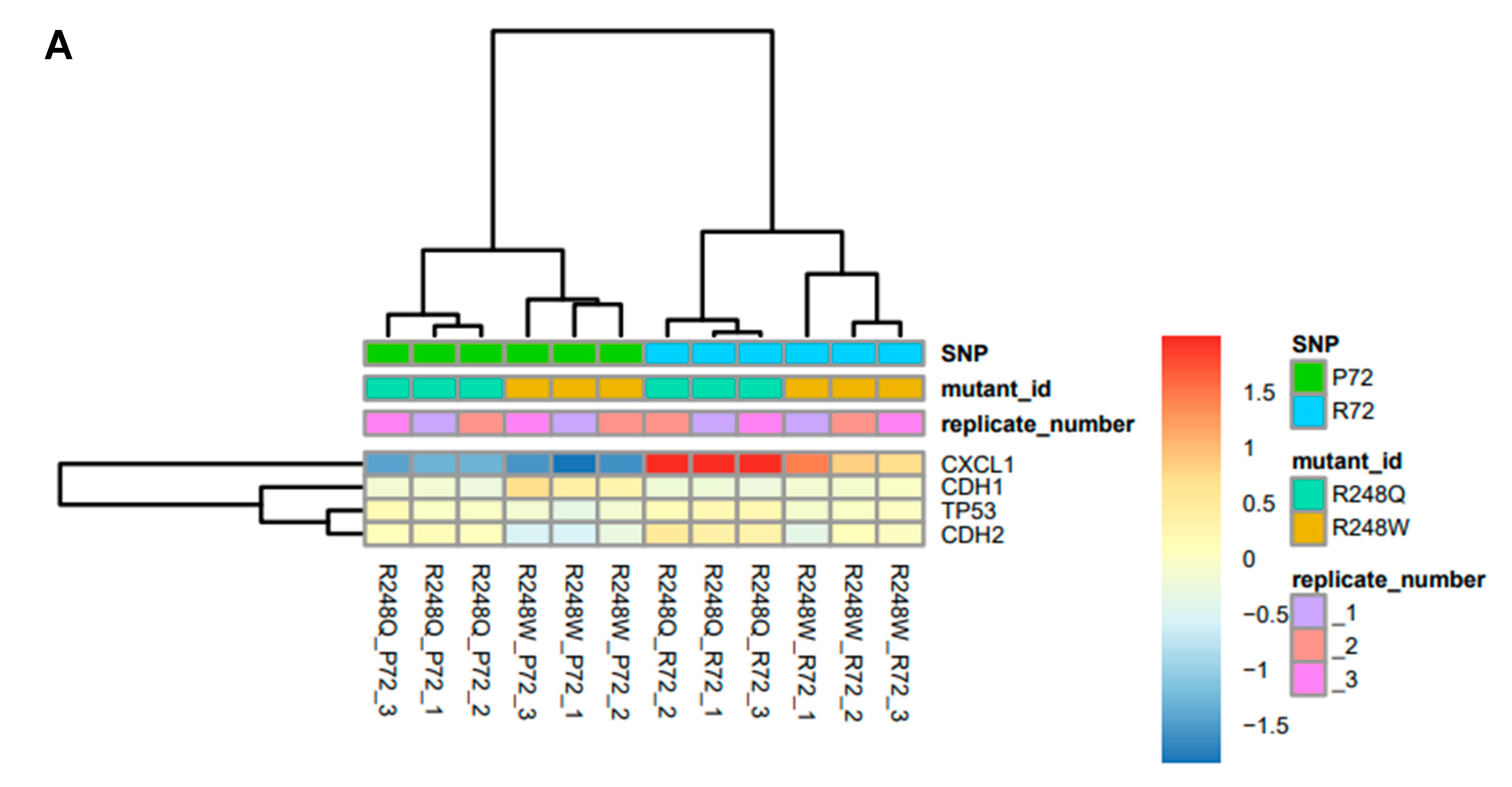
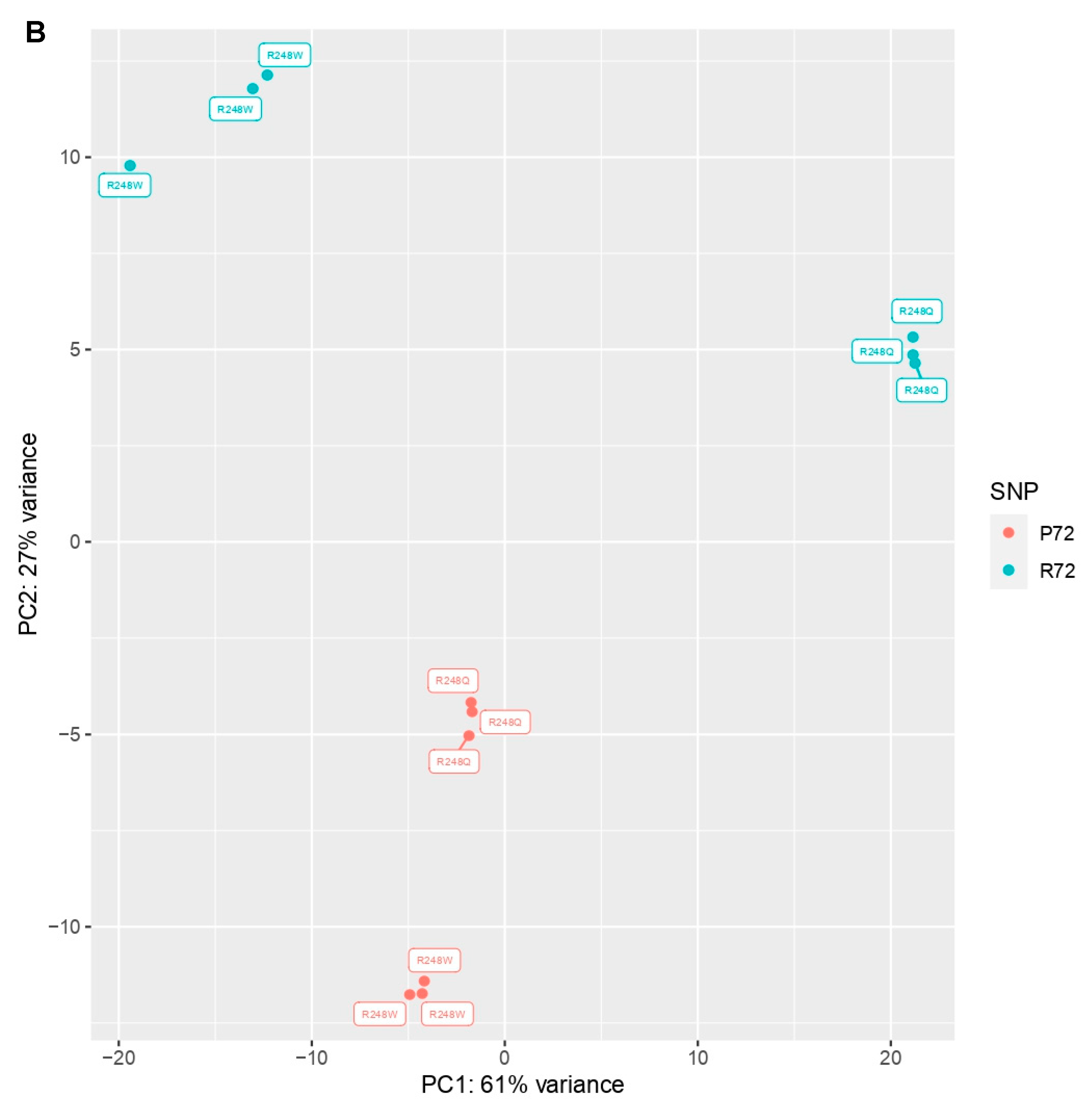
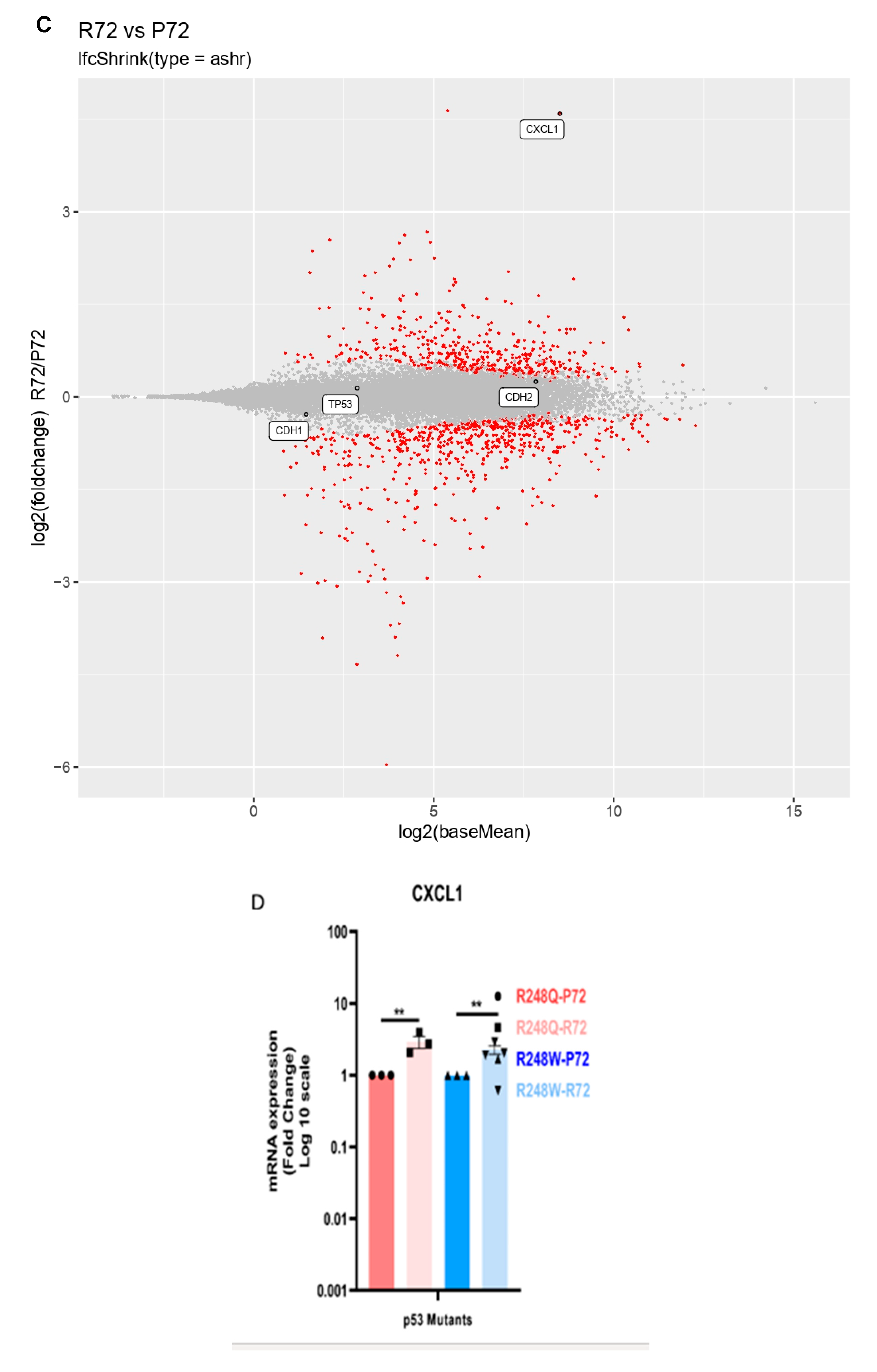
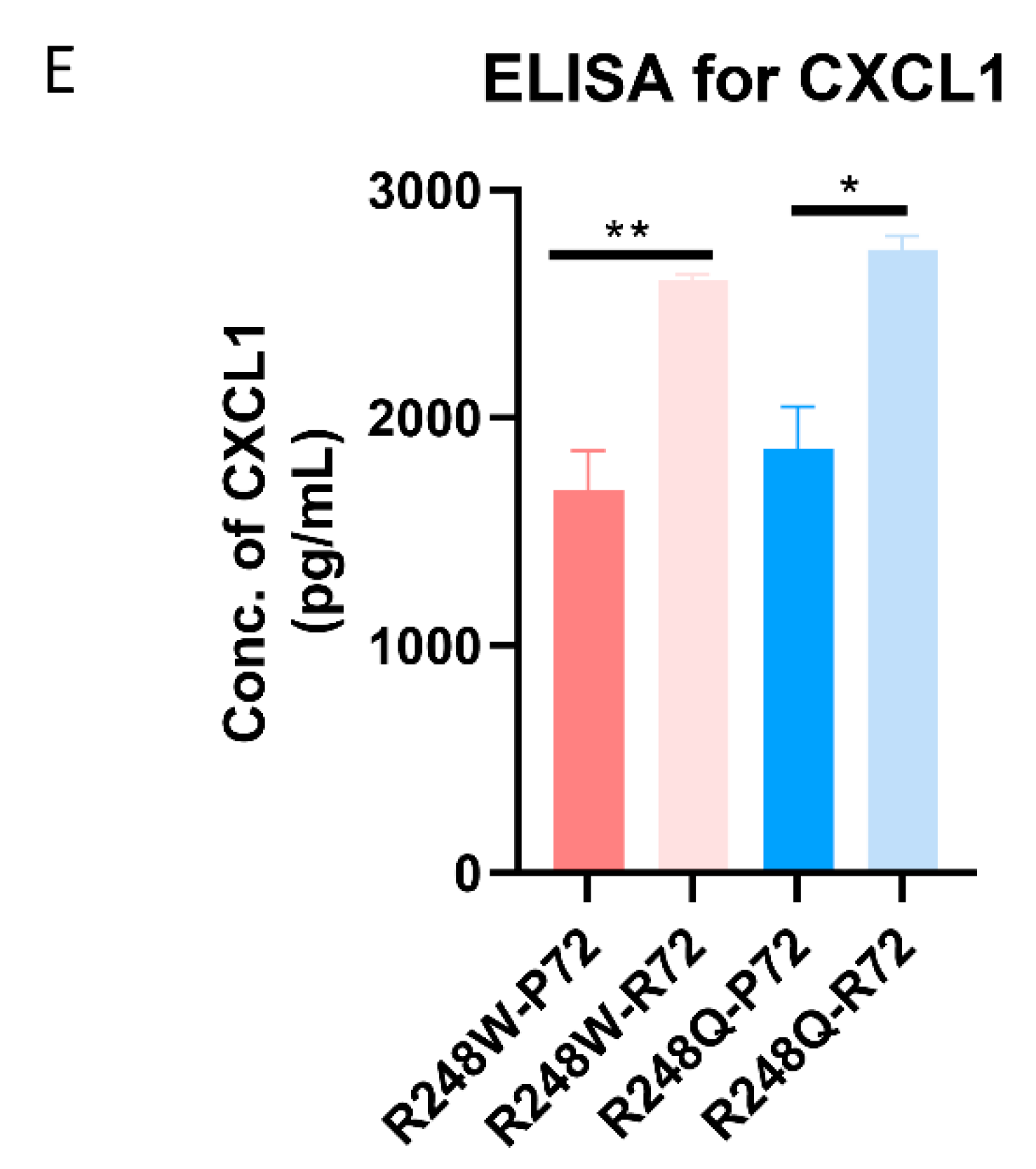
2.2. CXCL1 is Significantly Overexpressed in p53 Mutants with the R72 SNP
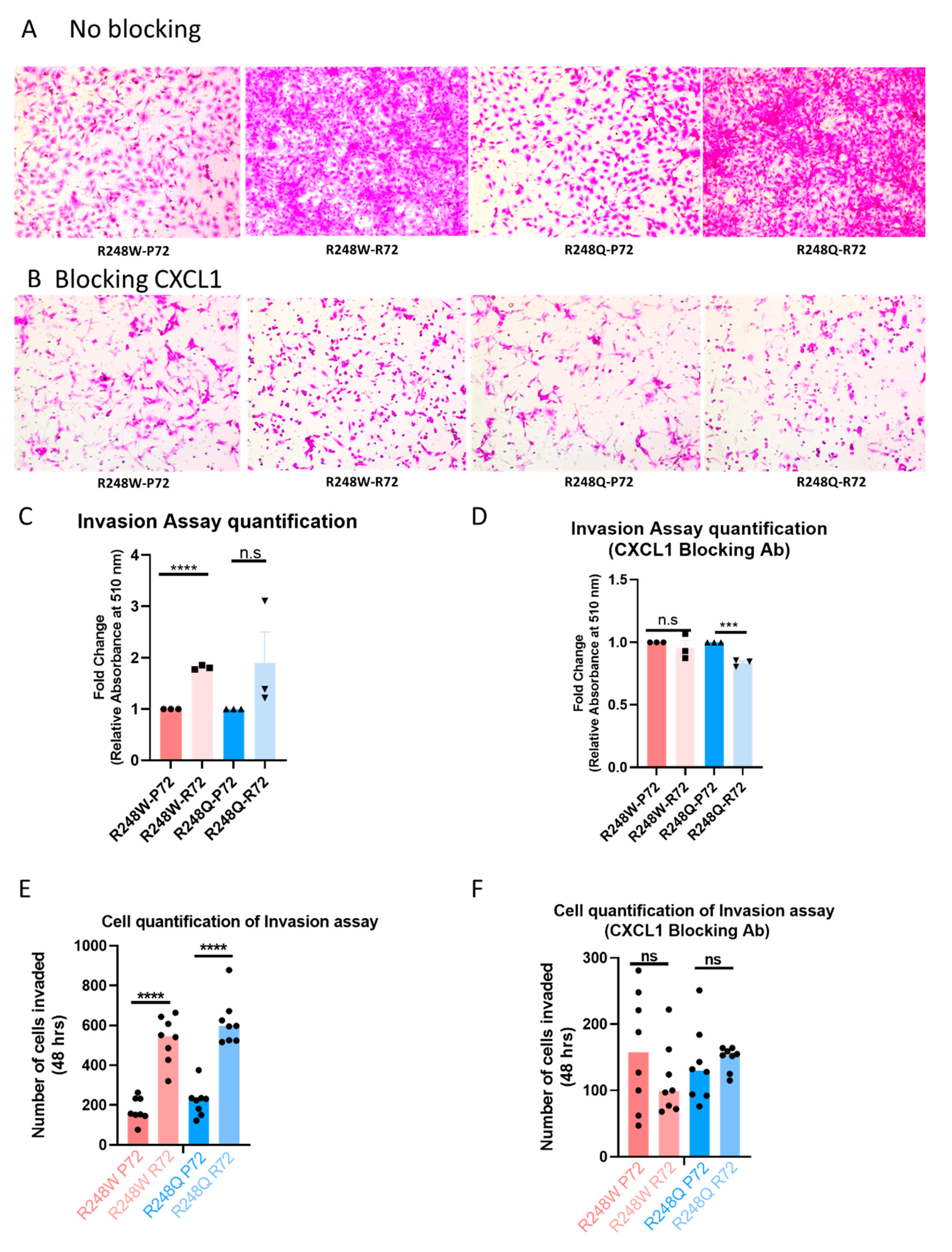
2.3. The P72R SNP Alters the Invasion Profile of Mutant p53 via CXCL1
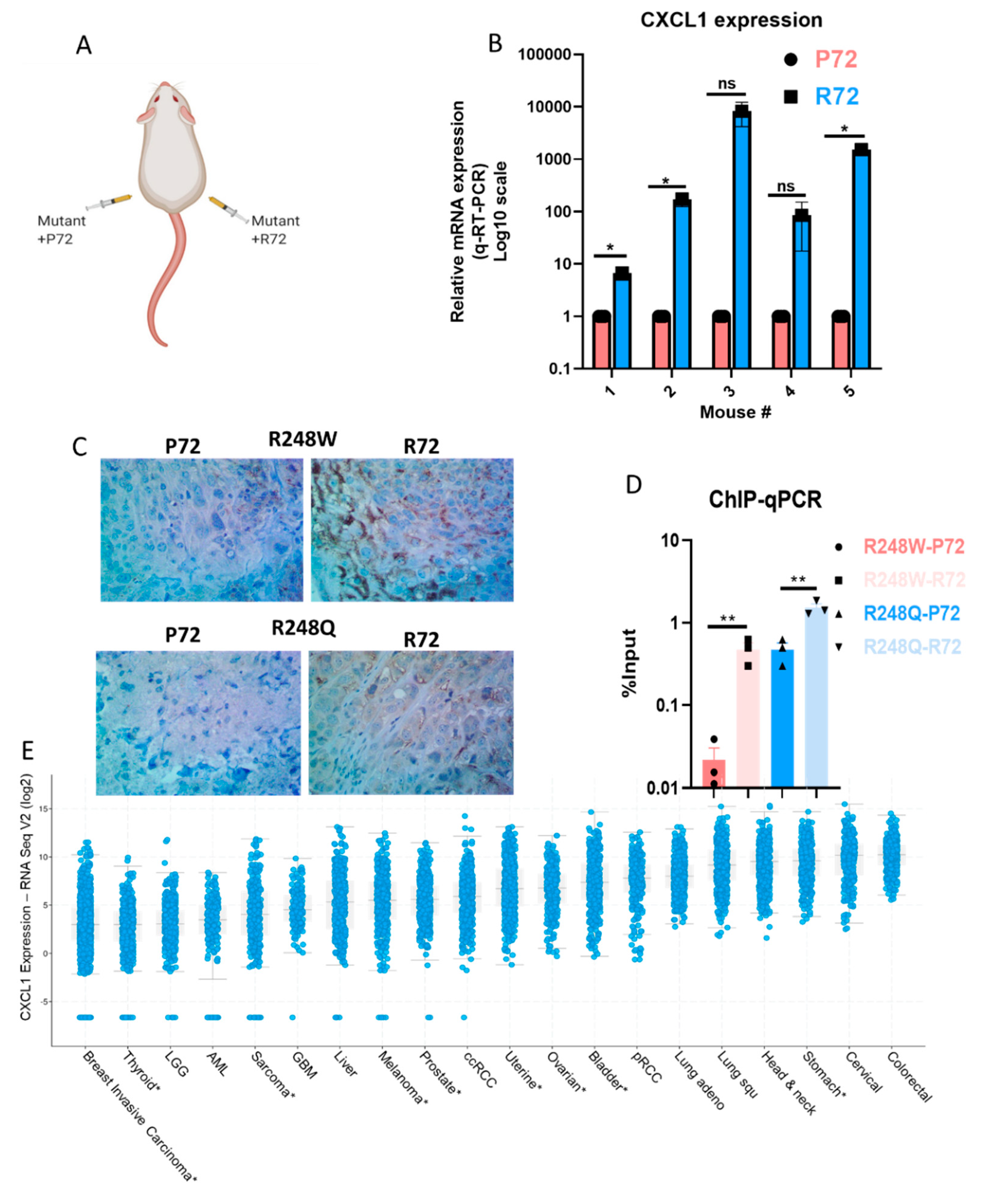
2.4. The R72 SNP Exhibits Higher Expression in Vivol and Enhances the Transactivation of CXCL1
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Plasmid and Lentiviral Particle Production
4.3. Antibodies
4.4. Gene Expression Analysis
4.5. Mouse Studies
4.6. Immunohistochemistry (IHC)
4.7. Flow Cytometry
4.8. Invasion Assay
4.9. Statistics and Reproducibility
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability Statement
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Borley, J.; Wilhelm-Benartzi, C.; Brown, R.; Ghaem-Maghami, S. Does tumour biology determine surgical success in the treatment of epithelial ovarian cancer? A systematic literature review. Brit. J. Cancer 2012, 107, 1069–1074. [Google Scholar] [CrossRef] [Green Version]
- Al-Alem, L.F.; Pandya, U.M.; Baker, A.T.; Bellio, C.; Zarrella, B.D.; Clark, J.; DiGloria, C.M.; Rueda, B.R. Ovarian cancer stem cells: What progress have we made? Int J. Biochem. Cell B 2019, 107, 92–103. [Google Scholar] [CrossRef]
- Bowtell, D.D.; Bohm, S.; Ahmed, A.A.; Aspuria, P.J.; Bast, R.C., Jr.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Yeung, T.L.; Leung, C.S.; Yip, K.P.; Au Yeung, C.L.; Wong, S.T.; Mok, S.C. Cellular and molecular processes in ovarian cancer metastasis. A Review in the Theme: Cell and Molecular Processes in Cancer Metastasis. Am. J. Physiol. Cell Physiol. 2015, 309, C444–C456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockinger, A.; Eger, A.; Wolf, J.; Beug, H.; Foisner, R. E-cadherin regulates cell growth by modulating proliferation-dependent beta-catenin transcriptional activity. J. Cell Biol. 2001, 154, 1185–1196. [Google Scholar] [CrossRef] [PubMed]
- Bolender, D.L.; Markwald, R.R. Epithelial-mesenchymal transformation in chick atrioventricular cushion morphogenesis. Scan. Electron. Microsc. 1979, 3, 313–321. [Google Scholar]
- Klymkowsky, M.W.; Savagner, P. Epithelial-Mesenchymal Transition A Cancer Researcher’s Conceptual Friend and Foe. Am. J. Pathol. 2009, 174, 1588–1593. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Liu, W.; Zheng, Y.; Wang, S.; Yang, B.; Li, M.; Song, J.; Zhang, F.; Zhang, X.; Wang, Q.; et al. CXCL1 derived from tumor-associated macrophages promotes breast cancer metastasis via activating NF-kappaB/SOX4 signaling. Cell Death Dis. 2018, 9, 880. [Google Scholar] [CrossRef] [PubMed]
- Dehne, N.; Mora, J.; Namgaladze, D.; Weigert, A.; Brune, B. Cancer cell and macrophage cross-talk in the tumor microenvironment. Curr. Opin. Pharmacol. 2017, 35, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Maolake, A.; Izumi, K.; Shigehara, K.; Natsagdorj, A.; Iwamoto, H.; Kadomoto, S.; Takezawa, Y.; Machioka, K.; Narimoto, K.; Namiki, M.; et al. Tumor-associated macrophages promote prostate cancer migration through activation of the CCL22-CCR4 axis. Oncotarget 2017, 8, 9739–9751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [Green Version]
- Manderson, E.N.; Presneau, N.; Provencher, D.; Mes-Masson, A.M.; Tonin, P.N. Comparative analysis of loss of heterozygosity of specific chromosome 3, 13, 17, and X loci and TP53 mutations in human epithelial ovarian cancer. Mol. Carcinogen. 2002, 34, 78–90. [Google Scholar] [CrossRef]
- Park, Y.R.; Kim, Y.M.; Lee, S.W.; Lee, H.Y.; Lee, G.E.; Lee, J.E.; Kim, Y.T. Optimization to detect TP53 mutations in circulating cell-free tumor DNA from patients with serous epithelial ovarian cancer. Obstet. Gynecol. Sci. 2018, 61, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Rechsteiner, M.; Zimmermann, A.K.; Wild, P.J.; Caduff, R.; von Teichman, A.; Fink, D.; Moch, H.; Noske, A. TP53 mutations are common in all subtypes of epithelial ovarian cancer and occur concomitantly with KRAS mutations in the mucinous type. Exp. Mol. Pathol. 2013, 95, 235–241. [Google Scholar] [CrossRef]
- Zhang, Y.; Cao, L.; Nguyen, D.; Lu, H. TP53 mutations in epithelial ovarian cancer. Transl. Cancer Res. 2016, 5, 650–663. [Google Scholar] [CrossRef]
- Edlund, K.; Larsson, O.; Ameur, A.; Bunikis, I.; Gyllensten, U.; Leroy, B.; Sundstrom, M.; Micke, P.; Botling, J.; Soussi, T. Data-driven unbiased curation of the TP53 tumor suppressor gene mutation database and validation by ultradeep sequencing of human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 9551–9556. [Google Scholar] [CrossRef] [Green Version]
- Brazdova, M.; Navratilova, L.; Tichy, V.; Nemcova, K.; Lexa, M.; Hrstka, R.; Pecinka, P.; Adamik, M.; Vojtesek, B.; Palecek, E.; et al. Preferential Binding of Hot Spot Mutant p53 Proteins to Supercoiled DNA In Vitro and in Cells. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Barnoud, T.; Parris, J.L.D.; Murphy, M.E. Common genetic variants in the TP53 pathway and their impact on cancer. J. Mol. Cell Biol. 2019, 11, 578–585. [Google Scholar] [CrossRef]
- Li, T.Y.; Kon, N.; Jiang, L.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell cycle arrest, apoptosis, and senescence. Cancer Res. 2013, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pim, D.; Banks, L. p53 polymorphic variants at codon 72 exert different effects on cell cycle progression. Int. J. Cancer 2004, 108, 196–199. [Google Scholar] [CrossRef]
- Thomas, M.; Kalita, A.; Labrecque, S.; Pim, D.; Banks, L.; Matlashewski, G. Two polymorphic variants of wild-type p53 differ biochemically and biologically. Mol. Cell Biol. 1999, 19, 1092–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumont, P.; Leu, J.I.J.; Della Pietra, A.C.; George, D.L.; Murphy, M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat. Genet. 2003, 33, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, D.; Samuels, Y.; Sullivan, A.; Zvelebil, M.; Breyssens, H.; Bisso, A.; Del Sal, G.; Syed, N.; Smith, P.; Gasco, M.; et al. iASPP preferentially binds p53 proline-rich region and modulates apoptotic function of codon 72 polymorphic p53. Nat. Genet. 2006, 38, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Azzam, G.; Wang, X.T.; Bell, D.; Murphy, M.E. CSF1 Is a Novel p53 Target Gene Whose Protein Product Functions in a Feed-Forward Manner to Suppress Apoptosis and Enhance p53-Mediated Growth Arrest. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Whibley, C.; Pharoah, P.D.P.; Hollstein, M. p53 polymorphisms: Cancer implications. Nat. Rev. Cancer 2009, 9, 95–107. [Google Scholar] [CrossRef]
- Kung, C.P.; Khaku, S.; Jennis, M.; Zhou, Y.; Murphy, M.E. Identification of TRIML2, a Novel p53 Target, that Enhances p53 SUMOylation and Regulates the Transactivation of Proapoptotic Genes. Mol. Cancer Res. 2015, 13, 250–262. [Google Scholar] [CrossRef] [Green Version]
- Kung, C.P.; Leu, J.I.; Basu, S.; Khaku, S.; Anokye-Danso, F.; Liu, Q.; George, D.L.; Ahima, R.S.; Murphy, M.E. The P72R Polymorphism of p53 Predisposes to Obesity and Metabolic Dysfunction. Cell Rep. 2016, 14, 2413–2425. [Google Scholar] [CrossRef] [Green Version]
- Basu, S.; Gnanapradeepan, K.; Barnoud, T.; Kung, C.P.; Tavecchio, M.; Scott, J.; Watters, A.; Chen, Q.; Kossenkov, A.V.; Murphy, M.E. Mutant p53 controls tumor metabolism and metastasis by regulating PGC-1alpha. Genes Dev. 2018, 32, 230–243. [Google Scholar] [CrossRef]
- Keeley, E.C.; Mehrad, B.; Strieter, R.M. CXC chemokines in cancer angiogenesis and metastases. Adv. Cancer Res. 2010, 106, 91–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strieter, R.M.; Belperio, J.A.; Burdick, M.D.; Sharma, S.; Dubinett, S.M.; Keane, M.P. CXC chemokines: Angiogenesis, immunoangiostasis, and metastases in lung cancer. Ann. N. Y. Acad. Sci. 2004, 1028, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.L.; Wang, C.S.; Huang, Y.H.; Tsai, M.M.; Liang, Y.; Lin, K.H. Overexpression of CXCL1 and its receptor CXCR2 promote tumor invasion in gastric cancer. Ann. Oncol. 2011, 22, 2267–2276. [Google Scholar] [CrossRef] [PubMed]
- Narendran, A.; Ganjavi, H.; Morson, N.; Connor, A.; Barlow, J.W.; Keystone, E.; Malkin, D.; Freedman, M.H. Mutant p53 in bone marrow stromal cells increases VEGF expression and supports leukemia cell growth. Exp. Hematol. 2003, 31, 693–701. [Google Scholar] [CrossRef]
- Schauer, I.G.; Zhang, J.; Xing, Z.; Guo, X.Q.; Mercado-Uribe, I.; Sood, A.K.; Huang, P.; Liu, J.S. Interleukin-1β Promotes Ovarian Tumorigenesis through a p53/NF-kappa B-Mediated Inflammatory Response in Stromal Fibroblasts. Neoplasia 2013, 15, 409–420. [Google Scholar] [CrossRef] [Green Version]
- Addadi, Y.; Moskovits, N.; Granot, D.; Lozano, G.; Carmi, Y.; Apte, R.N.; Neeman, M.; Oren, M. p53 Status in Stromal Fibroblasts Modulates Tumor Growth in an SDF1-Dependent Manner. Cancer Res. 2010, 70, 9650–9658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, N.; Marudamuthu, A.S.; Tsukasaki, Y.; Ikebe, M.; Fu, J.; Shetty, S. p53-and PAI-1-mediated induction of C-X-C chemokines and CXCR2: Importance in pulmonary inflammation due to cigarette smoke exposure. Am. J. Physiol. Lung C 2016, 310, L496–L506. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.S.; Chen, X.B. Identification of GRO1 as a Critical Determinant for Mutant p53 Gain of Function. J. Biol. Chem. 2009, 284, 12178–12187. [Google Scholar] [CrossRef] [Green Version]
- Fang, D.; Chen, H.; Zhu, J.Y.; Wang, W.; Teng, Y.; Ding, H.F.; Jing, Q.; Su, S.B.; Huang, S. Epithelial-mesenchymal transition of ovarian cancer cells is sustained by Rac1 through simultaneous activation of MEK1/2 and Src signaling pathways. Oncogene 2017, 36, 1546–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, P.; Karaayvaz, M.; Jia, N.; Kaneuchi, M.; Hamada, J.; Watari, H.; Sudo, S.; Ju, J.; Sakuragi, N. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene 2013, 32, 3286–3295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olszewski, M.B.; Pruszko, M.; Snaar-Jagalska, E.; Zylicz, A.; Zylicz, M. Diverse and cancer typespecific roles of the p53 R248Q gainoffunction mutation in cancer migration and invasiveness. Int. J. Oncol. 2019, 54, 1168–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Gotimer, K.; De Souza, C.; Tepper, C.G.; Karnezis, A.N.; Leiserowitz, G.S.; Chien, J.; Smith, L.H. Short-term organoid culture for drug sensitivity testing of high-grade serous carcinoma. Gynecol. Oncol. 2020. [Google Scholar] [CrossRef]
- Fang, P.; De Souza, C.; Minn, K.; Chien, J. Genome-scale CRISPR knockout screen identifies TIGAR as a modifier of PARP inhibitor sensitivity. Commun. Biol. 2019, 2, 335. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.W.; Lehnert, E.; Sethi, A.; Malhotra, R.; Kaushik, G.; Onder, Z.; Groves-Kirkby, N.; Mihajlovic, A.; DiGiovanna, J.; Srdic, M.; et al. The Cancer Genomics Cloud: Collaborative, Reproducible, and Democratized-A New Paradigm in Large-Scale Computational Research. Cancer Res. 2017, 77, e3–e6. [Google Scholar] [CrossRef] [Green Version]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Mounir, M.; Lucchetta, M.; Silva, T.C.; Olsen, C.; Bontempi, G.; Chen, X.; Noushmehr, H.; Colaprico, A.; Papaleo, E. New functionalities in the TCGAbiolinks package for the study and integration of cancer data from GDC and GTEx. PLoS Comput. Biol. 2019, 15, e1006701. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.S.; Mortazavi, A.; Myers, R.M.; Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science 2007, 316, 1497–1502. [Google Scholar] [CrossRef] [Green Version]
- Blecher-Gonen, R.; Barnett-Itzhaki, Z.; Jaitin, D.; Amann-Zalcenstein, D.; Lara-Astiaso, D.; Amit, I. High-throughput chromatin immunoprecipitation for genome-wide mapping of in vivo protein-DNA interactions and epigenomic states. Nat. Protoc. 2013, 8, 539–554. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.P.; Madden, J.A.; Neums, L.; Moulder, R.K.; Forrest, M.L.; Chien, J. Olaparib-induced Adaptive Response Is Disrupted by FOXM1 Targeting that Enhances Sensitivity to PARP Inhibition. Mol. Cancer Res. 2018, 16, 961–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Justus, C.R.; Leffler, N.; Ruiz-Echevarria, M.; Yang, L.V. In vitro cell migration and invasion assays. J. Vis. Exp. 2014. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Souza, C.; Madden, J.A.; Minn, D.; Kumar, V.E.; Montoya, D.J.; Nambiar, R.; Zhu, Z.; Xiao, W.-W.; Tahmassebi, N.; Kathi, H.; et al. The P72R Polymorphism in R248Q/W p53 Mutants Modifies the Mutant Effect on Epithelial to Mesenchymal Transition Phenotype and Cell Invasion via CXCL1 Expression. Int. J. Mol. Sci. 2020, 21, 8025. https://doi.org/10.3390/ijms21218025
De Souza C, Madden JA, Minn D, Kumar VE, Montoya DJ, Nambiar R, Zhu Z, Xiao W-W, Tahmassebi N, Kathi H, et al. The P72R Polymorphism in R248Q/W p53 Mutants Modifies the Mutant Effect on Epithelial to Mesenchymal Transition Phenotype and Cell Invasion via CXCL1 Expression. International Journal of Molecular Sciences. 2020; 21(21):8025. https://doi.org/10.3390/ijms21218025
Chicago/Turabian StyleDe Souza, Cristabelle, Jill A. Madden, Dennis Minn, Vigneshwari Easwar Kumar, Dennis J. Montoya, Roshni Nambiar, Zheng Zhu, Wen-Wu Xiao, Neeki Tahmassebi, Harikumara Kathi, and et al. 2020. "The P72R Polymorphism in R248Q/W p53 Mutants Modifies the Mutant Effect on Epithelial to Mesenchymal Transition Phenotype and Cell Invasion via CXCL1 Expression" International Journal of Molecular Sciences 21, no. 21: 8025. https://doi.org/10.3390/ijms21218025
APA StyleDe Souza, C., Madden, J. A., Minn, D., Kumar, V. E., Montoya, D. J., Nambiar, R., Zhu, Z., Xiao, W.-W., Tahmassebi, N., Kathi, H., Nelson, N., Karnezis, A. N., & Chien, J. (2020). The P72R Polymorphism in R248Q/W p53 Mutants Modifies the Mutant Effect on Epithelial to Mesenchymal Transition Phenotype and Cell Invasion via CXCL1 Expression. International Journal of Molecular Sciences, 21(21), 8025. https://doi.org/10.3390/ijms21218025