Evolution of Transmissible Gastroenteritis Virus (TGEV): A Codon Usage Perspective


Abstract
:1. Introduction
2. Results
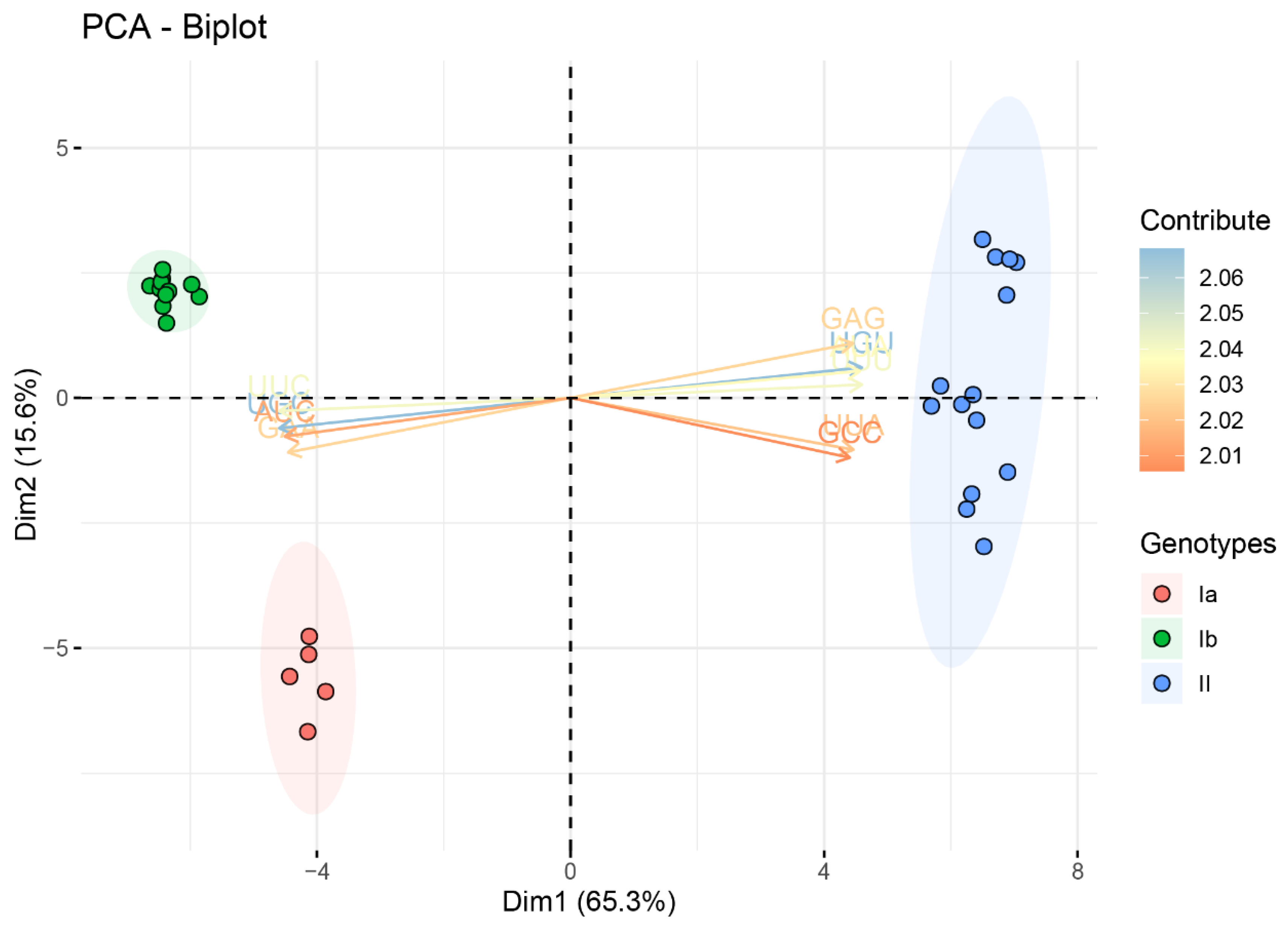
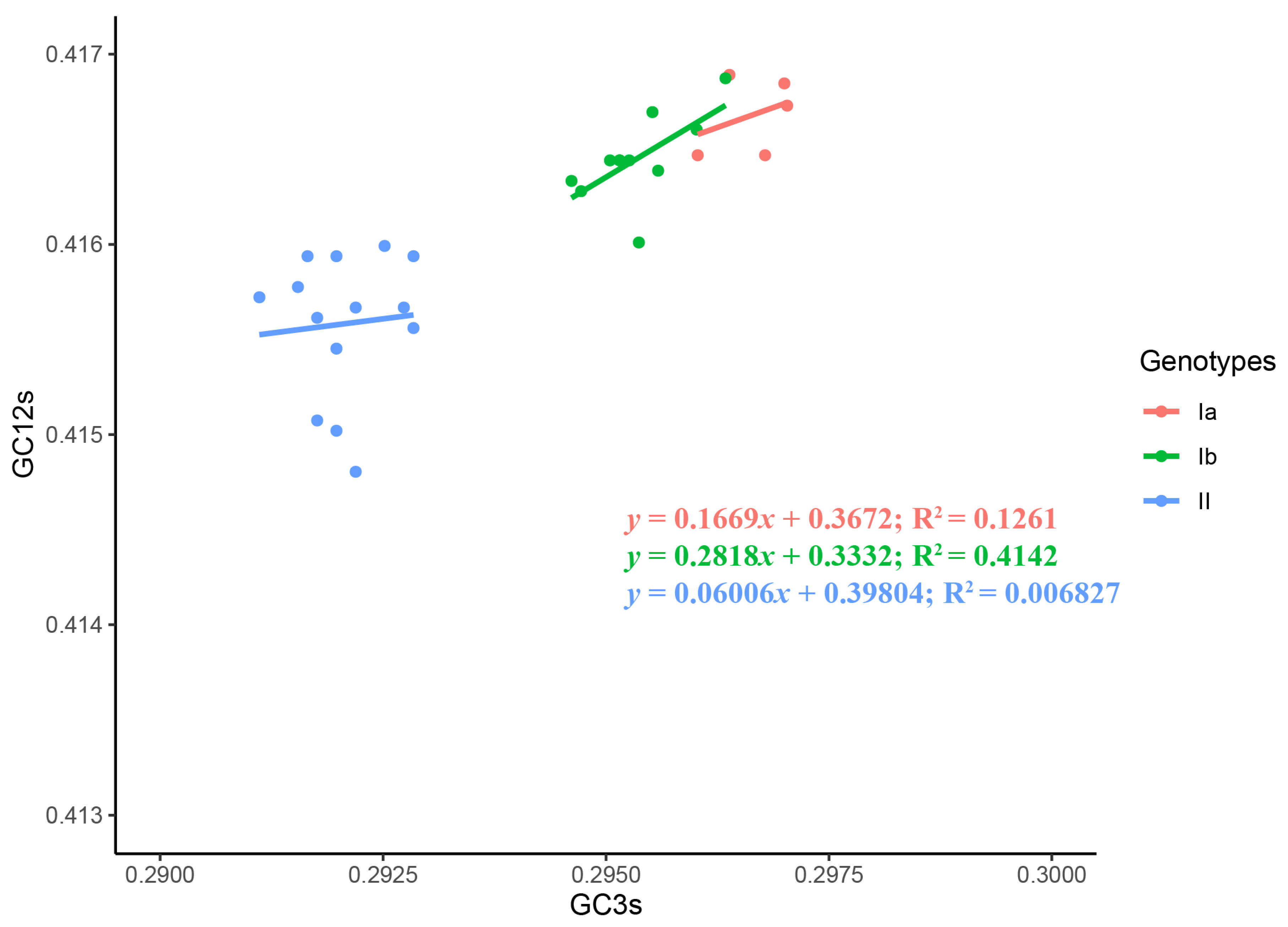
2.1. Distinct Genotypes
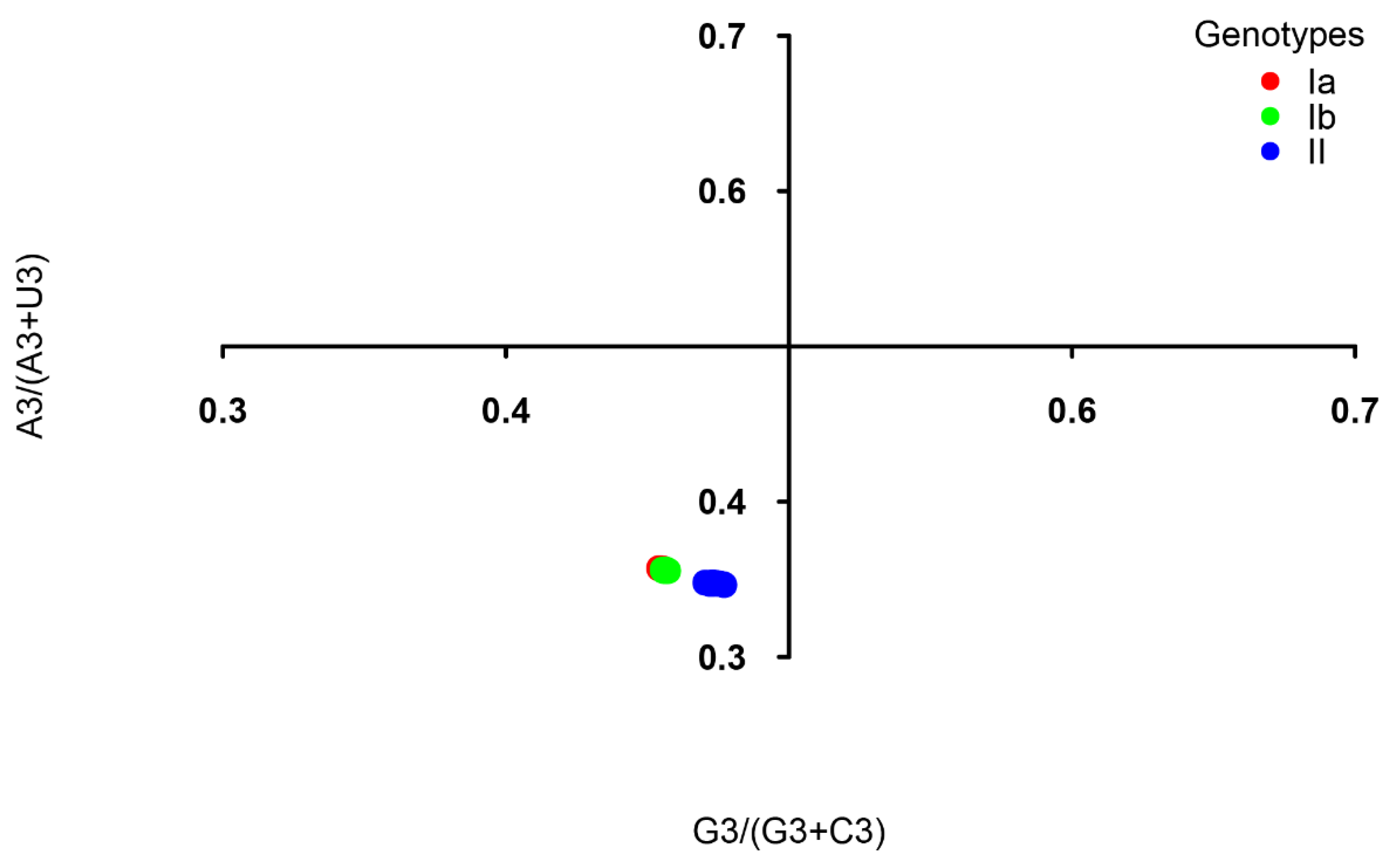
2.2. Nucleotide U Is the Most Frequent in the TGEV Coding Sequence
2.3. Genotype-Specific Codon Usage Pattern
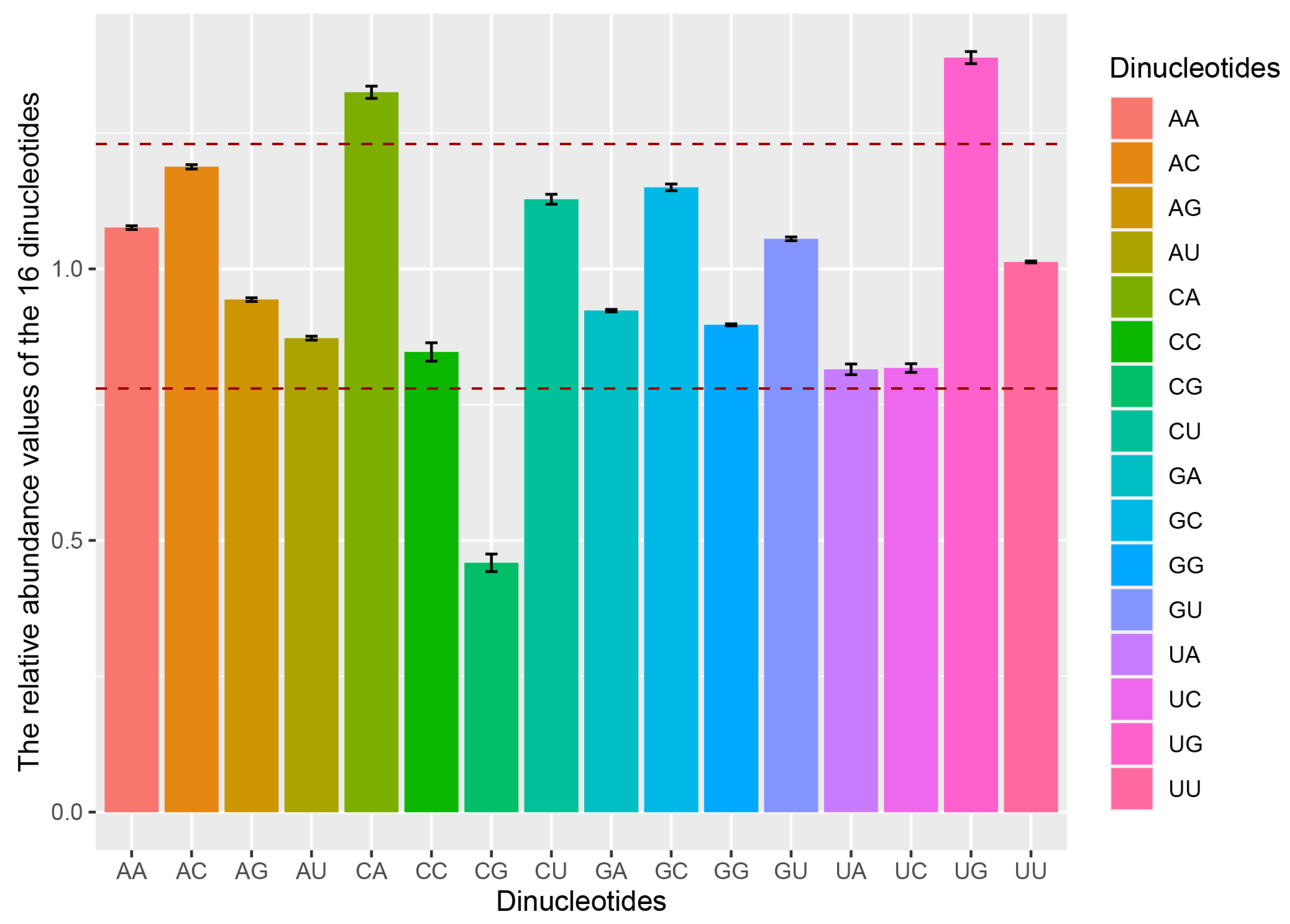
2.4. Dinucleotides Influence the Codon Usage Pattern of TGEV
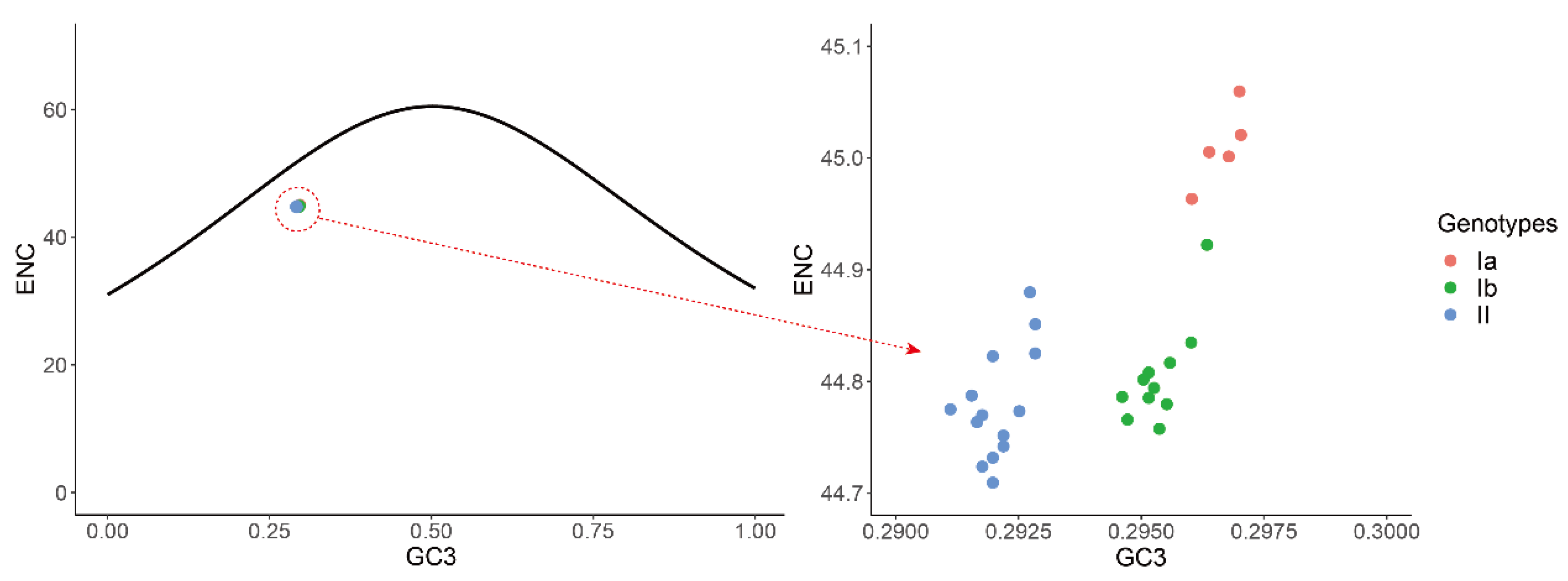
2.5. Effect of Mutation Pressure and Natural Selection on Codon Usage Bias
2.6. Differences in Adaptation of Genotypes toward the Host
3. Discussion
4. Materials and Methods
4.1. Sequence Data
4.2. Recombination Detection and Phylogenetic Analysis
4.3. Compositional Properties and Principal Parameters Analysis
4.4. Relative Synonymous Codon Usage
4.5. Principal Component Analysis
4.6. Relative Dinucleotide Abundance Analysis
4.7. Effective Number of Codons Analysis
4.8. ENC-Plot Analysis
4.9. Parity Rule 2 Analysis
4.10. Neutrality Analysis
4.11. Codon Adaptation Index Analysis
4.12. Relative Codon Deoptimization Index Analysis
4.13. Similarity Index Analysis
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, L.; Qiao, X.; Zhang, S.; Qin, Y.; Guo, T.; Hao, Z.; Sun, L.; Wang, X.; Wang, Y.; Jiang, Y. Porcine transmissible gastroenteritis virus nonstructural protein 2 contributes to inflammation via NF-κB activation. Virulence 2018, 9, 1685–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, R.D. Efficacy of a transmissible gastroenteritis coronavirus with an altered ORF-3 gene. Can. J. Vet. Res. 2001, 65, 28–32. [Google Scholar] [PubMed]
- Doyle, L.; Hutchings, L. A transmissible gastroenteritis in pigs. J. Am. Vet. Med. Assoc. 1946, 108, 257–259. [Google Scholar] [PubMed]
- Pritchard, C.; Paton, D.; Wibberley, G.; Ibata, G. Transmissible gastroenteritis and porcine epidemic diarrhoea in Britain. Vet. Rec. 1999, 144, 616–618. [Google Scholar] [CrossRef]
- Williams, R.; Esterhuysen, J.; Robinson, J. Pseudorabies and transmissible gastroenteritis: A serological survey in South Africa. Onderstepoort J. Vet. Rec. 1994, 61, 67–70. [Google Scholar]
- Cubero, M.; Leon, L.; Contreras, A.; Astorga, R.; Lanza, I.; Garcia, A. Transmissible gastroenteritis in pigs in south east Spain: Prevalence and factors associated with infection. Vet. Rec. 1993, 132, 238–241. [Google Scholar] [CrossRef]
- Hu, X.; Li, N.; Tian, Z.; Yin, X.; Qu, L.; Qu, J. Molecular characterization and phylogenetic analysis of transmissible gastroenteritis virus HX strain isolated from China. BMC Vet. Res. 2015, 11, 72. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Okada, K.; Ohshima, K. An outbreak of swine diarrhea of a new-type associated with coronavirus-like particles in Japan. Jpn. J. Vet. Sci. 1983, 45, 829–832. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Yue, X.; Cai, X.; Wang, S.; Liu, Y.; Yuan, C.; Cui, L.; Hua, X.; Yang, Z. Complete genome of transmissible gastroenteritis virus AYU strain isolated in Shanghai, China. J. Virol. 2012, 86, 11935. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Yu, Q.; Zhu, L.; Liu, H.; Zhao, S.; Gao, Q.; He, K.; Yang, Q. Complete genomic sequence of the coronavirus transmissible gastroenteritis virus SHXB isolated in China. Arch. Virol. 2014, 159, 2295–2302. [Google Scholar] [CrossRef] [Green Version]
- Laude, H.; Rasschaert, D.; Delmas, B.; Godet, M.; Gelfi, J.; Charley, B. Molecular biology of transmissible gastroenteritis virus. Vet. Microbiol. 1990, 23, 147–154. [Google Scholar] [CrossRef]
- Yount, B.; Curtis, K.M.; Baric, R.S. Strategy for systematic assembly of large RNA and DNA genomes: Transmissible gastroenteritis virus model. J. Virol. 2000, 74, 10600–10611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eleouet, J.-F.; Rasschaert, D.; Lambert, P.; Levy, L.; Vende, P.; Laude, H. Complete sequence (20 kilobases) of the polyprotein-encoding gene 1 of transmissible gastroenteritis virus. Virology 1995, 206, 817–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brian, D.; Baric, R. Coronavirus genome structure and replication. In Coronavirus Replication and Reverse Genetics; Springer: Berlin/Heidelberg, Germany, 2005; pp. 1–30. [Google Scholar]
- Heald-Sargent, T.; Gallagher, T. Ready, set, fuse! The coronavirus spike protein and acquisition of fusion competence. Viruses 2012, 4, 557–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F. Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delmas, B.; Rasschaert, D.; Godet, M.; Gelfi, J.; Laude, H. Four major antigenic sites of the coronavirus transmissible gastroenteritis virus are located on the amino-terminal half of spike glycoprotein S. J. Gen. Virol. 1990, 71, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, M.; Sanchez, C.; Enjuanes, L. Two amino acid changes at the N-terminus of transmissible gastroenteritis coronavirus spike protein result in the loss of enteric tropism. Virology 1997, 227, 378–388. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, C.M.; Izeta, A.; Sánchez-Morgado, J.M.; Alonso, S.; Sola, I.; Balasch, M.; Plana-Durán, J.; Enjuanes, L. Targeted recombination demonstrates that the spike gene of transmissible gastroenteritis coronavirus is a determinant of its enteric tropism and virulence. J. Virol. 1999, 73, 7607–7618. [Google Scholar] [CrossRef] [Green Version]
- Tung, F.Y.; Abraham, S.; Sethna, M.; Hung, S.-L.; Sethna, P.; Hogue, B.G.; Brian, D.A. The 9-kDa hydrophobic protein encoded at the 3′ end of the porcine transmissible gastroenteritis coronavirus genome is membrane-associated. Virology 1992, 186, 676–683. [Google Scholar] [CrossRef]
- Chen, F.; Knutson, T.P.; Rossow, S.; Saif, L.J.; Marthaler, D.G. Decline of transmissible gastroenteritis virus and its complex evolutionary relationship with porcine respiratory coronavirus in the United States. Sci. Rep. 2019, 9, 3953. [Google Scholar] [CrossRef] [Green Version]
- Pan, S.; Mou, C.; Wu, H.; Chen, Z. Phylogenetic and codon usage analysis of atypical porcine pestivirus (APPV). Virulence 2020, 11, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, C.; Govindarajan, S.; Minshull, J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004, 22, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Papamichail, D.; Coleman, J.R.; Skiena, S.; Wimmer, E. Reduction of the Rate of Poliovirus Protein Synthesis through Large-Scale Codon Deoptimization Causes Attenuation of Viral Virulence by Lowering Specific Infectivity. J. Virol. 2006, 80, 9687–9696. [Google Scholar] [CrossRef] [Green Version]
- Lauring, A.S.; Acevedo, A.; Cooper, S.B.; Andino, R. Codon usage determines the mutational robustness, evolutionary capacity, and virulence of an RNA virus. Cell Host Microbe 2012, 12, 623–632. [Google Scholar] [CrossRef] [Green Version]
- Costafreda, M.I.; Pérez-Rodriguez, F.J.; D’Andrea, L.; Guix, S.; Ribes, E.; Bosch, A.; Pintó, R.M. Hepatitis A virus adaptation to cellular shutoff is driven by dynamic adjustments of codon usage and results in the selection of populations with altered capsids. J. Virol. 2014, 88, 5029–5041. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Wang, N.; Tan, J.; Wang, R.; Yang, Y.; Li, G.; Guan, H.; Zheng, Y.; Shi, X.; Ye, R. Comprehensive codon usage analysis of porcine deltacoronavirus. Mol. Phylogenet. Evol. 2019, 141, 106618. [Google Scholar] [CrossRef]
- Cao, H.; Zhang, H. Synonymous Codon Usage Bias in Porcine Epidemic Diarrhea Virus. Isr. J. Vet. Med. 2013, 68, 185–189. [Google Scholar]
- Dutta, R.; Buragohain, L.; Borah, P. Analysis of codon usage of Severe Acute Respiratory Syndrome Corona Virus 2 (SARS-CoV-2) and its adaptability in dog. Virus Res. 2020, 198113. [Google Scholar] [CrossRef]
- Tort, F.L.; Castells, M.; Cristina, J. A comprehensive analysis of genome composition and codon usage patterns of emerging coronaviruses. Virus Res. 2020, 197976. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, Q.; Yuan, X.; Li, X.; Zhu, T.; Ma, Y.; Chen, J.-L. Analysis of the codon usage pattern in Middle East Respiratory Syndrome Coronavirus. Oncotarget 2017, 8, 110337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castells, M.; Victoria, M.; Colina, R.; Musto, H.; Cristina, J. Genome-wide analysis of codon usage bias in Bovine Coronavirus. Virol. J. 2017, 14, 115. [Google Scholar] [CrossRef] [Green Version]
- Brandão, P.E. Avian coronavirus spike glycoprotein ectodomain shows a low codon adaptation to Gallus gallus with virus-exclusive codons in strategic amino acids positions. J. Mol. Evol. 2012, 75, 19–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Cheng, X.; Virk, N.; Chen, W.; Ji, S.; Ji, S.; Sun, Y.; Wu, X. CpG usage in RNA viruses: Data and hypotheses. PLoS ONE 2013, 8, e74109. [Google Scholar] [CrossRef] [Green Version]
- Di Giallonardo, F.; Schlub, T.E.; Shi, M.; Holmes, E.C. Dinucleotide composition in animal RNA viruses is shaped more by virus family than by host species. J. Virol. 2017, 91, e02381-16. [Google Scholar] [CrossRef] [Green Version]
- Rima, B.K.; McFerran, N.V. Dinucleotide and stop codon frequencies in single-stranded RNA viruses. J. Gen. Virol. 1997, 78, 2859–2870. [Google Scholar] [CrossRef]
- Nambou, K.; Anakpa, M. Deciphering the co-adaptation of codon usage between respiratory coronaviruses and their human host uncovers candidate therapeutics for COVID-19. Infect. Genet. Evol. 2020, 85, 104471. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.-H. The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranwez, V.; Douzery, E.J.; Cambon, C.; Chantret, N.; Delsuc, F. MACSE v2: Toolkit for the alignment of coding sequences accounting for frameshifts and stop codons. Mol. Biol. Evol. 2018, 35, 2582–2584. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Charif, D.; Lobry, J.R. SeqinR 1.0-2: A contributed package to the R project for statistical computing devoted to biological sequences retrieval and analysis. In Structural Approaches to Sequence Evolution; Springer: Berlin/Heidelberg, Germany, 2007; pp. 207–232. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; Available online: https://www.R-project.org/ (accessed on 24 October 2020).
- Sharp, P.M.; Li, W.-H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef]
- Sharp, P.M.; Tuohy, T.M.; Mosurski, K.R. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.H.; Smith, D.K.; Rabadan, R.; Peiris, M.; Poon, L.L. Codon usage bias and the evolution of influenza A viruses. Codon Usage Biases of Influenza Virus. BMC Evol. Biol. 2010, 10, 253. [Google Scholar] [CrossRef] [Green Version]
- Wold, S.; Esbensen, K.; Geladi, P. Principal component analysis. Chemom. Intell. Lab. Syst. 1987, 2, 37–52. [Google Scholar] [CrossRef]
- Kassambara, A.; Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses. R Package Version 1.0.6. 2017. Available online: https://github.com/kassambara/factoextra (accessed on 24 October 2020).
- Elek, A.; Kuzman, M.; Vlahoviček, K. coRdon: Codon Usage Analysis and Prediction of Gene Expressivity. R Package Version 1.4.0. 2019. Available online: https://github.com/BioinfoHR/coRdon (accessed on 24 October 2020).
- Sueoka, N. Intrastrand parity rules of DNA base composition and usage biases of synonymous codons. J. Mol. Evol. 1995, 40, 318–325. [Google Scholar] [CrossRef]
- Sueoka, N. Translation-coupled violation of Parity Rule 2 in human genes is not the cause of heterogeneity of the DNA G+ C content of third codon position. Gene 1999, 238, 53–58. [Google Scholar] [CrossRef]
- Sueoka, N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653–2657. [Google Scholar] [CrossRef] [Green Version]
- Puigbò, P.; Bravo, I.G.; Garcia-Vallve, S. CAIcal: A combined set of tools to assess codon usage adaptation. Biol. Direct 2008, 3, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athey, J.; Alexaki, A.; Osipova, E.; Rostovtsev, A.; Santana-Quintero, L.V.; Katneni, U.; Simonyan, V.; Kimchi-Sarfaty, C. A new and updated resource for codon usage tables. BMC Bioinform. 2017, 18, 391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butt, A.M.; Nasrullah, I.; Qamar, R.; Tong, Y. Evolution of codon usage in Zika virus genomes is host and vector specific. Emerg. Microbes Infect. 2016, 5, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Khandia, R.; Singhal, S.; Kumar, U.; Ansari, A.I.; Tiwari, R.; Dhama, K.; Das, J.; Munjal, A.; Singh, R.K. Analysis of Nipah virus codon usage and adaptation to hosts. Front. Microbiol. 2019, 10, 886. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Zhang, J.; Sun, D.; Ma, Q.; Chen, H.; Ma, L.; Ding, Y.; Liu, Y. The distribution of synonymous codon choice in the translation initiation region of dengue virus. PLoS ONE 2013, 8, e77239. [Google Scholar] [CrossRef]
- Dinno, A. Dunn. Test: Dunn’s Test of Multiple Comparisons Using Rank Sums. R Package Version 1.3.5. 2017. Available online: https://CRAN.R-project.org/package=dunn.test (accessed on 24 October 2020).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Categories | Ia | Ib | II | All |
|---|---|---|---|---|
| A | 0.294 ± 0 | 0.294 ± 0 | 0.292 ± 0 | 0.293 ± 0.001 |
| C | 0.17 ± 0 | 0.169 ± 0 | 0.167 ± 0 | 0.168 ± 0.001 |
| G | 0.207 ± 0 | 0.207 ± 0 | 0.208 ± 0 | 0.207 ± 0 |
| U | 0.329 ± 0 | 0.33 ± 0 | 0.333 ± 0 | 0.331 ± 0.002 |
| A3 | 0.251 ± 0 | 0.251 ± 0 | 0.246 ± 0 | 0.249 ± 0.003 |
| C3 | 0.162 ± 0 | 0.161 ± 0 | 0.154 ± 0.001 | 0.158 ± 0.004 |
| G3 | 0.135 ± 0 | 0.135 ± 0 | 0.138 ± 0 | 0.136 ± 0.002 |
| U3 | 0.452 ± 0 | 0.454 ± 0 | 0.462 ± 0.001 | 0.457 ± 0.004 |
| GC | 0.377 ± 0 | 0.376 ± 0 | 0.374 ± 0 | 0.375 ± 0.001 |
| AU | 0.623 ± 0 | 0.624 ± 0 | 0.626 ± 0 | 0.625 ± 0.001 |
| GC1 | 0.461 ± 0 | 0.462 ± 0 | 0.46 ± 0 | 0.461 ± 0.001 |
| GC2 | 0.372 ± 0 | 0.371 ± 0 | 0.371 ± 0 | 0.371 ± 0.001 |
| GC12 | 0.417 ± 0 | 0.416 ± 0 | 0.416 ± 0 | 0.416 ± 0.001 |
| GC3 | 0.297 ± 0 | 0.295 ± 0.001 | 0.292 ± 0.001 | 0.294 ± 0.002 |
| ENC | 45.01 ± 0.035 | 44.805 ± 0.045 | 44.779 ± 0.05 | 44.827 ± 0.095 |
| Codon (Amino Acid) | Ia | Ib | II | All |
|---|---|---|---|---|
| GCA (Ala) | 1.299 ± 0.008 | 1.337 ± 0.005 | 1.267 ± 0.013 | 1.298 ± 0.034 |
| GCC (Ala) | 0.458 ± 0.006 | 0.396 ± 0.005 | 0.508 ± 0.009 | 0.459 ± 0.052 |
| GCG (Ala) | 0.154 ± 0.005 | 0.136 ± 0.003 | 0.164 ± 0.016 | 0.152 ± 0.017 |
| GCU (Ala) | 2.089 ± 0.003 | 2.13 ± 0.006 | 2.061 ± 0.014 | 2.091 ± 0.034 |
| AGA (Arg) | 2.745 ± 0.01 | 2.78 ± 0.01 | 2.738 ± 0.022 | 2.755 ± 0.026 |
| AGG (Arg) | 0.914 ± 0.002 | 0.873 ± 0.011 | 0.915 ± 0.013 | 0.899 ± 0.023 |
| CGA (Arg) | 0.209 ± 0.008 | 0.208 ± 0.008 | 0.222 ± 0.019 | 0.215 ± 0.016 |
| CGC (Arg) | 0.537 ± 0.009 | 0.491 ± 0.009 | 0.54 ± 0.012 | 0.522 ± 0.025 |
| CGG (Arg) | 0.209 ± 0.008 | 0.21 ± 0.005 | 0.232 ± 0.009 | 0.22 ± 0.014 |
| CGU (Arg) | 1.387 ± 0.008 | 1.438 ± 0.01 | 1.353 ± 0.017 | 1.39 ± 0.041 |
| AAC (Asn) | 0.699 ± 0.005 | 0.694 ± 0.003 | 0.669 ± 0.005 | 0.683 ± 0.014 |
| AAU (Asn) | 1.301 ± 0.005 | 1.306 ± 0.003 | 1.331 ± 0.005 | 1.317 ± 0.014 |
| GAC (Asp) | 0.68 ± 0.007 | 0.681 ± 0.005 | 0.64 ± 0.009 | 0.662 ± 0.022 |
| GAU (Asp) | 1.32 ± 0.007 | 1.319 ± 0.005 | 1.36 ± 0.009 | 1.338 ± 0.022 |
| UGC (Cys) | 0.64 ± 0.002 | 0.638 ± 0.002 | 0.549 ± 0.007 | 0.597 ± 0.046 |
| UGU (Cys) | 1.36 ± 0.002 | 1.362 ± 0.002 | 1.451 ± 0.007 | 1.403 ± 0.046 |
| CAA (Gln) | 1.229 ± 0.003 | 1.268 ± 0.004 | 1.26 ± 0.011 | 1.258 ± 0.016 |
| CAG (Gln) | 0.771 ± 0.003 | 0.732 ± 0.004 | 0.74 ± 0.011 | 0.742 ± 0.016 |
| GAA (Glu) | 1.485 ± 0.002 | 1.477 ± 0.004 | 1.425 ± 0.009 | 1.454 ± 0.029 |
| GAG (Glu) | 0.515 ± 0.002 | 0.523 ± 0.004 | 0.575 ± 0.009 | 0.546 ± 0.029 |
| GGA (Gly) | 0.739 ± 0.005 | 0.716 ± 0.003 | 0.705 ± 0.01 | 0.714 ± 0.014 |
| GGC (Gly) | 0.582 ± 0.005 | 0.573 ± 0.004 | 0.554 ± 0.013 | 0.566 ± 0.015 |
| GGG (Gly) | 0.116 ± 0 | 0.136 ± 0.002 | 0.154 ± 0.007 | 0.141 ± 0.015 |
| GGU (Gly) | 2.564 ± 0.01 | 2.575 ± 0.006 | 2.587 ± 0.017 | 2.579 ± 0.016 |
| CAC (His) | 0.51 ± 0 | 0.499 ± 0.007 | 0.452 ± 0.006 | 0.479 ± 0.027 |
| CAU (His) | 1.49 ± 0 | 1.501 ± 0.007 | 1.548 ± 0.006 | 1.521 ± 0.027 |
| AUA (Ile) | 0.691 ± 0.006 | 0.693 ± 0.003 | 0.759 ± 0.007 | 0.723 ± 0.034 |
| AUC (Ile) | 0.49 ± 0.007 | 0.488 ± 0.004 | 0.421 ± 0.011 | 0.457 ± 0.035 |
| AUU (Ile) | 1.819 ± 0.009 | 1.82 ± 0.003 | 1.821 ± 0.009 | 1.82 ± 0.008 |
| CUA (Leu) | 0.538 ± 0.008 | 0.562 ± 0.005 | 0.512 ± 0.01 | 0.535 ± 0.025 |
| CUC (Leu) | 0.555 ± 0.006 | 0.534 ± 0.005 | 0.552 ± 0.01 | 0.546 ± 0.012 |
| CUG (Leu) | 0.258 ± 0.009 | 0.283 ± 0.005 | 0.251 ± 0.007 | 0.264 ± 0.017 |
| CUU (Leu) | 2.147 ± 0.007 | 2.187 ± 0.004 | 2.155 ± 0.01 | 2.166 ± 0.018 |
| UUA (Leu) | 1.263 ± 0.007 | 1.224 ± 0.006 | 1.315 ± 0.012 | 1.273 ± 0.043 |
| UUG (Leu) | 1.239 ± 0.005 | 1.208 ± 0.003 | 1.216 ± 0.011 | 1.217 ± 0.013 |
| AAA (Lys) | 1.249 ± 0.002 | 1.221 ± 0.004 | 1.205 ± 0.005 | 1.218 ± 0.016 |
| AAG (Lys) | 0.751 ± 0.002 | 0.779 ± 0.004 | 0.795 ± 0.005 | 0.782 ± 0.016 |
| UUC (Phe) | 0.576 ± 0.003 | 0.584 ± 0.003 | 0.517 ± 0.008 | 0.551 ± 0.033 |
| UUU (Phe) | 1.424 ± 0.003 | 1.416 ± 0.003 | 1.483 ± 0.008 | 1.449 ± 0.033 |
| CCA (Pro) | 1.636 ± 0.005 | 1.64 ± 0.01 | 1.627 ± 0.007 | 1.633 ± 0.01 |
| CCC (Pro) | 0.274 ± 0.011 | 0.247 ± 0.004 | 0.274 ± 0.011 | 0.264 ± 0.016 |
| CCG (Pro) | 0.168 ± 0 | 0.197 ± 0.008 | 0.234 ± 0.011 | 0.209 ± 0.027 |
| CCU (Pro) | 1.921 ± 0.012 | 1.916 ± 0.005 | 1.865 ± 0.014 | 1.893 ± 0.029 |
| AGC (Ser) | 0.646 ± 0.004 | 0.665 ± 0.007 | 0.614 ± 0.008 | 0.638 ± 0.025 |
| AGU (Ser) | 1.624 ± 0.011 | 1.598 ± 0.004 | 1.648 ± 0.005 | 1.626 ± 0.024 |
| UCA (Ser) | 1.295 ± 0.011 | 1.299 ± 0.01 | 1.26 ± 0.011 | 1.28 ± 0.022 |
| UCC (Ser) | 0.457 ± 0.001 | 0.44 ± 0.006 | 0.455 ± 0.013 | 0.45 ± 0.012 |
| UCG (Ser) | 0.118 ± 0.005 | 0.123 ± 0.004 | 0.15 ± 0.009 | 0.135 ± 0.016 |
| UCU (Ser) | 1.861 ± 0.006 | 1.874 ± 0.014 | 1.873 ± 0.02 | 1.871 ± 0.017 |
| ACA (Thr) | 1.606 ± 0.016 | 1.633 ± 0.006 | 1.564 ± 0.005 | 1.596 ± 0.033 |
| ACC (Thr) | 0.4 ± 0.003 | 0.414 ± 0.005 | 0.392 ± 0.008 | 0.401 ± 0.012 |
| ACG (Thr) | 0.29 ± 0.008 | 0.257 ± 0.003 | 0.313 ± 0.009 | 0.288 ± 0.027 |
| ACU (Thr) | 1.704 ± 0.007 | 1.696 ± 0.005 | 1.732 ± 0.009 | 1.714 ± 0.018 |
| UAC (Tyr) | 0.772 ± 0.003 | 0.781 ± 0.004 | 0.728 ± 0.007 | 0.755 ± 0.026 |
| UAU (Tyr) | 1.228 ± 0.003 | 1.219 ± 0.004 | 1.272 ± 0.007 | 1.245 ± 0.026 |
| GUA (Val) | 0.785 ± 0.003 | 0.774 ± 0.005 | 0.744 ± 0.005 | 0.762 ± 0.018 |
| GUC (Val) | 0.652 ± 0.005 | 0.666 ± 0.005 | 0.631 ± 0.012 | 0.648 ± 0.019 |
| GUG (Val) | 0.651 ± 0.005 | 0.652 ± 0.003 | 0.65 ± 0.004 | 0.651 ± 0.004 |
| GUU (Val) | 1.911 ± 0.008 | 1.907 ± 0.003 | 1.975 ± 0.013 | 1.94 ± 0.035 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, S.; Wu, H.; Chen, Z. Evolution of Transmissible Gastroenteritis Virus (TGEV): A Codon Usage Perspective. Int. J. Mol. Sci. 2020, 21, 7898. https://doi.org/10.3390/ijms21217898
Cheng S, Wu H, Chen Z. Evolution of Transmissible Gastroenteritis Virus (TGEV): A Codon Usage Perspective. International Journal of Molecular Sciences. 2020; 21(21):7898. https://doi.org/10.3390/ijms21217898
Chicago/Turabian StyleCheng, Saipeng, Huiguang Wu, and Zhenhai Chen. 2020. "Evolution of Transmissible Gastroenteritis Virus (TGEV): A Codon Usage Perspective" International Journal of Molecular Sciences 21, no. 21: 7898. https://doi.org/10.3390/ijms21217898
APA StyleCheng, S., Wu, H., & Chen, Z. (2020). Evolution of Transmissible Gastroenteritis Virus (TGEV): A Codon Usage Perspective. International Journal of Molecular Sciences, 21(21), 7898. https://doi.org/10.3390/ijms21217898