Twisted Intramolecular Charge Transfer State of a “Push-Pull” Emitter


Abstract
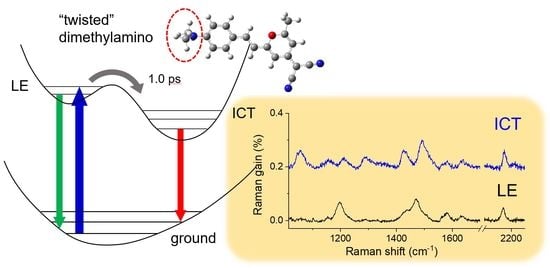
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
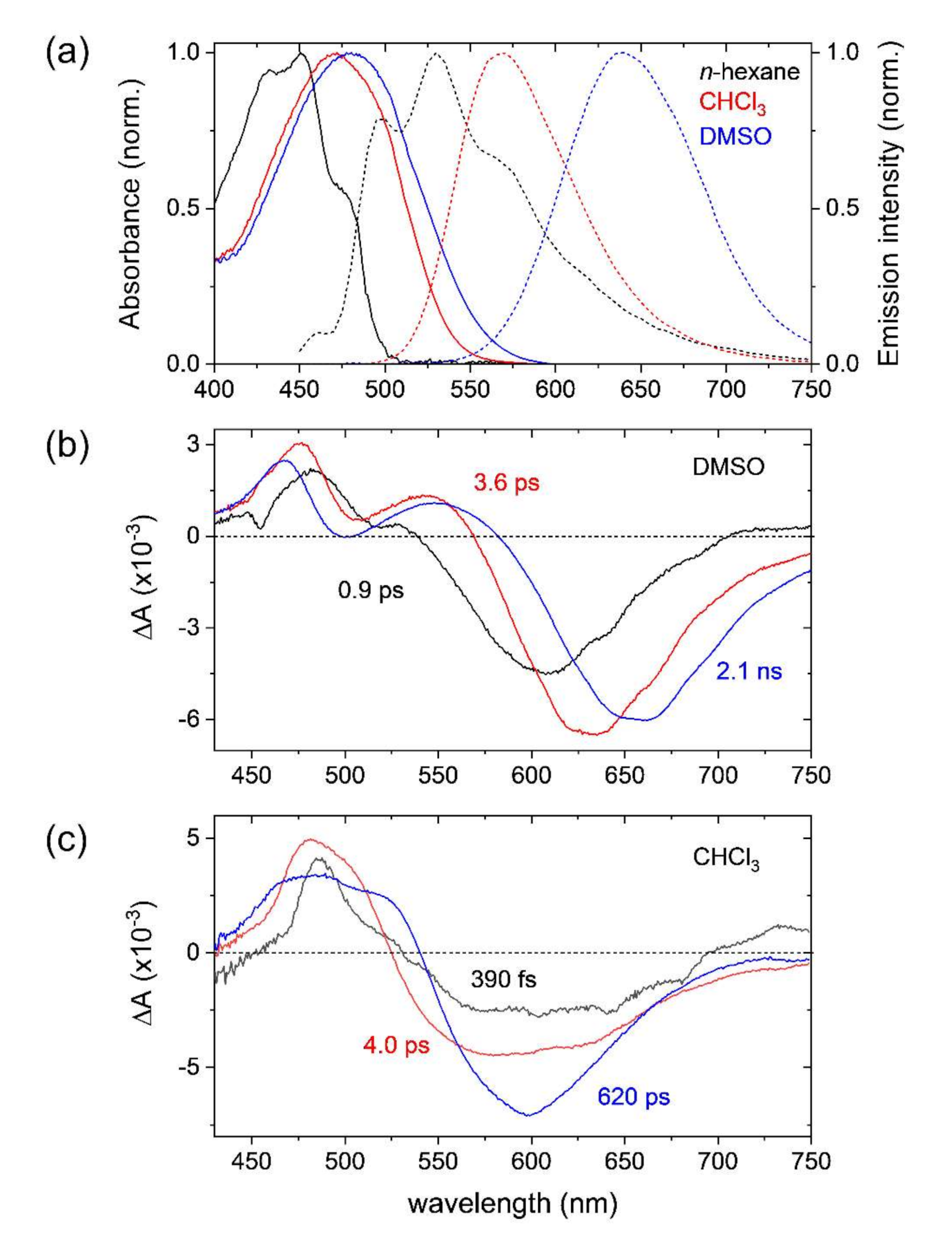
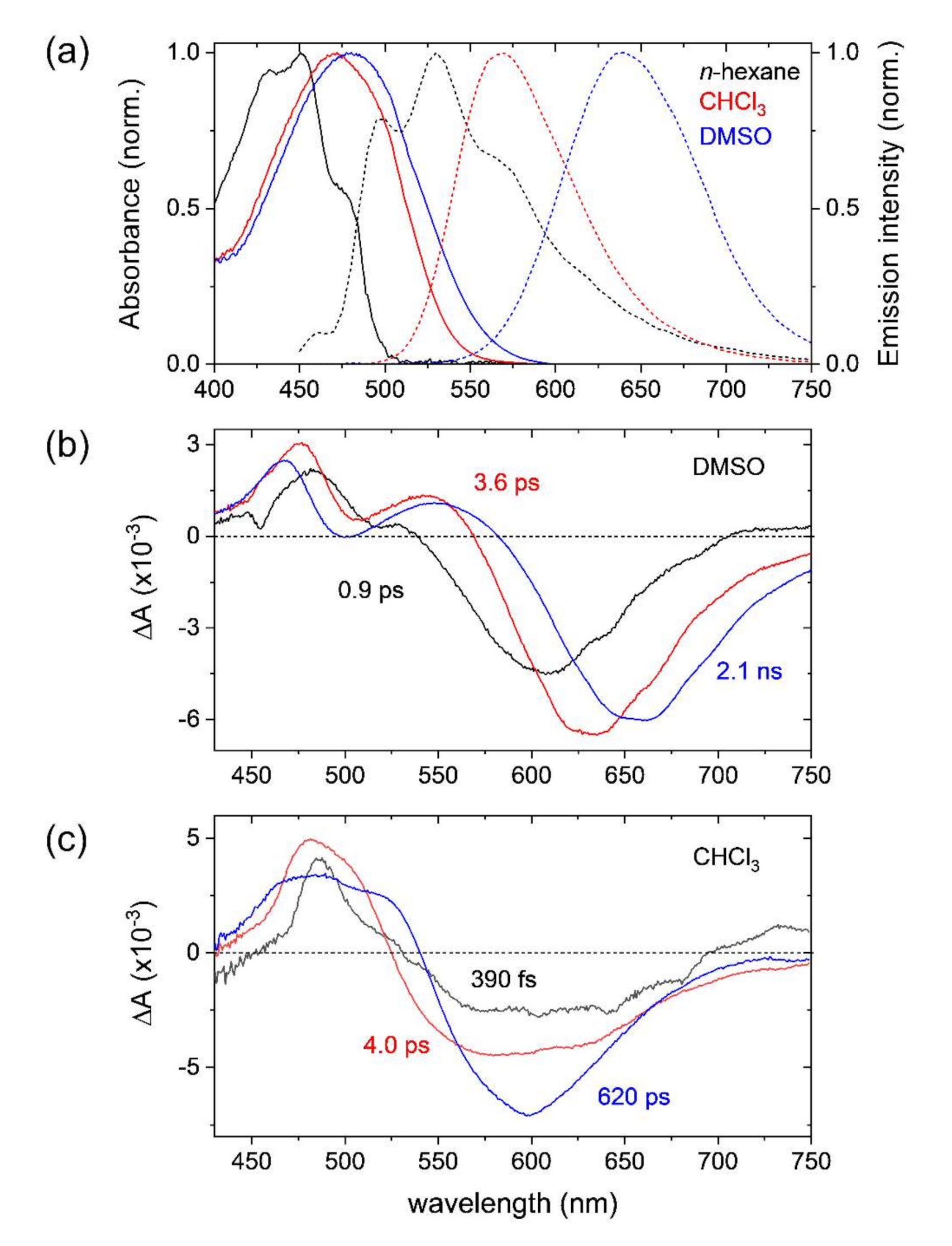
2.1. Steady-State Absorption and Emission Spectra of DCM
2.2. Transient Absorption Spectra and Kinetics of DCM
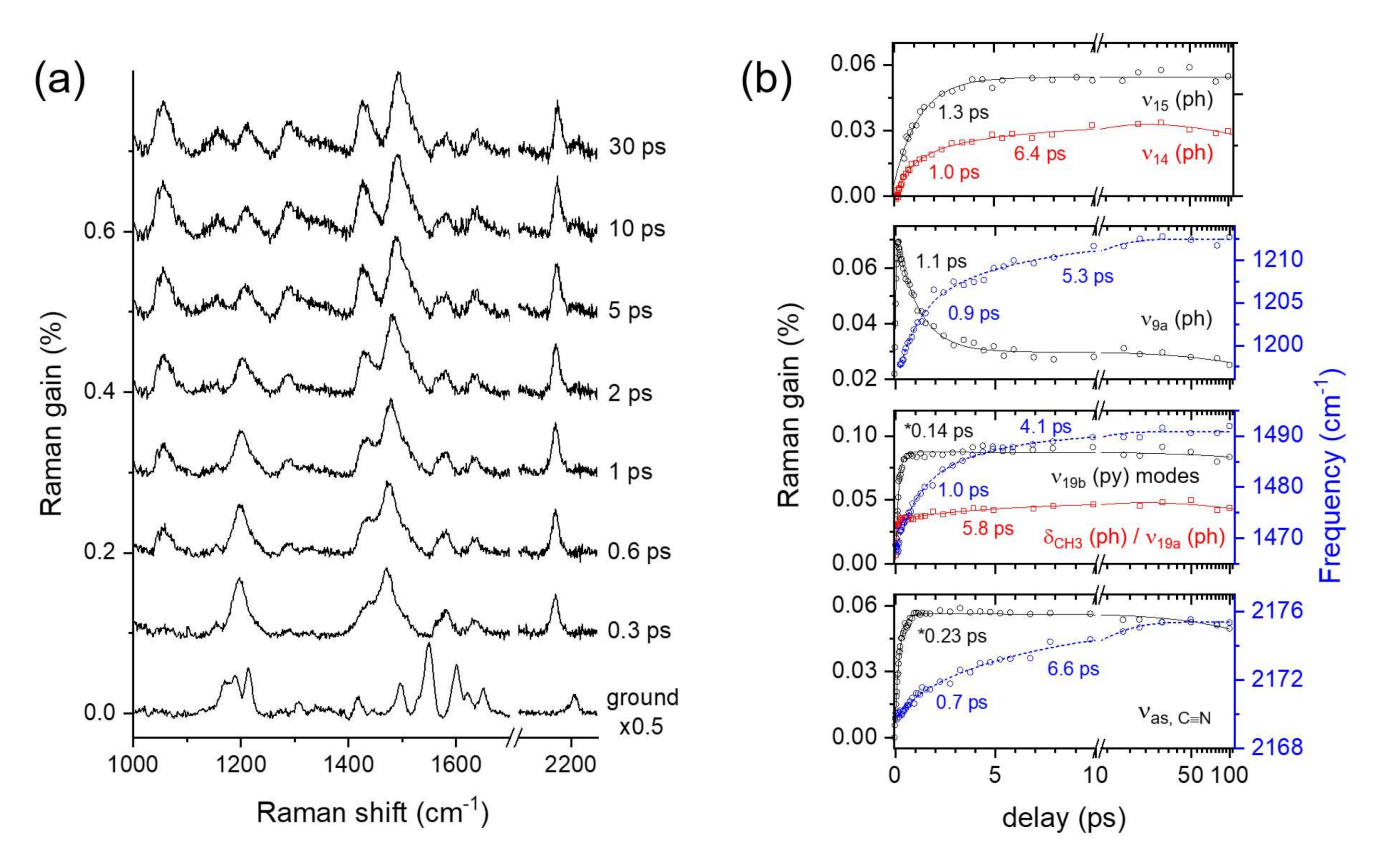
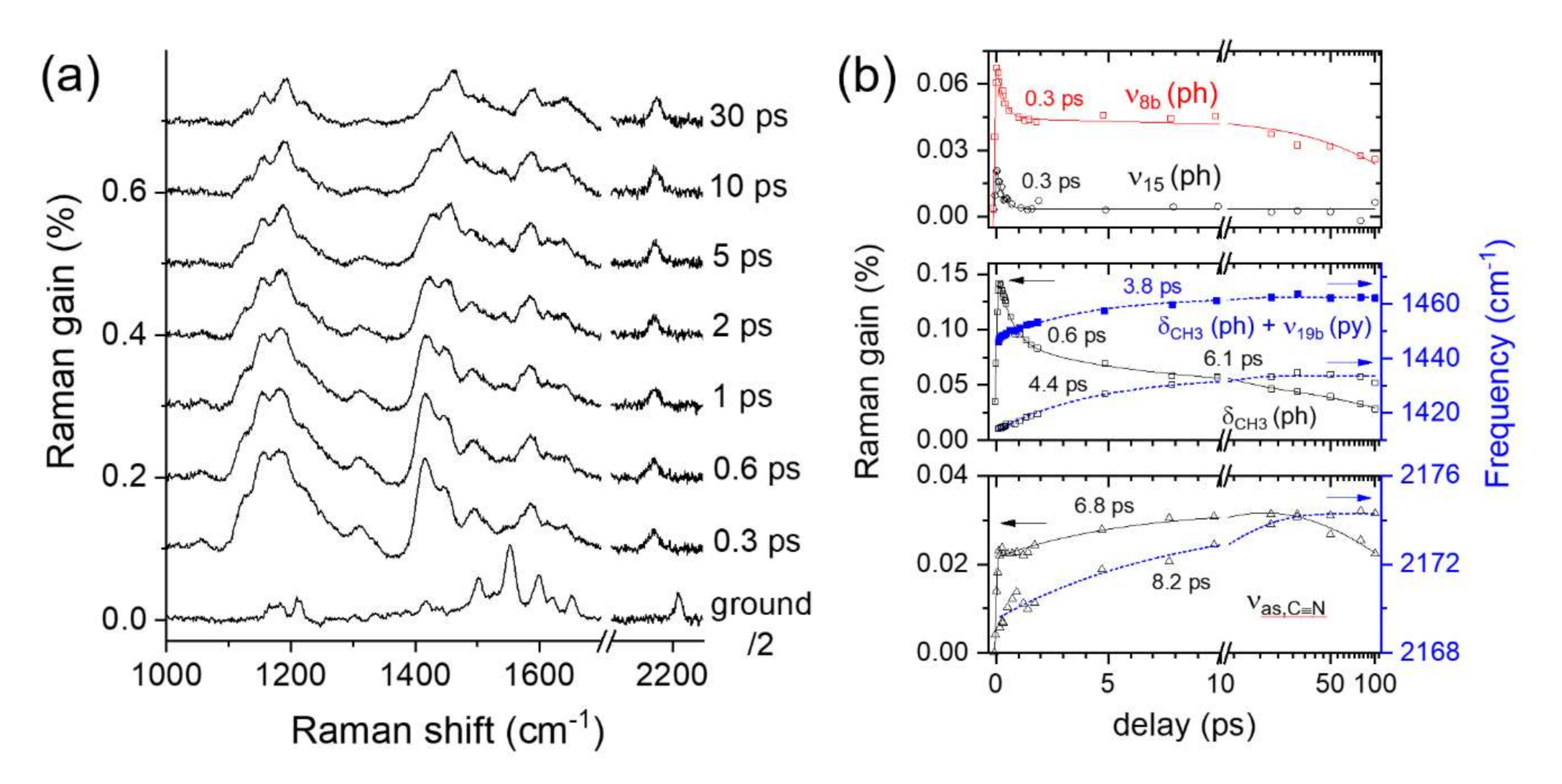
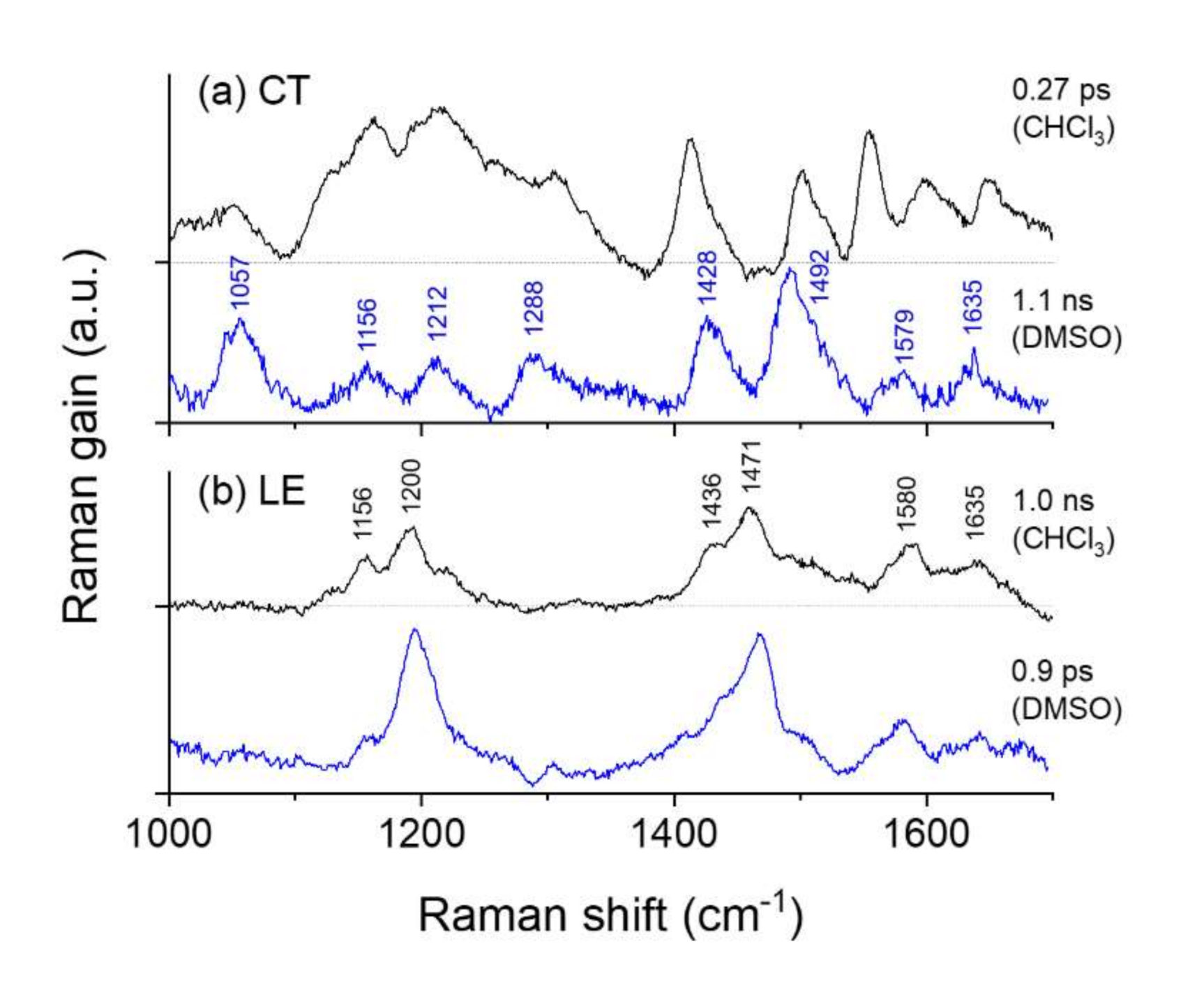
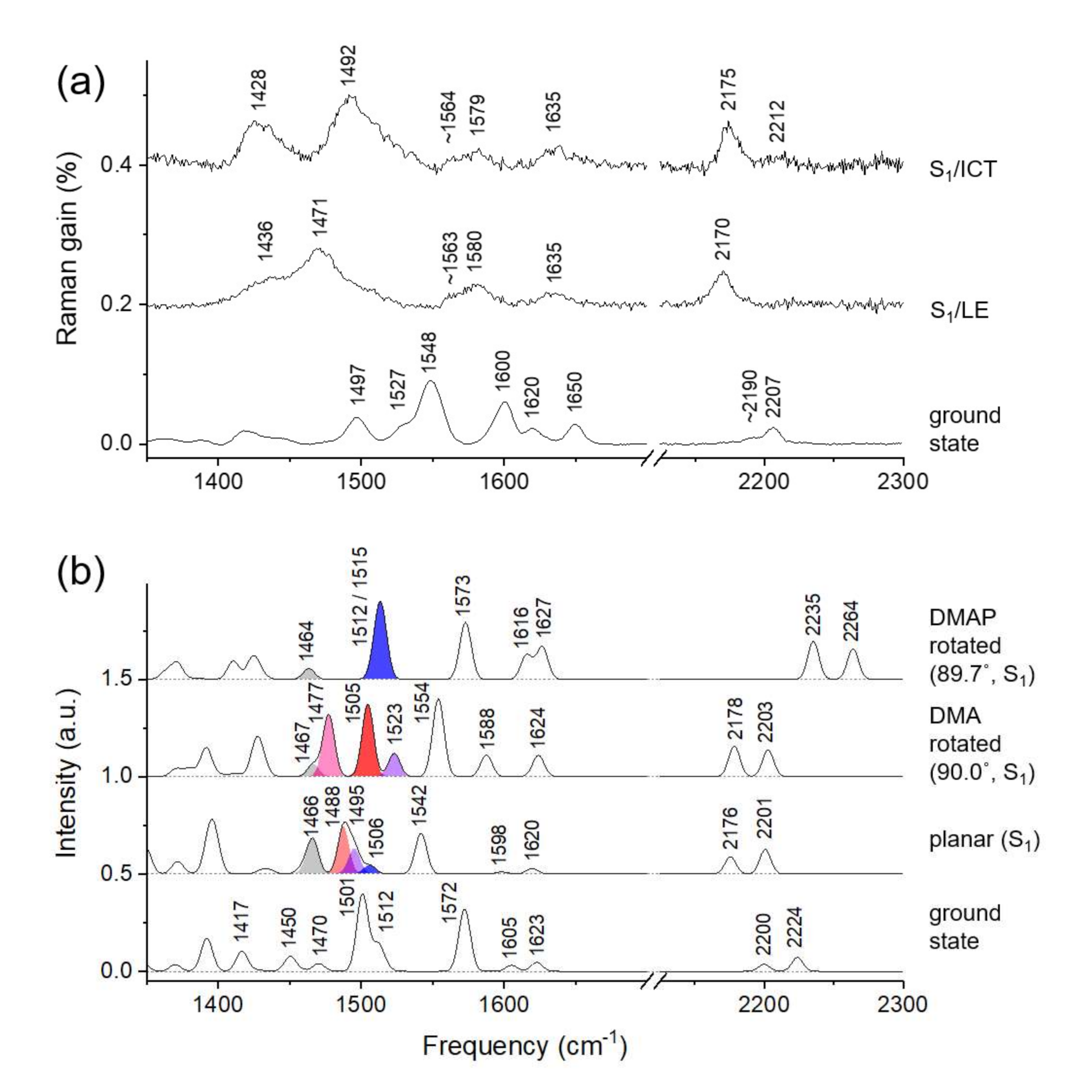
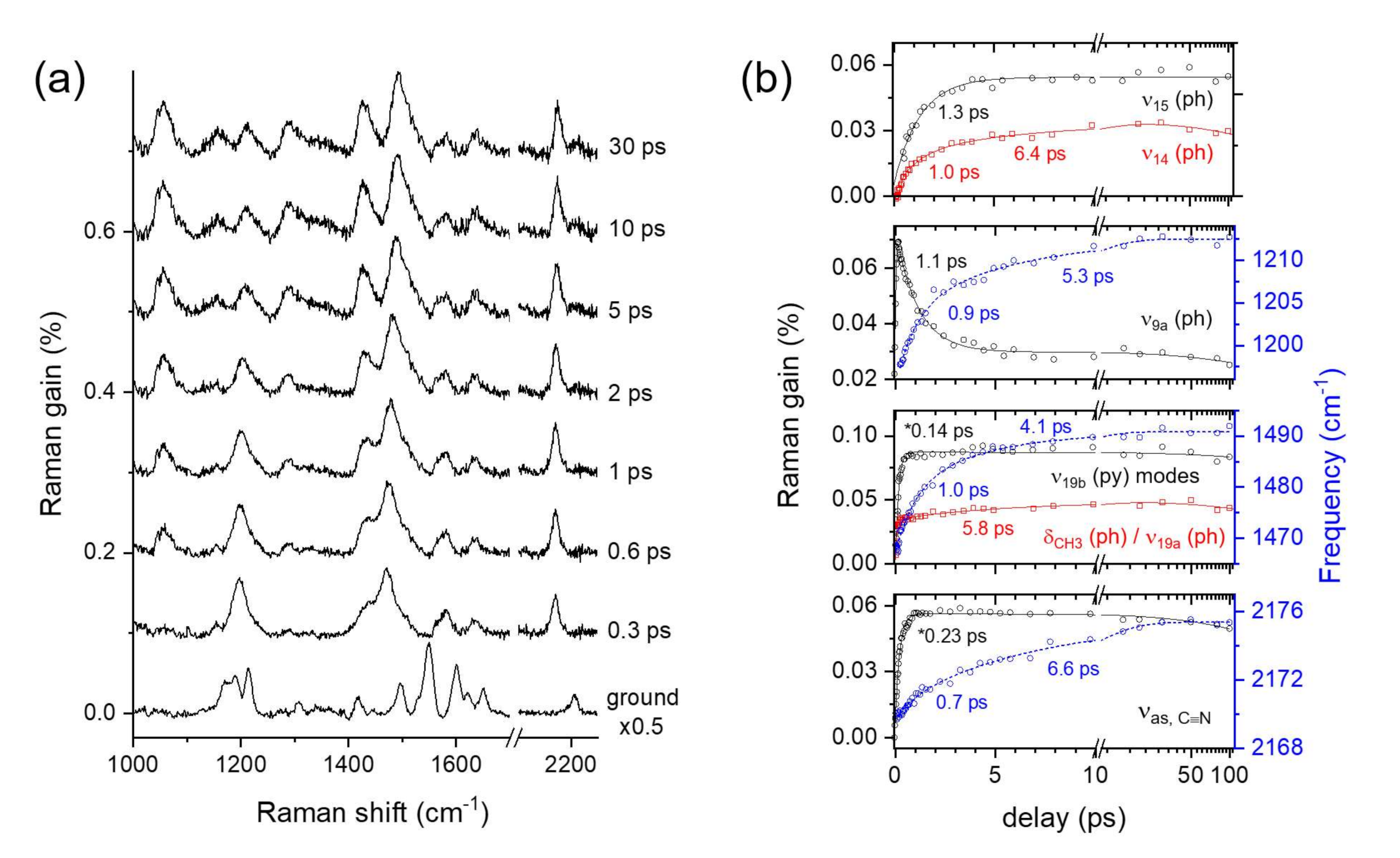
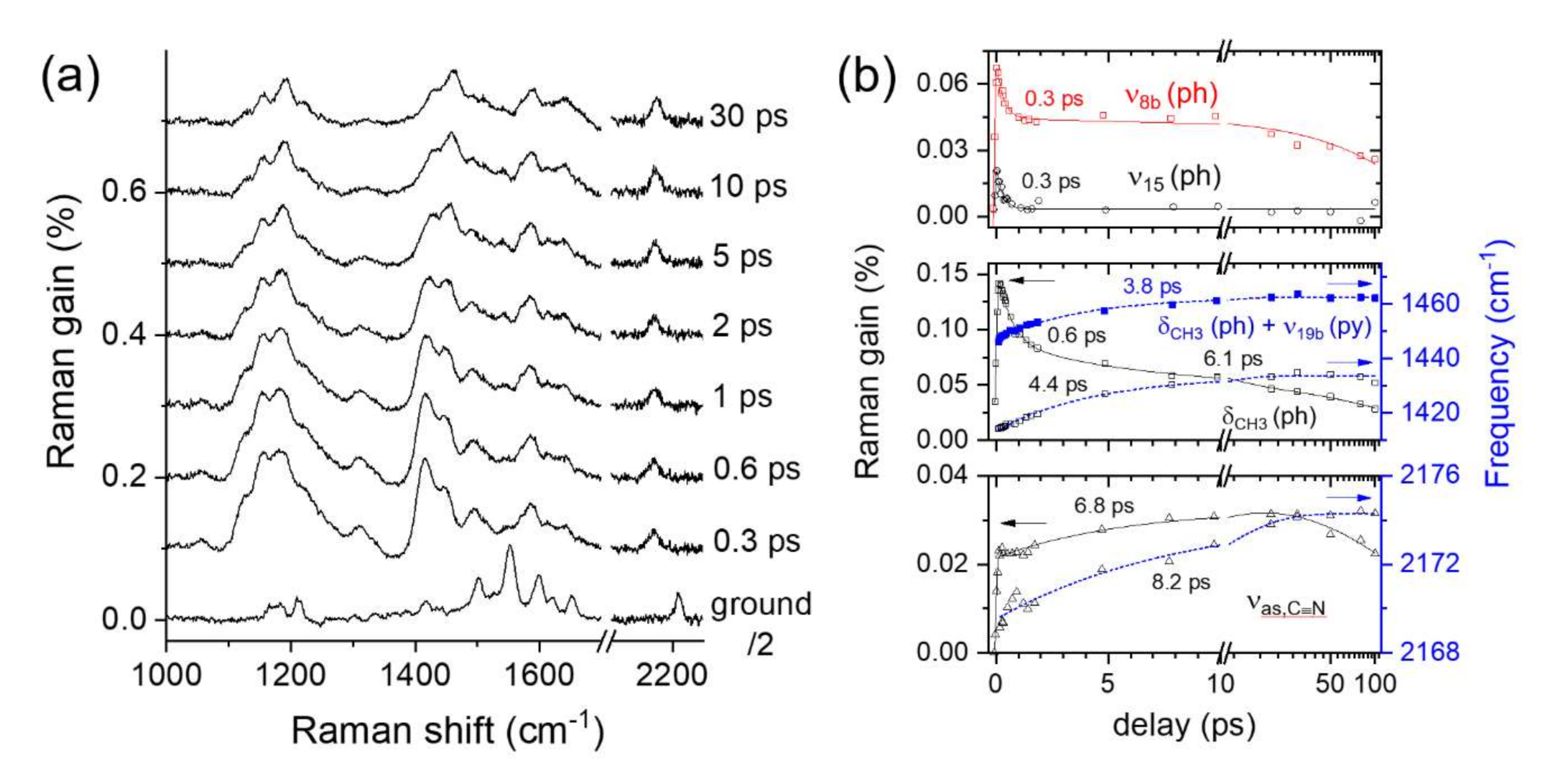
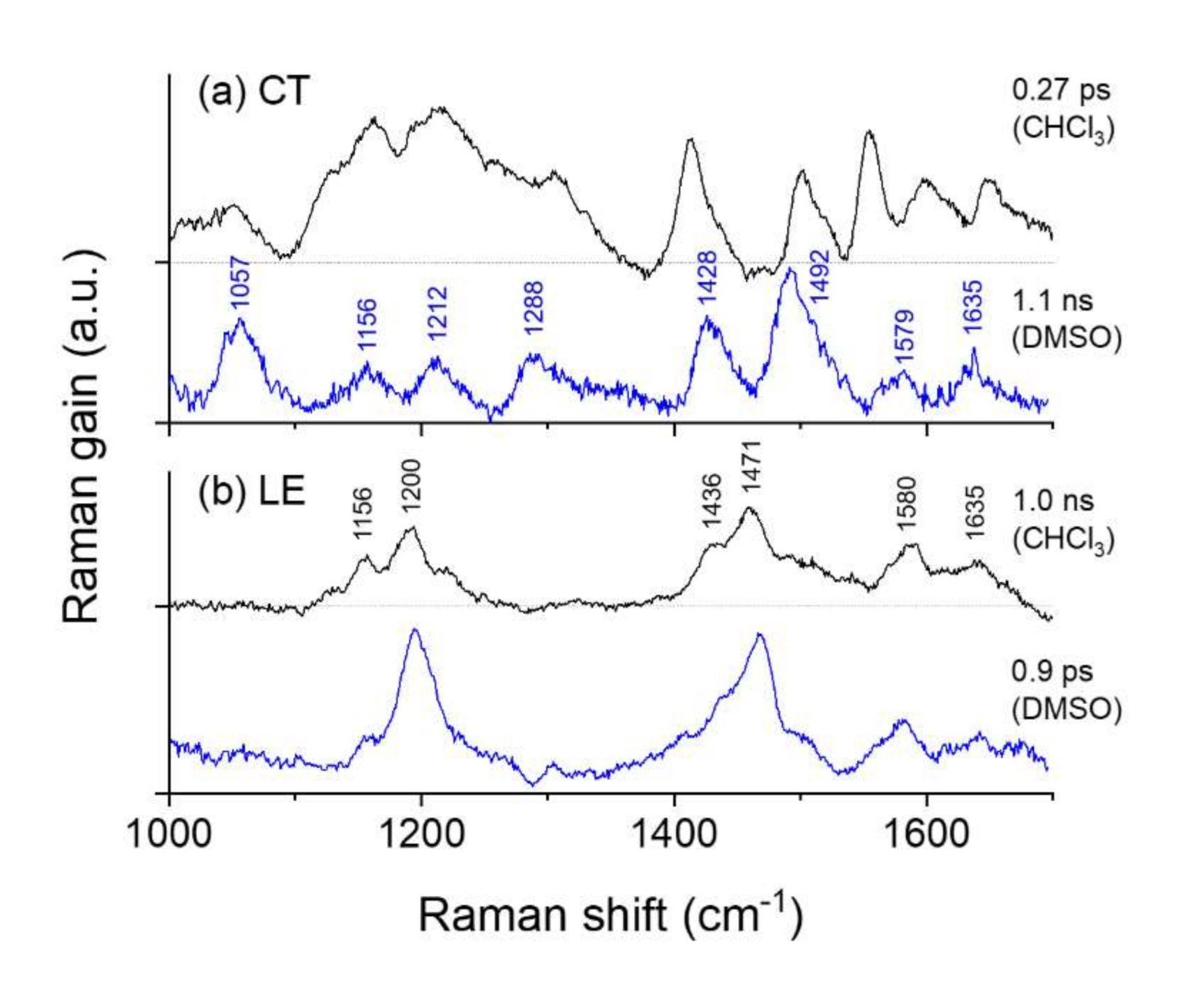
2.3. Time-Resolved Stimulated Raman Spectra of DCM
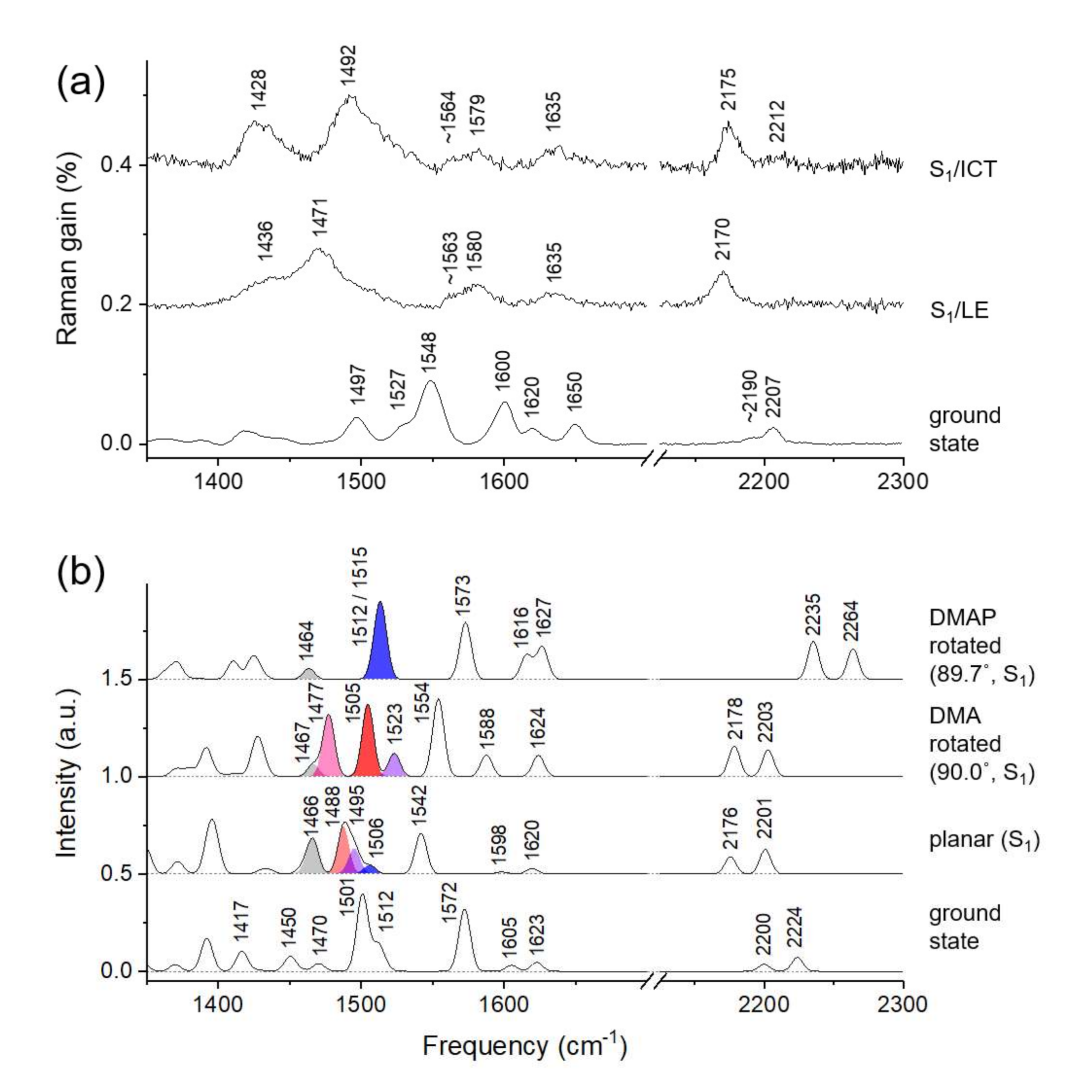
3. Discussion
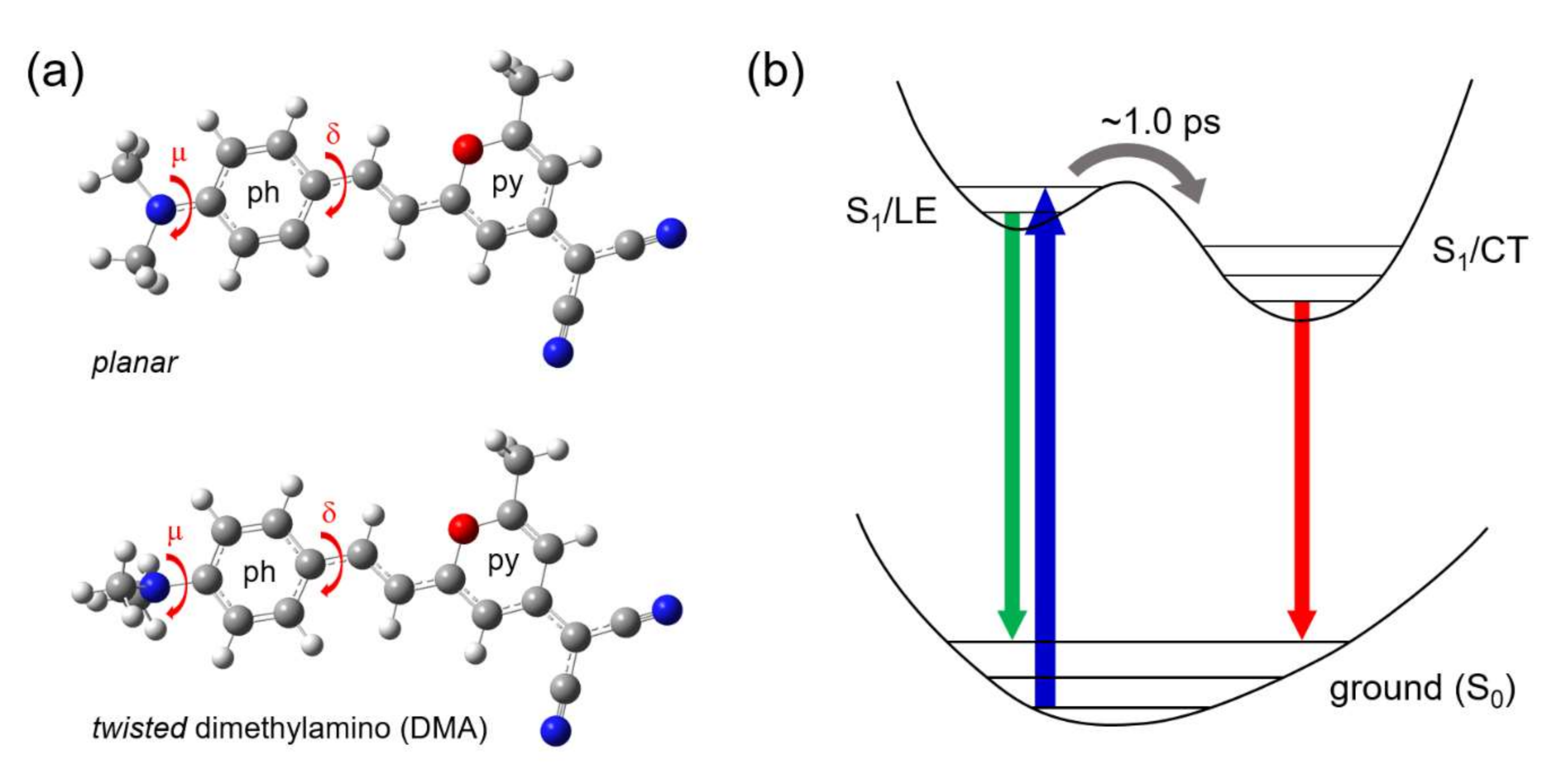
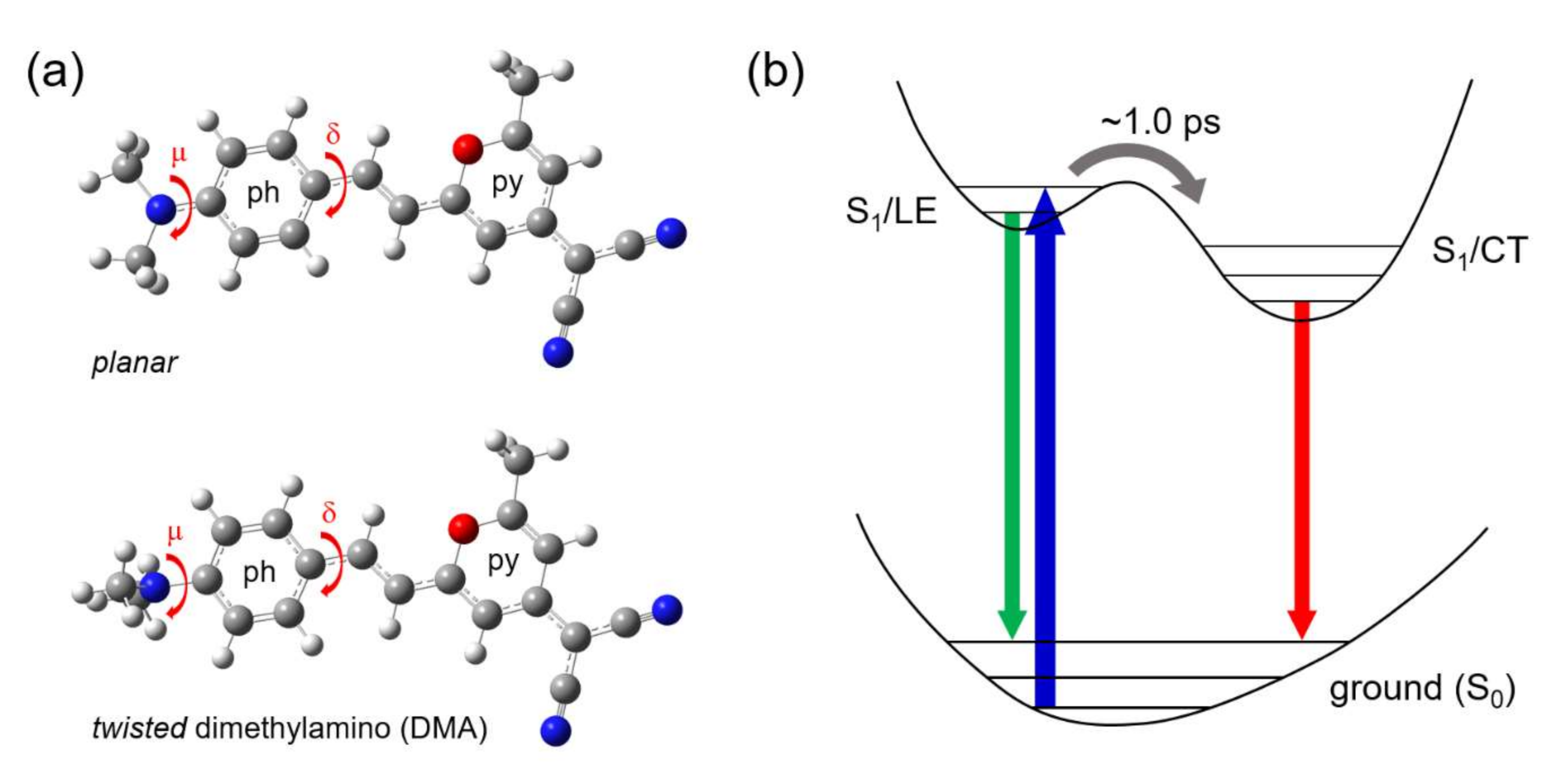
Twisted ICT State of DCM
4. Materials and Methods
4.1. Chemicals
4.2. Steady-State Absorption and Emission Measurements
4.3. Transient Absorption Setup
4.4. Femtosecond Stimulated Raman Setup
4.5. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DCM | 4-dicyanomethylene-2-methyl-6-(p-dimethylaminostyryl)-4H-pyran |
| LE | locally-excited |
| ICT | intramolecular charge transfer |
| DMA | dimethylamino |
| DMAP | dimethylaminophenyl |
| TICT | “twisted” ICT |
| PICT | “planar” ICT |
| CS INDO | conformation spectra-intermediate neglect of differential overlap |
| MRCI | multi references configuration interaction |
| CASSCF | complete active space self-consistent-field |
| DFT | density functional theory |
| TDDFT | time-dependent density functional theory |
| CT | charge-transferred |
| DMSO | dimethylsulfoxide |
| FSRS | femtosecond stimulated Raman spectroscopy |
| DMABN | 4-(dimethylamino)benzonitrile |
| CHCl3 | chloroform |
| EADS | evolution associated difference spectra |
| ph | phenyl |
| py | pyran |
| ps | picosecond |
| fs | femtosecond |
| PCM | polarized continuum model |
| CCD | charge-coupled device |
References
- Kilså, K.; Kajanus, J.; Macpherson, A.N.; Mårtensson, J.; Albinsson, B. Bridge-dependent electron transfer in porphyrin-based donor-bridge-acceptor systems. J. Am. Chem. Soc. 2001, 123, 3069–3080. [Google Scholar] [CrossRef]
- Staykov, A.; Nozaki, D.; Yoshizawa, K. Theoretical study of donor-pi-bridge-acceptor unimolecular electric rectifier. J. Phys. Chem. C 2007, 111, 11699–11705. [Google Scholar] [CrossRef]
- Marszalek, M.; Nagane, S.; Ichake, A.; Humphry-Baker, R.; Paul, V.; Zakeeruddin, S.M.; Grätzel, M. Tuning spectral properties of phenothiazine based donor–π–acceptor dyes for efficient dye-sensitized solar cells. J. Mater. Chem. 2012, 22, 889–894. [Google Scholar] [CrossRef]
- Huang, F.; Chen, K.-S.; Yip, H.-L.; Hau, S.K.; Acton, O.; Zhang, Y.; Luo, J.; Jen, A.K.-Y. Development of New Conjugated Polymers with Donor−π-Bridge−Acceptor Side Chains for High Performance Solar Cells. J. Am. Chem. Soc. 2009, 131, 13886–13887. [Google Scholar] [CrossRef] [PubMed]
- Wenger, O.S. How Donor-Bridge-Acceptor energetics influence electron tunneling dynamics and their distance dependences. Acc. Chem. Res. 2011, 44, 25–35. [Google Scholar] [CrossRef]
- Li, Y.; Liu, T.; Liu, H.; Tian, M.-Z.; Li, Y. Self-Assembly of Intramolecular Charge-Transfer Compounds into Functional Molecular Systems. Acc. Chem. Res. 2014, 47, 1186–1198. [Google Scholar] [CrossRef]
- Zhang, Q.; Kuwabara, H.; Potscavage, W.J.; Huang, S.; Hatae, Y.; Shibata, T.; Adachi, C. Anthraquinone-Based Intramolecular Charge-Transfer Compounds: Computational Molecular Design, Thermally Activated Delayed Fluorescence, and Highly Efficient Red Electroluminescence. J. Am. Chem. Soc. 2014, 136, 18070–18081. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Drummen, G.P.C.; Konishi, G.-i. Recent advances in twisted intramolecular charge transfer (TICT) fluorescence and related phenomena in materials chemistry. J. Mater. Chem. C 2016, 4, 2731–2743. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, Z.R.; Rotkiewicz, K.; Rettig, W. Structural Changes Accompanying Intramolecular Electron Transfer: Focus on Twisted Intramolecular Charge-Transfer States and Structures. Chem. Rev. 2003, 103, 3899–4032. [Google Scholar] [CrossRef]
- Martin, M.M.; Plaza, P.; Meyer, Y.H. Ultrafast intramolecular charge transfer in the merocyanine dye DCM. Chem. Phys. 1995, 192, 367–377. [Google Scholar] [CrossRef]
- Pommeret, S.; Gustavsson, T.; Naskrecki, R.; Baldacchino, G.; Mialocq, J.C. Femtosecond absorption and emission spectroscopy of the DCM laser dye. J. Mol. Liq. 1995, 64, 101–112. [Google Scholar] [CrossRef]
- Van Der Meulen, P.; Zhang, H.; Jonkman, A.M.; Glasbeek, M. Subpicosecond Solvation Relaxation of 4-(Dicyanomethylene)-2-methyl-6-(p-(dimethylamino) styryl)-4 H-pyran in Polar Liquids. J. Phys. Chem. 1996, 100, 5367–5373. [Google Scholar] [CrossRef]
- Gustavsson, T.; Baldacchino, G.; Mialocq, J.C.; Pommeret, S. A femtosecond fluorescence up-conversion study of the dynamic Stokes shift of the DCM dye molecule in polar and non-polar solvents. Chem. Phys. Lett. 1995, 236, 587–594. [Google Scholar] [CrossRef]
- Easter, D.C.; Baronavski, A.P. Ultrafast relaxation in the fluorescent state of the laser dye DCM. Chem. Phys. Lett. 1993, 201, 153–158. [Google Scholar] [CrossRef]
- Hammond, P.R. Laser dye DCM, its spectral properties, synthesis and comparison with other dyes in the red. Opt. Commun. 1979, 29, 331–333. [Google Scholar] [CrossRef]
- Marason, E.G. Laser dye DCM: CW, synchronously pumped, cavity pumped and single-frequency performance. Opt. Commun. 1981, 37, 56–58. [Google Scholar] [CrossRef]
- Petrova, P.K.; Ivanov, P.I.; Tomova, R.L. Color tunability in multilayer OLEDs based on DCM and DPVBi as emitting materials. J. Phys. Conf. Ser. 2014, 514, 012015. [Google Scholar] [CrossRef] [Green Version]
- Giebink, N.C.; Forrest, S.R. Quantum efficiency roll-off at high brightness in fluorescent and phosphorescent organic light emitting diodes. Phys. Rev. B 2008, 77, 235215. [Google Scholar] [CrossRef]
- Tian, Z.; Liu, Y.; Tian, B.; Zhang, J. Synthesis and proton-induced fluorescence “OFF-ON” switching of a new D-pi-A type pyran dye. Res. Chem. Intermed. 2015, 41, 525–533. [Google Scholar] [CrossRef]
- Guo, Z.; Zhao, P.; Zhu, W.; Huang, X.; Xie, Y.; Tian, H. Intramolecular charge-transfer process based on dicyanomethylene-4H-pyran derivative: An integrated operation of half-subtractor and comparator. J. Phys. Chem. C 2008, 112, 7047–7053. [Google Scholar] [CrossRef]
- Mandal, D.; Sen, S.; Bhattacharyya, K.; Tahara, T. Femtosecond study of solvation dynamics of DCM in micelles. Chem. Phys. Lett. 2002, 359, 77–82. [Google Scholar] [CrossRef]
- Lee, J.; Lee, S.; Jen, M.; Pang, Y. Metal-enhanced fluorescence: Wavelength-dependent ultrafast energy transfer. J. Phys. Chem. C 2015, 119, 23285–23291. [Google Scholar] [CrossRef]
- Rettig, W.; Majenz, W.; Majenzlfgang, W. Competing adiabatic photoreaction channels in stilbene derivatives. Chem. Phys. Lett. 1989, 154, 335–341. [Google Scholar] [CrossRef]
- Lapouyade, R.; Kuhn, A.; Letard, J.F.; Rettig, W. Multiple relaxation pathways in photoexcited dimethylaminonitro- and dimethylaminocyano-stilbenes. Chem. Phys. Lett. 1993, 208, 48–58. [Google Scholar] [CrossRef]
- Chang, C.W.; Kao, Y.T.; Diau, E.W.G. Fluorescence lifetime and nonradiative relaxation dynamics of DCM in nonpolar solvent. Chem. Phys. Lett. 2003, 374, 110–118. [Google Scholar] [CrossRef]
- Guo, H.; Zhang, X.; Aydin, M.; Xu, W.; Zhu, H.-r.; Akins, D.L. Spectroscopy and dynamics of DCM encapsulated in MCM-41 and Y zeolite mesoporous materials. J. Mol. Struct. 2004, 689, 153–158. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, R.; Cao, Z.; Zhang, Q. Intramolecular charge transfer and photoisomerization if the DCM styrene dye: A theoretical study. J. Theor. Comput. Chem. 2008, 7, 719–736. [Google Scholar] [CrossRef]
- Marguet, S.; Mialocq, J.C.; Millie, P.; Berthier, G.; Momicchioli, F. Intramolecular charge transfer and trans-cis isomerization of the DCM styrene dye in polar solvents. A CS INDO MRCI study. Chem. Phys. 1992, 160, 265–279. [Google Scholar] [CrossRef]
- Nabavi, S.H.; Khodabandeh, M.H.; Golbabaee, M.; Moshaii, A.; Davari, M.D. Excited states study reveals the twisted geometry induced large stokes shift in DCM fluorescent dye. J. Photochem. Photobiol. A 2018, 354, 127–138. [Google Scholar] [CrossRef]
- Petsalakis, I.D.; Georgiadou, D.G.; Vasilopoulou, M.; Pistolis, G.; Dimotikali, D.; Argitis, P.; Theodorakopoulos, G. Theoretical investigation on the effect of protonation on the absorption and emission spectra of two amine-group-bearing, red “push-pull” emitters, 4-dimethylamino-4’-nitrostilbene and 4-(dicyanomethylene)-2-methyl-6-p-(dimethylamino) styryl-4H-pyran, by DFT and TDDFT calculations. J. Phys. Chem. A 2010, 114, 5580–5587. [Google Scholar] [PubMed]
- Elliott, P.; Furche, F.; Burke, K. Excited states from time-dependent density functional theory. In Rev. Comput. Chem.; Lipkowitz, K.B., Cundari, T.R., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; pp. 91–165. [Google Scholar]
- Oliver, T.A.A.; Lewis, N.H.C.; Fleming, G.R. Correlating the motion of electrons and nuclei with two-dimensional electronic-vibrational spectroscopy. Proc. Natl. Acad. Sci. USA 2014, 111, 10061–10066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Tassle, A.J.; Prantil, M.A.; Fleming, G.R. Investigation of the excited state structure of DCM via ultrafast electronic pump/vibrational probe. J. Phys. Chem. B 2006, 110, 18989–18995. [Google Scholar] [CrossRef]
- Kovalenko, S.A.; Ernsting, N.P.; Ruthmann, J. Femtosecond hole-burning spectroscopy of the dye DCM in solution: The transition from the locally excited to a charge-transfer state. Chem. Phys. Lett. 1996, 258, 445–454. [Google Scholar] [CrossRef]
- Maciejewski, A.; Naskrecki, R.; Lorenc, M.; Ziolek, M.; Karolczak, J.; Kubicki, J.; Matysiak, M.; Szymanski, M. Transient absorption experimental set-up with femtosecond time resolution. Femto- and picosecond study of DCM molecule in cyclohexane and methanol solution. J. Mol. Struct. 2000, 555, 1–13. [Google Scholar] [CrossRef]
- Martin, M.M.; Plaza, P.; Changenet, P.; Meyer, Y. Investigation of excited-state charge transfer with structural change in compounds containing anilino subunits by subpicosecond spectroscopy. J. Photochem. Photobiol. A 1997, 105, 197–204. [Google Scholar] [CrossRef]
- Yoshizawa, M.; Kubo, M.; Kurosawa, M. Ultrafast photoisomerization in DCM dye observed by new femtosecond Raman spectroscopy. J. Lumin. 2000, 87–89, 739–741. [Google Scholar] [CrossRef]
- Karmakar, S.; Ambastha, A.; Jha, A.; Dharmadhikari, A.; Dharmadhikari, J.; Venkatramani, R.; Dasgupta, J. Transient Raman Snapshots of the Twisted Intramolecular Charge Transfer State in a Stilbazolium Dye. J. Phys. Chem. Lett. 2020, 11, 4842–4848. [Google Scholar] [CrossRef] [PubMed]
- McCamant, D.W.; Kukura, P.; Mathies, R.A. Femtosecond time-resolved stimulated Raman spectroscopy: Application to the ultrafast internal conversion in β-carotene. J. Phys. Chem. A 2003, 107, 8208–8214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukura, P.; McCamant, D.W.; Mathies, R.A. Femtosecond Time-Resolved Stimulated Raman Spectroscopy of the S(2) (1B(u)) Excited State of beta-Carotene. J. Phys. Chem. A 2004, 108, 5921–5925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oscar, B.G.; Liu, W.; Rozanov, N.D.; Fang, C. Ultrafast intermolecular proton transfer to a proton scavenger in an organic solvent. Phys. Chem. Chem. Phys. 2016, 18, 26151–26160. [Google Scholar] [CrossRef]
- Liu, W.; Wang, Y.; Tang, L.; Oscar, B.G.; Zhu, L.; Fang, C. Panoramic portrait of primary molecular events preceding excited state proton transfer in water. Chem. Sci. 2016, 7, 5484–5494. [Google Scholar] [CrossRef] [Green Version]
- Jen, M.; Lee, S.; Jeon, K.; Hussain, S.; Pang, Y. Ultrafast Intramolecular Proton Transfer of Alizarin Investigated by Femtosecond Stimulated Raman Spectroscopy. J. Phys. Chem. B 2017, 121, 4129–4136. [Google Scholar] [CrossRef] [PubMed]
- Jen, M.; Jeon, K.; Lee, S.; Hwang, S.; Chung, W.J.; Pang, Y. Ultrafast intramolecular proton transfer reactions and solvation dynamics of DMSO. Struct. Dyn. 2019, 6, 064901. [Google Scholar] [CrossRef] [Green Version]
- Hart, S.M.; Silva, W.R.; Frontiera, R.R. Femtosecond stimulated Raman evidence for charge-Transfer character in pentacene singlet fission. Chem. Sci. 2018, 9, 1242–1250. [Google Scholar] [CrossRef] [Green Version]
- Rhinehart, J.M.; Challa, J.R.; McCamant, D.W. Multimode charge-transfer dynamics of 4-(dimethylamino)benzonitrile probed with ultraviolet femtosecond stimulated Raman spectroscopy. J. Phys. Chem. B 2012, 116, 10522–10534. [Google Scholar] [CrossRef]
- Hoffman, D.P.; Mathies, R.A. Photoexcited structural dynamics of an azobenzene analog 4-nitro-4’-dimethylamino-azobenzene from femtosecond stimulated Raman. Phys. Chem. Chem. Phys. 2012, 14, 6298–6306. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.E.; Veldkamp, B.S.; Co, D.T.; Wasielewski, M.R. Vibrational Dynamics of a Perylene–Perylenediimide Donor–Acceptor Dyad Probed with Femtosecond Stimulated Raman Spectroscopy. J. Phys. Chem. Lett. 2012, 3, 2362–2366. [Google Scholar] [CrossRef]
- Hoffman, D.P.; Lee, O.P.; Millstone, J.E.; Chen, M.S.; Su, T.A.; Creelman, M.; Fréchet, J.M.J.; Mathies, R.A. Electron Transfer Dynamics of Triphenylamine Dyes Bound to TiO2 Nanoparticles from Femtosecond Stimulated Raman Spectroscopy. J. Phys. Chem. C 2013, 117, 6990–6997. [Google Scholar] [CrossRef]
- Hoffman, D.P.; Mathies, R.A. Femtosecond Stimulated Raman Exposes the Role of Vibrational Coherence in Condensed-Phase Photoreactivity. Acc. Chem. Res. 2016, 49, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, R.; Hamada, N.; Abe, K.; Yoshizawa, M. Ultrafast hydrogen-bonding dynamics in the electronic excited state of photoactive yellow protein revealed by femtosecond stimulated Raman spectroscopy. J. Phys. Chem. B 2012, 116, 14768–14775. [Google Scholar] [CrossRef]
- Hall, C.R.; Conyard, J.; Heisler, I.A.; Jones, G.; Frost, J.; Browne, W.R.; Feringa, B.L.; Meech, S.R. Ultrafast Dynamics in Light-Driven Molecular Rotary Motors Probed by Femtosecond Stimulated Raman Spectroscopy. J. Am. Chem. Soc. 2017, 139, 7408–7414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukura, P.; McCamant, D.W.; Mathies, R.A. Femtosecond stimulated Raman spectroscopy. Annu. Rev. Phys. Chem. 2007, 58, 461–488. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Mialocq, J.C. Ground state and singlet excited state of laser dye DCM: Dipole moments and solvent induced spectral shifts. Opt. Commun. 1987, 64, 264–268. [Google Scholar] [CrossRef]
- Van Stokkum, I.H.M.; Larsen, D.S.; Van Grondelle, R. Global and target analysis of time-resolved spectra. Biochim. Biophys. Acta 2004, 1657, 82–104. [Google Scholar] [CrossRef] [Green Version]
- Kwok, W.-M.; George, M.W.; Grills, D.C.; Ma, C.; Matousek, P.; Parker, A.W.; Phillips, D.; Toner, W.T.; Towrie, M. Direct Observation of a Hydrogen-Bonded Charge-Transfer State of 4-Dimethylaminobenzonitrile in Methanol by Time-Resolved IR Spectroscopy. Angew. Chem. Int. Ed. 2003, 42, 1826–1830. [Google Scholar] [CrossRef]
- Kwok, W.M.; Ma, C.; George, M.W.; Grills, D.C.; Matousek, P.; Parker, A.W.; Phillips, D.; Toner, W.T.; Towrie, M. Solvent effects on the charge transfer excited states of 4-dimethylaminobenzonitrile (DMABN) and 4-dimethylamino-3,5-dimethylbenzonitrile (TMABN) studied by time-resolved infrared spectroscopy: A direct observation of hydrogen bonding interactions. Photochem. Photobiol. Sci. 2007, 6, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Levinson, N.M.; Fried, S.D.; Boxer, S.G. Solvent-induced infrared frequency shifts in aromatic nitriles are quantitatively described by the vibrational stark effect. J. Phys. Chem. B 2012, 116, 10470–10476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stsiapura, V.I.; Maskevich, A.A.; Kuzmitsky, V.A.; Turoverov, K.K.; Kuznetsova, I.M. Computational study of thioflavin T torsional relaxation in the excited state. J. Phys. Chem. A 2007, 111, 4829–4835. [Google Scholar] [CrossRef]
- Singh, C.; Ghosh, R.; Mondal, J.A.; Palit, D.K. Excited state dynamics of a push-pull stilbene: A femtosecond transient absorption spectroscopic study. J. Photochem. Photobiol. A 2013, 263, 50–60. [Google Scholar] [CrossRef]
- Chiu, C.-C.; Chen, W.-C.; Cheng, P.-Y. Excited-state vibrational relaxation and deactivation dynamics of trans-4-(N, N-dimethylamino)-4′-nitrostilbene in nonpolar solvents studied by ultrafast time-resolved broadband fluorescence spectroscopy. J. Photochem. Photobiol. A 2015, 310, 26–32. [Google Scholar] [CrossRef]
- Ghosh, R.; Nandi, A.; Palit, D.K. Solvent sensitive intramolecular charge transfer dynamics in the excited states of 4-N, N-dimethylamino-4′-nitrobiphenyl. Phys. Chem. Chem. Phys. 2016, 18, 7661–7671. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Jen, M.; Lee, G.; Pang, Y. Structural Changes of Nitroaromatic Molecules during the Intramolecular Charge Transfer. In Proceedings of the Frontiers in Optics/Laser Science, Washington, DC, USA, 17 September 2020; p. FTh2D.5. [Google Scholar]
- Lee, D.; Lee, J.; Song, J.; Jen, M.; Pang, Y. Homogeneous silver colloidal substrates optimal for metal-enhanced fluorescence. Phys. Chem. Chem. Phys. 2019, 21, 11599–11607. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Song, J.; Lee, D.; Pang, Y. Metal-enhanced fluorescence and excited state dynamics of carotenoids in thin polymer films. Sci. Rep. 2019, 9, 3551. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Lee, S.; Pang, Y. Excited-State Dynamics of Carotenoids Studied by Femtosecond Transient Absorption Spectroscopy. Bull. Korean Chem. Soc. 2014, 35, 851–857. [Google Scholar] [CrossRef] [Green Version]
- Snellenburg, J.J.; Laptenok, S.P.; Seger, R.; Mullen, K.M.; Stokkun, I.H.M. Glotaran: A Java -Based Graphical User Interface for the R Package TIMP. J. Stat. Softw. 2012, 49, 1–22. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.; Jen, M.; Pang, Y. Twisted Intramolecular Charge Transfer State of a “Push-Pull” Emitter. Int. J. Mol. Sci. 2020, 21, 7999. https://doi.org/10.3390/ijms21217999
Lee S, Jen M, Pang Y. Twisted Intramolecular Charge Transfer State of a “Push-Pull” Emitter. International Journal of Molecular Sciences. 2020; 21(21):7999. https://doi.org/10.3390/ijms21217999
Chicago/Turabian StyleLee, Sebok, Myungsam Jen, and Yoonsoo Pang. 2020. "Twisted Intramolecular Charge Transfer State of a “Push-Pull” Emitter" International Journal of Molecular Sciences 21, no. 21: 7999. https://doi.org/10.3390/ijms21217999
APA StyleLee, S., Jen, M., & Pang, Y. (2020). Twisted Intramolecular Charge Transfer State of a “Push-Pull” Emitter. International Journal of Molecular Sciences, 21(21), 7999. https://doi.org/10.3390/ijms21217999