Dual Effect of Soloxolone Methyl on LPS-Induced Inflammation In Vitro and In Vivo

 , and
, and
Abstract
1. Introduction
2. Results
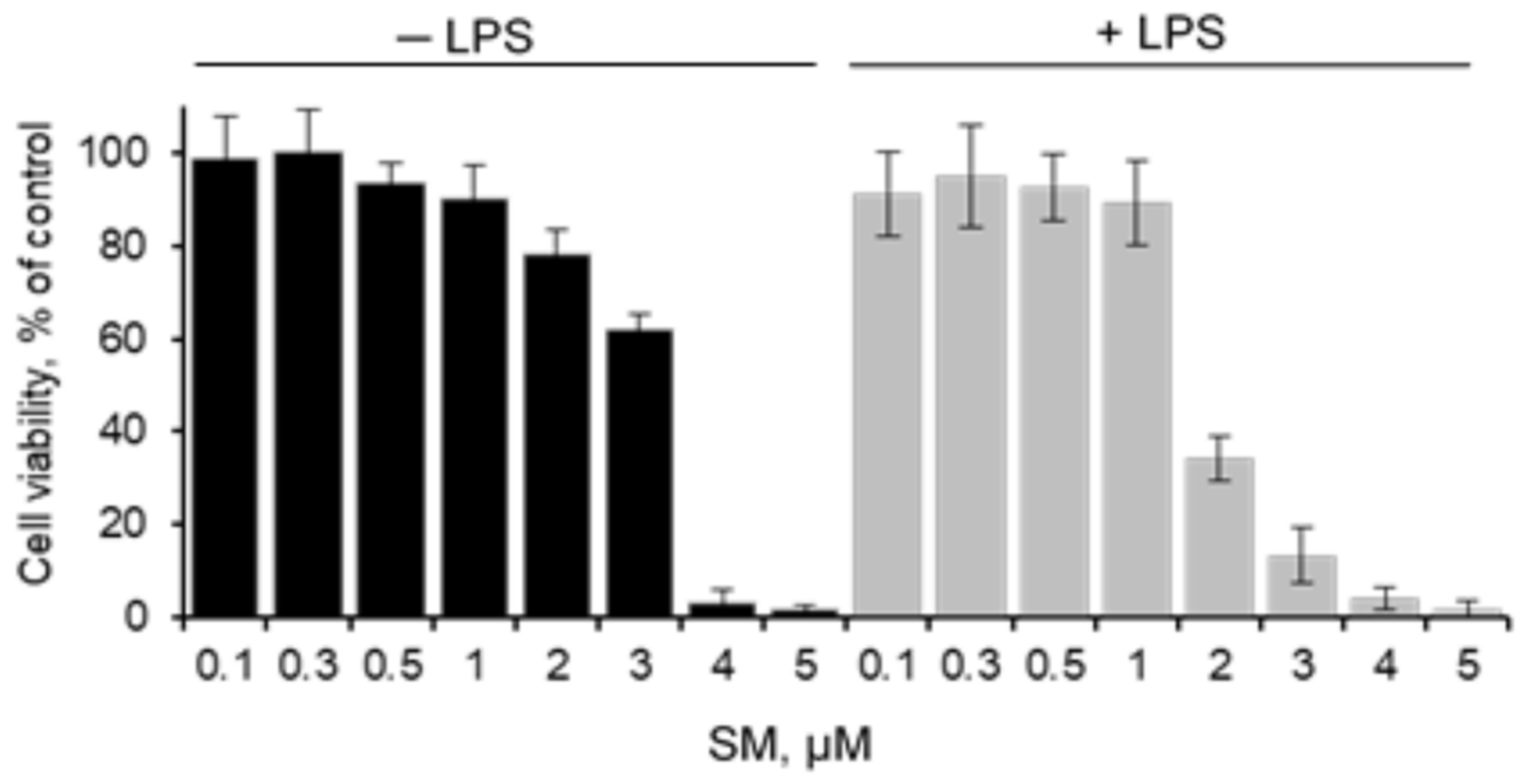
2.1. Cytotoxicity of SM on RAW 264.7
2.2. Effects of SM on Cellular Redox Imbalance and NO Production in RAW 264.7 Macrophages
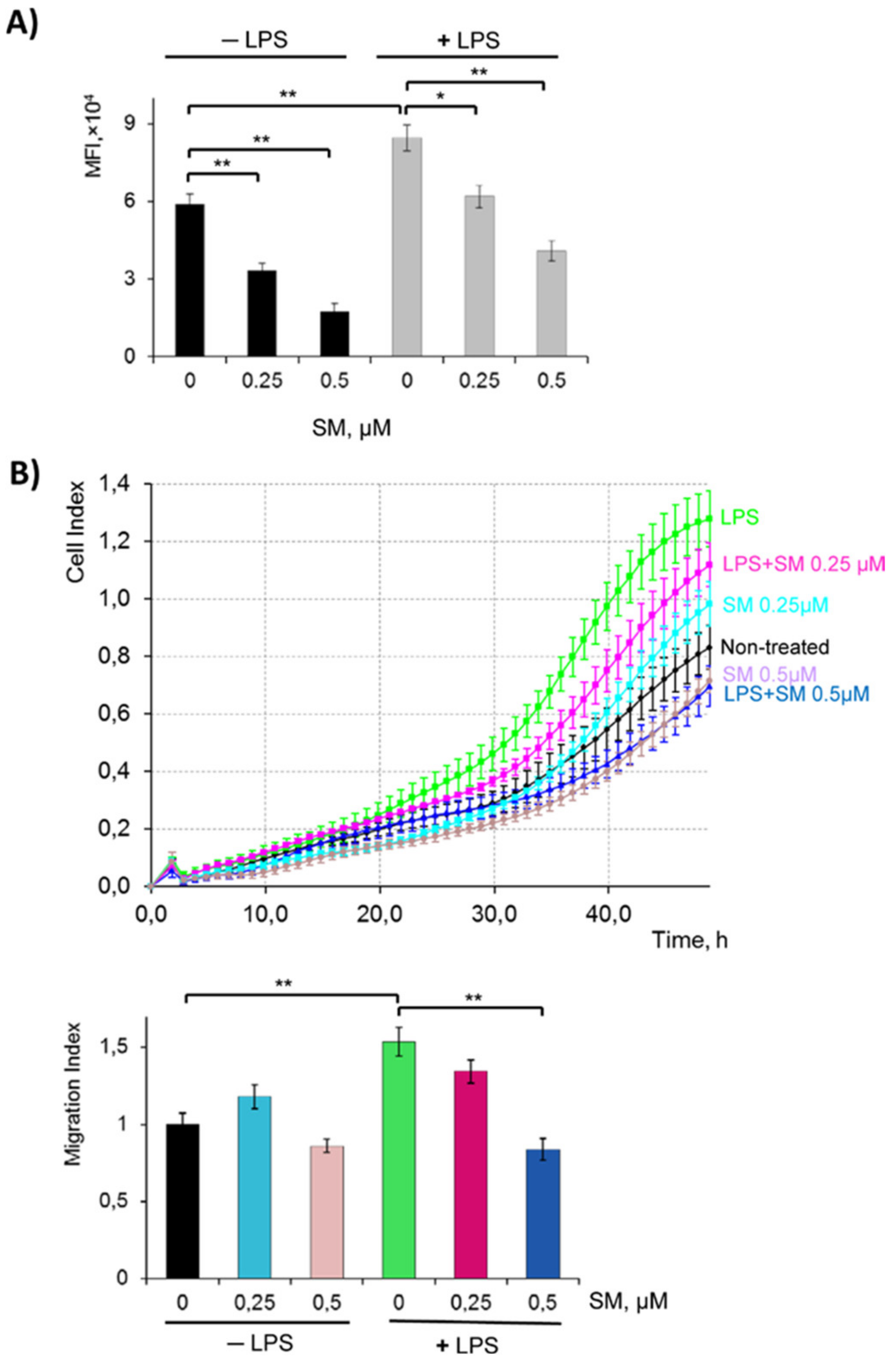
2.3. SM Inhibits the Phagocytic and Migratory Activity of Macrophages
2.4. Effect of SM on Pro-Inflammatory Cytokines in LPS, Poly(I:C), and IFN-γ-Stimulated RAW 264.7 Cells
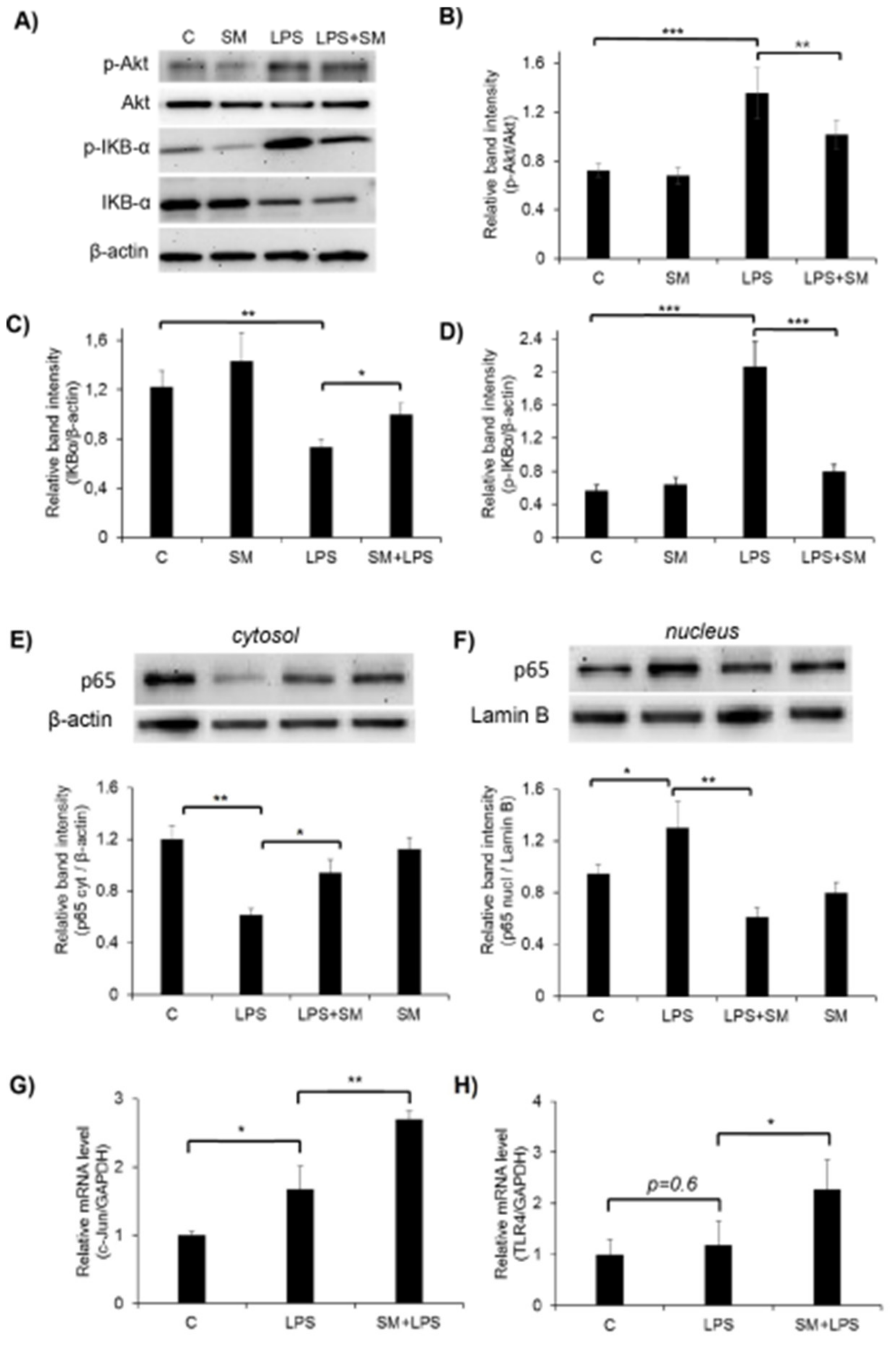
2.5. Effect of SM on the Activation of the Akt, NF-κB, and AP-1 Signaling Pathways and TLR4 in LPS-Induced RAW264.7 Cells
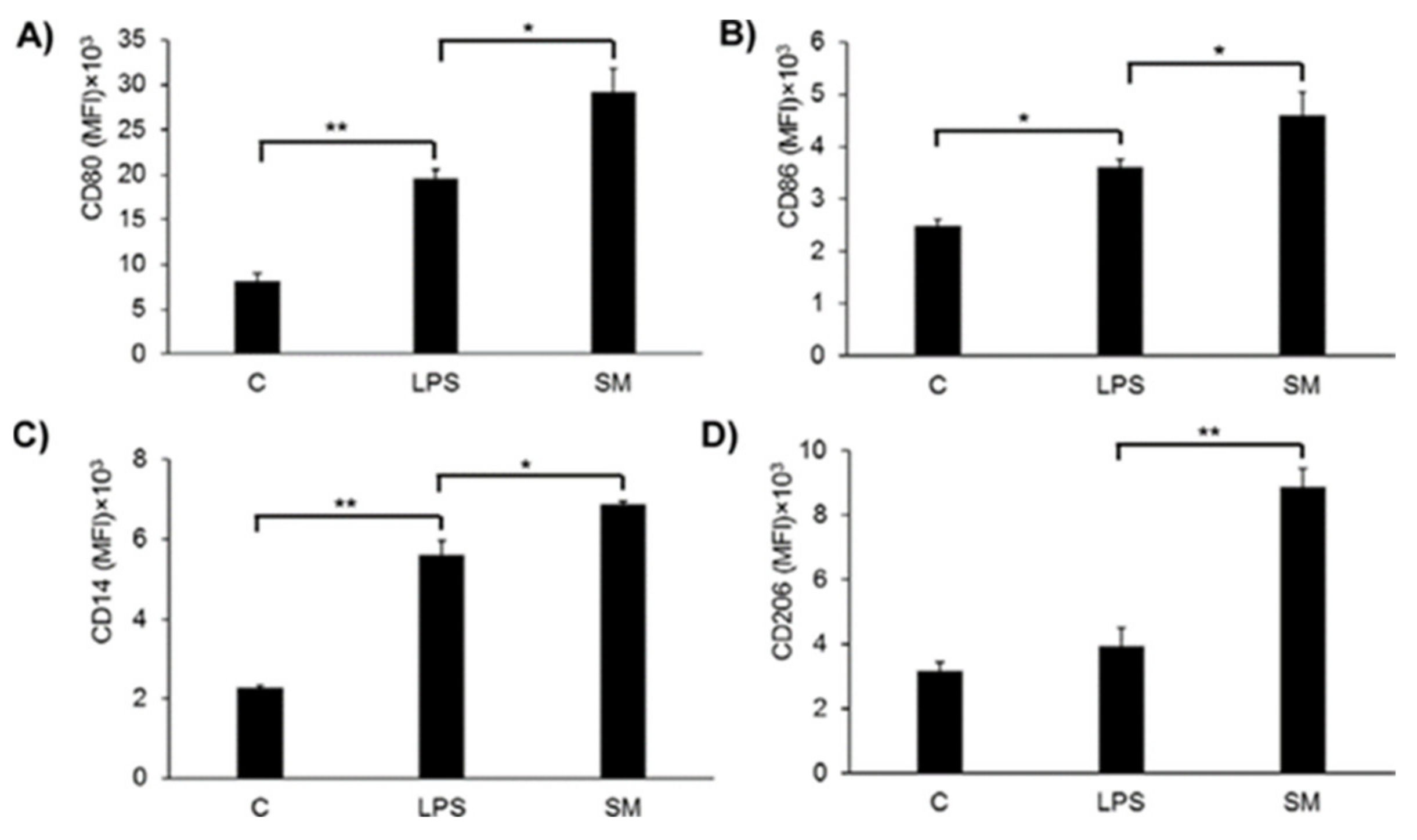
2.6. Effects of SM on Macrophage Surface Marker Expression
2.7. Anti-Inflammatory Effect of SM In Vivo
2.7.1. LPS-Induced Endotoxemia
2.7.2. Carrageenan-Induced Peritonitis
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Semisynthetic Triterpenoids SM
4.2. Assessment of Cell Viability
4.3. Measurement of Cell Migration Using xCelligence Technology
4.4. Phagocytic Uptake
4.5. Measurement of NO Production
4.6. ELISA for Pro-Inflammatory Cytokines
4.7. Real-Time Quantitative PCR (RT-qPCR) Assay for mRNA Levels
- iNOS forward, 5′-AAGGTCTACGTTCAGGACATC-3′;
- iNOS reverse, 5′-AGAAATAGTCTTCCACCTGCT-3′;
- HO-1 forward, 5′-ACAGATGGCGTCACTTCGT-3′;
- HO-1 reverse, 5′-GTGAGGACCCACTGGAGGA-3′;
- IL-6 forward, 5′-CCGGAGAGGAGACTTCACAG-3′;
- IL-6 reverse, 5′-TCCACGATTTCCCAGAGAAC-3′;
- TNF-α forward, 5′-TCAGCCTCTTCTCATTCCTG-3′;
- TNF-α reverse, 5′-TGAAGAGAACCTGGGAGTAG-3′;
- IL-1β forward, 5′-TGCAGAGTTCCCCAACTGGTACATC -3′;
- IL-1β reverse, 5′-GTGCTGCCTAATGTCCCCTTGAATC -3′;
- cJUN forward, 5′-ACGACCTTCTACGACGATGC-3′;
- cJUN reverse, 5′-CCAGGTTCAAGGTCATGCTC-3′;
- TLR4 forward, 5′-AGATCTGAGCTTCAACCC-3′;
- TLR4 reverse, 5′-AGTCCAGAGAAACTTCCTG-3′;
- GAPDH forward, 5′-ACCCCCAATGTGTCCGTCGT-3′;
- GAPDH reverse, 5′-TACTCCTTGGAGGCCATGTA-3′
4.8. Protein Isolation and Western Blot Analysis
4.9. Determination of Intracellular ROS
4.10. Glutathione Estimation
4.11. Evaluation of CD14, CD206, CD80, and CD86 Expression
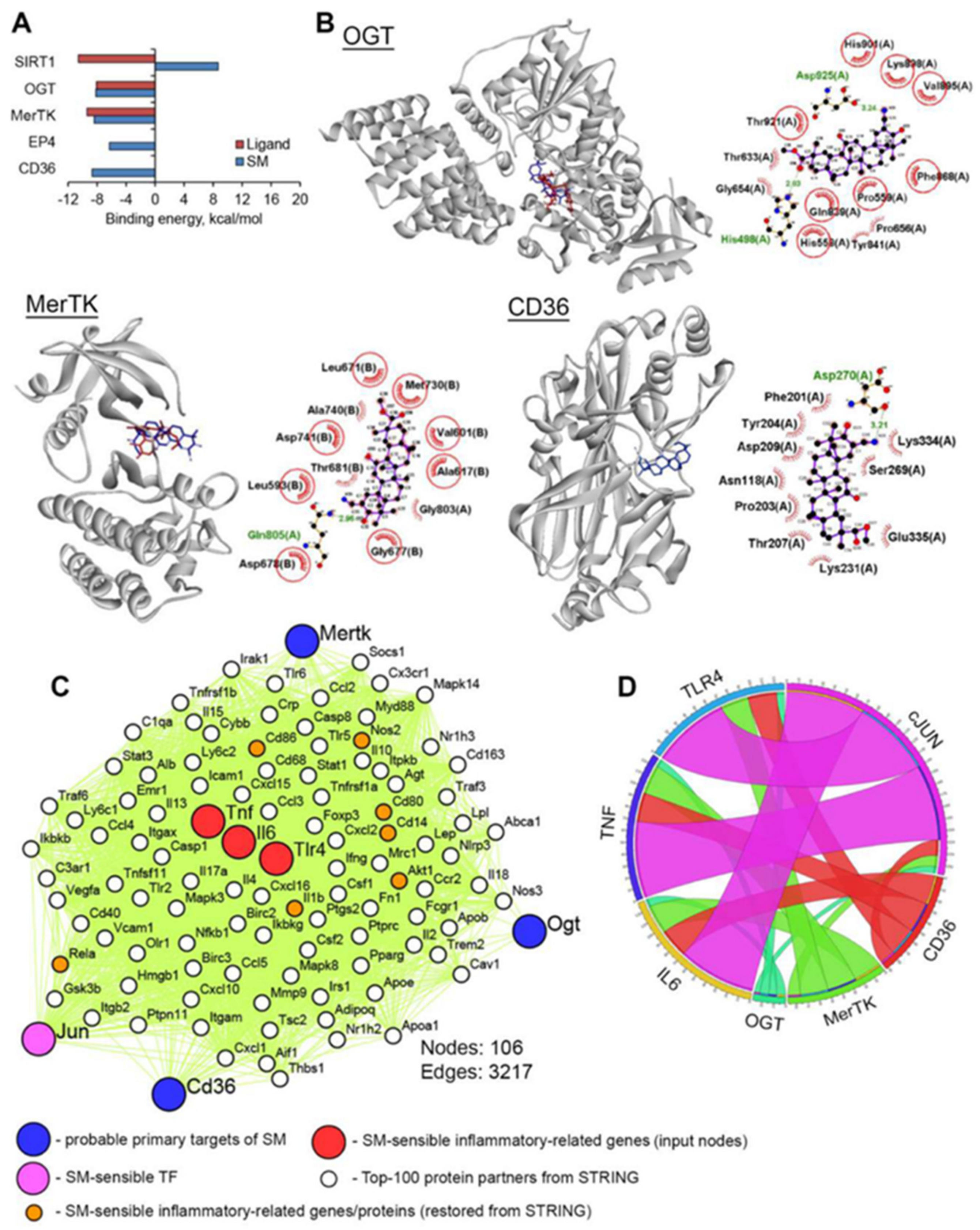
4.12. Molecular Docking
4.13. PPI Network Reconstruction
4.14. Mice
4.15. LPS-Induced Endotoxemia
4.16. Carrageenan-Induced Peritonitis
4.17. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aderem, A.; Ulevitch, R.J. Toll-like receptors in the induction of the innate immune response. Nature 2000, 406, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6. [Google Scholar] [CrossRef]
- Colin, S.; Chinetti-Gbaguidi, G.; Staels, B. Macrophage phenotypes in atherosclerosis. Immunol. Rev. 2014, 262, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.J.; Rosenthal, N. Preparing the ground for tissue regeneration: From mechanism to therapy. Nat. Med. 2014, 20, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Misawa, T.; Satoh, T.; Saitoh, T. Macrophages control innate inflammation. Diabetes Obes. Metab. 2013, 15, 10–18. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Invest. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Novak, M.L.; Koh, T.J. Macrophage phenotypes during tissue repair. J. Leukoc. Biol. 2013, 93, 875–881. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Chinetti-Gbaguidi, G.; Colin, S.; Staels, B. Macrophage subsets in atherosclerosis. Nat. Rev. Cardiol. 2015, 12, 10–17. [Google Scholar] [CrossRef]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef]
- Gensel, J.C.; Zhang, B. Macrophage activation and its role in repair and pathology after spinal cord injury. Brain Res. 2015, 1619, 1–11. [Google Scholar] [CrossRef]
- Graff, J.W.; Dickson, A.M.; Clay, G.; McCaffrey, A.P.; Wilson, M.E. Identifying functional microRNAs in macrophages with polarized phenotypes. J. Biol. Chem. 2012, 287, 21816–21825. [Google Scholar] [CrossRef]
- Cuschieri, J.; Maier, R.V. Oxidative stress, lipid rafts, and macrophage reprogramming. Antioxidants Redox Signal. 2007, 9, 1485–1497. [Google Scholar] [CrossRef]
- Lucas, K.; Maes, M. Role of the toll like receptor (TLR) radical cycle in chronic inflammation: Possible treatments targeting the TLR4 pathway. Mol. Neurobiol. 2013, 48, 190–204. [Google Scholar] [CrossRef]
- Leiro, J.; Álvarez, E.; Arranz, J.A.; Laguna, R.; Uriarte, E.; Orallo, F. Effects of cis -resveratrol on inflammatory murine macrophages: Antioxidant activity and down-regulation of inflammatory genes. J. Leukoc. Biol. 2004, 75, 1156–1165. [Google Scholar] [CrossRef]
- Su, Y.W.; Chiou, W.F.; Chao, S.H.; Lee, M.H.; Chen, C.C.; Tsai, Y.C. Ligustilide prevents LPS-induced iNOS expression in RAW 264.7 macrophages by preventing ROS production and down-regulating the MAPK, NF-κB and AP-1 signaling pathways. Int. Immunopharmacol. 2011, 11, 1166–1172. [Google Scholar] [CrossRef]
- Hong, S.H.; Jeong, H.K.; Han, M.H.; Park, C.; Choi, Y.H. Esculetin suppresses lipopolysaccharide-induced inflammatory mediators and cytokines by inhibiting nuclear factor-κB translocation in RAW 264.7 macrophages. Mol. Med. Rep. 2014, 10, 3241–3246. [Google Scholar] [CrossRef]
- Murakami, A.; Ohigashi, H. Targeting NOX, INOS and COX-2 in inflammatory cells: Chemoprevention using food phytochemicals. Int. J. Cancer 2007, 121, 2357–2363. [Google Scholar] [CrossRef]
- Yang, G.Y.; Taboada, S.; Liao, J. Induced nitric oxide synthase as a major player in the oncogenic transformation of inflamed tissue. Methods Mol. Biol. 2009, 512, 119–156. [Google Scholar]
- Klapproth, J.M.A.; Sasaki, M. Bacterial induction of proinflammatory cytokines in inflammatory bowel disease. Inflamm. Bowel Dis. 2010, 16, 2173–2179. [Google Scholar] [CrossRef]
- Striz, I.; Brabcova, E.; Kolesar, L.; Sekerkova, A. Cytokine networking of innate immunity cells: A potential target of therapy. Clin. Sci. 2014, 126, 593–612. [Google Scholar] [CrossRef]
- Endale, M.; Park, S.C.; Kim, S.S.H.S.; Kim, S.S.H.S.; Yang, Y.; Cho, J.Y.; Rhee, M.H. Quercetin disrupts tyrosine-phosphorylated phosphatidylinositol 3-kinase and myeloid differentiation factor-88 association, and inhibits MAPK/AP-1 and IKK/NF-κB-induced inflammatory mediators production in RAW 264.7 cells. Immunobiology 2013, 218, 1452–1467. [Google Scholar] [CrossRef]
- Huang, B.P.; Lin, C.H.; Chen, H.M.; Lin, J.T.; Cheng, Y.F.; Kao, S.H. AMPK activation inhibits expression of proinflammatory mediators through downregulation of PI3K/p38 MAPK and NF-κB signaling in murine macrophages. DNA Cell Biol. 2015, 34, 133–141. [Google Scholar] [CrossRef]
- Jung, J.S.; Choi, M.J.; Lee, Y.Y.; Moon, B.I.; Park, J.S.; Kim, H.S. Suppression of lipopolysaccharide-induced neuroinflammation by morin via MAPK, PI3K/Akt, and PKA/HO-1 signaling pathway modulation. J. Agric. Food Chem. 2017, 65, 373–382. [Google Scholar] [CrossRef]
- Nakajima, S.; Kitamura, M. Bidirectional regulation of NF-κB by reactive oxygen species: A role of unfolded protein response. Free Radic. Biol. Med. 2013, 65, 162–174. [Google Scholar] [CrossRef]
- Huang, Y.; Li, W.; Su, Z.Y.; Kong, A.N.T. The complexity of the Nrf2 pathway: Beyond the antioxidant response. J. Nutr. Biochem. 2015, 26, 1401–1413. [Google Scholar] [CrossRef]
- Proniewski, B.; Kij, A.; Sitek, B.; Kelley, E.E.; Chlopicki, S. Multiorgan development of oxidative and nitrosative stress in LPS-induced endotoxemia in C57BL/6 mice: DHE-based in vivo approach. Oxid. Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef]
- Liang, Y.; Pan, B.; Alam, H.B.; Deng, Q.; Wang, Y.Y.; Chen, E.; Liu, B.; Tian, Y.; Williams, A.M.; Duan, X.; et al. Inhibition of peptidylarginine deiminase alleviates LPS-induced pulmonary dysfunction and improves survival in a mouse model of lethal endotoxemia. Eur. J. Pharmacol. 2018, 833, 432–440. [Google Scholar] [CrossRef]
- Domscheit, H.; Hegeman, M.A.; Carvalho, N.; Spieth, P.M. Molecular Dynamics of Lipopolysaccharide-Induced Lung Injury in Rodents. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef]
- Kumari, A.; Dash, D.; Singh, R. Curcumin inhibits lipopolysaccharide (LPS)-induced endotoxemia and airway inflammation through modulation of sequential release of inflammatory mediators (TNF-α and TGF-β1) in murine model. Inflammopharmacology 2017, 25, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Elmarakby, A.A.; Ibrahim, A.S.; Katary, M.A.; Elsherbiny, N.M.; El-Shafey, M.; Abd-Elrazik, A.M.; Abdelsayed, R.A.; Maddipati, K.R.; Al-Shabrawey, M. A dual role of 12/15-lipoxygenase in LPS-induced acute renal inflammation and injury. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2019, 1864, 1669–1680. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zeng, X.; Guo, F.; Huang, R.; Feng, Y.; Ma, L.; Zhou, L.; Fu, P. Anti-inflammatory pyranochalcone derivative attenuates LPS-induced acute kidney injury via inhibiting TLR4/NF-κB pathway. Molecules 2017, 22, 1683. [Google Scholar] [CrossRef]
- Khan, H.A.; Ahmad, M.Z.; Khan, J.A.; Arshad, M.I. Crosstalk of liver immune cells and cell death mechanisms in different murine models of liver injury and its clinical relevance. Hepatobiliary Pancreat. Dis. Int. 2017, 16, 245–256. [Google Scholar] [CrossRef]
- Liu, J.; Du, S.; Kong, Q.; Zhang, X.; Jiang, S.; Cao, X.; Li, Y.; Li, C.; Chen, H.; Ding, Z.; et al. HSPA12A attenuates lipopolysaccharide-induced liver injury through inhibiting caspase-11-mediated hepatocyte pyroptosis via PGC-1α-dependent acyloxyacyl hydrolase expression. Cell Death Differ. 2020. [Google Scholar] [CrossRef]
- Nava Catorce, M.; Gevorkian, G. LPS-induced Murine Neuroinflammation Model: Main Features and Suitability for Pre-clinical Assessment of Nutraceuticals. Curr. Neuropharmacol. 2016, 14, 155–164. [Google Scholar] [CrossRef]
- Li, Y.; Li, H.; Liu, S.; Pan, P.; Su, X.; Tan, H.; Wu, D.; Zhang, L.; Song, C.; Dai, M.; et al. Pirfenidone ameliorates lipopolysaccharide-induced pulmonary inflammation and fibrosis by blocking NLRP3 inflammasome activation. Mol. Immunol. 2018, 99, 134–144. [Google Scholar] [CrossRef]
- Pfalzgraff, A.; Weindl, G. Intracellular Lipopolysaccharide Sensing as a Potential Therapeutic Target for Sepsis. Trends Pharmacol. Sci. 2019, 40, 187–197. [Google Scholar] [CrossRef]
- Hayashi, T.; Suzuki, K. Changes of Expression of the Protein C Pathway Components in LPSInduced Endotoxemia–Implication for Sepsis. Cardiovasc. Hematol. Disord. Targets 2015, 15, 2–9. [Google Scholar] [CrossRef]
- Kwiatkowska, K.; Ciesielska, A. Lipid-mediated regulation of pro-inflammatory responses induced by lipopolysaccharide. Postepy Biochem. 2018. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Qiu, J.J.; Chen, B.; Lin, Y.; Chen, Y.Y.; Xie, G.; Qiu, J.J.; Tong, H.; Jiang, D. Long non-coding RNA NEAT1 plays an important role in sepsis-induced acute kidney injury by targeting miR-204 and modulating the NF-κB pathway. Int. Immunopharmacol. 2018, 59, 252–260. [Google Scholar] [CrossRef]
- Shah, R.; Reyes-Ordillo, K.; Cheng, Y.; Varatharajalu, R.; Ibrahim, J.; Lakshman, M.R. Thymosin β 4 prevents oxidative stress, inflammation, and fibrosis in ethanol- and lps-induced liver injury in mice. Oxid. Med. Cell. Longev. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Kosyreva, A.M.; Makarova, O.V.; Kakturskiy, L.V.; Mikhailova, L.P.; Boltovskaya, M.N.; Rogov, K.A. Sex differences of inflammation in target organs, induced by intraperitoneal injection of lipopolysaccharide, depend on its dose. J. Inflamm. Res. 2018, 11, 431–445. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Honda, T.; Rounds, B.V.; Bore, L.; Finlay, H.J.; Favaloro, F.G.; Suh, N.; Wang, Y.; Sporn, M.B.; Gribble, G.W. Synthetic oleanane and ursane triterpenoids with modified rings A and C: A series of highly active inhibitors of nitric oxide production in mouse macrophages. J. Med. Chem. 2000, 43, 4233–4246. [Google Scholar] [CrossRef]
- Honda, T.; Honda, Y.; Favaloro, F.G.; Gribble, G.W.; Suh, N.; Place, A.E.; Rendi, M.H.; Sporn, M.B. A novel dicyanotriterpenoid, 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-onitrile, active at picomolar concentrations for inhibition of nitric oxide production. Bioorganic Med. Chem. Lett. 2002, 12, 1027–1030. [Google Scholar] [CrossRef]
- Place, A.E.; Suh, N.; Williams, C.R.; Risingsong, R.; Honda, T.; Honda, Y.; Gribble, G.W.; Leesnitzer, L.M.; Stimmel, J.B.; Willson, T.M.; et al. The novel synthetic triterpenoid, CDDO-imidazolide, inhibits inflammatory response and tumor growth in vivo. Clin. Cancer Res. 2003, 9, 2798–2806. [Google Scholar]
- Samudio, I.; Konopleva, M.; Pelicano, H.; Huang, P.; Frolova, O.; Bornmann, W.; Ying, Y.; Evans, R.; Contractor, R.; Andreeff, M. A novel mechanism of action of methyl-2-cyano-3,12 dioxoolean-1,9 diene-28-oate: Direct permeabilization of the inner mitochondrial membrane to inhibit electron transport and induce apoptosis. Mol. Pharmacol. 2006, 69, 1182–1193. [Google Scholar] [CrossRef]
- Liby, K.; Voong, N.; Williams, C.R.; Risingsong, R.; Royce, D.B.; Honda, T.; Gribble, G.W.; Sporn, M.B.; Letterio, J.J. The synthetic triterpenoid CDDO-Imidazolide suppresses STAT phosphorylation and induces apoptosis in myeloma and lung cancer cells. Clin. Cancer Res. 2006, 12, 4288–4293. [Google Scholar] [CrossRef]
- Melichar, B.; Konopleva, M.; Hu, W.; Melicharova, K.; Andreeff, M.; Freedman, R.S. Growth-inhibitory effect of a novel synthetic triterpenoid, 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid, on ovarian carcinoma cell lines not dependent on peroxisome proliferator-activated receptor-γ expression. Gynecol. Oncol. 2004, 93, 149–154. [Google Scholar] [CrossRef]
- Nassiri Asl, M.; Hosseinzadeh, H. Review of pharmacological effects of glycyrrhiza sp. and its bioactive compounds. Phyther. Res. 2008, 22, 709–724. [Google Scholar]
- Wang, Z.Y.; Nixon, D.W. Licorice and cancer. Nutr. Cancer 2001, 39, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Logashenko, E.B.; Salomatina, O.V.; Markov, A.V.; Korchagina, D.V.; Salakhutdinov, N.F.; Tolstikov, G.A.; Vlassov, V.V.; Zenkova, M.A. Synthesis and Pro-Apoptotic Activity of Novel Glycyrrhetinic Acid Derivatives. ChemBioChem 2011, 12, 784–794. [Google Scholar] [CrossRef]
- Salomatina, O.V.; Markov, A.V.; Logashenko, E.B.; Korchagina, D.V.; Zenkova, M.A.; Salakhutdinov, N.F.; Vlassov, V.V.; Tolstikov, G.A. Synthesis of novel 2-cyano substituted glycyrrhetinic acid derivatives as inhibitors of cancer cells growth and NO production in LPS-activated J-774 cells. Bioorganic Med. Chem. 2014, 22, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Markov, A.V.; Sen’Kova, A.V.; Warszycki, D.; Salomatina, O.V.; Salakhutdinov, N.F.; Zenkova, M.A.; Logashenko, E.B. Soloxolone methyl inhibits influenza virus replication and reduces virus-induced lung inflammation. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Jun, M.S.; Ha, Y.M.; Kim, H.S.H.J.; Jang, H.J.; Kim, Y.M.; Lee, Y.S.; Kim, H.S.H.J.; Seo, H.G.; Lee, J.H.; Lee, S.H.; et al. Anti-inflammatory action of methanol extract of Carthamus tinctorius involves in heme oxygenase-1 induction. J. Ethnopharmacol. 2011, 133, 524–530. [Google Scholar] [CrossRef]
- Motterlini, R.; Foresti, R.; Bassi, R.; Green, C.J. Curcumin, an antioxidant and anti-inflammatory agent, induces heme oxygenase-1 and protects endothelial cells against oxidative stress. Free Radic. Biol. Med. 2000, 28, 1303–1312. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Soares, M.P.; Yamashita, K.; Bach, F.H. Heme oxygenase-1: Unleashing the protective properties of heme. Trends Immunol. 2003, 24, 449–455. [Google Scholar] [CrossRef]
- Li, Q.F.; Zhu, Y.S.; Jiang, H.; Xu, H.; Sun, Y. Heme oxygenase-1 mediates the anti-inflammatory effect of isoflurane preconditioning in LPS-stimulated macrophages. Acta Pharmacol. Sin. 2009, 30, 228–234. [Google Scholar] [CrossRef][Green Version]
- Markov, A.V.; Kel, A.E.; Salomatina, O.V.; Salakhutdinov, N.F.; Zenkova, M.A.; Logashenko, E.B. Deep insights into the response of human cervical carcinoma cells to a new cyano enone-bearing triterpenoid soloxolone methyl: A transcriptome analysis. Oncotarget 2019, 10, 5267–5297. [Google Scholar] [CrossRef][Green Version]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-S.; Pi, S.-H.; Lee, Y.-M.; Lee, S.-I.; Kim, E.-C. The Anti-Inflammatory Role of Heme Oxygenase-1 in Lipopolysaccharide and Cytokine-Stimulated Inducible Nitric Oxide Synthase and Nitric Oxide Production in Human Periodontal Ligament Cells. J. Periodontol. 2009, 80, 2045–2055. [Google Scholar] [CrossRef]
- Ashino, T.; Yamanaka, R.; Yamamoto, M.; Shimokawa, H.; Sekikawa, K.; Iwakura, Y.; Shioda, S.; Numazawa, S.; Yoshida, T. Negative feedback regulation of lipopolysaccharide-induced inducible nitric oxide synthase gene expression by heme oxygenase-1 induction in macrophages. Mol. Immunol. 2008, 45, 2106–2115. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.D.; Riches, D.W.H. IFN-γ + LPS induction of iNOS is modulated by ERK, JNK/SAPK, and p38mapk in a mouse macrophage cell line. Am. J. Physiol.-Cell Physiol. 2001, 280. [Google Scholar] [CrossRef]
- Tajima, T.; Murata, T.; Aritake, K.; Urade, Y.; Hirai, H.; Nakamura, M.; Ozaki, H.; Hori, M. Lipopolysaccharide induces macrophage migration via prostaglandin D 2 and prostaglandin E2. J. Pharmacol. Exp. Ther. 2008, 326, 493–501. [Google Scholar] [CrossRef]
- Lawrence, T.; Willoughby, D.A.; Gilroy, D.W. Anti-inflammatory lipid mediators and insights into the resolution of inflammation. Nat. Rev. Immunol. 2002, 2, 787–795. [Google Scholar] [CrossRef]
- Androulidaki, A.; Iliopoulos, D.; Arranz, A.; Doxaki, C.; Zacharioudaki, V.; Margioris, A.N.; Tsichlis, P.N.; Tsatsanis, C. Akt1 controls macrophage response to LPS by regulating microRNAs. Immunity 2010, 31, 220–231. [Google Scholar] [CrossRef]
- McGuire, V.A.; Gray, A.; Monk, C.E.; Santos, S.G.; Lee, K.; Aubareda, A.; Crowe, J.; Ronkina, N.; Schwermann, J.; Batty, I.H.; et al. Cross Talk between the Akt and p38 Pathways in Macrophages Downstream of Toll-Like Receptor Signaling. Mol. Cell. Biol. 2013, 33, 4152–4165. [Google Scholar] [CrossRef]
- Medzhitov, R.; Horng, T. Transcriptional control of the inflammatory response. Nat. Rev. Immunol. 2009, 343, 692–703. [Google Scholar] [CrossRef]
- Smale, S.T.; Natoli, G. Transcriptional Control of Inflammatory Responses. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef]
- Ahmed, A.U.; Williams, B.R.G.; Hannigan, G.E. Transcriptional activation of inflammatory genes: Mechanistic insight into selectivity and diversity. Biomolecules 2015, 5, 3087–3111. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Baig, M.S. NOS1 mediates AP1 nuclear translocation and inflammatory response. Biomed. Pharmacother. 2018, 102, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Markov, A.V.; Sen’kova, A.V.; Zenkova, M.A.; Logashenko, E.B. Novel Glycyrrhetinic Acid Derivative Soloxolone Methyl Inhibits the Inflammatory Response and Tumor Growth in vivo. Mol. Biol. 2018, 52, 262–268. [Google Scholar] [CrossRef]
- Wang, Y.F.; Lee, G.L.; Huang, Y.H.; Kuo, C.C. sn-1,2-diacylglycerols protect against lethal endotoxemia by controlling systemic inflammation. Immunobiology 2016, 221, 1309–1318. [Google Scholar] [CrossRef]
- Chen, N.; Liu, D.; Soromou, L.W.; Sun, J.; Zhong, W.; Guo, W.; Huo, M.; Li, H.; Guan, S.; Chen, Z.; et al. Paeonol suppresses lipopolysaccharide-induced inflammatory cytokines in macrophage cells and protects mice from lethal endotoxin shock. Fundam. Clin. Pharmacol. 2014, 28, 268–276. [Google Scholar] [CrossRef]
- Mizokami, S.S.; Hohmann, M.S.N.; Staurengo-Ferrari, L.; Carvalho, T.T.; Zarpelon, A.C.; Possebon, M.I.; De Souza, A.R.; Veneziani, R.C.S.; Arakawa, N.S.; Casagrande, R.; et al. Pimaradienoic acid inhibits carrageenan-induced inflammatory leukocyte recruitment and edema in mice: Inhibition of oxidative stress, nitric oxide and cytokine production. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Barth, C.R.; Funchal, G.A.; Luft, C.; de Oliveira, J.R.; Porto, B.N.; Donadio, M.V.F. Carrageenan-induced inflammation promotes ROS generation and neutrophil extracellular trap formation in a mouse model of peritonitis. Eur. J. Immunol. 2016, 46, 964–970. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.K.; Patra, M.C.; Shin, H.J.; Gui, X.; Achek, A.; Panneerselvam, S.; Kim, D.J.; Song, S.J.; Hong, R.; Kim, K.S.; et al. A cell-penetrating peptide blocks Toll-like receptor-mediated downstream signaling and ameliorates autoimmune and inflammatory diseases in mice. Exp. Mol. Med. 2019, 51. [Google Scholar] [CrossRef]
- Cicinskas, E.; Kalitnik, A.A.; Karetin, Y.A.; Mohan Ram, M.S.G.; Achary, A.; Kravchenko, A.O. Immunomodulating Properties of Carrageenan from Tichocarpus crinitus. Inflammation 2020. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zhou, E.; Wei, Z.; Song, X.; Liu, Z.; Wang, T.; Wang, W.; Zhang, N.; Liu, G.; Yang, Z. Glycyrrhizin inhibits lipopolysaccharide-induced inflammatory response by reducing TLR4 recruitment into lipid rafts in RAW264.7 cells. Biochim. Biophys. Acta-Gen. Subj. 2014, 1840, 1755–1764. [Google Scholar] [CrossRef]
- Feng, X.; Jia, A. Protective effect of carvacrol on acute lung injury induced by lipopolysaccharide in mice. Inflammation 2014, 37, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Soromou, L.W.; Chen, N.; Jiang, L.; Huo, M.; Wei, M.; Chu, X.; Millimouno, F.M.; Feng, H.; Sidime, Y.; Deng, X. Astragalin attenuates lipopolysaccharide-induced inflammatory responses by down-regulating NF-κB signaling pathway. Biochem. Biophys. Res. Commun. 2012, 419, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Kao, T.C.; Wu, C.H.; Yen, G.C. Glycyrrhizic acid and 18β-glycyrrhetinic acid recover glucocorticoid resistance via PI3K-induced AP1, CRE and NFAT activation. Phytomedicine 2013, 20, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, C.C. Inflammatory response of macrophages in infection. Hepatobiliary Pancreat. Dis. Int. 2014, 13, 138–152. [Google Scholar] [CrossRef]
- Chawla, A.; Nguyen, K.D.; Goh, Y.P.S. Macrophage-mediated inflammation in metabolic disease. Nat. Rev. Immunol. 2011, 11, 738–749. [Google Scholar] [CrossRef]
- Bach-Rojecky, L.; Vadunec, D.; Žunić, K.; Kurija, J.; Šipicki, S.; Gregg, R.; Mikula, I.; Primorac, D. Continuing war on pain: A personalized approach to the therapy with nonsteroidal anti-inflammatory drugs and opioids. Per. Med. 2019, 16, 171–184. [Google Scholar] [CrossRef]
- Dunster, J.L. The macrophage and its role in inflammation and tissue repair: Mathematical and systems biology approaches. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 87–99. [Google Scholar] [CrossRef]
- Klessens, C.Q.F.; Zandbergen, M.; Wolterbeek, R.; Bruijn, J.A.; Rabelink, T.J.; Bajema, I.M.; Ijpelaar, D.H.T. Macrophages in diabetic nephropathy in patients with type 2 diabetes. Nephrol. Dial. Transplant. 2017, 32, 1322–1329. [Google Scholar] [CrossRef]
- Griffin, T.M.; Scanzello, C.R. Innate inflammation and synovial macrophages in osteoarthritis pathophysiology. Clin. Exp. Rheumatol. 2019, 37, 57–63. [Google Scholar]
- Maurya, M.R.; Gupta, S.; Li, X.; Fahy, E.; Dinasarapu, A.R.; Sud, M.; Brown, H.A.; Glass, C.K.; Murphy, R.C.; Russell, D.W.; et al. Analysis of inflammatory and lipid metabolic networks across RAW264.7 and thioglycolate-elicited macrophages. J. Lipid Res. 2013, 54, 2525–2542. [Google Scholar] [CrossRef]
- Rouzer, C.A.; Jacobs, A.T.; Nirodi, C.S.; Kingsley, P.J.; Morrow, J.D.; Marnett, L.J. RAW264.7 cells lack prostaglandin-dependent autoregulation of tumor necrosis factor-α secretion. J. Lipid Res. 2005, 46, 1027–1037. [Google Scholar] [CrossRef] [PubMed]
- Asehnoune, K.; Strassheim, D.; Mitra, S.; Kim, J.Y.; Abraham, E. Involvement of Reactive Oxygen Species in Toll-Like Receptor 4-Dependent Activation of NF-κB. J. Immunol. 2004, 172, 2522–2529. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, J.; Biswas, S.; Datta, A. Mode of Action of Endotoxin: Role of Free Radicals and Antioxidants. Curr. Med. Chem. 2005, 11, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Chauveau, C.; Rémy, S.; Royer, P.J.; Hill, M.; Tanguy-Royer, S.; Hubert, F.X.; Tesson, L.; Brion, R.; Beriou, G.; Gregoire, M.; et al. Heme oxygenase-1 expression inhibits dendritic cell maturation and proinflammatory function but conserves IL-10 expression. Blood 2005, 106, 1694–1702. [Google Scholar] [CrossRef]
- Xia, Z.-W.; Zhong, W.-W.; Xu, L.-Q.; Sun, J.-L.; Shen, Q.-X.; Wang, J.-G.; Shao, J.; Li, Y.-Z.; Yu, S.-C. Heme Oxygenase-1-Mediated CD4 + CD25 high Regulatory T Cells Suppress Allergic Airway Inflammation. Am. Assoc. Immunol. 2006, 177, 5936–5945. [Google Scholar]
- Chaudhari, N.; Talwar, P.; Parimisetty, A.; d’Hellencourt, C.L.; Ravanan, P. A molecular web: Endoplasmic reticulum stress, inflammation, and oxidative stress. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef]
- Wiesel, P.; Patel, A.P.; DiFonzo, N.; Marria, P.B.; Sim, C.U.; Pellacani, A.; Maemura, K.; LeBlanc, B.W.; Marino, K.; Doerschuk, C.M.; et al. Endotoxin-induced mortality is related to increased oxidative stress and end-organ dysfunction, not refractory hypotension, in heme oxygenase-1-deficient mice. Circulation 2000, 102, 3015–3022. [Google Scholar] [CrossRef]
- Kapturczak, M.H.; Wasserfall, C.; Brusko, T.; Campbell-Thompson, M.; Ellis, T.M.; Atkinson, M.A.; Agarwal, A. Heme oxygenase-1 modulates early inflammatory responses: Evidence from the heme oxygenase-1-deficient mouse. Am. J. Pathol. 2004, 165, 1045–1053. [Google Scholar] [CrossRef]
- Zhang, X.; Shan, P.; Qureshi, S.; Homer, R.; Medzhitov, R.; Noble, P.W.; Lee, P.J. Cutting Edge: TLR4 Deficiency Confers Susceptibility to Lethal Oxidant Lung Injury. J. Immunol. 2005, 175, 4834–4838. [Google Scholar] [CrossRef]
- Allijn, I.E.; Brinkhuis, R.P.; Storm, G.; Schiffelers, R.M. Anti-Inflammatory Properties of Plant Derived Natural Products – A Systematic Review. Curr. Med. Chem. 2019, 26, 4506–4536. [Google Scholar] [CrossRef]
- Markov, A.V.; Sen’kova, A.V.; Salomatina, O.V.; Logashenko, E.B.; Korchagina, D.V.; Salakhutdinov, N.F.; Zenkova, M.A. Trioxolone methyl, a novel cyano enone-bearing 18βH-glycyrrhetinic acid derivative, ameliorates dextran sulphate sodium-induced colitis in mice. Molecules 2020, 25, 2406. [Google Scholar] [CrossRef]
- Robinson, N.; Ganesan, R.; Hegedűs, C.; Kovács, K.; Kufer, T.A.; Virág, L. Programmed necrotic cell death of macrophages: Focus on pyroptosis, necroptosis, and parthanatos. Redox Biol. 2019, 26, 101239. [Google Scholar] [CrossRef]
- Chen, T.; Mou, Y.; Tan, J.; Wei, L.; Qiao, Y.; Wei, T.; Xiang, P.; Peng, S.; Zhang, Y.; Huang, Z.; et al. The protective effect of CDDO-Me on lipopolysaccharide-induced acute lung injury in mice. Int. Immunopharmacol. 2015, 25, 55–64. [Google Scholar] [CrossRef] [PubMed]
- MOU, Y.; JIAN, Y.L.; CHEN, T.; HUANG, Z.J.; QIAO, Y.X.; PENG, S.X.; ZHANG, D.Y.; JI, H.; ZHANG, Y.H. Synthesis and evaluation of 2-cyano-3, 12-dioxooleana-1, 9(11)-en-28-oate-13β, 28-olide as a potent anti-inflammatory agent for intervention of LPS-induced acute lung injury. Chin. J. Nat. Med. 2017, 15, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Abdalrahman, A.; Lai, Y.; Janicki, J.S.; Ward, K.W.; Meyer, C.J.; Wang, X.L.; Tang, D.; Cui, T. Dihydro-CDDO-trifluoroethyl amide suppresses inflammatory responses in macrophages via activation of Nrf2. Biochem. Biophys. Res. Commun. 2014, 444, 555–561. [Google Scholar] [CrossRef]
- Pei, X.; Zhang, X.J.; Chen, H.M. Bardoxolone treatment alleviates lipopolysaccharide (LPS)-induced acute lung injury through suppressing inflammation and oxidative stress regulated by Nrf2 signaling. Biochem. Biophys. Res. Commun. 2019, 516, 270–277. [Google Scholar] [CrossRef] [PubMed]
- To, C.; Liby, K.T.; Risingsong, R.; Royce, D.B.; Ringelberg, C.S.; Williams, C.R.; Sporn, M.B. Dimethyl fumarate and the oleanane triterpenoids, CDDO-imidazolide and CDDO-methyl ester, both activate the Nrf2 pathway but have opposite effects in the A/J model of lung carcinogenesis. Carcinogenesis 2015, 36, 769–781. [Google Scholar] [CrossRef]
- Xiang, P.; Chen, T.; Mou, Y.; Wu, H.; Xie, P.; Lu, G.; Gong, X.; Hu, Q.; Zhang, Y.; Ji, H. NZ suppresses TLR4/NF-κB signalings and NLRP3 inflammasome activation in LPS-induced RAW264.7 macrophages. Inflamm. Res. 2015, 64, 799–808. [Google Scholar] [CrossRef]
- Yang, N.; Tang, Q.; Qin, W.; Li, Z.; Wang, D.; Zhang, W.; Cao, X.; Lu, Y.; Ge, X.; Sun, H.; et al. Treatment of obesity-related inflammation with a novel synthetic pentacyclic oleanane triterpenoids via modulation of macrophage polarization. EBioMedicine 2019, 45, 473–486. [Google Scholar] [CrossRef]
- Tran, T.A.; McCoy, M.K.; Sporn, M.B.; Tansey, M.G. The synthetic triterpenoid CDDO-methyl ester modulates microglial activities, inhibits TNF production, and provides dopaminergic neuroprotection. J. Neuroinflamm. 2008, 5. [Google Scholar] [CrossRef]
- Thimmulappa, R.K.; Scollick, C.; Traore, K.; Yates, M.; Trush, M.A.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem. Biophys. Res. Commun. 2006, 351, 883–889. [Google Scholar] [CrossRef]
- Thimmulappa, R.K.; Fuchs, R.J.; Malhotra, D.; Scollick, C.; Traore, K.; Bream, J.H.; Trush, M.A.; Liby, K.T.; Sporn, M.B.; Kensler, T.W.; et al. Preclinical evaluation of targeting the Nrf2 pathway by triterpenoids (CDDO-Im and CDDO-Me) for protection from LPS-induced inflammatory response and reactive oxygen species in human peripheral blood mononuclear cells and neutrophils. Antioxidants Redox Signal. 2007, 9, 1963–1970. [Google Scholar] [CrossRef]
- Speen, A.; Jones, C.; Patel, R.; Shah, H.; Nallasamy, P.; Brooke, E.A.S.; Zhu, H.; Li, Y.R.; Jia, Z. Mechanisms of CDDO-imidazolide-mediated cytoprotection against acrolein-induced neurocytotoxicity in SH-SY5Y cells and primary human astrocytes. Toxicol. Lett. 2015, 238, 32–42. [Google Scholar] [CrossRef]
- Yang, L.; Calingasan, N.Y.; Thomas, B.; Charturvedi, R.K.; Kiaei, M.; Wille, E.J.; Liby, K.T.; Williams, C.; Royce, D.; Risingson, R.; et al. Neuroprotective effects of the triterpenoid, CDDO methyl amide, a potent inducer of Nrf2-mediated transcription. PLoS ONE 2009, 4, e5757. [Google Scholar] [CrossRef]
- Vergadi, E.; Ieronymaki, E.; Lyroni, K.; Vaporidi, K.; Tsatsanis, C. Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J. Immunol. 2017, 198, 1006–1014. [Google Scholar] [CrossRef]
- Xie, Q.W.; Kashiwabara, Y.; Nathan, C. Role of transcription factor NF-κB/Rel in induction of nitric oxide synthase. J. Biol. Chem. 1994, 269, 4705–4708. [Google Scholar]
- Liby, K.T.; Sporn, M.B. Synthetic Oleanane Triterpenoids: Multifunctional Drugs with a Broad Range of Applications for Prevention and Treatment of Chronic Disease. Pharmacol. Rev. 2012, 64, 972–1003. [Google Scholar] [CrossRef]
- Wu, T.T.; Chen, T.L.; Chen, R.M. Lipopolysaccharide triggers macrophage activation of inflammatory cytokine expression, chemotaxis, phagocytosis, and oxidative ability via a toll-like receptor 4-dependent pathway: Validated by RNA interference. Toxicol. Lett. 2009, 191, 195–202. [Google Scholar] [CrossRef]
- Kim, J.E.; Park, H.; Lee, J.E.; Kang, T.C. CDDO-Me Inhibits Microglial Activation and Monocyte Infiltration by Abrogating NFκB- and p38 MAPK-Mediated Signaling Pathways Following Status Epilepticus. Cells 2020, 9, 1123. [Google Scholar] [CrossRef]
- Suzuki, E.; Umezawa, K. Inhibition of macrophage activation and phagocytosis by a novel NF-κB inhibitor, dehydroxymethylepoxyquinomicin. Biomed. Pharmacother. 2006, 60, 578–586. [Google Scholar] [CrossRef]
- To, C.; Kulkarni, S.; Pawson, T.; Honda, T.; Gribble, G.W.G.W.; Sporn, M.B.M.B.; Wrana, J.L.J.L.; Di Guglielmo, G.M.G.M. The synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid-imidazolide alters transforming growth factor β-dependent signaling and cell migration by affecting the cytoskeleton and the polarity complex. J. Biol. Chem. 2008, 283, 11700–11713. [Google Scholar] [CrossRef]
- To, C.; Shilton, B.H.; Di Guglielmo, G.M. Synthetic triterpenoids target the Arp2/3 complex and inhibit branched actin polymerization. J. Biol. Chem. 2010, 285, 27944–27957. [Google Scholar] [CrossRef]
- May, R.C.; Caron, E.; Hall, A.; Machesky, L.M. Involvement of the Arp2/3 complex in phagocytosis mediated by FcγR or CR3. Nat. Cell Biol. 2000, 2, 246–248. [Google Scholar] [CrossRef]
- Wittmann, T.; Desai, A. Microtubule cytoskeleton: A new twist at the end. Curr. Biol. 2005, 15, R126–R129. [Google Scholar] [CrossRef]
- Rosales, C.; Uribe-Querol, E. Phagocytosis: A Fundamental Process in Immunity. Biomed Res. Int. 2017, 2017. [Google Scholar] [CrossRef]
- Ball, M.S.; Shipman, E.P.; Kim, H.; Liby, K.T.; Pioli, P.A. CDDO-Me redirects activation of breast tumor associated macrophages. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef]
- Harvey, C.J.; Thimmulappa, R.K.; Singh, A.; Blake, D.J.; Ling, G.; Wakabayashi, N.; Fujii, J.; Myers, A.; Biswal, S. Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic. Biol. Med. 2009, 46, 443–453. [Google Scholar] [CrossRef]
- Lee, M.S.; Kim, Y.-J. Signaling Pathways Downstream of Pattern-Recognition Receptors and Their Cross Talk. Annu. Rev. Biochem. 2007, 76, 447–480. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Lin, C.C.; Jou, M.J.; Hsiao, L.D.; Yang, C.M. RTA 408 inhibits interleukin-1β-induced MMP-9 expression via suppressing protein kinase-dependent NF-κB and AP-1 activation in rat brain astrocytes. Int. J. Mol. Sci. 2019, 20, 2826. [Google Scholar] [CrossRef]
- Wang, X.; Bynum, J.A.; Stavchansky, S.; Bowman, P.D. Cytoprotection of human endothelial cells against oxidative stress by 1-[2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole (CDDO-Im): Application of systems biology to understand the mechanism of action. Eur. J. Pharmacol. 2014, 734, 122–131. [Google Scholar] [CrossRef]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of human macrophage polarization in inflammation during infectious diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- Porcheray, F.; Viaud, S.; Rimaniol, A.C.; Léone, C.; Samah, B.; Dereuddre-Bosquet, N.; Dormont, D.; Gras, G. Macrophage activation switching: An asset for the resolution of inflammation. Clin. Exp. Immunol. 2005, 142, 481–489. [Google Scholar] [CrossRef]
- Wang, L.X.; Zhang, S.X.; Wu, H.J.; Rong, X.L.; Guo, J. M2b macrophage polarization and its roles in diseases. J. Leukoc. Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Bailey, W.M.; Braun, K.J.; Gensel, J.C. Age decreases macrophage IL-10 expression: Implications for functional recovery and tissue repair in spinal cord injury. Exp. Neurol. 2015, 273, 83–91. [Google Scholar] [CrossRef]
- Chen, C.; Gault, A.; Shen, L.; Nabavi, N. Molecular cloning and expression of early T cell costimulatory molecule-1 and its characterization as B7-2 molecule. J. Immunol. 1994, 152, 4929–4936. [Google Scholar] [PubMed]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Ajmo, J.M.; Rogers, C.Q.; Liang, X.; Le, L.; Murr, M.M.; Peng, Y.; You, M. Role of SIRT1 in regulation of LPS- or two ethanol metabolites-induced TNF-α production in cultured macrophage cell lines. Am. J. Physiol.-Gastrointest. Liver Physiol. 2009, 296, G1047–G1053. [Google Scholar] [CrossRef]
- Lin, Q.Q.; Geng, Y.W.; Jiang, Z.W.; Tian, Z.J. SIRT1 regulates lipopolysaccharide-induced CD40 expression in renal medullary collecting duct cells by suppressing the TLR4-NF-κB signaling pathway. Life Sci. 2017, 170, 100–107. [Google Scholar] [CrossRef]
- Li, X.; Gong, W.; Wang, H.; Li, T.; Attri, K.S.; Lewis, R.E.; Kalil, A.C.; Bhinderwala, F.; Powers, R.; Yin, G.; et al. O-GlcNAc Transferase Suppresses Inflammation and Necroptosis by Targeting Receptor-Interacting Serine/Threonine-Protein Kinase 3. Immunity 2019, 50, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Lu, H.Y.; Jiang, A.G.; Wu, H.M.; Fang, L.; Lv, Y.X. MerTK Downregulates Lipopolysaccharide-Induced Inflammation Through SOCS1 Protein but Does Not Affect Phagocytosis of Escherichia coli in Macrophages. Inflammation 2019, 42, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Camenisch, T.D.; Koller, B.H.; Earp, H.S.; Matsushima, G.K. A novel receptor tyrosine kinase, Mer, inhibits TNF-alpha production and lipopolysaccharide-induced endotoxic shock. J. Immunol. 1999, 162, 3498–3503. [Google Scholar]
- Treffkorn, L.; Scheibe, R.; Maruyama, T.; Dieter, P. PGE 2 exerts its effect on the LPS-induced release of TNF-α, ET-1, IL-lα, IL-6 and IL-10 via the EP2 and EP4 receptor in rat liver macrophages. Prostaglandins Other Lipid Mediat. 2004, 74, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Murakami, A.; Fujimura, Y.; Tachibana, H.; Yamada, K.; Masuda, D.; Hirano, K.; Yamashita, S.; Ohigashi, H. Aggregated Ursolic Acid, a Natural Triterpenoid, Induces IL-1β Release from Murine Peritoneal Macrophages: Role of CD36. J. Immunol. 2007, 178, 4854–4864. [Google Scholar] [CrossRef]
- Clarke, A.J.; Hurtado-Guerrero, R.; Pathak, S.; Schüttelkopf, A.W.; Borodkin, V.; Shepherd, S.M.; Ibrahim, A.F.M.; Van Aalten, D.M.F. Structural insights into mechanism and specificity of O-GlcNAc transferase. EMBO J. 2008, 27, 2780–2788. [Google Scholar] [CrossRef]
- Lazarus, M.B.; Nam, Y.; Jiang, J.; Sliz, P.; Walker, S. Structure of human O-GlcNAc transferase and its complex with a peptide substrate. Nature 2011, 469, 564–569. [Google Scholar] [CrossRef]
- Balupuri, A.; Balasubramanian, P.K.; Cho, S.J. Molecular modeling study on Mer kinase inhibitors using 3D-QSAR and docking approaches. Med. Chem. Res. 2015, 24, 3730–3742. [Google Scholar] [CrossRef]
- Banesh, S.; Ramakrishnan, V.; Trivedi, V. Mapping of phosphatidylserine recognition region on CD36 ectodomain. Arch. Biochem. Biophys. 2018, 660, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S.; Suzuki, J.; Segawa, K.; Fujii, T. Exposure of phosphatidylserine on the cell surface. Cell Death Differ. 2016, 23, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Febbraio, M.; Hajjar, D.P.; Silverstein, R.L. CD36: A class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Invest. 2001, 108, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Auletta, J.J.; Alabran, J.L.; Kim, B.G.; Meyer, C.J.; Letterio, J.J. The synthetic triterpenoid, CDDO-Me, modulates the proinflammatory response to in vivo lipopolysaccharide challenge. J. Interf. Cytokine Res. 2010, 30, 497–508. [Google Scholar] [CrossRef]
- Morris, C.J. Carrageenan-induced paw edema in the rat and mouse. Methods Mol. Biol. 2003, 225, 115–121. [Google Scholar] [PubMed]
- Phumsuay, R.; Muangnoi, C.; Wasana, P.W.D.; Hasriadi; Vajragupta, O.; Rojsitthisak, P.; Towiwat, P. Molecular insight into the anti-inflammatory effects of the curcumin ester prodrug curcumin diglutaric acid in vitro and in vivo. Int. J. Mol. Sci. 2020, 21, 5700. [Google Scholar] [CrossRef]
- Souto, A.L.; Tavares, J.F.; Da Silva, M.S.; De Diniz, M.F.F.M.; De Athayde-Filho, P.F.; Barbosa Filho, J.M. Anti-inflammatory activity of alkaloids: An update from 2000 to 2010. Molecules 2011, 16, 8515–8534. [Google Scholar] [CrossRef]
- Dickson, K.; Lehmann, C. Inflammatory response to different toxins in experimental sepsis models. Int. J. Mol. Sci. 2019, 20, 4341. [Google Scholar] [CrossRef]
- Chousterman, B.G.; Swirski, F.K.; Weber, G.F. Cytokine storm and sepsis disease pathogenesis. Semin. Immunopathol. 2017, 39, 517–528. [Google Scholar] [CrossRef]
- Dienz, O.; Rud, J.G.; Eaton, S.M.; Lanthier, P.A.; Burg, E.; Drew, A.; Bunn, J.; Suratt, B.T.; Haynes, L.; Rincon, M. Essential role of IL-6 in protection against H1N1 influenza virus by promoting neutrophil survival in the lung. Mucosal Immunol. 2012, 5, 258–266. [Google Scholar] [CrossRef]
- Yang, M.L.; Wang, C.T.; Yang, S.J.; Leu, C.H.; Chen, S.H.; Wu, C.L.; Shiau, A.L. IL-6 ameliorates acute lung injury in influenza virus infection. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Ladel, C.H.; Blum, C.; Dreher, A.; Reifenberg, K.; Kopf, M.; Kaufmann, S.H.E. Lethal tuberculosis in interleukin-6-deficient mutant mice. Infect. Immun. 1997, 65, 4843–4849. [Google Scholar] [CrossRef] [PubMed]
- van der Poll, T.; Keogh, C.V.V.; Guirao, X.; Buurman, W.A.A.; Kopf, M.; Lowry, S.F.F. Interleukin-6 gene-deficient mice show impaired defense against pneumococcal pneumonia. J. Infect. Dis. 1997, 176, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, R.; Janssen, W.; Altmann, C.; Andrés-Hernando, A.; Okamura, K.; Vandivier, R.W.; Ahuja, N.; Faubel, S. Intratracheal IL-6 Protects against Lung Inflammation in Direct, but Not Indirect, Causes of Acute Lung Injury in Mice. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Voiriot, G.; Razazi, K.; Amsellem, V.; Tran Van Nhieu, J.; Abid, S.; Adnot, S.; Mekontso Dessap, A.; Maitre, B. Interleukin-6 displays lung anti-inflammatory properties and exerts protective hemodynamic effects in a double-hit murine acute lung injury. Respir. Res. 2017, 18. [Google Scholar] [CrossRef]
- Kobayashi, T.; Tanaka, K.; Fujita, T.; Umezawa, H.; Amano, H.; Yoshioka, K.; Naito, Y.; Hatano, M.; Kimura, S.; Tatsumi, K.; et al. Bidirectional role of IL-6 signal in pathogenesis of lung fibrosis. Respir. Res. 2015, 16. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Scardoni, G.; Tosadori, G.; Pratap, S.; Spoto, F.; Laudanna, C. Finding the shortest path with PesCa: A tool for network reconstruction. F1000Research 2015, 4, 484. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Name | Relation to LPS-Induced Inflammation | Ref. |
|---|---|---|---|
| SIRT1 | Sirtuin 1 | Sirtinol (SIRT1 inhibitor) or knocking down SIRT1 by SIRT1 shRNA augmented LPS-induced TNF-α release by immortalized rat Kupffer RKC1 cells | [141] |
| Down-regulation of SIRT1 expression through inhibition of SIRT1 activity using Ex527 and sirtinol enhanced LPS-induced TLR4 expression in rat renal inner medullary collecting duct cells | [142] | ||
| OGT | O-Linked N-acetylglucosamine (GlcNAc) transferase | OSMI-1 (selective inhibitor of OGT) significantly enhanced LPS-induced expression of IL-6 and TNF-α in murine bone marrow-derived macrophages | [143] |
| MerTK | MER receptor tyrosine kinase | MerTK-specific blocking antibody promoted LPS-induced production of TNF-α, IL-6, and IL1β in RAW264.7 cells | [144] |
| Macrophages isolated from MerTK knockdown (MerTKKD) mice have been shown to be hypersensitive to bacterial LPS | [145] | ||
| EP4 | Prostaglandin E receptor 4 | EP2 agonist ONO-AE1-259 enhanced LPS-induced release of IL-6 by primary rat liver macrophages | [146] |
| CD36 | Thrombospondin receptor | Direct binding of ursolic acid to CD36 induced release of IL-1β from murine peritoneal macrophages | [147] |
| Protein | PDB ID | Centre | Size | ||||
|---|---|---|---|---|---|---|---|
| x | y | z | x | y | z | ||
| SIRT1 | 4I5I | 43.323 | −20.576 | 18.462 | 20 | 20 | 20 |
| OGT | 6MA3 | −1.55 | −43.134 | 16.031 | 22 | 20 | 20 |
| MerTK | 5U6C | −4.614 | 17.118 | −18.308 | 20 | 20 | 20 |
| EP4 | 5YWY | −42.732 | −44.411 | 0.0 | 22 | 18 | 20 |
| CD36 | 5LGD | −43.253 | −26.463 | 25.028 | 50 | 50 | 70 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markov, A.V.; Sen’kova, A.V.; Babich, V.O.; Odarenko, K.V.; Talyshev, V.A.; Salomatina, O.V.; Salakhutdinov, N.F.; Zenkova, M.A.; Logashenko, E.B. Dual Effect of Soloxolone Methyl on LPS-Induced Inflammation In Vitro and In Vivo. Int. J. Mol. Sci. 2020, 21, 7876. https://doi.org/10.3390/ijms21217876
Markov AV, Sen’kova AV, Babich VO, Odarenko KV, Talyshev VA, Salomatina OV, Salakhutdinov NF, Zenkova MA, Logashenko EB. Dual Effect of Soloxolone Methyl on LPS-Induced Inflammation In Vitro and In Vivo. International Journal of Molecular Sciences. 2020; 21(21):7876. https://doi.org/10.3390/ijms21217876
Chicago/Turabian StyleMarkov, Andrey V., Aleksandra V. Sen’kova, Valeriya O. Babich, Kirill V. Odarenko, Vadim A. Talyshev, Oksana V. Salomatina, Nariman F. Salakhutdinov, Marina A. Zenkova, and Evgeniya B. Logashenko. 2020. "Dual Effect of Soloxolone Methyl on LPS-Induced Inflammation In Vitro and In Vivo" International Journal of Molecular Sciences 21, no. 21: 7876. https://doi.org/10.3390/ijms21217876
APA StyleMarkov, A. V., Sen’kova, A. V., Babich, V. O., Odarenko, K. V., Talyshev, V. A., Salomatina, O. V., Salakhutdinov, N. F., Zenkova, M. A., & Logashenko, E. B. (2020). Dual Effect of Soloxolone Methyl on LPS-Induced Inflammation In Vitro and In Vivo. International Journal of Molecular Sciences, 21(21), 7876. https://doi.org/10.3390/ijms21217876