Statins Stimulate New Myocyte Formation After Myocardial Infarction by Activating Growth and Differentiation of the Endogenous Cardiac Stem Cells

 ,
,  ,
,  ,
,  ,
,
Abstract
1. Introduction
2. Results
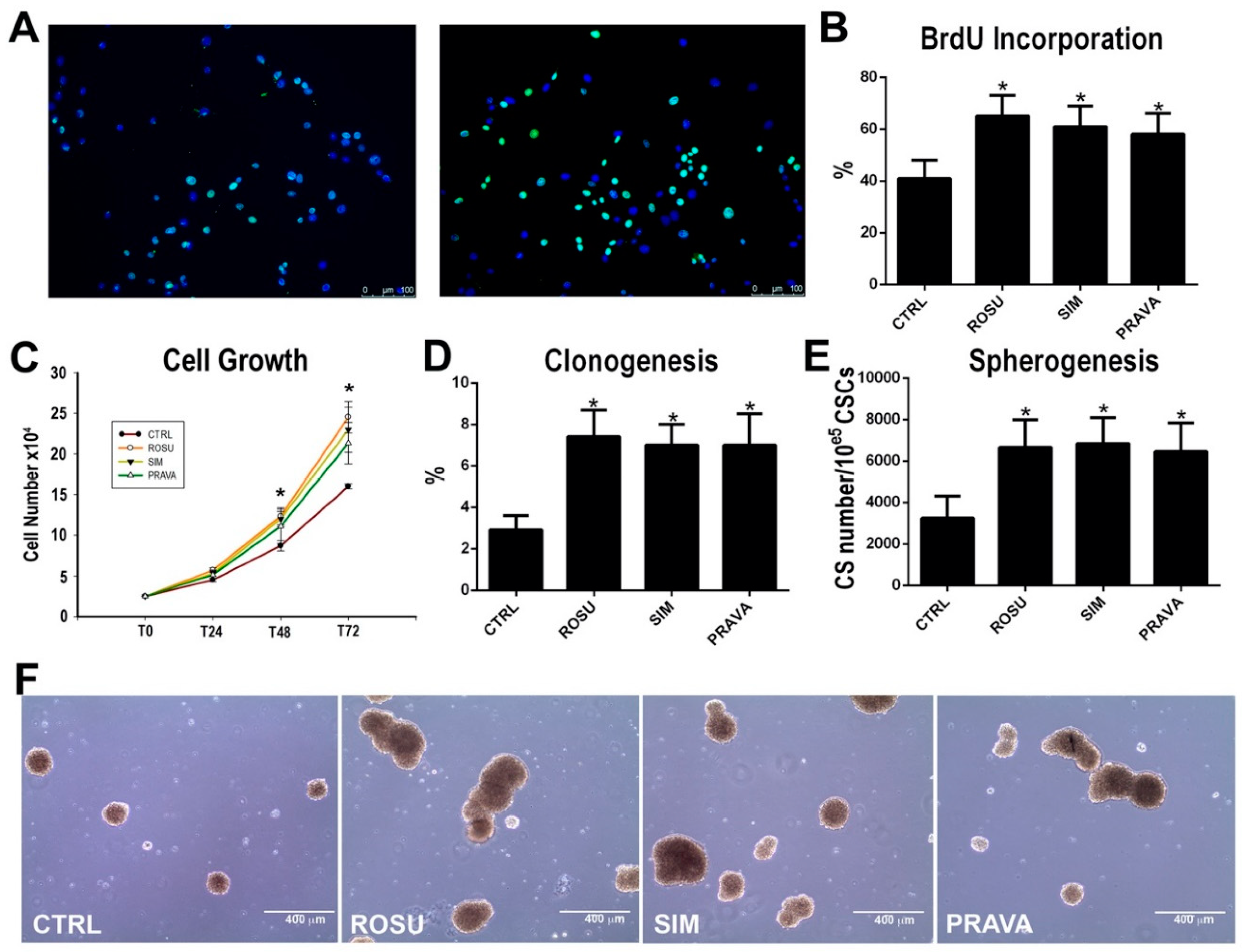
2.1. HMG-CoA Reductase Inhibitors Enhance Rat Cardiac Stem Cell Growth, Clonogenesis and Spherogenesis In Vitro
2.2. HMG-CoA Reductase Inhibitors Activate the Protein Kinase Akt and Promote CSC Survival In Vitro
2.3. HMG-CoA Reductase Inhibitors Foster Myogenic Commitment of CSCs In Vitro
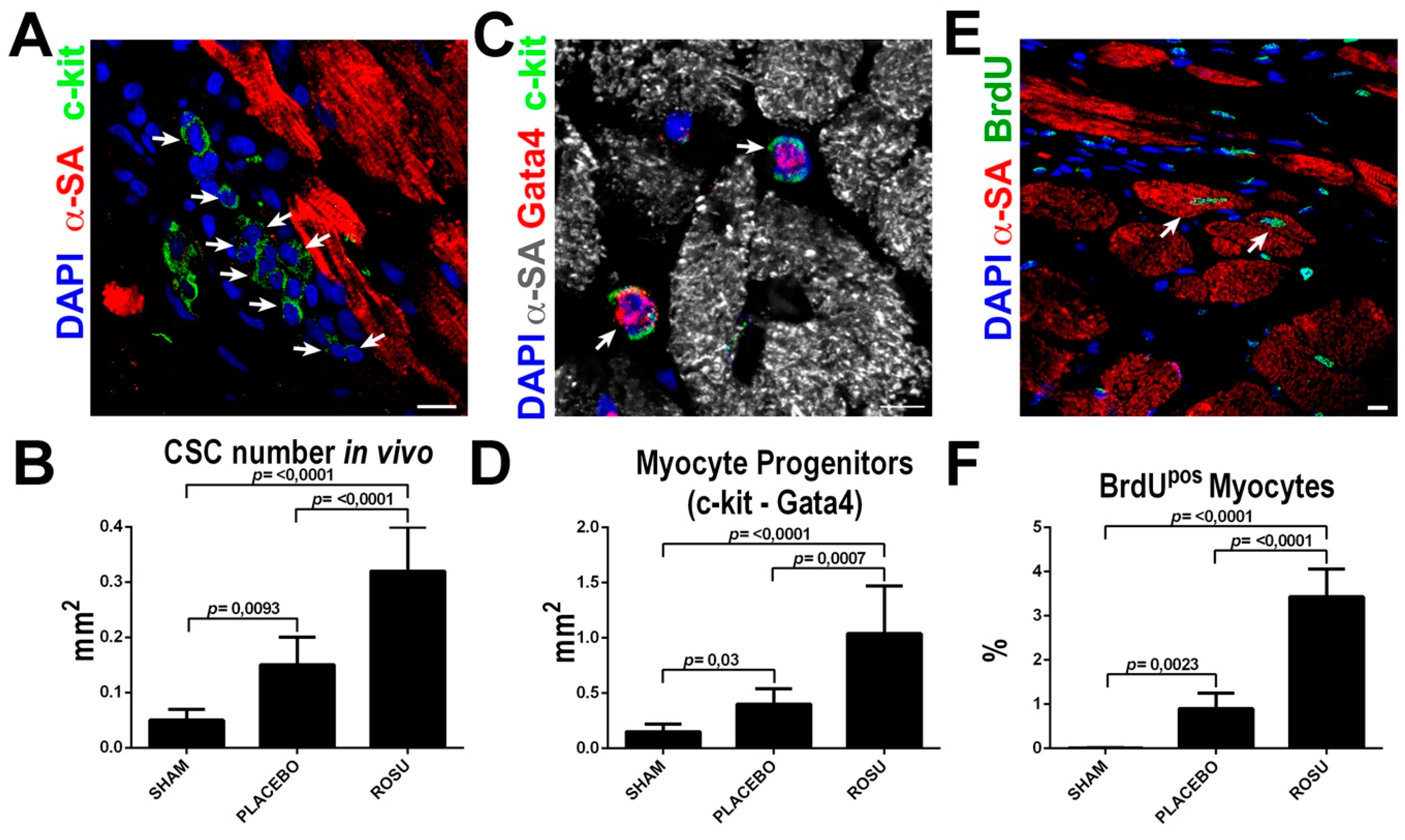
2.4. HMG-CoA Reductase Inhibitors Increase CSC Number and New Myocyte Formation After Myocardial Infarction
2.5. HMG-CoA Reductase Inhibitors Ameliorate the In Vitro and In Vivo Regenerative Defect of CSCs from c-Kit Haploinsufficient Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Cell Reagents
4.3. CSC Isolation and Culture
4.4. CSC Clonogenic and Spherogenesis Assay In Vitro
4.5. Cardiac Differentiation Potential and Cardiosphere Myogenic Differentiation Assay In Vitro
4.6. Proliferation and Apoptosis Assay In Vitro
4.7. Immunocytochemistry
4.8. Quantitative RT-PCR (qRT-PCR)
4.9. Western Blot Analysis
4.10. Myocardial Infarction Procedure
4.11. Echocardiography
4.12. Tissue Harvesting, Histology and Immunohistochemistry
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CSCs | Cardiac Stem Cells |
| MI | Myocardial Infarction |
| HMG-CoA | 3-hydroxy-3-methyl-glutaryl-coenzyme |
| LV | Left ventricle |
References
- Beltrami, A.P.; Barlucchi, L.; Torella, D.; Baker, M.; Limana, F.; Chimenti, S.; Kasahara, H.; Rota, M.; Musso, E.; Urbanek, K.; et al. Adult Cardiac Stem Cells Are Multipotent and Support Myocardial Regeneration. Cell 2003, 114, 763–776. [Google Scholar] [CrossRef]
- Dai, D.-F.; Chen, T.; Johnson, S.C.; Szeto, H.; Rabinovitch, P.S. Cardiac aging: From molecular mechanisms to significance in human health and disease. Antioxid. Redox Signal. 2012, 16, 1492–1526. [Google Scholar] [CrossRef] [PubMed]
- Torella, D.; Rota, M.; Nurzynska, D.; Musso, E.; Monsen, A.; Shiraishi, I.; Zias, E.; Walsh, K.; Rosenzweig, A.; Sussman, M.A.; et al. Cardiac stem cell and myocyte aging, heart failure, and insulin-like growth factor-1 overexpression. Circ. Res. 2004, 94, 514–524. [Google Scholar] [CrossRef] [PubMed]
- van Berlo, J.H.; Kanisicak, O.; Maillet, M.; Vagnozzi, R.J.; Karch, J.; Lin, S.-C.J.; Middleton, R.C.; Marbán, E.; Molkentin, J.D. c-kit+ cells minimally contribute cardiomyocytes to the heart. Nature 2014, 509, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.-L.; Molkentin, J.D. The Elusive Progenitor Cell in Cardiac Regeneration: Slip Slidin’ Away. Circ. Res. 2017, 120, 400–406. [Google Scholar] [CrossRef] [PubMed]
- van Berlo, J.H.; Molkentin, J.D. Most of the Dust Has Settled. Circ. Res. 2016, 118, 17–19. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Han, M.; Zhang, Z.; Li, Y.; Huang, X.; Liu, X.; Pu, W.; Zhao, H.; Wang, Q.-D.; Nie, Y.; et al. Reassessment of c-Kit+ Cells for Cardiomyocyte Contribution in Adult Heart. Circulation 2019, 140, 164–166. [Google Scholar] [CrossRef]
- Vagnozzi, R.J.; Sargent, M.A.; Lin, S.-C.J.; Palpant, N.J.; Murry, C.E.; Molkentin, J.D. Genetic Lineage Tracing of Sca-1 + Cells Reveals Endothelial but Not Myogenic Contribution to the Murine Heart. Circulation 2018, 138, 2931–2939. [Google Scholar] [CrossRef]
- Sultana, N.; Zhang, L.; Yan, J.; Chen, J.; Cai, W.; Razzaque, S.; Jeong, D.; Sheng, W.; Bu, L.; Xu, M.; et al. Resident c-kit+ cells in the heart are not cardiac stem cells. Nat. Commun. 2015, 6, 8701. [Google Scholar] [CrossRef]
- Vicinanza, C.; Aquila, I.; Cianflone, E.; Scalise, M.; Marino, F.; Mancuso, T.; Fumagalli, F.; Giovannone, E.D.; Cristiano, F.; Iaccino, E.; et al. Kitcre knock-in mice fail to fate-map cardiac stem cells. Nature 2018, 555, E1–E5. [Google Scholar] [CrossRef]
- Ellison, G.M.; Vicinanza, C.; Smith, A.J.; Aquila, I.; Leone, A.; Waring, C.D.; Henning, B.J.; Stirparo, G.G.; Papait, R.; Scarfò, M.; et al. Adult c-kitpos Cardiac Stem Cells Are Necessary and Sufficient for Functional Cardiac Regeneration and Repair. Cell 2013, 154, 827–842. [Google Scholar] [CrossRef] [PubMed]
- Vicinanza, C.; Aquila, I.; Scalise, M.; Cristiano, F.; Marino, F.; Cianflone, E.; Mancuso, T.; Marotta, P.; Sacco, W.; Lewis, F.C.; et al. Adult cardiac stem cells are multipotent and robustly myogenic: c-kit expression is necessary but not sufficient for their identification. Cell Death Differ. 2017, 24, 2101–2116. [Google Scholar] [CrossRef] [PubMed]
- Aquila, I.; Cianflone, E.; Scalise, M.; Marino, F.; Mancuso, T.; Filardo, A.; Smith, A.J.; Cappetta, D.; De Angelis, A.; Urbanek, K.; et al. c-kit Haploinsufficiency impairs adult cardiac stem cell growth, myogenicity and myocardial regeneration. Cell Death Dis. 2019, 10, 436. [Google Scholar] [CrossRef]
- Messina, E.; De Angelis, L.; Frati, G.; Morrone, S.; Chimenti, S.; Fiordaliso, F.; Salio, M.; Battaglia, M.; Latronico, M.V.G.; Coletta, M.; et al. Isolation and Expansion of Adult Cardiac Stem Cells From Human and Murine Heart. Circ. Res. 2004, 95, 911–921. [Google Scholar] [CrossRef]
- Smith, R.R.; Barile, L.; Cho, H.C.; Leppo, M.K.; Hare, J.M.; Messina, E.; Giacomello, A.; Abraham, M.R.; Marban, E. Regenerative Potential of Cardiosphere-Derived Cells Expanded From Percutaneous Endomyocardial Biopsy Specimens. Circulation 2007, 115, 896–908. [Google Scholar] [CrossRef]
- Valente, M.; Nascimento, D.S.; Cumano, A.; Pinto-do-Ó, P. Sca-1 + Cardiac Progenitor Cells and Heart-Making: A Critical Synopsis. Stem Cells Dev. 2014, 23, 2263–2273. [Google Scholar] [CrossRef]
- Linke, A.; Muller, P.; Nurzynska, D.; Casarsa, C.; Torella, D.; Nascimbene, A.; Castaldo, C.; Cascapera, S.; Bohm, M.; Quaini, F.; et al. Stem cells in the dog heart are self-renewing, clonogenic, and multipotent and regenerate infarcted myocardium, improving cardiac function. Proc. Natl. Acad. Sci. USA 2005, 102, 8966–8971. [Google Scholar] [CrossRef] [PubMed]
- Torella, D.; Ellison, G.M.; Torella, M.; Vicinanza, C.; Aquila, I.; Iaconetti, C.; Scalise, M.; Marino, F.; Henning, B.J.; Lewis, F.C.; et al. Carbonic anhydrase activation is associated with worsened pathological remodeling in human ischemic diabetic cardiomyopathy. J. Am. Heart Assoc. 2014, 3, e000434. [Google Scholar] [CrossRef]
- Savi, M.; Bocchi, L.; Rossi, S.; Frati, C.; Graiani, G.; Lagrasta, C.; Miragoli, M.; Di Pasquale, E.; Stirparo, G.G.; Mastrototaro, G.; et al. Antiarrhythmic effect of growth factor-supplemented cardiac progenitor cells in chronic infarcted heart. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1622–H1648. [Google Scholar] [CrossRef]
- De Angelis, A.; Piegari, E.; Cappetta, D.; Russo, R.; Esposito, G.; Ciuffreda, L.P.; Ferraiolo, F.A.V.; Frati, C.; Fagnoni, F.; Berrino, L.; et al. SIRT1 activation rescues doxorubicin-induced loss of functional competence of human cardiac progenitor cells. Int. J. Cardiol. 2015, 189, 30–44. [Google Scholar] [CrossRef]
- Piegari, E.; Russo, R.; Cappetta, D.; Esposito, G.; Urbanek, K.; Dell’Aversana, C.; Altucci, L.; Berrino, L.; Rossi, F.; De Angelis, A. MicroRNA-34a regulates doxorubicin-induced cardiotoxicity in rat. Oncotarget 2016, 7, 62312–62326. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.; Reith, C.; Emberson, J.; Armitage, J.; Baigent, C.; Blackwell, L.; Blumenthal, R.; Danesh, J.; Smith, G.D.; DeMets, D.; et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet 2016, 388, 2532–2561. [Google Scholar] [CrossRef]
- Schmidt-Lucke, C.; Fichtlscherer, S.; Rössig, L.; Kämper, U.; Dimmeler, S. Improvement of endothelial damage and regeneration indexes in patients with coronary artery disease after 4 weeks of statin therapy. Atherosclerosis 2010, 211, 249–254. [Google Scholar] [CrossRef]
- Indolfi, C.; Di Lorenzo, E.; Perrino, C.; Stingone, A.M.; Curcio, A.; Torella, D.; Cittadini, A.; Cardone, L.; Coppola, C.; Cavuto, L.; et al. Hydroxymethylglutaryl coenzyme A reductase inhibitor simvastatin prevents cardiac hypertrophy induced by pressure overload and inhibits p21ras activation. Circulation 2002, 106, 2118–2124. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lefer, D.J. Statins as Potent Antiinflammatory Drugs. Circulation 2002, 106, 2041–2042. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Tangney, C.C.; Casey, L.C. Inhibition of proinflammatory cytokine production by pravastatin. Lancet 1999, 353, 983–984. [Google Scholar] [CrossRef]
- Pignatelli, P.; Carnevale, R.; Di Santo, S.; Bartimoccia, S.; Nocella, C.; Vicario, T.; Loffredo, L.; Angelico, F.; Violi, F. Rosuvastatin reduces platelet recruitment by inhibiting NADPH oxidase activation. Biochem. Pharmacol. 2012, 84, 1635–1642. [Google Scholar] [CrossRef]
- Sanguigni, V.; Pignatelli, P.; Lenti, L.; Ferro, D.; Bellia, A.; Carnevale, R.; Tesauro, M.; Sorge, R.; Lauro, R.; Violi, F. Short-term treatment with atorvastatin reduces platelet CD40 ligand and thrombin generation in hypercholesterolemic patients. Circulation 2005, 111, 412–419. [Google Scholar] [CrossRef]
- Assmus, B.; Urbich, C.; Aicher, A.; Hofmann, W.K.; Haendeler, J.; Rössig, L.; Spyridopoulos, I.; Zeiher, A.M.; Dimmeler, S. HMG-CoA reductase inhibitors reduce senescence and increase proliferation of endothelial progenitor cells via regulation of cell cycle regulatory genes. Circ. Res. 2003, 92, 1049–1055. [Google Scholar] [CrossRef]
- Llevadot, J.; Murasawa, S.; Kureishi, Y.; Uchida, S.; Masuda, H.; Kawamoto, A.; Walsh, K.; Isner, J.M.; Asahara, T. HMG-CoA reductase inhibitor mobilizes bone marrow--derived endothelial progenitor cells. J. Clin. Investig. 2001, 108, 399–405. [Google Scholar] [CrossRef]
- Dimmeler, S.; Aicher, A.; Vasa, M.; Mildner-Rihm, C.; Adler, K.; Tiemann, M.; Rütten, H.; Fichtlscherer, S.; Martin, H.; Zeiher, A.M. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. J. Clin. Investig. 2001, 108, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Leri, A. Aging and disease as modifiers of efficacy of cell therapy. Circ. Res. 2008, 102, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.; Lewis, F.C.; Aquila, I.; Waring, C.D.; Nocera, A.; Agosti, V.; Nadal-Ginard, B.; Torella, D.; Ellison, G.M. Isolation and characterization of resident endogenous c-Kit+ cardiac stem cells from the adult mouse and rat heart. Nat. Protoc. 2014, 9, 1662–1681. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Wang, K.; Li, B.; Xie, Y.; Xia, N.; Li, M.; Gao, G. Statin rosuvastatin inhibits apoptosis of human coronary artery endothelial cells through upregulation of the JAK2/STAT3 signaling pathway. Mol. Med. Rep. 2020, 22, 2052–2062. [Google Scholar] [CrossRef] [PubMed]
- Di Siena, S.; Gimmelli, R.; Nori, S.L.; Barbagallo, F.; Campolo, F.; Dolci, S.; Rossi, P.; Venneri, M.A.; Giannetta, E.; Gianfrilli, D.; et al. Activated c-Kit receptor in the heart promotes cardiac repair and regeneration after injury. Cell Death Dis. 2016, 7, e2317. [Google Scholar] [CrossRef][Green Version]
- Bernex, F.; De Sepulveda, P.; Kress, C.; Elbaz, C.; Delouis, C.; Panthier, J.J. Spatial and temporal patterns of c-kit-expressing cells in WlacZ/+ and WlacZ/WlacZ mouse embryos. Development 1996, 122, 3023–3033. [Google Scholar]
- Lennartsson, J.; Rönnstrand, L. Stem cell factor receptor/c-Kit: From basic science to clinical implications. Physiol. Rev. 2012, 92, 1619–1649. [Google Scholar] [CrossRef]
- Aquila, I.; Marino, F.; Cianflone, E.; Marotta, P.; Torella, M.; Mollace, V.; Indolfi, C.; Nadal-Ginard, B.; Torella, D. The use and abuse of Cre/Lox recombination to identify adult cardiomyocyte renewal rate and origin. Pharmacol. Res. 2018, 127, 116–128. [Google Scholar] [CrossRef]
- Cimini, M.; Fazel, S.; Zhuo, S.; Xaymardan, M.; Fujii, H.; Weisel, R.D.; Li, R.-K. c-kit dysfunction impairs myocardial healing after infarction. Circulation 2007, 116, I77–I82. [Google Scholar] [CrossRef]
- Ye, L.; Zhang, E.Y.; Xiong, Q.; Astle, C.M.; Zhang, P.; Li, Q.; From, A.H.L.; Harrison, D.E.; Zhang, J.J. Aging Kit Mutant Mice Develop Cardiomyopathy. PLoS ONE 2012, 7, e33407. [Google Scholar] [CrossRef]
- Fazel, S.; Cimini, M.; Chen, L.; Li, S.; Angoulvant, D.; Fedak, P.; Verma, S.; Weisel, R.D.; Keating, A.; Li, R.-K. Cardioprotective c-kit+ cells are from the bone marrow and regulate the myocardial balance of angiogenic cytokines. J. Clin. Investig. 2006, 116, 1865–1877. [Google Scholar] [CrossRef] [PubMed]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic Effects of Statins on the Cardiovascular System. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, M.D.; Tavori, H.; Fazio, S. PCSK9: From Basic Science Discoveries to Clinical Trials. Circ. Res. 2018, 122, 1420–1438. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Yang, Y.-J.; Yang, T.; Qian, H.-Y. Statins and stem cell modulation. Ageing Res. Rev. 2013, 12, 1–7. [Google Scholar] [CrossRef]
- Cai, A.; Qiu, R.; Li, L.; Zheng, D.; Dong, Y.; Yu, D.; Huang, Y.; Rao, S.; Zhou, Y.; Mai, W. Atorvastatin Treatment of Rats with Ischemia-Reperfusion Injury Improves Adipose-Derived Mesenchymal Stem Cell Migration and Survival via the SDF-1a/CXCR-4 Axis. PLoS ONE 2013, 8, e79100. [Google Scholar] [CrossRef]
- Cohen, K.S.; Cheng, S.; Larson, M.G.; Cupples, L.A.; McCabe, E.L.; Wang, Y.A.; Ngwa, J.S.; Martin, R.P.; Klein, R.J.; Hashmi, B.; et al. Circulating CD34+ progenitor cell frequency is associated with clinical and genetic factors. Blood 2013, 121, e50–e56. [Google Scholar] [CrossRef]
- Erbs, S.; Beck, E.B.; Linke, A.; Adams, V.; Gielen, S.; Kränkel, N.; Möbius-Winkler, S.; Höllriegel, R.; Thiele, H.; Hambrecht, R.; et al. High-dose rosuvastatin in chronic heart failure promotes vasculogenesis, corrects endothelial function, and improves cardiac remodeling--results from a randomized, double-blind, and placebo-controlled study. Int. J. Cardiol. 2011, 146, 56–63. [Google Scholar] [CrossRef]
- Suzuki, G.; Iyer, V.; Cimato, T.; Canty, J.M., Jr. Pravastatin improves function in hibernating myocardium by mobilizing CD133+ and cKit+ bone marrow progenitor cells and promoting myocytes to reenter the growth phase of the cardiac cell cycle. Circ. Res. 2009, 104, 255–264. [Google Scholar] [CrossRef]
- Carson, R.A.; Rudine, A.C.; Tally, S.J.; Franks, A.L.; Frahm, K.A.; Waldman, J.K.; Silswal, N.; Burale, S.; Phan, J.V.; Chandran, U.R.; et al. Statins impact primary embryonic mouse neural stem cell survival, cell death, and fate through distinct mechanisms. PLoS ONE 2018, 13, e0196387. [Google Scholar] [CrossRef]
- Marotta, P.; Cianflone, E.; Aquila, I.; Vicinanza, C.; Scalise, M.; Marino, F.; Mancuso, T.; Torella, M.; Indolfi, C.; Torella, D. Combining cell and gene therapy to advance cardiac regeneration. Expert Opin. Biol. Ther. 2018, 18, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Cianflone, E.; Aquila, I.; Scalise, M.; Marotta, P.; Torella, M.; Nadal-Ginard, B.; Torella, D. Molecular basis of functional myogenic specification of Bona Fide multipotent adult cardiac stem cells. Cell Cycle 2018, 17, 927–946. [Google Scholar] [CrossRef] [PubMed]
- Cianflone, E.; Torella, M.; Biamonte, F.; De Angelis, A.; Urbanek, K.; Costanzo, F.S.; Rota, M.; Ellison-Hughes, G.M.; Torella, D. Targeting Cardiac Stem Cell Senescence to Treat Cardiac Aging and Disease. Cells 2020, 9, 1558. [Google Scholar] [CrossRef]
- Cianflone, E.; Torella, M.; Chimenti, C.; De Angelis, A.; Beltrami, A.P.; Urbanek, K.; Rota, M.; Torella, D. Adult Cardiac Stem Cell Aging: A Reversible Stochastic Phenomenon? Oxid. Med. Cell. Longev. 2019, 2019, 5813147. [Google Scholar] [CrossRef]
- Lewis-McDougall, F.C.; Ruchaya, P.J.; Domenjo-Vila, E.; Shin Teoh, T.; Prata, L.; Cottle, B.J.; Clark, J.E.; Punjabi, P.P.; Awad, W.; Torella, D.; et al. Aged-senescent cells contribute to impaired heart regeneration. Aging Cell 2019, 18, e12931. [Google Scholar] [CrossRef]
- Martin, G.; Duez, H.; Blanquart, C.; Berezowski, V.; Poulain, P.; Fruchart, J.C.; Najib-Fruchart, J.; Glineur, C.; Staels, B. Statin-induced inhibition of the Rho-signaling pathway activates PPARalpha and induces HDL apoA-I. J. Clin. Investig. 2001, 107, 1423–1432. [Google Scholar] [CrossRef]
- Kureishi, Y.; Luo, Z.; Shiojima, I.; Bialik, A.; Fulton, D.; Lefer, D.J.; Sessa, W.C.; Walsh, K. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat. Med. 2000, 6, 1004–1010. [Google Scholar] [CrossRef]
- Ota, H.; Eto, M.; Kano, M.R.; Kahyo, T.; Setou, M.; Ogawa, S.; Iijima, K.; Akishita, M.; Ouchi, Y. Induction of endothelial nitric oxide synthase, SIRT1, and catalase by statins inhibits endothelial senescence through the Akt pathway. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2205–2211. [Google Scholar] [CrossRef]
- Torella, D.; Indolfi, C.; Nadal-Ginard, B. Generation of new cardiomyocytes after injury: De novo formation from resident progenitors vs. replication of pre-existing cardiomyocytes. Ann. Transl. Med. 2015, 3, S8. [Google Scholar] [CrossRef]
- Nadal-Ginard, B.; Ellison, G.M.; Torella, D. Absence of evidence is not evidence of absence: Pitfalls of cre knock-ins in the c-Kit locus. Circ. Res. 2014, 115, 415–418. [Google Scholar] [CrossRef]
- Marino, F.; Scalise, M.; Cianflone, E.; Mancuso, T.; Aquila, I.; Agosti, V.; Torella, M.; Paolino, D.; Mollace, V.; Nadal-Ginard, B.; et al. Role of c-Kit in Myocardial Regeneration and Aging. Front. Endocrinol. 2019, 10, 371. [Google Scholar] [CrossRef] [PubMed]
- Meor Anuar Shuhaili, M.F.R.; Samsudin, I.N.; Stanslas, J.; Hasan, S.; Thambiah, S.C. Effects of Different Types of Statins on Lipid Profile: A Perspective on Asians. Int. J. Endocrinol. Metab. 2017, 15, e43319. [Google Scholar] [CrossRef]
- Kapur, N.K.; Musunuru, K. Clinical efficacy and safety of statins in managing cardiovascular risk. Vasc. Health Risk Manag. 2008, 4, 341–353. [Google Scholar] [CrossRef]
- Xu, Z.; Okamoto, H.; Akino, M.; Onozuka, H.; Matsui, Y.; Tsutsui, H. Pravastatin attenuates left ventricular remodeling and diastolic dysfunction in angiotensin II-induced hypertensive mice. J. Cardiovasc. Pharmacol. 2008, 51, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Chang, G.; Liu, J.; Sun, G.; Liu, L.; Qin, S.; Zhang, D. Simvastatin ameliorates ventricular remodeling via the TGF-β1 signaling pathway in rats following myocardial infarction. Mol. Med. Rep. 2016, 13, 5093–5101. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Klein, S.; Seidler, B.; Kettenberger, A.; Sibaev, A.; Rohn, M.; Feil, R.; Allescher, H.-D.; Vanderwinden, J.-M.; Hofmann, F.; Schemann, M.; et al. Interstitial cells of Cajal integrate excitatory and inhibitory neurotransmission with intestinal slow-wave activity. Nat. Commun. 2013, 4, 1630. [Google Scholar] [CrossRef] [PubMed]
- Scalise, M.; Torella, M.; Marino, F.; Ravo, M.; Giurato, G.; Vicinanza, C.; Cianflone, E.; Mancuso, T.; Aquila, I.; Salerno, L.; et al. Atrial myxomas arise from multipotent cardiac stem cells. Eur. Heart J. 2020, ehaa156. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, A.; Cappetta, D.; Piegari, E.; Rinaldi, B.; Ciuffreda, L.P.; Esposito, G.; Ferraiolo, F.A.V.; Rivellino, A.; Russo, R.; Donniacuo, M.; et al. Long-term administration of ranolazine attenuates diastolic dysfunction and adverse myocardial remodeling in a model of heart failure with preserved ejection fraction. Int. J. Cardiol. 2016, 217, 69–79. [Google Scholar] [CrossRef]
- Cappetta, D.; Esposito, G.; Coppini, R.; Piegari, E.; Russo, R.; Ciuffreda, L.P.; Rivellino, A.; Santini, L.; Rafaniello, C.; Scavone, C.; et al. Effects of ranolazine in a model of doxorubicin-induced left ventricle diastolic dysfunction. Br. J. Pharmacol. 2017, 174, 3696–3712. [Google Scholar] [CrossRef]
- Mancuso, T.; Barone, A.; Salatino, A.; Molinaro, C.; Marino, F.; Scalise, M.; Torella, M.; De Angelis, A.; Urbanek, K.; Torella, D.; et al. Unravelling the Biology of Adult Cardiac Stem Cell-Derived Exosomes to Foster Endogenous Cardiac Regeneration and Repair. Int. J. Mol. Sci. 2020, 21, 3725. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GENE | SPECIES | ID NUMBER |
|---|---|---|
| Gapdh | Mouse | Mm99999915_g1 |
| Mef2c | Mouse | Mm01340842_m1 |
| Nkx2.5 | Mouse | Mm01309813_s1 |
| Gata4 | Mouse | Mm00484689_m1 |
| c-kit | Mouse | Mm00445212_m1 |
| Myh7 | Mouse | Mm01319006_g1 |
| Myl2 | Mouse | Mm00440384_m1 |
| Actc1 | Mouse | Mm01333821_m1 |
| TnnT2 | Mouse | Mm01290256_m1 |
| Gapdh | Rat | Rn01775763_g1 |
| Gata4 | Rat | Rn01530459_m1 |
| Mef2c | Rat | Rn01494046_m1 |
| Tnnt2 | Rat | Rn01483694_m1 |
| Myh7 | Rat | Rn01488777_g1 |
| Myo6 | Rat | Rn01521319_m1 |
| Actc1 | Rat | Rn01513700_g1 |
| ANTIGEN | ANTIBODY ID | COMPANY | APPLICATION 1 |
|---|---|---|---|
| AKT | 9272 | Cell Signaling | WB |
| pAKT | 4058S | Cell Signaling | WB |
| WGA | Invitrogen | IH | |
| MF20 | Ab_2147781 | DSHB | IH |
| c-kit | AF 1356 | R&D System | IH |
| GATA4 | sc-9053 | SantaCruz Biotech | IH |
| GFP | Rockland Immunochemicals | IH | |
| α-Actinin | sc-17809 | Santa Cruz Biotech | IH |
| cTNI | H170 | Santa Cruz Biotech | IH |
| BrdU | Roche | IH | |
| α-SARC | A2172 | SIGMA | IH |
| TdT | 9272 | Roche | IH |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cianflone, E.; Cappetta, D.; Mancuso, T.; Sabatino, J.; Marino, F.; Scalise, M.; Albanese, M.; Salatino, A.; Parrotta, E.I.; Cuda, G.; et al. Statins Stimulate New Myocyte Formation After Myocardial Infarction by Activating Growth and Differentiation of the Endogenous Cardiac Stem Cells. Int. J. Mol. Sci. 2020, 21, 7927. https://doi.org/10.3390/ijms21217927
Cianflone E, Cappetta D, Mancuso T, Sabatino J, Marino F, Scalise M, Albanese M, Salatino A, Parrotta EI, Cuda G, et al. Statins Stimulate New Myocyte Formation After Myocardial Infarction by Activating Growth and Differentiation of the Endogenous Cardiac Stem Cells. International Journal of Molecular Sciences. 2020; 21(21):7927. https://doi.org/10.3390/ijms21217927
Chicago/Turabian StyleCianflone, Eleonora, Donato Cappetta, Teresa Mancuso, Jolanda Sabatino, Fabiola Marino, Mariangela Scalise, Michele Albanese, Alessandro Salatino, Elvira Immacolata Parrotta, Giovanni Cuda, and et al. 2020. "Statins Stimulate New Myocyte Formation After Myocardial Infarction by Activating Growth and Differentiation of the Endogenous Cardiac Stem Cells" International Journal of Molecular Sciences 21, no. 21: 7927. https://doi.org/10.3390/ijms21217927
APA StyleCianflone, E., Cappetta, D., Mancuso, T., Sabatino, J., Marino, F., Scalise, M., Albanese, M., Salatino, A., Parrotta, E. I., Cuda, G., De Angelis, A., Berrino, L., Rossi, F., Nadal-Ginard, B., Torella, D., & Urbanek, K. (2020). Statins Stimulate New Myocyte Formation After Myocardial Infarction by Activating Growth and Differentiation of the Endogenous Cardiac Stem Cells. International Journal of Molecular Sciences, 21(21), 7927. https://doi.org/10.3390/ijms21217927