Genetic Landscape of Common Epilepsies: Advancing towards Precision in Treatment

 ,
,  ,
,
Abstract
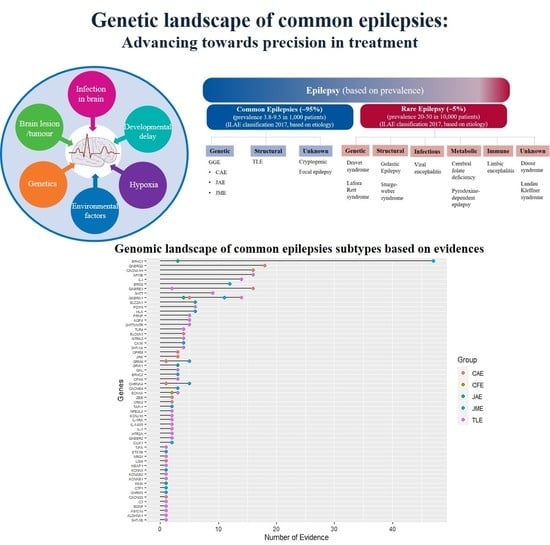
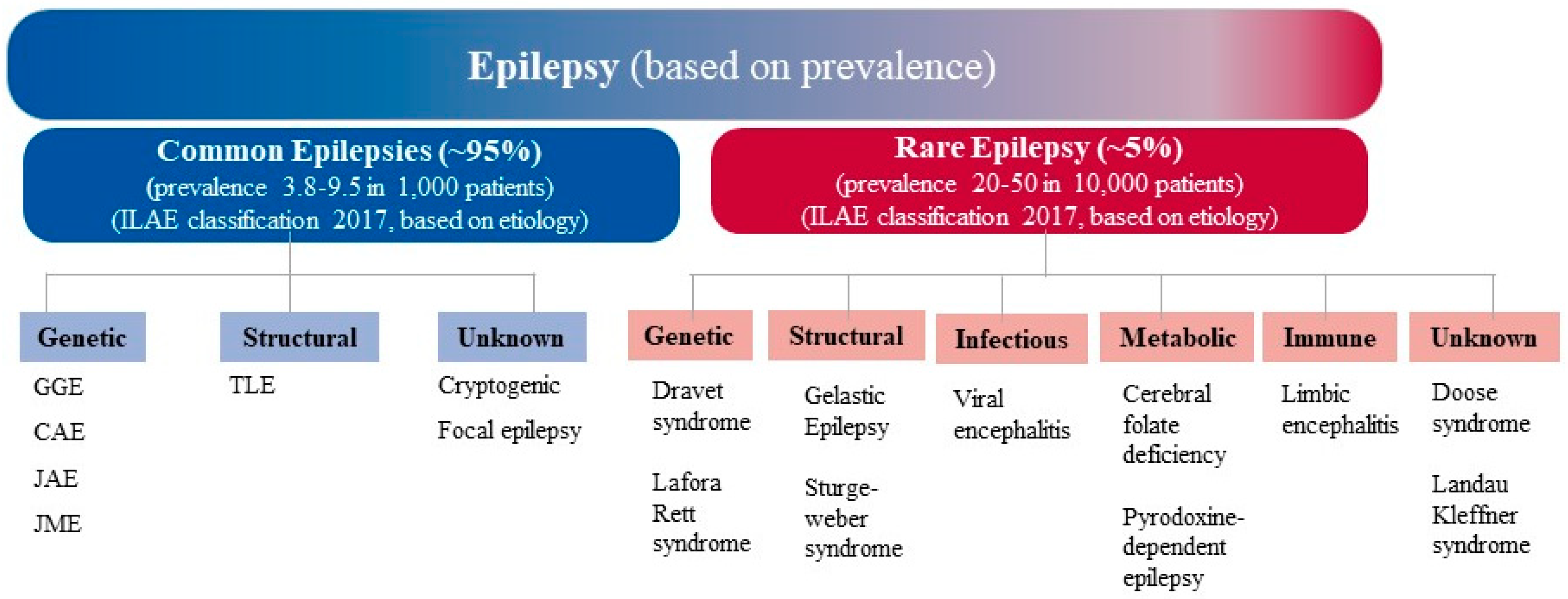
1. Introduction
2. Genetic Studies of Common Epilepsies
2.1. Genetic Generalized Epilepsy (GGE)
2.1.1. Childhood Absence Epilepsy (CAE)
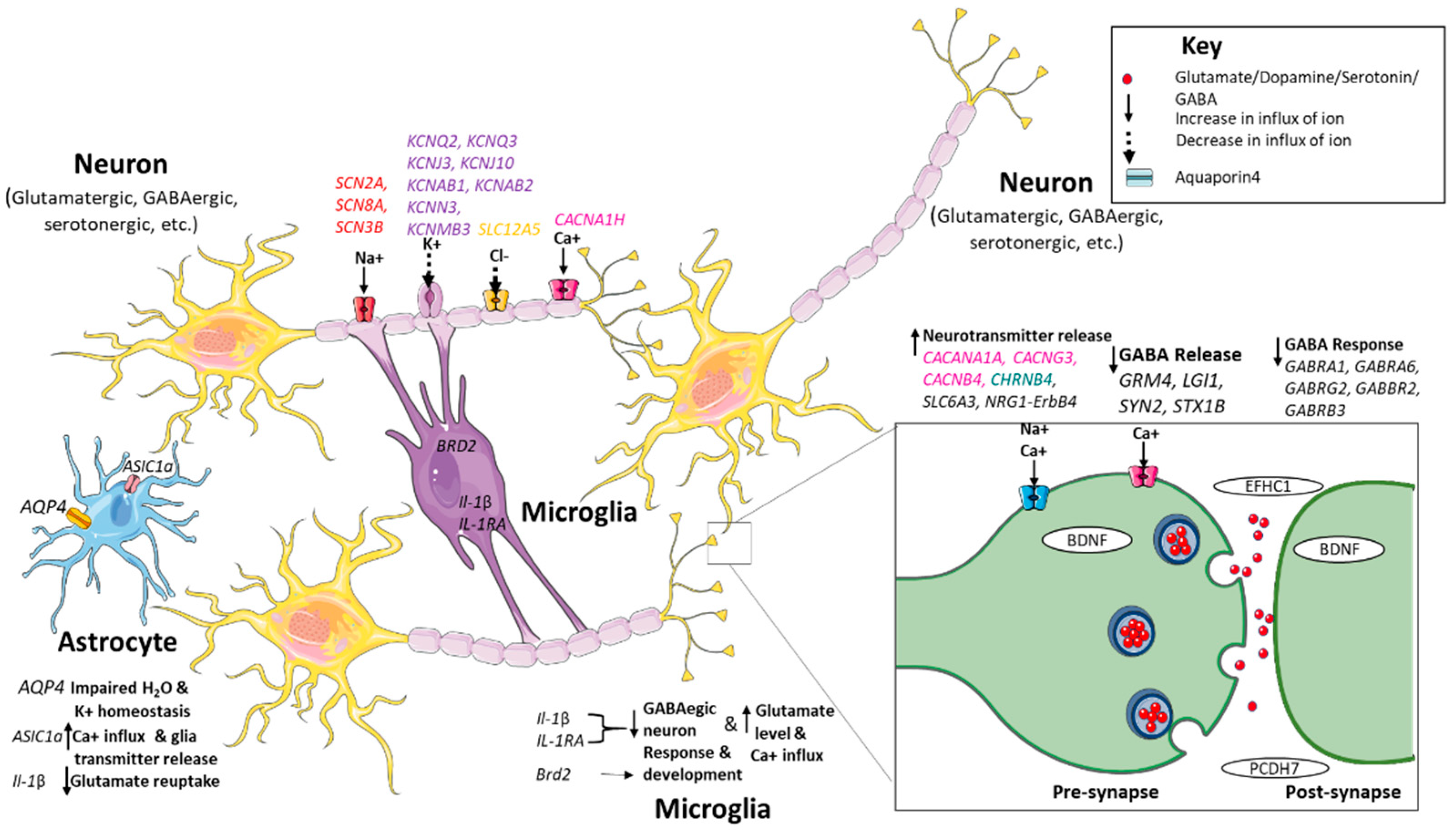
Voltage-Gated Ion Channels
Calcium Channel Genes
Ligand-Gated Channels
Solute Carrier Transporters
Unclassified
2.1.2. Juvenile Myoclonic Epilepsy (JME)
Potassium Ion Channels

{kind=link}
{kind=link}
{kind=link}
{kind=link}
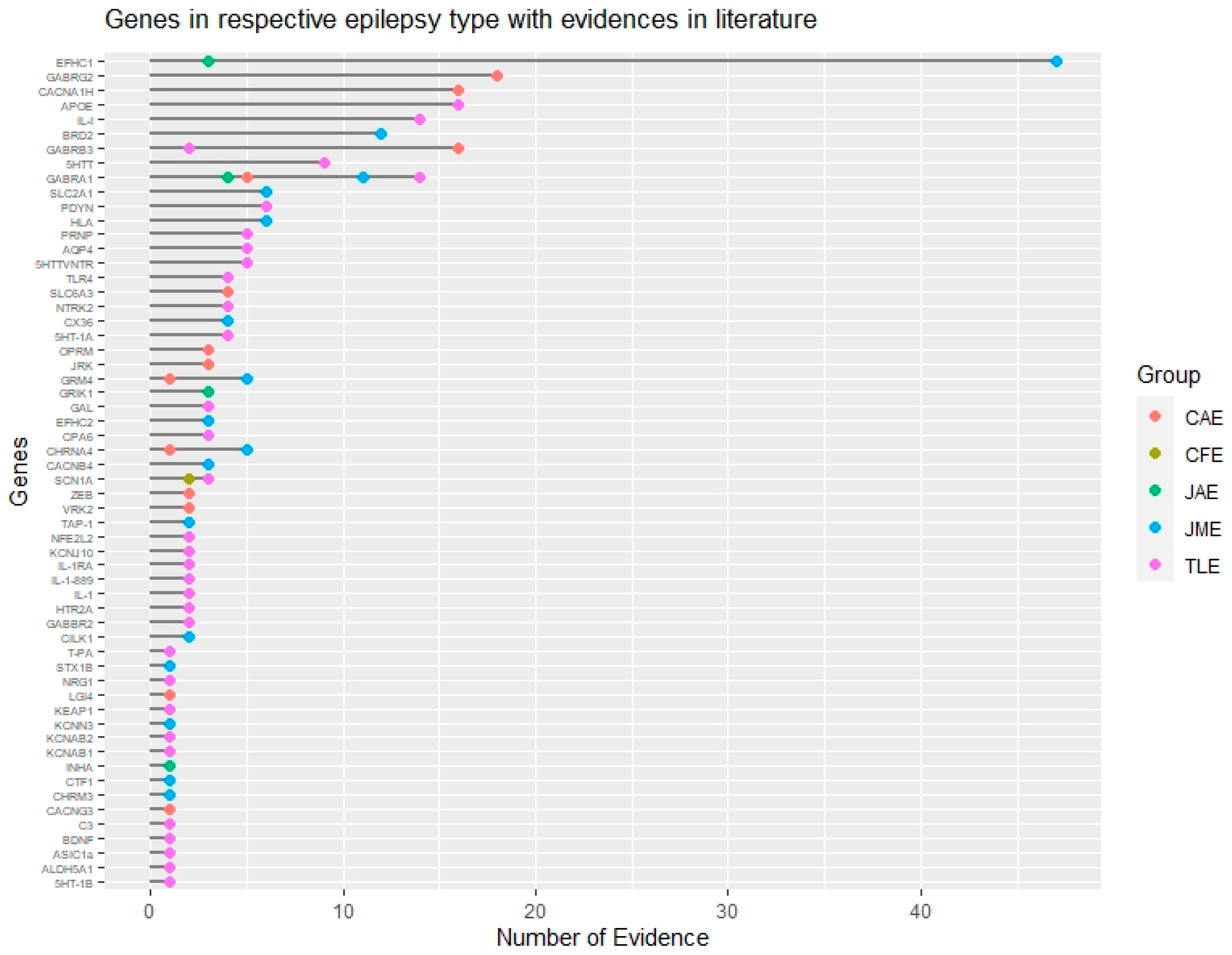{kind=link}
| Sl. No. | Gene/Loci | Gene Family | SNP/Genetic Variant | Risk Allele | Protein Change | GMAF | p-Value | OR (CI) | Country | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| GGE | ||||||||||
| 1 | SLC4A3 | Solute carrier transporter family | 2600G/A | A | A867D | - | 0.021 | 1.48 (1.03–2.14) | Germany | [75] |
| 2 | CACNA1A | Calcium channel | SNP 8 (SNP in exon 8) | A | - | - | 0.00033 | 1.8 (1.3–2.4) | USA | [76] |
| 3 | CHRNA4 | Acetylcholine receptor | rs1044396 | T | S543S | 0.323 | 0.0126 | 4.9 (1.71–14.04) | Taiwan | [77] |
| rs1044394 | T | - | 0.136 | 0.02 | 3.57 (1.31–9.72) | Germany | [52] | |||
| 4 | D18S474 locus/18q12 | - | - | D18S474 8- and 9- | - | - | <0.001 | - | Italy | [78] |
| 5 | GABRA6 | GABA A receptor | rs3219151 | T | - | 0.43112 | <0.001 | 3.6 (2.1–5.9) | South India | [79] |
| 6 | GABARG | GABA A receptor | rs211037 | T | - | 0.371605 | 0.004 | 7.36 | Egypt | [80] |
| 7 | GRIK | Glutamate receptor | GRIK tetra-nucleotide polymorphism | 9 repeat allele | - | - | 0.004 | 1.26 (1.08–1.47) | Germany | [81] |
| 8 | GRM4 | Glutamate receptor | rs9380405 | T | - | 0.69 | 0.003 | - | Germany | [51] |
| rs937039 | G | - | 0.257987 | 0.0038 | - | |||||
| rs2451334 | A | - | 0.163339 | 0.0118 | - | |||||
| rs2029461-rs2451334-rs745501-rs2499697-rs937039 | TGTAA | - | 0.402157,0.163339,0.330471, 0.078474, 0.25798 | 0.0069 | 3.54 (1.42–8.83) | Jordan | [82] | |||
| 9 | Haptoglobin (Hp) | - | Hp*1 | Hp*1 | - | - | <0.001 (Hp*1/*1 vs. other types of epilepsy vs. controls in individuals containing *B/*B genotype in ACP1 | 0.72 (0.011–0.58) | Italy | [83] |
| 10 | KCNJ3 | Potassium channel gene | T1038C | T | - | - | 0.051 | 1.4 (1.0–1.9) | UK | [84] |
| 11 | KCNJ10 | Potassium channel gene | rs1130183 | C | R271C | 0.014776 | 0.03 | 0.69 (0.50–0.95) | Germany | [85] |
| 12 | KCNMB3 | Potassium channel gene | delA750 | - | - | - | 0.016 | 1.52 (1.05–2.21) | Germany | [86] |
| 13 | KCNQ2 | Potassium channel gene | rs1801545 | C | 0.069286 | 0.01 | 1.62 (1.12–2.34) | Germany | [74] | |
| 14 | KCNQ3 | Potassium channel gene | rs74582884 | A | P574S | 0.0015 | 0.008 | - | India | |
| 15 | ME2 | Enzyme | rs674351- rs584087 - rs585344 - rs608781 - rs642698 - rs674210 - rs645088 - rs649224 - rs654136 | A-A-C-A-G-A-C-C-A | - | 0.287141, 0.269169, 0.270168, 0.164736, 0.260783, 0.270767, 0.272364, 0.15615 | - | 6.1 (2.9–12.7) | New York | [87] |
| 16 | MTHFR | Methylene tetrahydrofolate reductase | rs1801133/677C>T | T | A222V | 0.24 | 0.01 | 2.26 (1.13–4.5) | Scotland | [88] |
| 17 | COPZ2 | Coatomer subunit zeta-2 | rs72823592 | A | - | 0.103834 | 9.3 × 10–9 | 0.77 (0.71–0.83) | Austria, Belgium, Denmark, Germany and the Netherlands | [89] |
| 18 | SCN1A | Sodium channel gene | rs8191987 | G | - | 0.225439 | 0.03 | - | UK | [90] |
| rs16851381 | G | - | 0.166134 | 0.05 | - | |||||
| rs2298771 | G | - | 0.2115 | 0.002 | - | |||||
| 19 | SCN2A | Sodium channel gene | rs2060199 | A | - | 0.53 | 0.04 | - | ||
| rs935403 | - | - | - | 0.03 | - | |||||
| rs3943809 | G | - | 0.202 | 0.04 | - | |||||
| 20 | SCN8A | Sodium channel gene | rs303777 | A | - | 0.297724 | 0.007 | - | ||
| 21 | SLC12A5 | Transporter | c.3145 C > T | T | R1049C | - | 0.044 | 9.61 (0.8–503.6) | Canada | [91] |
| 22 | SYN II | Membrane trafficking | rs37733634 | - | - | - | <0.001 | 2.57 (2.0–3.2) | South India | [79] |
| rs3773364 | G | - | 0.269968 | 0.02 | 1.55 (1.06–2.26) | North India | [87] | |||
| 23 | TAP-1A | Transporter | 333Val-637Asp | - | 333V-637D | - | 0.02 | 0.47 | Tunisia | [92] |
| 24 | TAP-1C | Transporter | 333Ile-637Asp | - | 333I-637D | - | 0.03 | 4.3 | ||
| 25 | VAMP2 | Vesicle-associated membrane protein (VAMP)/synaptobrevin family | 26 bp Ins/Del | ins/del* | - | - | 0.042 | 0.474 (0.230–0.978) | Turkey | [93] |
| 26 | SYT11 | Synaptotagmin XI | 33-bp repeats in promoter region | C | - | - | <0.001 | 2.317 (1.503–3.573) | ||
| 27 | VRK2 | Vesicle-associated membrane protein (VAMP)/synaptobrevin family | rs13026414 | T | - | 0.224641 | 2.5 × 10–8 | - | Finland, USA, Australia, Canada, Belgium, UK, Republic of Ireland, China, Hong Kong, USA, Canada | [94] |
| 28 | - | - | rs2947349 | C | - | 0.28 | 1 x 10–8 | 1.23 (1.16–1.31) | ||
| 29 | MMP8 | Proteinase | rs1939012 | T | - | 0.385184 | 2 x 10–8 | 1.02 (1.07–1.17) | ||
| 30 | PCDH7 | Cell adhesion molecule | rs1044352 | G | - | 0.473243 | 2 x 10–7 | 1.14 (1.08–1.22) | ||
| 31 | - | - | rs55670112 | C | - | 0.474441 | 6 x 10–8 | 0.47 (1.10–1.26) | ||
| CAE | ||||||||||
| 32 | CACNA1H | Calcium channel gene | rs9934839 | G | R603R | 0.396565 | <0.0001 | - | China | [31] |
| rs2745150 | T | - | 0.090256 | <0.0001 | - | |||||
| rs8044363 | C | - | 0.332668 | 0.0242 | - | [25] | ||||
| rs8043905 | A | - | 0.330871 | 0.015 | - | |||||
| rs9934839 | G | R603R | 0.3966 | 0.012 | 3.367 (1.307–8.671) | |||||
| rs3751664 | T | - | 0.046725 | 0.025 | 1.760 (1.074– 2.886) | |||||
| c.937A>G | G | M313V | - | 0.01 | 0.070 (0.008–0.619) | |||||
| rs119454947 | A | F161L | 0 | - | - | [30,32] | ||||
| rs119454949 | A | V831M | 0.00002 | - | - | |||||
| 33 | CACNG3 | Calcium channel gene | rs447292-rs4787924-rs2239341-rs1494550-c-597delT- rs965830-rs2214437-rs2238500 and rs4787924, rs965830, rs2214437 | A, G, A, T, T, G, A | - | 0.348442, 0.46865, 0.277356, 0.240815, 0.470847, 0.470647, 0.427316 | <0.005 | - | UK, France, Germany, Austria, the Netherlands, Denmark, Sweden, Finland and Italy | [26] |
| 34 | CHRNA4 | Acetylcholine receptor | CfoI bp595 | T | - | - | 0.0397 | 3.57 (1.16–11.02) | Germany | [52] |
| 35 | GRM4 | Glutamate receptor | rs2499697 | C | - | 0.92 | 0.0021 | 2.36 (1.34–4.15) | Germany | [51] |
| rs2451357 | G | - | 0.87 | 0.0466 | - | |||||
| 36 | GABRA1 | GABA A receptor | rs1581220270 | c.975del | - | - | - | - | Germany | [95] |
| 37 | GABRB3 | GABA A receptor | rs25409 | T | P11S | 0.002 | - | - | Mexico | [46] |
| 38 | GABRG2 | GABA A receptor | rs1561645243 | G | - | - | - | - | Germany | [96] |
| 39 | JRK/JH8 | Nucleic acid binding | Thr456Met | 456Met | T456M | - | - | - | France, Switzerland, Italy and the UK | [97] |
| 40 | LGI4 | Leucine-rich repeat LGI | c.1914GC-AT | GC/GC | - | - | 0.024 | 2.57 (1.24–5.33) | Germany, Belgium, Turkey | [62] |
| 41 | OPRM1 | Opioid receptor | rs1799971 | G | N40D | 0.2234 | 0.019 | 2.03 (1.12–3.68) | Germany | [57] |
| 42 | SCL6A3/DAT1 | Transporter | 40 base pair VNTR polymorphism | Nine-copy allele | - | - | 0.002 | 2.258 (1.32–3.85) | Germany | [59] |
| 43 | VRK2, ACTG1P22 | Vaccinia-related kinase | rs12185644 | C | - | 0.35643 | 5 x 10–10 | - | Caucasian, Asian and African-American | [13] |
| 44 | ZEB2 | Zfh1 | rs13020210 | G | - | 0.311302 | 2 x 10–8 | - | ||
| JME | ||||||||||
| 45 | BRD2 | Nucleic acid binding | rs3918149 | A | - | 0.161542 | 0.043 | 1.93(1.01–3.70) | Ireland | [98] |
| rs3918149 | A | - | 0.161542 | 0.001 | 2.63(1.42–4.87) | UK | ||||
| rs3918149 | A | - | 0.161542 | - | 2.8(1.19–6.64) | North America | [99] | |||
| rs516535 | G | - | 0.390775 | - | 2.05(1.00–4.22) | |||||
| rs635688 | T | - | 0.390775 | - | 2.16(1.05–4.42) | |||||
| rs206674 | G | - | 0.003994 | - | 2.51(1.20–5.24) | |||||
| rs206787 | - | - | 0.390575 | - | 2.21(1.08–4.52) | |||||
| rs3918149 | A | - | 0.161542 | - | 2.8(1.19–6.64) | |||||
| rs206777 | G | - | 0.369409 | - | 2.29(1.11–4.71) | |||||
| rs497058 | T | - | 0.389776 | - | 2.08(1.01–4.28) | |||||
| 46 | CHRNA4 | Acetylcholine receptor | rs45442394 | T | - | 0.021166 | 0.029 | 1.914 (1.057–3.467) | Poland | [100] |
| 47 | CACNB4 | Calcium channel gene | R482X | T | R482X | <0.006 | - | - | United state | [101] |
| 48 | CTF1 | Serum cardiotrophin-1 | rs1046276 | T | - | 0.4113 | 3 × 10–11 | - | Europe | [13] |
| 49 | CHRM3 | Cholinergic receptor | rs12059546 | G | - | 0.3298 | 4.1 × 10–8 | 1.42 | Europe | [89] |
| 50 | CILK1 | Kinase | rs376111440 | T | R632 | 0.000032 (GnomAD) | - | - | United States, Mexico, Honduras, Brazil and Japan | [102] |
| rs55932059 | A | A615T | 0 (ALFA) | - | - | |||||
| rs1554169267 | G | K220E | - | - | ||||||
| rs765078446 | C | K305T | 0.000033 | - | - | |||||
| 51 | CX36 | Connexin | rs3743123 | TT | S196S | 0.308906 | 0.0195 | 1.62(1.02–2.57) | Germany | [103] |
| rs3743123 | TT | S196S | 0.308906 | 0.017 | 4.3 (1.5–12.3) | UK, Denmark, France, Greece, Portugal and Sweden | [104] | |||
| 52 | EFHC1 | Signal transduction molecule | rs137852778 | T | D234Y | 0.00002 | - | - | Japan | [105] |
| rs137852776 | C | F229L | 0.0018 | - | - | |||||
| rs137852777 | A | D210N | 0.000058 | - | - | |||||
| rs149055334 | A | P77T | 0.002349 | - | - | |||||
| rs79761183 | A | R221H | 0.009585 | - | - | |||||
| 53 | EFHC2 | Signal transduction molecule | rs2208592) | T | S430Y | 0.085298 | 0.03 | 2.17 (1.06–4.43) | Germany | [106] |
| 54 | GABRA1 | GABA A receptor | rs121434579 | - | A322D | - | - | - | Canada | [107] |
| 55 | GRM4 | Glutamate receptor | rs9380405 | T | - | 0.310304 | 0.0106 | 1.33 (1.07–1.65) | Germany | [51] |
| rs4711374 | T | - | 0.301717 | 0.0266 | - | |||||
| rs1466650 | T | - | 0.404153 | 0.042 | - | |||||
| rs11753413 | T | - | - | 0.0294 | - | |||||
| rs2029461 | G | - | 0.402157 | 0.0204 | - | |||||
| rs2029461 | AG | - | 0.402157 | 0.005 | 1.641 (1.238–2.175) | India | [108] | |||
| rs2029461-rs2451334-rs745501-rs2499697-rs937039 | ACAAA | - | 0.402157, 0.163339, 0.330471, 0.0784747 0.257987 | <0.0001 | 0.4907 (0.3475–0.6927) | |||||
| ACACA | - | 0.00047 | 2.5490 (1.5119–4.2973) | |||||||
| ATAAG | - | <0.0001 | 4.8533 (2.2672–10.3895) | |||||||
| GCACA | - | 0.004525 | 0.4125 (0.227–0.7495) | |||||||
| 56 | KCNJ10 | Potassium channel gene | rs1130183 | T | R271C | 0.014776 | 0.011 | 0.59 (0.37–0.95) | Germany | [85] |
| 57 | KCNQ2 | Potassium channel gene | rs1801545 | C | - | 0.069286 | 0.022 | - | Germany | [74] |
| 58 | hSKCa3 | Potassium channel gene | polyglutamine CAG tract | CAG16 | - | - | 0.018 | 1.198 | South India | [77] |
| CAG18 | - | - | 0.019 | 1.178 | ||||||
| CAG19 | - | - | <0.00001 | 0.514 | ||||||
| 59 | HLA | HLA complex | HLA-DRB1*1301-1302 | DRB1* 1301-1302 | - | - | <0.017 | 6.6 | USA | [109] |
| HLA-DQB1*0603-0604 | DQB1* 0603-0604 | - | - | <0.005 | 13.8 | |||||
| HLA-DQ1 | DQ1 | - | - | <0.01 | - | Japan | [110] | |||
| HLA-DQ3 | DQ3 | - | - | <0.02 | - | |||||
| HLA_DRW13 | DRW13 | - | - | 0.002 | 4.85 (1.70–14.0) | Saudi Arabia | [111] | |||
| 60 | SLC2A1 | Transporter | rs387907313 | T | R232C | 0.000008 | - | - | Italy | [93] |
| 61 | TAP-1 | Transporter | 333Val-637Asp | - | 333V-637D | - | 0.007 | 2.61 (1.27–5.33) | France | [112] |
| Transporter | 333Ile-637Gly | - | 333I-637G | - | 0.02 | 2.30 (1.11–4.77) | ||||
| Transporter | 333Val-637Asp | - | 333V-637D | - | 0.04 | 0.4 | Tunisia | [92] | ||
| Transporter | 333Ile-637Asp | - | 333I-637D | - | 0.03 | 6.36 | ||||
| 62 | STX1B | Membrane trafficking | rs1046276 | T | - | 0.411342 | 3 × 10–11 | - | Finland, USA, Australia, Canada, Belgium, UK, Republic of Ireland, China, Hong Kong, Canada | [13] |
| JAE | ||||||||||
| 63 | INHA | - | rs7588807 | G | - | 0.4722 | - | - | Turkey | [113] |
| TLE | ||||||||||
| 64 | APOE | Apolipoproteins | ApoE-epsilon-4 | epsilon4 | - | - | 0.004 | - | Australia | [114] |
| ApoE-epsilon-4 | epsilon4 | - | - | >0.05 | 1.06 (0.38–2.95) | Turkish | [115] | |||
| 65 | ASC1a | Sodium channel | rs844347 | A | - | 0.224641 | 0.004 | 1.516 (1.142–2.013) | China | [116] |
| rs844347 | A | - | 0.224641 | 0.002 | 1.628 (1.193–2.222) | |||||
| 66 | ALDH5A1 | Enzyme | rs1883415 | C | - | 0.289537 | 0.0019 | - | Germany | [117] |
| 67 | AQP4 | Water channel | ss119336753, ss119336754 and rs1058424 | - | - | 0.228834 | <0.05 | - | Norway | [118] |
| 68 | BDNF | Nucleic acid binding | rs6265 | A | V66M (M66 protective) | 0.201278 | 0.012 | 1.21 (1.04–1.41) | China | [119] |
| rs6265 | A | V66M (M66 protective) | 0.201278 | 0.636 | 0.636 | Brazil (Caucasian, African, African descent, Asian) | [120] | |||
| rs6265 | A | V66M (M66 protective) | 0.201278 | 0.355 | - | Europe | [121] | |||
| C240T | T | S80I | - | 0.022 | - | Japan | [13] | |||
| 69 | CALHM1 | Calcium channel | rs2986017 | A | - | 0.128 | 0.072 | 1.37 (0.96–1.96) | China | [122] |
| rs11191692 | A | - | 0.298522 | 0.003 | 1.35 (1.103–1.653) | China | [123] | |||
| rs11191692–rs729211–rs2986016–rs2986017 | G, G, G, T | - | 0.298522, 0.363618, 0.119808, 0.127995 | 0.0029 | 2.09 (1.27–3.42) | |||||
| rs11191692–rs729211–rs2986016–rs2986017 | G, A, G, C | - | 0.298522, 0.363618, 0.119808, 0.127995 | 0.0106 | 0.7 (0.53–0.92) | |||||
| 70 | C3 | Immune | Dinucleotide (CA) repeat | - | - | - | <0.05 | - | Spain | [124] |
| 71 | CPA6 | Enzyme | rs114402678 | T | A270V | 0.00359 | - | - | Morocco | [125] |
| rs61738009 | A | G267R | 0.001398 | - | - | |||||
| 72 | GABBR1 | GABA A receptor | G1465A | A/G | - | - | <000.1 | - | Italy | [126] |
| G1465A (with drug-resistant TLE) | A | - | - | 0.003 | 6.47 (2.02–20.76) | |||||
| G1465A | A/G | - | - | <0.0001 | 10.01 (3.98–25.18) | Argentina | [127] | |||
| 73 | GABRB3 | GABA A receptor | rs4906902 | - | - | 0.199681 | 0.5498 | Germany | [117] | |
| 74 | GABBR2 | GABA A receptor | rs967932 | A | - | 0.157149 | 0.018 | 1.305 (1.048–1.624) | China | [128] |
| 75 | GAL | Galanin | C116A | - | A39E | - | - | - | Geneva | [129] |
| 76 | 5-HTR2A | Serotonin receptor | rs6314 | T | - | 0.074681 | 0.006 | - | Italy | [130] |
| 77 | 5-HTTVNTR | Serotonin receptor | 10-repeat allele | - | - | - | 0.0187 | 1.55 (1.07–2.26) | China | [131] |
| 78 | 5-HT1A | Serotonin receptor | rs6295 | C | - | 0.45 | 0.048 | 2.77 (1.01–7.63) | Brazil | [132] |
| 79 | 5HT-1B | Serotonin receptor | rs6296/G861C | G | - | 0.33 | 0.0385 | 1.574 (1.031–2.402) | Croatia | [129] |
| 80 | 5-HTTVNTR | Serotonin receptor | - | - | - | - | 0.0145 | RR= 0.21 (0.07–0.65) | Croatia | [133] |
| 81 | IL-1α | Signaling molecule | rs1800587/IL-1α–889 allele | A | - | 0.278 | 0.018 | - | Europe (Caucasian) | [134] |
| 82 | KEAP1 | Kelch-like ECH-associated protein 1 | rs1048290 | G | - | 0.491813 | 0.04 | 0.41 (0.20–0.84) | China | [135] |
| 83 | KCNAB1 | Potassium channel gene | rs2280032 | A | - | 0.479233 | 0.028 | 1.84 (1.07–3.18) | Italy | [136] |
| Potassium channel gene | rs992353 | C | - | 0.459265 | 0.0058 | 2.25 (1.26–4.04) | ||||
| 84 | KCNAB2 | Potassium channel gene | rs1386956-rs9816126-rs728382-rs4679773-rs9876870-rs3755631-rs2280561-rs1546750-rs17352408-rs2720281-rs429513-rs2280299-rs2280032-rs992353 | GGGGCCTGGGGTTG | 0.30611, 0.344249, 0.401358, 0.483826, 0.421326, 0.216653, 0.433706, 0.382987, 0.228634, 0.464058, 0.307308, 0.479233, 0.459265 | 0.028 | 12.24 (1.32–113.05) | Italy | [136] | |
| 85 | KCNJ10 | Potassium channel gene | rs17375748 | - | - | 0.024 | 0.025 | - | Norway | [118] |
| rs1186685 | - | - | 0.1829 | 0.009 | - | |||||
| rs4656873 | - | - | 0.1787 | 0.019 | - | |||||
| rs1186679 | - | - | 0.1759 | 0.021 | - | |||||
| rs1890532 | - | - | 0.1905 | 0.041 | - | |||||
| rs946420 | - | - | 0.1771 | 0.024 | - | |||||
| rs2820585 | - | - | 0.1771 | 0.02 | - | |||||
| 86 | NTRK2 | Receptor | rs10868235 | T | - | 0.351837 | 0.01 | 1.9 (1.17–3.09) | Brazil | [137] |
| 87 | NRG1 | Signaling molecule | rs35753505 | C | - | - | 0.026 | 0.082 (0.082 (0.009–0.746)) | China | [138] |
| 88 | NFE2L2 | Basic leucine zipper (bZIP) proteins | rs7557529–rs35652124–rs6706649–rs6721961–rs2886161– rs1806649–rs2001350–rs10183914–rs2706110 | A, A, G, C, A, G, A, G, G | - | 0.395168, 0.375599, 0.1451, 0.063299, 0.33631,0.105232, 0.128, 0.23, 0.331669 | 0.03 | 7.11 (1.53–32.98) | China | [135] |
| rs2706110 | A | - | 0.331669 | 0.03 | 1.95 (1.06–3.58) | |||||
| 89 | PDYN | Signaling molecule | - | L-allele | - | - | 0.005 | - | Austria | [139] |
| - | L-allele | - | - | 0.061 | - | Italy | [140] | |||
| - | L- allele | - | - | 0.163 | 1.6 (0.82–3.31) | Europe (Caucasian) | [134] | |||
| 90 | PRNP | Prion protein | rs1799990 | G | M129V | 0.266 | 0.021 | 2.527 (1.11–5.75) | Italy | [141] |
| Asn171Ser | - | N171S | <0.0001 | Brazil | [107] | |||||
| 91 | TLR4 | Receptor | rs4986790 | G | - | 0.059904 | 0.512 | 1.964 (0.176–21.90) | Europe (Caucasian) | [142] |
| 92 | t-PA | Tissue plasminogen activator | rs2020918 | T | - | - | 0.006 | 2.008 (1.223–3.298) | China | [143] |
| rs4646972 | 311 bp deletion | - | - | 0.000 | 2.007 (1.418–2.840) | China | ||||
| 93 | SCN1A | Sodium channel gene | rs7587026 | A | - | 0.212061 | 4 × 10–8 | 1.24 (1.15–1.34) | Finland, USA, Belgium, UK, Switzerland, Austria, Republic of Ireland, Australia, Italy, the Netherlands, Portugal, Germany | [144] |
| rs3812718 | T | - | 0.493411 | 0.0001 | 1.67 (1.28–2.16) | South India | [76] | |||
Signal Transduction Molecule
Nucleic Acid Binding
GABA A Receptor
2.1.3. Juvenile Absence Epilepsy (JAE) and Epilepsy with Generalized Tonic–Clonic Seizures (EGTCS)
2.2. Temporal Lobe Epilepsy (TLE)
2.2.1. Sodium Ion Channels
2.2.2. Calcium Homeostasis Modulator 1 (CALHM1)
2.2.3. γ- Aminobutyric Acid B Receptor 1 (GABBR1)
2.2.4. Aquaporin
2.2.5. Serotonin Transporter (5-HTT)
2.2.6. Prodynorphin (PDYN)
2.2.7. Acid-Sensing Ion Channel Subunit 1 (ASIC1)
2.2.8. Apolipoprotein E (ApoE)
2.2.9. Neurotrophic Receptor Tyrosine Kinase 2 (NTRK2)
2.3. Cryptogenic Focal Epilepsy (CFE)
3. Genetic Burden of Rare and Common Variants in Common Epilepsies
4. Copy Number Variants (CNVs) in Epilepsy
5. Clinical Implication and Relevance of Genetic Findings
5.1. Prognosis
5.2. Diagnosis
5.3. Pharmacogenomics
6. Conclusions and Future Direction
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Epilepsy Factsheet. WHO Webpage. 2020. Available online: https://www.who.int/news-room/fact-sheets/detail/epilepsy (accessed on 16 October 2020).
- Beghi, E. The Epidemiology of Epilepsy. Neuroepidemiology 2019, 54, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G. Rare and common variants: Twenty arguments. Nat. Rev. Genet 2011, 13, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Buono, R.J. Genome wide association studies (GWAS) and common forms of human epilepsy. Epilepsy Behav. 2013, 28, S63–S65. [Google Scholar] [CrossRef]
- Weber, Y.G.; Lerche, H. Genetic mechanisms in idiopathic epilepsies. Dev. Med. Child Neurol. 2008, 50, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Prasad, D.K.V.; Satyanarayana, U.; Munshi, A. Genetics of idiopathic generalized epilepsy: An overview. Neurol. India 2013, 61, 572. [Google Scholar] [CrossRef]
- Lennox, W.G. The heredity of epilepsy as told by relatives and twins. J. Am. Med. Assoc. 1951, 146, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Myers, C.T.; Mefford, H.C. Advancing epilepsy genetics in the genomic era. Genome Med. 2015, 7, 91. [Google Scholar] [CrossRef]
- Alhusaini, S.; Whelan, C.D.; Sisodiya, S.M.; Thompson, P.M. Quantitative magnetic resonance imaging traits as endophenotypes for genetic mapping in epilepsy. Neuroimage Clin. 2016, 12, 526–534. [Google Scholar] [CrossRef][Green Version]
- Gottesman, I.I.; Gould, T.D. The Endophenotype Concept in Psychiatry: Etymology and Strategic Intentions. Am. J. Psychiatry 2003, 160, 636–645. [Google Scholar] [CrossRef]
- Kasperavičiūtė, D.; Catarino, C.B.; Heinzen, E.L.; Depondt, C.; Cavalleri, G.L.; Caboclo, L.O.; Tate, S.K.; Jamnadas-Khoda, J.; Chinthapalli, K.; Clayton, L.M.; et al. Common genetic variation and susceptibility to partial epilepsies: A genome-wide association study. Brain 2010, 133, 2136–2147. [Google Scholar] [CrossRef]
- Guo, Y.; Baum, L.W.; Sham, P.C.; Wong, V.; Ng, P.W.; Lui, C.H.T.; Sin, N.C.; Tsoi, T.H.; Tang, C.S.; Kwan, J.S.; et al. Two-stage genome-wide association study identifies variants in CAMSAP1L1 as susceptibility loci for epilepsy in Chinese. Hum. Mol. Genet. 2011, 21, 1184–1189. [Google Scholar] [CrossRef] [PubMed]
- The International League against Epilepsy Consortium on Complex Epilepsies Genome-wide mega-analysis identifies 16 loci and highlights diverse biological mechanisms in the common epilepsies. Nat. Commun. 2018, 9, 5269. [CrossRef] [PubMed]
- Scheffer, I.E.; Berkovic, S.; Meletti, S.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Commission on Classification and Terminology of the International League against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Commission on Classification and Terminology of the International League against Epilepsy. Epilepsia 1989, 30, 389–399. [Google Scholar] [CrossRef]
- Wallace, R.H.; Wang, D.W.; Singh, R.; Scheffer, I.E., Jr.; George, A.L.; Phillips, H.A.; Saar, K.; Reis, A.; Johnson, E.W.; Sutherland, G.R.; et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel ß1 subunit gene SCN1B. Nat. Genet. 1998, 19, 366–370. [Google Scholar] [CrossRef]
- McNamara, J.O. Emerging insights into the genesis of epilepsy. Nature 1999, 399, A15–A22. [Google Scholar] [CrossRef] [PubMed]
- Steinlein, O.K. Genetic mechanisms that underlie epilepsy. Nat. Rev. Neurosci. 2004, 5, 400–408. [Google Scholar] [CrossRef]
- Staley, K. Molecular mechanisms of epilepsy. Nat. Neurosci. 2015, 18, 367–372. [Google Scholar] [CrossRef]
- Mullen, S.A.; Berkovic, S.F.; Commission, T.I.G. Genetic generalized epilepsies. Epilepsia 2018, 59, 1148–1153. [Google Scholar] [CrossRef]
- Sander, T.; Schulz, H.; Saar, K.; Gennaro, E.; Riggio, M.C.; Bianchi, A.; Zara, F.; Luna, D.; Bulteau, C.; Kaminska, A.; et al. Genome search for susceptibility loci of common idiopathic generalised epilepsies. Hum. Mol. Genet. 2000, 9, 1465–1472. [Google Scholar] [CrossRef]
- Crunelli, V.; LeResche, N. Childhood absence epilepsy: Genes, channels, neurons and networks. Nat. Rev. Neurosci. 2002, 3, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Foundation, E. Childhood Absence Epilepsy. Available online: https://www.epilepsy.com/learn/types-epilepsy-syndromes/childhood-absence-epilepsy (accessed on 16 October 2020).
- Özlem, Y. Genes and molecular mechanisms involved in the epileptogenesis of idiopathic absence epilepsies. Seizure 2012, 21, 79–86. [Google Scholar]
- Liang, J.; Zhang, Y.; Chen, Y.; Wang, J.; Pan, H.; Wu, H.; Xu, K.; Liu, X.; Jiang, Y.; Shen, Y.; et al. Common Polymorphisms in theCACNA1HGene Associated with Childhood Absence Epilepsy in Chinese Han Population. Ann. Hum. Genet. 2006, 71, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Everett, K.; Chioza, B.A.; Aicardi, J.; Aschauer, H.N.; Brouwer, O.; Callenbach, P.M.; Covanis, A.; Dooley, J.; Dulac, O.; Durner, M.; et al. Linkage and mutational analysis of CLCN2 in childhood absence epilepsy. Epilepsy Res. 2007, 75, 145–153. [Google Scholar] [CrossRef]
- Oyrer, J.; Maljevic, S.; Scheffer, I.E.; Berkovic, S.F.; Petrou, S.; Reid, C.A. Ion Channels in Genetic Epilepsy: From Genes and Mechanisms to Disease-Targeted Therapies. Pharm. Rev. 2017, 70, 142–173. [Google Scholar] [CrossRef]
- Alexander, S.P.; Fabbro, D.; Kelly, E.; Marrion, N.; Peters, J.A.; Benson, H.E.; Faccenda, E.; Pawson, A.J.; Sharman, J.L.; Southan, C.; et al. The Concise Guide to Pharmacology 2015/16: Enzymes. Br. J. Pharm. 2015, 172, 6024–6109. [Google Scholar] [CrossRef]
- Feng, T.; Kalyaanamoorthy, S.; Barakat, K. L-Type Calcium Channels: Structure and Functions. Ion Channels Health Sick. 2018, 77305. [Google Scholar] [CrossRef]
- Chen, Y.; Lu, J.; Pan, H.; Zhang, Y.; Wu, H.; Xu, K.; Liu, X.; Jiang, Y.; Bao, X.; Yao, Z.; et al. Association between genetic variation ofCACNA1H and childhood absence epilepsy. Ann. Neurol. 2003, 54, 239–243. [Google Scholar] [CrossRef]
- Liang, J.; Zhang, Y.; Wang, J.; Pan, H.; Wu, H.; Xu, K.; Liu, X.; Jiang, Y.; Shen, Y.; Wu, X.-R. New variants in the CACNA1H gene identified in childhood absence epilepsy. Neurosci. Lett. 2006, 406, 27–32. [Google Scholar] [CrossRef]
- Khosravani, H.; Altier, C.; Simms, B.; Hamming, K.S.; Snutch, T.P.; Mezeyova, J.; McRory, J.E.; Zamponi, G.W. Gating Effects of Mutations in the Cav3.2 T-type Calcium Channel Associated with Childhood Absence Epilepsy. J. Biol. Chem. 2004, 279, 9681–9684. [Google Scholar] [CrossRef]
- Meldrum, B.S. The role of glutamate in epilepsy and other CNS disorders. Neurology 1994, 44, 14–23. [Google Scholar]
- Rogawski, M.A.; Donevan, S.D. AMPA receptors in epilepsy and as targets for antiepileptic drugs. Adv. Neurol. 1999, 79, 947–963. [Google Scholar] [PubMed]
- Rogawski, M.A. AMPA receptors as a molecular target in epilepsy therapy. Acta Neurol. Scand. Suppl. 2013, 127, 9–18. [Google Scholar] [CrossRef]
- Gallagher, M.J.; Song, L.; Arain, F.; Macdonald, R.L. The Juvenile Myoclonic Epilepsy GABAA Receptor 1 Subunit Mutation A322D Produces Asymmetrical, Subunit Position-Dependent Reduction of Heterozygous Receptor Currents and 1 Subunit Protein Expression. J. Neurosci. 2004, 24, 5570–5578. [Google Scholar] [CrossRef] [PubMed]
- Ewatanabe, M.; Efukuda, A. Development and regulation of chloride homeostasis in the central nervous system. Front. Cell. Neurosci. 2015, 9, 371. [Google Scholar] [CrossRef] [PubMed]
- Bowser, D.N.; Wagner, D.A.; Czajkowski, C.; Cromer, B.A.; Parker, M.W.; Wallace, R.H.; Harkin, L.A.; Mulley, J.C.; Marini, C.; Berkovic, S.F.; et al. Altered kinetics and benzodiazepine sensitivity of a GABAA receptor subunit mutation [ 2(R43Q)] found in human epilepsy. Proc. Natl. Acad. Sci. USA 2002, 99, 15170–15175. [Google Scholar] [CrossRef]
- Wallace, R.H.; Marini, C.; Petrou, S.; Harkin, L.A.; Bowser, D.N.; Panchal, R.G.; Williams, D.A.; Sutherland, G.R.; Mulley, J.C.; Scheffer, I.E.; et al. Mutant GABA A receptor γ2-subunit in childhood absence epilepsy and febrile seizures. Nat. Genet. 2001, 28, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Sancar, F.; Czajkowski, C. A GABAA receptor mutation linked to human epilepsy (γ2R43Q) impairs cell surface expression of αβγ receptors. J. Biol. Chem. 2004, 279, 47034–47039. [Google Scholar] [CrossRef]
- Kang, J.; Macdonald, R.L. The GABAA Receptor 2 Subunit R43Q Mutation Linked to Childhood Absence Epilepsy and Febrile Seizures Causes Retention of 1 2 2S Receptors in the Endoplasmic Reticulum. J. Neurosci. 2004, 24, 8672–8677. [Google Scholar] [CrossRef]
- Frugier, G.; Coussen, F.; Emerit, M.B.; Garret, M.; Giraud, M.-F.; Odessa, M.-F.; Boué-Grabot, É. A γ2(R43Q) Mutation, Linked to Epilepsy in Humans, Alters GABAA Receptor Assembly and Modifies Subunit Composition on the Cell Surface*. J. Biol. Chem. 2006, 282, 3819–3828. [Google Scholar] [CrossRef]
- Chaumont, S.; André, C.; Perrais, D.; Boué-Grabot, É.; Taly, A.; Garret, M. Agonist-dependent Endocytosis of γ-Aminobutyric Acid Type A (GABAA) Receptors Revealed by a γ2(R43Q) Epilepsy Mutation. J. Biol. Chem. 2013, 288, 28254–28265. [Google Scholar] [CrossRef] [PubMed]
- Feucht, M.; Fuchs, K.; Pichlbauer, E.; Hornik, K.; Scharfetter, J.; Goessler, R.; Füreder, T.; Cvetkovic, N.; Sieghart, W.; Kasper, S.; et al. Possible association between childhood absence epilepsy and the gene encoding GABRB3. Biol. Psychiatry 1999, 46, 997–1002. [Google Scholar] [CrossRef]
- Urak, L.; Feucht, M.; Fathi, N.; Hornik, K.; Fuchs, K. A GABRB3 promoter haplotype associated with childhood absence epilepsy impairs transcriptional activity. Hum. Mol. Genet. 2006, 15, 2533–2541. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Olsen, R.W.; Medina, M.T.; Schwartz, E.; Alonso, M.E.; Durón, R.M.; Castro-Ortega, R.; Martinez-Juarez, I.E.; Pascual-Castroviejo, I.; Machado-Salas, J.; et al. Hyperglycosylation and Reduced GABA Currents of Mutated GABRB3 Polypeptide in Remitting Childhood Absence Epilepsy. Am. J. Hum. Genet. 2008, 82, 1249–1261. [Google Scholar] [CrossRef]
- Delahanty, R.J.; Kang, J.Q.; Brune, C.W.; Kistner, E.O.; Courchesne, E.; Cox, N.J.; Cook, E.H.; Macdonald, R.L.; Sutcliffe, J.S. Maternal transmission of a rare GABRB3 signal peptide variant is associated with autism. Mol. Psychiatry 2009, 16, 86–96. [Google Scholar] [CrossRef]
- Thomsen, C.; Dalby, N.O. Roles of metabotropic glutamate receptor subtypes in modulation of pentylenetetrazole-induced seizure activity in mice. Neuropharmacology 1998, 37, 1465–1473. [Google Scholar] [CrossRef]
- Ngomba, R.; Ferraguti, F.; Badura, A.; Citraro, R.; Santolini, I.; Battaglia, G.; Bruno, V.; De Sarro, G.; Simonyi, A.; Van Luijtelaar, G.; et al. Positive allosteric modulation of metabotropic glutamate 4 (mGlu4) receptors enhances spontaneous and evoked absence seizures. Neuropharmacology 2008, 54, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Snead, O.C.; Banerjee, P.K.; Burnham, M.; Hampson, D. Modulation of Absence Seizures by the GABAA Receptor: A Critical Role for Metabotropic Glutamate Receptor 4 (mGluR4). J. Neurosci. 2000, 20, 6218–6224. [Google Scholar] [CrossRef]
- Muhle, H.; Von Spiczak, S.; Gaus, V.; Kara, S.; Helbig, I.; Hampe, J.; Franke, A.; Weber, Y.; Lerche, H.; Kleefuss-Lie, A.A.; et al. Role of GRM4 in idiopathic generalized epilepsies analysed by genetic association and sequence analysis. Epilepsy Res. 2010, 89, 319–326. [Google Scholar] [CrossRef]
- Steinlein, O.; Sander, T.; Stoodt, J.; Kretz, R.; Janz, D.; Propping, P. Possible association of a silent polymorphism in the neuronal nicotinic acetylcholine receptor subunit α4 with common idiopathic generalized epilepsies. Am. J. Med Genet. 1997, 74, 445–449. [Google Scholar] [CrossRef]
- Vaccarino, A.L.; Olson, G.A.; Olson, R.D.; Kastin, A.J. Endogenous opiates: 1998. Peptides 1999, 20, 1527–1574. [Google Scholar] [CrossRef]
- Lasoń, W.; Przewłocka, B.; Coenen, A.; Przewłocki, R.; Van Luijtelaar, G. Effects of μ and δ opioid receptor agonists and antagonists on absence epilepsy in WAG/Rij rats. Neuropharmacology 1994, 33, 161–166. [Google Scholar] [CrossRef]
- Przewłocka, B.; Lasoń, W.; Turchan, J.; De Bruin, N.M.W.J.; Van Luijtelaar, G.; Przewłocki, R.; Coenen, A. Anatomical and functional aspects of μ opioid receptors in epileptic WAG/Rij rats. Epilepsy Res. 1998, 29, 167–173. [Google Scholar] [CrossRef]
- Dua, A.K.; Pinsky, C.; Labella, F.S. MU- and Delta-opioid receptor-mediated epileptoid responses in morphine-dependent and non-dependent rats. Electroencephalogr. Clin. Neurophysiol. 1985, 61, 569–572. [Google Scholar] [CrossRef]
- Sander, T.; Berlin, W.; Gscheidel, N.; Wendel, B.; Janz, D.; Hoehe, M.R. Genetic variation of the human μ-opioid receptor and susceptibility to idiopathic absence epilepsy. Epilepsy Res. 2000, 39, 57–61. [Google Scholar] [CrossRef]
- Barratt, C.; Lai, T.; Fisniku, L.; Moran, N.; Nashef, L.; Valentin, A.; Asherson, P.; Makoff, A. No Association of Single Nucleotide Polymorphisms in the Opioid Receptor Subunit Gene with Idiopathic Generalized Epilepsy. Epilepsia 2006, 47, 1728–1731. [Google Scholar] [CrossRef]
- Sander, T.; Berlin, W.; Ostapowicz, A.; Samochowiec, J.; Gscheidel, N.; Hoehe, M. Variation of the genes encoding the human glutamate EAAT2, serotonin and dopamine transporters and Susceptibility to idiopathic generalized epilepsy. Epilepsy Res. 2000, 41, 75–81. [Google Scholar] [CrossRef]
- Sander, T.; Harms, H.; Podschus, J.; Finckh, U.; Nickel, B.; Rolfs, A.; Rommelspacher, H.; Schmidt, L.G. Allelic association of a dopamine transporter gene polymorphism in alcohol dependence with withdrawal seizures or delirium. Biol. Psychiatry 1997, 41, 299–304. [Google Scholar] [CrossRef]
- Szot, P.; Reigel, C.E.; White, S.S.; Veith, R.C. Alterations in mRNA expression of systems that regulate neurotransmitter synaptic content in seizure-naive genetically epilepsy-prone rat (GEPR): Transporter proteins and rate-limiting synthesizing enzymes for norepinephrine, dopamine and serotonin. Mol. Brain Res. 1996, 43, 233–245. [Google Scholar] [CrossRef]
- Gu, W.; Sander, T.; Becker, T.; Steinlein, O.K. Genotypic association of exonic LGI4 polymorphisms and childhood absence epilepsy. Neurogenetics 2004, 5, 41–44. [Google Scholar] [CrossRef]
- Genton, P.; Thomas, P.; Trenité, D.G.K.-N.; Medina, M.T.; Salas-Puig, J. Clinical aspects of juvenile myoclonic epilepsy. Epilepsy Behav. 2013, 28, S8–S14. [Google Scholar] [CrossRef] [PubMed]
- Gilsoul, M.; Grisar, T.; Delgado-Escueta, A.V.; De Nijs, L.; Lakaye, B. Subtle Brain Developmental Abnormalities in the Pathogenesis of Juvenile Myoclonic Epilepsy. Front. Cell. Neurosci. 2019, 13, 433. [Google Scholar] [CrossRef] [PubMed]
- Striano, P.; Nobile, C. The genetic basis of juvenile myoclonic epilepsy. Lancet Neurol. 2018, 17, 493–495. [Google Scholar] [CrossRef]
- Delgado-Escueta, A.V. Advances in genetics of juvenile myoclonic epilepsies. Epilepsy Curr. 2007, 7, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, B.P.; Marinho, C.R.M.; Marques, T.E.B.S.; Angelo, L.K.G.; Malta, M.V.D.S.; Duzzioni, M.; De Castro, O.W.; Leite, J.P.; Barbosa, F.T.; Gitaí, D.L.G. Genetic susceptibility in Juvenile Myoclonic Epilepsy: Systematic review of genetic association studies. PLoS ONE 2017, 12, e0179629. [Google Scholar] [CrossRef]
- Medina, M.T.; Duron, R.; Alonso, M.E. Novel mutations in myoclonin1/EFHC1 in families from Honduras, Mexico and Japan. Epilepsia 2005, 46, 156. [Google Scholar]
- Ma, S.; Blair, M.A.; Abou-Khalil, B.; Lagrange, A.H.; Gurnett, C.A.; Hedera, P. Mutations in the GABRA1 and EFHC1 genes are rare in familial juvenile myoclonic epilepsy. Epilepsy Res. 2006, 71, 129–134. [Google Scholar] [CrossRef]
- De Nijs, L.; Wolkoff, N.; Coumans, B.; Delgado-Escueta, A.V.; Grisar, T.; Lakaye, B. Mutations of EFHC1, linked to juvenile myoclonic epilepsy, disrupt radial and tangential migrations during brain development. Hum. Mol. Genet. 2012, 21, 5106–5117. [Google Scholar] [CrossRef] [PubMed]
- Noctor, S.C.; Flint, A.C.; Weissman, T.A.; Dammerman, R.S.; Kriegstein, A.R. Neurons derived from radial glial cells establish radial units in neocortex. Nat. Cell Biol. 2001, 409, 714–720. [Google Scholar] [CrossRef]
- Woermann, F.G.; Free, S.L.; Koepp, M.J.; Sisodiya, S.M.; Duncan, J.S. Abnormal cerebral structure in juvenile myoclonic epilepsy demonstrated with voxel-based analysis of MRI. Brain 1999, 122, 2101–2108. [Google Scholar] [CrossRef] [PubMed]
- De Nijs, L.; Leon, C.; Nguyen, L.; LoTurco, J.J.; Delgado-Escueta, A.V.; Grisar, T.; Lakaye, B. EFHC1 interacts with microtubules to regulate cell division and cortical development. Nat. Neurosci. 2009, 12, 1266–1274. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, B.A.; Waldegger, S.; Heinzinger, J.; Hahn, A.; Kurlemann, G.; Fiedler, B.; Eberhard, F.; Muhle, H.; Stephani, U.; Garkisch, S.; et al. KCNQ2 and KCNQ3 mutations contribute to different idiopathic epilepsy syndromes. Neurology 2008, 71, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Toliat, M.R.; Heils, A.; Leschik, G.; Becker, C.; Rüschendorf, F.; Rohde, K.; Mundlos, S.; Nürnberg, P. Association of the 867Asp variant of the human anion exchanger 3 gene with common subtypes of idiopathic generalized epilepsy. Epilepsy Res. 2002, 51, 249–255. [Google Scholar] [CrossRef]
- Balan, S.; Vellichirammal, N.N.; Banerjee, M.; Radhakrishnan, K. Failure to find association between febrile seizures and SCN1A rs3812718 polymorphism in south Indian patients with mesial temporal lobe epilepsy and hippocampal sclerosis. Epilepsy Res. 2012, 101, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-C.; Chou, I.-C.; Tsai, C.-H.; Wan, L.; Shu, Y.-A.; Tsai, Y.; Lin, C.-C.; Tsai, F.-J. Association of idiopathic generalized epilepsy with polymorphisms in the neuronal nicotinic acetylcholine receptor subunits. J. Clin. Lab. Anal. 2007, 21, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, P.; Rizzo, R.; Gagliano, A.; Palmarino, M.; Volzone, A.; Arpino, C.; Curatolo, P. Association between D18S474 locus on chromosome 18q12 and idiopathic generalized epilepsy. Brain Dev. 2007, 29, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Prasad, D.; Shaheen, U.; Satyanarayana, U.; Prabha, T.S.; Jyothy, A.; Munshi, A. Association of GABRA6 1519 T > C (rs3219151) and Synapsin II (rs37733634) gene polymorphisms with the development of idiopathic generalized epilepsy. Epilepsy Res. 2014, 108, 1267–1273. [Google Scholar] [CrossRef]
- El Ella, S.S.A.; Tawfik, M.A.; El-Fotoh, W.M.M.A.; Soliman, O.A.M. The genetic variant “C588T” of GABARG2 is linked to childhood idiopathic generalized epilepsy and resistance to antiepileptic drugs. Seizure 2018, 60, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Hildmann, T.; Kretz, R.; Fürst, R.; Sailer, U.; Bauer, G.; Schmitz, B.; Beck-Mannagetta, G.; Wienker, T.F.; Janz, D. Allelic association of juvenile absence epilepsy with a GluR5 kainate receptor gene (GRIK1) polymorphism. Am. J. Med Genet. 1997, 74, 416–421. [Google Scholar] [CrossRef]
- Al-Eitan, L.; Al-Dalalah, I.M.; Aljamal, H. Effects of GRM4, SCN2A and SCN3B polymorphisms on antiepileptic drugs responsiveness and epilepsy susceptibility. Saudi Pharm. J. 2019, 27, 731–737. [Google Scholar] [CrossRef]
- Gloria-Bottini, F.; Lucarelli, P.; Saccucci, P.; Cozzoli, E.; Cerminara, C.; Curatolo, P.; Bottini, E. Genetic Polymorphism and Idiopathic Generalized Epilepsy. Evidence of Interaction between Haptoglobin and ACP1 Systems. Neuropediatrics 2008, 39, 357–358. [Google Scholar] [CrossRef] [PubMed]
- Chioza, B.A.; Osei-Lah, A.; Wilkie, H.; Nashef, L.; McCormick, D.; Asherson, P.; Makoff, A. Suggestive evidence for association of two potassium channel genes with different idiopathic generalised epilepsy syndromes. Epilepsy Res. 2002, 52, 107–116. [Google Scholar] [CrossRef]
- Lenzen, K.; Heils, A.; Lorenz, S.; Hempelmann, A.; Hofels, S.; Lohoff, F.; Schmitz, B.; Sander, T. Supportive evidence for an allelic association of the human KCNJ10 potassium channel gene with idiopathic generalized epilepsy. Epilepsy Res. 2005, 63, 113–118. [Google Scholar] [CrossRef]
- Lorenz, S.; Heils, A.; Kasper, J.M.; Sander, T. Allelic association of a truncation mutation of theKCNMB3 gene with idiopathic generalized epilepsy. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2006, 144, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Lakhan, R.; Kalita, J.; Misra, U.; Kumari, R.; Mittal, B. Association of intronic polymorphism rs3773364 A>G in synapsin-2 gene with idiopathic epilepsy. Synapse 2010, 64, 403–408. [Google Scholar] [CrossRef]
- Dean, J.; Robertson, Z.; Reid, V.; Wang, Q.; Hailey, H.; Moore, S.; Rasalam, A.D.; Turnpenny, P.; Lloyd, D.; Shaw, D.; et al. A high frequency of the MTHFR 677C>T polymorphism in Scottish women with epilepsy: Possible role in pathogenesis. Seizure 2008, 17, 269–275. [Google Scholar] [CrossRef][Green Version]
- Epicure, C.; Steffens, M.; Leu, C.; Ruppert, A.-K.; Zara, F.; Striano, S.; Robbiano, A.; Capovilla, G.; Tinuper, P.; Gambardella, A.; et al. Genome-wide association analysis of genetic generalized epilepsies implicates susceptibility loci at 1q43, 2p16.1, 2q22.3 and 17q21.32. Hum. Mol. Genet. 2012, 21, 5359–5372. [Google Scholar] [CrossRef] [PubMed]
- Makoff, A.; Lai, T.; Barratt, C.; Valentin, A.; Moran, N.; Asherson, P.; Nashef, L. High-density SNP screen of sodium channel genes by haplotype tagging and DNA pooling for association with idiopathic generalized epilepsy. Epilepsia 2010, 51, 694–698. [Google Scholar] [CrossRef]
- Kahle, K.T.; Merner, N.D.; Friedel, P.; Silayeva, L.; Liang, B.; Khanna, A.; Shang, Y.; Lachance-Touchette, P.; Bourassa, C.; Levert, A.; et al. Genetically encoded impairment of neuronal KCC 2 cotransporter function in human idiopathic generalized epilepsy. EMBO Rep. 2014, 15, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Layouni, S.; Chouchane, L.; Malafosse, A.; Dogui, M. Dimorphism ofTAP-1gene in Caucasian with juvenile myoclonic epilepsy and in Tunisian with idiopathic generalized epilepsies. Int. J. Immunogenet. 2010, 37, 117–123. [Google Scholar] [CrossRef]
- Striano, P.; Weber, Y.G.; Toliat, M.R.; Schubert, J.; Leu, C.; Chaimana, R.; Baulac, S.; Guerrero, R.; LeGuern, E.; Lehesjoki, A.-E.; et al. GLUT1 mutations are a rare cause of familial idiopathic generalized epilepsy. Neurology 2012, 78, 557–562. [Google Scholar] [CrossRef] [PubMed]
- International League against Epilepsy Consortium on Complex Epilepsies Genetic determinants of common epilepsies: A meta-analysis of genome-wide association studies. Lancet Neurol. 2014, 13, 893–903. [CrossRef]
- Maljevic, S.; Krampfl, K.; Cobilanschi, J.; Tilgen, N.; Beyer, S.; Weber, Y.G.; Schlesinger, F.; Ursu, D.; Melzer, W.; Cossette, P.; et al. A mutation in the GABAAreceptor α1-subunit is associated with absence epilepsy. Ann. Neurol. 2006, 59, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Kananura, C.; Haug, K.; Sander, T.; Runge, U.; Gu, W.; Hallmann, K.; Rebstock, J.; Heils, A.; Steinlein, O.K. A Splice-Site Mutation in GABRG2 Associated With Childhood Absence Epilepsy and Febrile Convulsions. Arch. Neurol. 2002, 59, 1137–1141. [Google Scholar] [CrossRef]
- Moore, T.; Hecquet, S.; McLellann, A.; Ville, D.; Grid, D.; Picard, F.; Moulard, B.; Asherson, P.; Makoff, A.; McCormick, D.; et al. Polymorphism analysis of JRK/JH8, the human homologue of mouse jerky, and description of a rare mutation in a case of CAE evolving to JME. Epilepsy Res. 2001, 46, 157–167. [Google Scholar] [CrossRef]
- Cavalleri, G.L.; Walley, N.M.; Soranzo, N.; Mulley, J.; Doherty, C.P.; Kapoor, A.; Depondt, C.; Lynch, J.M.; Scheffer, I.E.; Heils, A.; et al. A Multicenter Study of BRD2 as a Risk Factor for Juvenile Myoclonic Epilepsy. Epilepsia 2007, 48, 706–712. [Google Scholar] [CrossRef]
- Pal, D.K.; Evgrafov, O.V.; Tabares, P.; Zhang, F.; Durner, M.; Greenberg, D.A. BRD2 (RING3) Is a Probable Major Susceptibility Gene for Common Juvenile Myoclonic Epilepsy. Am. J. Hum. Genet. 2003, 73, 261–270. [Google Scholar] [CrossRef]
- Rozycka, A.; Steinborn, B.; Trzeciak, W.H. The 1674+11C>T polymorphism of CHRNA4 is associated with juvenile myoclonic epilepsy. Seizure 2009, 18, 601–603. [Google Scholar] [CrossRef][Green Version]
- Escayg, A.; De Waard, M.; Lee, D.D.; Bichet, D.; Wolf, P.; Mayer, T.; Johnston, J.; Baloh, R.; Sander, T.; Meisler, M.H. Coding and Noncoding Variation of the Human Calcium-Channel β4-Subunit Gene CACNB4 in Patients with Idiopathic Generalized Epilepsy and Episodic Ataxia. Am. J. Hum. Genet. 2000, 66, 1531–1539. [Google Scholar] [CrossRef]
- Bailey, J.N.; De Nijs, L.; Bai, D.; Suzuki, T.; Miyamoto, H.; Tanaka, M.; Patterson, C.; Lin, Y.-C.; Medina, M.T.; Alonso, M.E.; et al. Variant Intestinal-Cell Kinase in Juvenile Myoclonic Epilepsy. N. Engl. J. Med. 2018, 378, 1018–1028. [Google Scholar] [CrossRef]
- Hempelmann, A.; Heils, A.; Sander, T. Confirmatory evidence for an association of the connexin-36 gene with juvenile myoclonic epilepsy. Epilepsy Res. 2006, 71, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Mas, C.; Taske, N.; Deutsch, S.; Guipponi, M.; Thomas, P.; Covanis, A.; Friis, M.; Kjeldsen, M.J.; Pizzolato, G.P.; Villemure, J.-G.; et al. Association of the connexin36 gene with juvenile myoclonic epilepsy. J. Med. Genet. 2004, 41, e93. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Miyamoto, H.; Nakahari, T.; Inoue, I.; Suemoto, T.; Jiang, B.; Hirota, Y.; Itohara, S.; Saido, T.C.; Tsumoto, T.; et al. Efhc1 deficiency causes spontaneous myoclonus and increased seizure susceptibility. Hum. Mol. Genet. 2009, 18, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Sander, T.; Heils, A.; Lenzen, K.P.; Steinlein, O.K. A new EF-hand containing gene EFHC2 on Xp11.4: Tentative evidence for association with juvenile myoclonic epilepsy. Epilepsy Res. 2005, 66, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Walz, R.; Castro, R.M.; Velasco, T.R.; Alexandre, V.; Lopes, M.H.; Leite, J.P.; Santos, A.C.; Assirati, J.A.; Wichert-Ana, L.; Terra-Bustamante, V.C.; et al. Surgical outcome in mesial temporal sclerosis correlates with prion protein gene variant. Neurology 2003, 61, 1204–1210. [Google Scholar] [CrossRef]
- Parihar, R.; Mishra, R.; Singh, S.K.; Jayalakshmi, S.; Mehndiratta, M.M.; Ganesh, S. Association of the GRM4 gene variants with juvenile myoclonic epilepsy in an Indian population. J. Genet. 2014, 93, 193–197. [Google Scholar] [CrossRef]
- Greenberg, D.A.; Durner, M.; Shinnar, S.; Resor, S.; Rosenbaum, D.; Klotz, I.; Dicker, E.; Keddache, M.; Zhou, G.; Yang, X.; et al. Association of HLA class II alleles in patients with juvenile myoclonic epilepsy compared with patients with other forms of adolescent-onset generalized epilepsy. Neurology 1996, 47, 750–755. [Google Scholar] [CrossRef]
- Yamamoto, S.; Yamamoto, J.; Kotani, K.; Shimizu, A. A study of the association between Japanese juvenile myoclonic epilepsy patients and HLA class II antigens. Psychiatry Clin. Neurosci. 1995, 49, S286–S288. [Google Scholar] [CrossRef]
- Obeid, T.; Rab, M.O.G.; Daif, A.K.; Panayiotopoulos, C.P.; Halim, K.; Bahakim, H.; Bamgboye, E. Is HLA-DRW 13 (W6) Associated with Juvenile Myoclonic Epilepsy in Arab Patients? Epilepsia 1994, 35, 319–321. [Google Scholar] [CrossRef]
- Layouni, S.; Buresi, C.; Thomas, P.; Malafosse, A.; Dogui, M. BRD2 and TAP-1 genes and juvenile myoclonic epilepsy. Neurol. Sci. 2009, 31, 53–56. [Google Scholar] [CrossRef]
- Yalcin, O.; Baykan, B.; Agan, K.; Yapici, Z.; Yalçın, D.; Dizdarer, G.; Turkdogan, D.; Özkara, Ç.; Ünalp, A.; Uluduz, D.; et al. An association analysis at 2q36 reveals a new candidate susceptibility gene for juvenile absence epilepsy and/or absence seizures associated with generalized tonic-clonic seizures. Epilepsia 2011, 52, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Briellmann, R.S.; Torn-Broers, Y.; Busuttil, B.E.; Major, B.J.; Kalnins, R.M.; Olsen, M.; Jackson, G.D.; Frauman, A.G.; Berkovic, S.F. APOE 4 genotype is associated with an earlier onset of chronic temporal lobe epilepsy. Neurol. 2000, 55, 435–437. [Google Scholar] [CrossRef] [PubMed]
- Yeni, S.N.; Ozkara, C.; Buyru, N.; Baykara, O.; Hanoğlu, L.; Karaagac, N.; Ozyurt, E.; Uzan, M. Association between APOE polymorphisms and mesial temporal lobe epilepsy with hippocampal sclerosis. Eur. J. Neurol. 2005, 12, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Lv, R.-J.; He, J.-S.; Fu, Y.-H.; Zhang, Y.-Q.; Shao, X.-Q.; Wu, L.-W.; Lu, Q.; Jin, L.-R.; Liu, H. ASIC1a polymorphism is associated with temporal lobe epilepsy. Epilepsy Res. 2011, 96, 74–80. [Google Scholar] [CrossRef]
- Pernhorst, K.; Raabe, A.; Niehusmann, P.; Van Loo, K.M.; Grote, A.; Hoffmann, P.; Cichon, S.; Sander, T.; Schoch, S.; Becker, A.J. Promoter Variants Determine γ-Aminobutyric Acid Homeostasis-Related Gene Transcription in Human Epileptic Hippocampi. J. Neuropathol. Exp. Neurol. 2011, 70, 1080–1088. [Google Scholar] [CrossRef]
- Heuser, K.; Nagelhus, E.A.; Taubøll, E.; Indahl, U.; Berg, P.R.; Lien, S.; Nakken, S.; Gjerstad, L.; Ottersen, O.P. Variants of the genes encoding AQP4 and Kir4.1 are associated with subgroups of patients with temporal lobe epilepsy. Epilepsy Res. 2010, 88, 55–64. [Google Scholar] [CrossRef]
- Shen, N.; Zhu, X.; Lin, H.; Li, J.; Li, L.; Niu, F.; Liu, A.; Wu, X.; Wang, Y.; Liu, Y. Role of BDNF Val66Met functional polymorphism in temporal lobe epilepsy. Int. J. Neurosci. 2015, 126, 436–441. [Google Scholar] [CrossRef]
- Alcantara, J.A.; Vincentiis, S.; Santos, B.; Kerr, D.; De Paula, V.; Alessi, R.; Linden, H.; Chaim, T.; Serpa, M.H.; Busatto, G.; et al. BDNF Val66Met polymorphism is not related with temporal lobe epilepsy caused by hippocampal sclerosis in Brazilian population. Seizure 2018, 60, 159–162. [Google Scholar] [CrossRef]
- Lohoff, F.W.; Ferraro, T.N.; Dahl, J.P.; Hildebrandt, M.A.; Scattergood, T.M.; O’Connor, M.J.; Sperling, M.R.; Dlugos, D.J.; Berrettini, W.H.; Buono, R.J. Lack of association between variations in the brain-derived neurotrophic factor (BDNF) gene and temporal lobe epilepsy. Epilepsy Res. 2005, 66, 59–62. [Google Scholar] [CrossRef]
- Li, X.; Wang, Y.; Gu, J.; Meng, Q.; Gao, Y.; Zhao, H.; Yin, Z. No association between polymorphisms in the calcium homeostasis modulator 1 gene and mesial temporal lobe epilepsy risk in a Chinese population. Seizure 2014, 23, 231–233. [Google Scholar] [CrossRef]
- Lv, R.-J.; He, J.-S.; Fu, Y.-H.; Shao, X.-Q.; Wu, L.-W.; Lu, Q.; Jin, L.-R.; Liu, H. A polymorphism in CALHM1 is associated with temporal lobe epilepsy. Epilepsy Behav. 2011, 20, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Jamali, S.; Salzmann, A.; Perroud, N.; Ponsole-Lenfant, M.; Cillario, J.; Roll, P.; Roeckel-Trevisiol, N.; Crespel, A.; Balzar, J.; Schlachter, K.; et al. Functional Variant in Complement C3 Gene Promoter and Genetic Susceptibility to Temporal Lobe Epilepsy and Febrile Seizures. PLoS ONE 2010, 5, e12740. [Google Scholar] [CrossRef] [PubMed]
- Salzmann, A.; Guipponi, M.; Lyons, P.J.; Fricker, L.D.; Sapio, M.; Lambercy, C.; Buresi, C.; Bencheikh, B.O.A.; Lahjouji, F.; Ouazzani, R.; et al. Carboxypeptidase A6 gene (CPA6) mutations in a recessive familial form of febrile seizures and temporal lobe epilepsy and in sporadic temporal lobe epilepsy. Hum. Mutat. 2011, 33, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, A.; Manna, I.; Labate, A.; Chifari, R.; La Russa, A.; Serra, P.; Cittadella, R.; Bonavita, S.; Andreoli, V.; Lepiane, E.; et al. GABA(B) receptor 1 polymorphism (G1465A) is associated with temporal lobe epilepsy. Neurology 2003, 60, 560–563. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, M.; Levy, E.M.; Consalvo, D.; Mordoh, J.; Kochen, S. GABABR1 (G1465A) gene variation and temporal lobe epilepsy controversy: New evidence. Seizure 2008, 17, 567–571. [Google Scholar] [CrossRef][Green Version]
- Wang, X.; Sun, W.; Zhu, X.; Li, L.; Wu, X.; Lin, H.; Zhu, S.; Liu, A.; Du, T.; Liu, Y.; et al. Association between the γ-aminobutyric acid type B receptor 1 and 2 gene polymorphisms and mesial temporal lobe epilepsy in a Han Chinese population. Epilepsy Res. 2008, 81, 198–203. [Google Scholar] [CrossRef]
- Guipponi, M.; Chentouf, A.; Webling, K.E.; Freimann, K.; Crespel, A.; Nobile, C.; Lemke, J.R.; Hansen, J.; Dorn, T.; Lesca, G.; et al. Galanin pathogenic mutations in temporal lobe epilepsy. Hum. Mol. Genet. 2015, 24, 3082–3091. [Google Scholar] [CrossRef]
- Manna, I.; Labate, A.; Mumoli, L.; Palamara, G.; Ferlazzo, E.; Aguglia, U.; Quattrone, A.; Gambardella, A. A Functional Genetic Variation of the 5-HTR2A Receptor Affects Age at Onset in Patients with Temporal Lobe Epilepsy. Ann. Hum. Genet. 2012, 76, 277–282. [Google Scholar] [CrossRef]
- Li, J.; Lin, H.; Zhu, X.; Li, L.; Wang, X.; Sun, W.; Wu, X.; Liu, A.; Niu, F.; Wang, Y.; et al. Association study of functional polymorphisms in serotonin transporter gene with temporal lobe epilepsy in Han Chinese population. Eur. J. Neurol. 2011, 19, 351–353. [Google Scholar] [CrossRef]
- Schenkel, L.C.; Bragatti, J.A.; Becker, J.A.; Torres, C.M.; Martin, K.C.; De Souza, A.C.; Manfro, G.G.; Leistner-Segal, S.; Bianchin, M.M. Serotonin gene polymorphisms and psychiatry comorbidities in temporal lobe epilepsy. Epilepsy Res. 2012, 99, 260–266. [Google Scholar] [CrossRef]
- Manna, I.; Labate, A.; Gambardella, A.; Forabosco, P.; La Russa, A.; Le Piane, E.; Aguglia, U.; Quattrone, A. Serotonin transporter gene (5-Htt): Association analysis with temporal lobe epilepsy. Neurosci. Lett. 2007, 421, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Salzmann, A.; Perroud, N.; Crespel, A.; Lambercy, C.; Malafosse, A. Candidate genes for temporal lobe epilepsy: A replication study. Neurol. Sci. 2008, 29, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yin, X.; Liu, L.; Tao, H.; Zhou, H.; Ma, G.; Cui, L.; Li, Y.; Zhang, S.; Xu, Z.; et al. Association of KEAP1 and NFE2L2 polymorphisms with temporal lobe epilepsy and drug resistant epilepsy. Gene 2015, 571, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Busolin, G.; Malacrida, S.; Bisulli, F.; Striano, P.; Di Bonaventura, C.; Egeo, G.; Pasini, E.; Cianci, V.; Ferlazzo, E.; Bianchi, A.; et al. Association of intronic variants of the KCNAB1 gene with lateral temporal epilepsy. Epilepsy Res. 2011, 94, 110–116. [Google Scholar] [CrossRef]
- Torres, C.M.; Siebert, M.; Bock, H.; Mota, S.M.; Krammer, B.R.; Duarte, J.Á.; Bragatti, J.A.; Castan, J.U.; De Castro, L.A.; Saraiva-Pereira, M.L.; et al. NTRK2 (TrkB gene) variants and temporal lobe epilepsy: A genetic association study. Epilepsy Res. 2017, 137, 1–8. [Google Scholar] [CrossRef]
- Zhu, W.-Y.; Jiang, P.; He, X.; Cao, L.-J.; Zhang, L.-H.; Dang, R.-L.; Tang, M.-M.; Xue, Y.; Li, H.-D. Contribution of NRG1 Gene Polymorphisms in Temporal Lobe Epilepsy. J. Child Neurol. 2015, 31, 271–276. [Google Scholar] [CrossRef]
- Stogmann, E.; Zimprich, A.; Baumgartner, C.; Aull-Watschinger, S.; Höllt, V.; Zimprich, F. A functional polymorphism in the prodynorphin gene promotor is associated with temporal lobe epilepsy. Ann. Neurol. 2002, 51, 260–263. [Google Scholar] [CrossRef]
- Bovo, G.; Diani, E.; Bisulli, F.; Di Bonaventura, C.; Striano, P.; Gambardella, A.; Ferlazzo, E.; Egeo, G.; Mecarelli, O.; Elia, M.; et al. Analysis of LGI1 promoter sequence, PDYN and GABBR1 polymorphisms in sporadic and familial lateral temporal lobe epilepsy. Neurosci. Lett. 2008, 436, 23–26. [Google Scholar] [CrossRef]
- Labate, A.; Manna, I.; Gambardella, A.; Le Piane, E.; La Russa, A.; Condino, F.; Cittadella, R.; Aguglia, U.; Quattrone, A. Association between the M129V variant allele of PRNP gene and mild temporal lobe epilepsy in women. Neurosci. Lett. 2007, 421, 1–4. [Google Scholar] [CrossRef]
- Manna, I.; Labate, A.; Mumoli, L.; Ferlazzo, E.; Aguglia, U.; Quattrone, A.; Gambardella, A. No evidence for a role of the coding variant of the Toll-like receptor 4 gene in temporal lobe epilepsy. Seizure 2013, 22, 791–793. [Google Scholar] [CrossRef][Green Version]
- Han, W.; Jiang, P.; Guo, Y.; Xu, P.; Dang, R.; Li, G.; He, X.; Liao, D.; Yan, G. Role of t-PA and PAI-1 variants in temporal lobe epilepsy in Chinese Han population. BMC Neurol. 2019, 19, 13. [Google Scholar] [CrossRef] [PubMed]
- Kasperavičiūtė, D.; Catarino, C.B.; Matarin, M.; Leu, C.; Novy, J.; Tostevin, A.; Leal, B.; Hessel, E.V.S.; Hallmann, K.; Hildebrand, M.S.; et al. Epilepsy, hippocampal sclerosis and febrile seizures linked by common genetic variation around SCN1A. Brain 2013, 136, 3140–3150. [Google Scholar] [CrossRef]
- Suzuki, T.; Delgado-Escueta, A.V.; Aguan, K.; Alonso, M.E.; Shi, J.; Hara, Y.; Nishida, M.; Numata, T.; Medina, M.T.; Takeuchi, T.; et al. Mutations in EFHC1 cause juvenile myoclonic epilepsy. Nat. Genet. 2004, 36, 842–849. [Google Scholar] [CrossRef]
- Loucks, C.M.; Park, K.; Walker, D.S.; McEwan, A.H.; Timbers, T.A.; Ardiel, E.L.; Grundy, L.J.; Li, C.; Johnson, J.-L.; Kennedy, J.; et al. EFHC1, implicated in juvenile myoclonic epilepsy, functions at the cilium and synapse to modulate dopamine signaling. eLife 2019, 8, 37271. [Google Scholar] [CrossRef]
- De Nijs, L.; Lakaye, B.; Coumans, B.; Léon, C.; Ikeda, T.; Delgado-Escueta, A.V.; Grisar, T.; Chanas, G. EFHC1, a protein mutated in juvenile myoclonic epilepsy, associates with the mitotic spindle through its N-terminus. Exp. Cell Res. 2006, 312, 2872–2879. [Google Scholar] [CrossRef]
- Woermann, F.G.; Sisodiya, S.M.; Free, S.L.; Duncan, J.S. Quantitative MRI in patients with idiopathic generalized epilepsy. Evidence of widespread cerebral structural changes. Brain 1998, 121, 1661–1667. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Library. Juvenile Myoclonic Epilepsy: Genes; National Library of Medicine (US), Genetics Home Reference Bethesda (MD), The Library: Bethesda, MD, USA, 2015. [Google Scholar]
- Saegusa, H.; Kurihara, T.; Zong, S.; Minowa, O.; Kazuno, A.-A.; Han, W.; Matsuda, Y.; Yamanaka, H.; Osanai, M.; Noda, T.; et al. Altered pain responses in mice lacking alpha 1E subunit of the voltage-dependent Ca2+ channel. Proc. Natl. Acad. Sci. USA 2000, 97, 6132–6137. [Google Scholar] [CrossRef]
- Kanno, T.; Kanno, Y.; Siegel, R.M.; Jang, M.K.; Lenardo, M.J.; Ozato, K. Selective Recognition of Acetylated Histones by Bromodomain Proteins Visualized in Living Cells. Mol. Cell 2004, 13, 33–43. [Google Scholar] [CrossRef]
- Crowley, T.; Brunori, M.; Rhee, K.; Wang, X.; Wolgemuth, D.J. Change in nuclear-cytoplasmic localization of a double-bromodomain protein during proliferation and differentiation of mouse spinal cord and dorsal root ganglia. Dev. Brain Res. 2004, 149, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Gyuris, A.; Donovan, D.J.; Seymour, K.A.; Lovasco, L.A.; Smilowitz, N.R.; Halperin, A.L.P.; Klysik, J.E.; Freiman, R.N. The chromatin-targeting protein Brd2 is required for neural tube closure and embryogenesis. Biochim. Biophys. Acta (Bba) Bioenerg. 2009, 1789, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Velíšek, L.; Shang, E.; Velíšková, J.; Chachua, T.; Macchiarulo, S.; Maglakelidze, G.; Wolgemuth, D.J.; Greenberg, D.A. GABAergic Neuron Deficit As An Idiopathic Generalized Epilepsy Mechanism: The Role Of BRD2 Haploinsufficiency In Juvenile Myoclonic Epilepsy. PLoS ONE 2011, 6, e23656. [Google Scholar] [CrossRef]
- Pathak, S.; Miller, J.; Morris, E.C.; Stewart, W.C.L.; Greenberg, D.A. DNA methylation of the BRD 2 promoter is associated with juvenile myoclonic epilepsy in Caucasians. Epilepsia 2018, 59, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Schulz, H.; Ruppert, A.; Zara, F.; Madia, F.; Iacomino, M.; Vari, M.S.; Balagura, G.; Minetti, C.; Striano, P.; Bianchi, A.; et al. No evidence for a BRD 2 promoter hypermethylation in blood leukocytes of Europeans with juvenile myoclonic epilepsy. Epilepsia 2019, 60, e31–e36. [Google Scholar] [CrossRef] [PubMed]
- Cossette, P.; Liu, L.; Brisebois, K.; Dong, H.; Lortie, A.; Vanasse, M.; Saint-Hilaire, J.-M.; Carmant, L.; Verner, A.; Lu, W.-Y.; et al. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat. Genet. 2002, 31, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Cossette, P. Channelopathies and juvenile myoclonic epilepsy. Epilepsia 2010, 51, 30–32. [Google Scholar] [CrossRef]
- Gallagher, M.J.; Ding, L.; Maheshwari, A.; Macdonald, R.L. The GABAA receptor 1 subunit epilepsy mutation A322D inhibits transmembrane helix formation and causes proteasomal degradation. Proc. Natl. Acad. Sci. USA 2007, 104, 12999–13004. [Google Scholar] [CrossRef]
- Ding, L.; Feng, H.; Macdonald, R.L.; Botzolakis, E.J.; Hu, N.; Gallagher, M.J. GABAA receptor α1 subunit mutation A322D associated with autosomal dominant juvenile myoclonic epilepsy reduces the expression and alters the composition of wild type GABAA receptors. J. Biol. 2010, 285, 26390–26405. [Google Scholar]
- Macdonald, R.L.; Gallagher, M.J.; Feng, H.-J.; Kang, J. GABAA receptor epilepsy mutations. Biochem. Pharm. 2004, 68, 1497–1506. [Google Scholar] [CrossRef]
- Dibbens, L.M.; Feng, H.-J.; Richards, M.C.; Harkin, L.A.; Hodgson, B.L.; Scott, D.; Jenkins, M.; Petrou, S.; Sutherland, G.R.; Scheffer, I.E.; et al. GABRD encoding a protein for extra or peri-synaptic GABAA receptors is a susceptibility locus for generalized epilepsies. Hum. Mol. Genet. 2004, 13, 1315–1319. [Google Scholar] [CrossRef]
- Bhat, M.A.; Guru, S.A.; Mir, R.; Waza, A.A.; Zuberi, M.; Sumi, M.P.; Bodeliwala, S.; Puri, V.; Saxena, A. Association of GABAA Receptor Gene with Epilepsy Syndromes. J. Mol. Neurosci. 2018, 65, 141–153. [Google Scholar] [CrossRef]
- Lenzen, K.P.; Heils, A.; Lorenz, S.; Hempelmann, A.; Sander, T. Association analysis of the Arg220His variation of the human gene encoding the GABA δ subunit with idiopathic generalized epilepsy. Epilepsy Res. 2005, 65, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Bando, S.Y.; Alegro, M.C.; Amaro, E.; Silva, A.V.; Castro, L.H.M.; Wen, H.-T.; Lima, L.D.A.; Brentani, H.P.; Moreira-Filho, C.A. Hippocampal CA3 Transcriptome Signature Correlates with Initial Precipitating Injury in Refractory Mesial Temporal Lobe Epilepsy. PLoS ONE 2011, 6, e26268. [Google Scholar] [CrossRef] [PubMed]
- Bymaster, F.P.; Carter, P.A.; Yamada, M.; Gomeza, J.; Wess, J.; Hamilton, S.E.; Nathanson, N.M.; McKinzie, D.L.; Felder, C.C. Role of specific muscarinic receptor subtypes in cholinergic parasympathomimetic responses, in vivophosphoinositide hydrolysis, and pilocarpine-induced seizure activity. Eur. J. Neurosci. 2003, 17, 1403–1410. [Google Scholar] [CrossRef]
- Vlaskamp, D.R.; Rump, P.; Callenbach, P.M.; Vos, Y.J.; Sikkema-Raddatz, B.; Van Ravenswaaij-Arts, C.M.; Brouwer, O.F.; Van Ravenswaaij, C.M. Haploinsufficiency of the STX1B gene is associated with myoclonic astatic epilepsy. Eur. J. Paediatr. Neurol. 2016, 20, 489–492. [Google Scholar] [CrossRef]
- Kang, J.Q. Defects at the crossroads of GABAergic signaling in generalized genetic epilepsies. Epilepsy Res. 2017, 137, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Pearl, P.L. Epilepsy syndromes in childhood. Continuum 2018, 24, 186–209. [Google Scholar] [CrossRef] [PubMed]
- Stogmann, E.; Lichtner, P.; Baumgartner, C.; Bonelli, S.; Assem-Hilger, E.; Leutmezer, F.; Schmied, M.; Hotzy, C.; Strom, T.M.; Meitinger, T.; et al. Idiopathic generalized epilepsy phenotypes associated with different EFHC1 mutations. Neurology 2006, 67, 2029–2031. [Google Scholar] [CrossRef] [PubMed]
- Van Luijtelaar, E.; Budziszewska, B.; Jaworska-Feil, L.; Ellis, J.; Coenen, A.; Lasoń, W. The ovarian hormones and absence epilepsy: A long-term EEG study and pharmacological effects in a genetic absence epilepsy model. Epilepsy Res. 2001, 46, 225–239. [Google Scholar] [CrossRef]
- Jenz, D. Epilepsy with grand mal on awakening and sleep-waking cycle. Clin. Neurophysiol. 2000, 111, S103–S110. [Google Scholar] [CrossRef]
- Lee, C.G.; Lee, J.; Lee, M. Multi-gene panel testing in Korean patients with common genetic generalized epilepsy syndromes. PLoS ONE 2018, 13, e0199321. [Google Scholar] [CrossRef]
- Maurer-Morelli, C.V.; Secolin, R.; Morita, M.E.; Domingues, R.R.; Marchesini, R.B.; Santos, N.F.; Kobayashi, E.; Cendes, F.; Lopes-Cendes, I. A Locus Identified on Chromosome18P11.31 is Associated with Hippocampal Abnormalities in a Family with Mesial Temporal Lobe Epilepsy. Front. Neurol. 2012, 3, 124. [Google Scholar] [CrossRef]
- Catterall, W.A. Sodium channel mutations and epilepsy. InJasper’s Basic Mechanisms of the Epilepsies 4th edition 2012. National Center for Biotechnology Information (US). National Center for Biotechnology Information 2012. Available online: https://www.ncbi.nlm.nih.gov/books/NBK98185/ (accessed on 16 October 2020).
- Marban, E.; Yamagishi, T.; Tomaselli, G.F. Structure and function of voltage-gated sodium channels. J. Physiol. 1998, 508, 647–657. [Google Scholar] [CrossRef]
- Bhat, M.A.; Guru, S.A.; Mir, R.; Waza, A.A.; Zuberi, M.; Sumi, M.; Bodeliwala, S.; Samadhiya, A.; Puri, V.; Saxena, A. Role of SCN1A and SCN2A Gene Polymorphisms in Epilepsy Syndromes-A Study from India. J. Neurol. Neurosci. 2018, 9, 338. [Google Scholar] [CrossRef]
- Heinzen, E.L.; Yoon, W.; Tate, S.K.; Sen, A.; Wood, N.; Sisodiya, S.M.; Goldstein, D.B. Nova2 Interacts with a Cis-Acting Polymorphism to Influence the Proportions of Drug-Responsive Splice Variants of SCN1A. Am. J. Hum. Genet. 2007, 80, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Dreses-Werringloer, U.; Vingtdeux, V.; Zhao, H.; Chandakkar, P.; Davies, P.; Marambaud, P. CALHM1 controls the Ca2+-dependent MEK, ERK, RSK and MSK signaling cascade in neurons. J. Cell Sci. 2013, 126, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- DeLorenzo, R.J.; Sun, D.A.; Deshpande, L.S. Erratum to “Cellular mechanisms underlying acquired epilepsy: The calcium hypothesis of the induction and maintenance of epilepsy. Pharmacol. Ther. 2006, 111, 288–325. [Google Scholar] [CrossRef] [PubMed]
- Dreses-Werringloer, U.; Lambert, J.-C.; Vingtdeux, V.; Zhao, H.; Vais, H.; Siebert, A.; Jain, A.; Koppel, J.; Rovelet-Lecrux, A.; Hannequin, D.; et al. A Polymorphism in CALHM1 Influences Ca2+ Homeostasis, Aβ Levels, and Alzheimer’s Disease Risk. Cell 2008, 133, 1149–1161. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J. Epilepsy and Cognitive Impairments in Alzheimer Disease. Arch. Neurol. 2009, 66, 435–440. [Google Scholar] [CrossRef]
- Westmark, C.J.; Westmark, P.R.; Beard, A.M.; Hildebrandt, S.M.; Malter, J.S. Seizure Susceptibility and Mortality in Mice that Over-Express Amyloid Precursor Protein. Int. J. Clin. Exp. Pathol. 2008, 1, 157–168. [Google Scholar]
- MacKenzie, I.R.; Miller, L.A. Senile plaques in temporal lobe epilepsy. Acta Neuropathol. 1994, 87, 504–510. [Google Scholar] [CrossRef]
- Raza, M.; Pal, S.; Rafiq, A.; DeLorenzo, R.J. Long-term alteration of calcium homeostatic mechanisms in the pilocarpine model of temporal lobe epilepsy. Brain Res. 2001, 903, 1–12. [Google Scholar] [CrossRef]
- Minkeviciene, R.; Rheims, S.; Dobszay, M.B.; Zilberter, M.; Hartikainen, J.; Fülöp, L.; Penke, B.; Zilberter, Y.; Harkany, T.; Pitkänen, A.; et al. Amyloid β-Induced Neuronal Hyperexcitability Triggers Progressive Epilepsy. J. Neurosci. 2009, 29, 3453–3462. [Google Scholar] [CrossRef] [PubMed]
- Billinton, A.; Baird, V.H.; Thom, M.; Duncan, J.S.; Upton, N.; Bowery, N.G. GABAB(1) mRNA expression in hippocampal sclerosis associated with human temporal lobe epilepsy. Mol. Brain Res. 2001, 86, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Cavalleri, G.L.; Lynch, J.M.; Depondt, C.; Burley, M.-W.; Wood, N.W.; Sisodiya, S.M.; Goldstein, D.B. Failure to replicate previously reported genetic associations with sporadic temporal lobe epilepsy: Where to from here? Brain 2005, 128, 1832–1840. [Google Scholar] [CrossRef]
- Ma, S.; Abou-Khalil, B.; Sutcliffe, J.S.; Haines, J.L.; Hedera, P. The GABBR1 locus and the G1465A variant is not associated with temporal lobe epilepsy preceded by febrile seizures. BMC Med Genet. 2005, 6, 13. [Google Scholar] [CrossRef]
- Salzmann, A.; Moulard, B.; Crespel, A.; Buresi, C.; Malafosse, A.; Baldy-Moulinier, M. GABAB Receptor 1 Polymorphism (G1465A) and Temporal Lobe Epilepsy. Epilepsia 2005, 46, 931–933. [Google Scholar] [CrossRef]
- Tan, N.C.K.; Heron, S.E.; Scheffer, I.E.; Berkovic, S.F.; Mulley, J.C. Is Variation in the GABA(B) Receptor 1 Gene Associated with Temporal Lobe Epilepsy? Epilepsia 2005, 46, 778–780. [Google Scholar] [CrossRef]
- Stogmann, E.; Zimprich, A.; Baumgartner, C.; Gleiss, A.; Zimprich, F. Lack of Association between a GABAB Receptor 1 Gene Polymorphism and Temporal Lobe Epilepsy. Epilepsia 2006, 47, 437–439. [Google Scholar] [CrossRef]
- Ren, L.; Jin, L.; Zhang, B.; Jia, Y.; Wu, L.; Shen, Y. Lack of GABABR1 gene variation (G1465A) in a Chinese population with temporal lobe epilepsy. Seizure 2005, 14, 611–613. [Google Scholar] [CrossRef]
- Nagelhus, E.A.; Mathiisen, T.; Ottersen, O. Aquaporin-4 in the central nervous system: Cellular and subcellular distribution and coexpression with KIR4.1. Neuroscience 2004, 129, 905–913. [Google Scholar] [CrossRef]
- Amiry-Moghaddam, M.; Williamson, A.; Palomba, M.; Eid, T.; De Lanerolle, N.C.; Nagelhus, E.A.; Adams, M.E.; Froehner, S.C.; Agre, P.; Ottersen, O.P. Delayed K+ clearance associated with aquaporin-4 mislocalization: Phenotypic defects in brains of -syntrophin-null mice. Proc. Natl. Acad. Sci. USA 2003, 100, 13615–13620. [Google Scholar] [CrossRef] [PubMed]
- Binder, D.K.; Yao, X.; Zador, Z.; Sick, T.J.; Verkman, A.S.; Manley, G.T. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia 2006, 53, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.J.; Hsu, M.S.; Seldin, M.M.; Arellano, J.L.; Binder, D.K. Decreased expression of the glial water channel aquaporin-4 in the intrahippocampal kainic acid model of epileptogenesis. Exp. Neurol. 2012, 235, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Binder, D.K.; Nagelhus, E.A.; Ottersen, O.P. Aquaporin-4 and epilepsy. Glia 2012, 60, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Eid, T.; Lee, T.-S.W.; Thomas, M.J.; Amiry-Moghaddam, M.; Bjørnsen, L.P.; Spencer, D.D.; Agre, P.; Ottersen, O.P.; De Lanerolle, N.C. Loss of perivascular aquaporin 4 may underlie deficient water and K+ homeostasis in the human epileptogenic hippocampus. Proc. Natl. Acad. Sci. USA 2005, 102, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Lesch, K.-P.; Mössner, R. Genetically driven variation in serotonin uptake: Is there a link to affective spectrum, neurodevelopmental, and neurodegenerative disorders? Biol. Psychiatry 1998, 44, 179–192. [Google Scholar] [CrossRef]
- Kauffman, M.; Consalvo, D.; González-Morón, D.; Aguirre, F.; D’Alessio, L.; Kochen, S. Serotonin transporter gene variation and refractory mesial temporal epilepsy with hippocampal sclerosis. Epilepsy Res. 2009, 85, 231–234. [Google Scholar] [CrossRef]
- Hrvoje, H.; Jasminka, S.; Lipa, C.-S.; Vida, D.; Branimir, J. Association of serotonin transporter promoter (5-HTTLPR) and intron 2 (VNTR-2) polymorphisms with treatment response in temporal lobe epilepsy. Epilepsy Res. 2010, 91, 35–38. [Google Scholar] [CrossRef]
- Schenkel, L.C.; Bragatti, J.A.; Torres, C.M.; Martin, K.C.; Manfro, G.G.; Leistner-Segal, S.; Bianchin, M.M. Serotonin transporter gene (5HTT) polymorphisms and temporal lobe epilepsy. Epilepsy Res. 2011, 95, 152–157. [Google Scholar] [CrossRef]
- Che, F.; Wei, Y.; Heng, X.; Fu, Q.; Jiang, J. Association between serotonin transporter gene polymorphisms and non-lesional temporal lobe epilepsy in a Chinese Han population★. Neural Regen. Res. 2010, 5, 1270–1273. [Google Scholar]
- Stefulj, J.; Bordukalo-Niksic, T.; Hecimovic, H.; Demarin, V.; Jernej, B. Epilepsy and serotonin (5HT): Variations of 5HT-related genes in temporal lobe epilepsy. Neurosci. Lett. 2010, 478, 29–31. [Google Scholar] [CrossRef] [PubMed]
- Zimprich, A.; Kraus, J.; Wöltje, M.; Mayer, P.; Rauch, E.; Hollt, V. An allelic variation in the human prodynorphin gene promoter alters stimulus-induced expression. J. Neurochem. 2000, 74, 472–477. [Google Scholar] [CrossRef]
- Gambardella, A.; Manna, I.; Labate, A.; Chifari, R.; Serra, P.; La Russa, A.; Lepiane, E.; Cittadella, R.; Andreoli, V.; Sasanelli, F.; et al. Prodynorphin gene promoter polymorphism and temporal lobe epilepsy. Epilepsia 2003, 44, 1255–1256. [Google Scholar] [CrossRef] [PubMed]
- Tilgen, N.; Rebstock, J.; Horvath, S.; Propping, P.; Elger, C.E.; Heils, A. Prodynorphin gene promoter polymorphism and temporal lobe epilepsy. Ann. Neurol. 2003, 53, 280–281. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Ouyang, T.-H.; Zhou, Q.; Kang, H.-C.; Zhu, S. Prodynorphin gene promoter polymorphism and temporal lobe epilepsy: A meta-analysis. Acta Acad. Med. Wuhan 2015, 35, 635–639. [Google Scholar] [CrossRef]
- Waldmann, R.; Champigny, G.; Bassilana, F.; Heurteaux, C.; Lazdunski, M. A proton-gated cation channel involved in acid-sensing. Nat. Cell Biol. 1997, 386, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kalbacher, H.; Gründer, S. The Tarantula Toxin Psalmotoxin 1 Inhibits Acid-sensing Ion Channel (ASIC) 1a by Increasing Its Apparent H+ Affinity. J. Gen. Physiol. 2005, 126, 71–79. [Google Scholar] [CrossRef]
- Wu, H.; Wang, C.; Liu, B.; Li, H.; Zhang, Y.; Dong, S.; Gao, G.; Zhang, H. Altered Expression Pattern of Acid-Sensing Ion Channel Isoforms in Piriform Cortex After Seizures. Mol. Neurobiol. 2015, 53, 1782–1793. [Google Scholar] [CrossRef]
- Yang, F.; Sun, X.; Ding, Y.; Ma, H.; Yang, T.O.; Ma, Y.; Wei, D.; Li, W.; Xu, T.; Jiang, W. Astrocytic acid-sensing ion channel 1a contributes to the development of chronic epileptogenesis. Sci. Rep. 2016, 6, 1–4. [Google Scholar] [CrossRef]
- Ziemann, A.E.; Schnizler, M.K.; Albert, G.W.; Severson, M.A.; Iii, M.A.H.; Welsh, M.J.; Wemmie, J.A. Seizure termination by acidosis depends on ASIC1a. Nat. Neurosci. 2008, 11, 816–822. [Google Scholar] [CrossRef]
- Gambardella, A.; Aguglia, U.; Cittadella, R.; Romeo, N.; Sibilia, G.; Lepiane, E.; Messina, D.; Manna, I.; Oliveri, R.L.; Zappia, M.; et al. Apolipoprotein E polymorphisms and the risk of nonlesional temporal lobe epilepsy. Epilepsia 1999, 40, 1804–1807. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, A.; Aguglia, U.; Chifari, R.; Labate, A.; Manna, I.; Serra, P.; Romeo, N.; Sibilia, G.; Lepiane, E.; La Russa, A.; et al. ApoE Epsilon4 Allele and Disease Duration Affect Verbal Learning in Mild Temporal Lobe Epilepsy. Epilepsia 2005, 46, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, M.; Consalvo, D.; Moron, D.G.; Lereis, V.P.; Kochen, S. ApoE ɛ4 genotype and the age at onset of temporal lobe epilepsy: A case–control study and meta-analysis. Epilepsy Res. 2010, 90, 234–239. [Google Scholar] [CrossRef]
- Busch, R.M.; Lineweaver, T.T.; Naugle, R.I.; Kim, K.H.; Gong, Y.; Tilelli, C.Q.; Prayson, R.A.; Bingaman, W.; Najm, I.M.; Diaz-Arrastia, R. ApoE- 4 is associated with reduced memory in long-standing intractable temporal lobe epilepsy. Neurol. 2007, 68, 409–414. [Google Scholar] [CrossRef]
- Chapin, J.S.; Busch, R.; Janigro, D.; Dougherty, M.; Tilelli, C.Q.; Lineweaver, T.T.; Naugle, R.I.; Diaz-Arrastia, R.; Najm, I.M. APOE ɛ4 is associated with postictal confusion in patients with medically refractory temporal lobe epilepsy. Epilepsy Res. 2008, 81, 220–224. [Google Scholar] [CrossRef][Green Version]
- Kauffman, M.; Pereira-De-Silva, N.; Consalvo, D.; Kochen, S. ApoE ɛ4 is not associated with posictal confusion in patients with mesial temporal lobe epilepsy with hippocampal sclerosis. Epilepsy Res. 2009, 85, 311–313. [Google Scholar] [CrossRef]
- Fu, Y.-H.; Lv, R.-J.; Jin, L.-R.; Lu, Q.; Shao, X.-Q.; He, J.-S.; Wu, L.-W.; Zhang, L.-S.; Hu, H.-G. Association of apolipoprotein E polymorphisms with temporal lobe epilepsy in a Chinese Han population. Epilepsy Res. 2010, 91, 253–259. [Google Scholar] [CrossRef]
- Li, Z.; Ding, C.; Gong, X.; Wang, X.; Cui, T. Apolipoprotein E ε4 Allele was Associated with Nonlesional Mesial Temporal Lobe Epilepsy in Han Chinese Population. Medicine 2016, 95, e2894. [Google Scholar] [CrossRef]
- Polvikoski, T.; Sulkava, R.; Haltia, M.; Kainulainen, K.; Vuorio, A.; Verkkoniemi, A.; Niinistö, L.; Halonen, P.; Kontula, K. Apolipoprotein E, Dementia, and Cortical Deposition of β-Amyloid Protein. N. Engl. J. Med. 1995, 333, 1242–1248. [Google Scholar] [CrossRef]
- Sheng, J.G.; Boop, F.A.; Mrak, R.E.; Griffin, W.S.T. Increased Neuronal β-Amyloid Precursor Protein Expression in Human Temporal Lobe Epilepsy: Association with Interleukin-1α Immunoreactivity. J. Neurochem. 2002, 63, 1872–1879. [Google Scholar] [CrossRef]
- Kodam, A.; Ourdev, D.; Maulik, M.; Hariharakrishnan, J.; Banerjee, M.; Wang, Y.; Kar, S. A role for astrocyte-derived amyloid β peptides in the degeneration of neurons in an animal model of temporal lobe epilepsy. Brain Pathol. 2018, 29, 28–44. [Google Scholar] [CrossRef] [PubMed]
- Nakagawara, A.; Liu, X.-G.; Ikegaki, N.; White, P.S.; Yamashiro, D.J.; Nycum, L.M.; Biegel, J.A.; Brodeur, G.M. Cloning and chromosomal localization of the human TRK-B tyrosine kinase receptor gene (NTRK2). Genomics 1995, 25, 538–546. [Google Scholar] [CrossRef]
- Wirrell, E.C.; Grossardt, B.R.; So, E.L.; Nickels, K.C. A population-based study of long-term outcomes of cryptogenic focal epilepsy in childhood: Cryptogenic epilepsy is probably not symptomatic epilepsy. Epilepsia 2011, 52, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Harkin, L.A.; McMahon, J.M.; Iona, X.; Dibbens, L.; Pelekanos, J.T.; Zuberi, S.M.; Sadleir, L.G.; Andermann, E.; Gill, D.; Farrell, K.; et al. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain 2007, 130, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Zucca, C.; Redaelli, F.; Epifanio, R.; Zanotta, N.; Romeo, A.; Lodi, M.; Veggiotti, P.; Airoldi, G.; Panzeri, C.; Romaniello, R.; et al. Cryptogenic Epileptic Syndromes Related to SCN1A. Arch. Neurol. 2008, 65, 489–494. [Google Scholar] [CrossRef]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nat. Cell Biol. 2009, 461, 747–753. [Google Scholar] [CrossRef]
- Allen, A.S.; Bellows, S.T.; Berkovic, S.F.; Bridgers, J.; Burgess, R.; Cavalleri, G.; Chung, S.-K.; Cossette, P.; Delanty, N.; Dlugos, D.; et al. Ultra-rare genetic variation in common epilepsies: A case-control sequencing study. Lancet Neurol. 2017, 16, 135–143. [Google Scholar] [CrossRef]
- Bennett, C.A.; Petrovski, S.; Oliver, K.L.; Berkovic, S.F. ExACtly zero or once: A clinically helpful guide to assessing genetic variants in mild epilepsies. Neurol. Genet. 2017, 3, 4. [Google Scholar] [CrossRef]
- May, P.; Girard, A.; Harrer, M.; Bobbili, D.R.; Schubert, J.; Wolking, S.; Becker, F.; Lachance-Touchette, P.; Meloche, C.; Gravel, M.; et al. Rare coding variants in genes encoding GABAA receptors in genetic generalised epilepsies: An exome-based case-control study. Lancet Neurol. 2018, 17, 699–708. [Google Scholar] [CrossRef]
- Feng, Y.-C.A.; Howrigan, D.P.; Abbott, L.E.; Tashman, K.; Cerrato, F.; Singh, T.; Heyne, H.; Byrnes, A.; Churchhouse, C.; Watts, N.; et al. Ultra-Rare Genetic Variation in the Epilepsies: A Whole-Exome Sequencing Study of 17,606 Individuals. Am. J. Hum. Genet. 2019, 105, 267–282. [Google Scholar] [CrossRef]
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA 2009, 106, 9362–9367. [Google Scholar] [CrossRef] [PubMed]
- Leu, C.; Stevelink, R.; Smith, A.W.; Goleva, S.B.; Kanai, M.; Ferguson, L.; Campbell, C.; Kamatani, Y.; Okada, Y.; Sisodiya, S.M.; et al. Polygenic burden in focal and generalized epilepsies. Brain 2019, 142, 3473–3481. [Google Scholar] [CrossRef] [PubMed]
- Speed, D.; O’Brien, T.J.; Palotie, A.; Shkura, K.; Marson, A.G.; Balding, D.J.; Johnson, M.R. Describing the genetic architecture of epilepsy through heritability analysis. Brain 2014, 137, 2680–2689. [Google Scholar] [CrossRef] [PubMed]
- Ellis, C.A.; Petrovski, S.; Berkovic, S.F. Epilepsy genetics: Clinical impacts and biological insights. Lancet Neurol. 2020, 19, 93–100. [Google Scholar] [CrossRef]
- Martin, A.R.; Gignoux, C.R.; Walters, R.K.; Wojcik, G.L.; Neale, B.M.; Gravel, S.; Daly, M.J.; Bustamante, C.D.; Kenny, E.E. Human Demographic History Impacts Genetic Risk Prediction across Diverse Populations. Am. J. Hum. Genet. 2017, 100, 635–649. [Google Scholar] [CrossRef]
- Poduri, A.; Sheidley, B.R.; Shostak, S.; Ottman, R. Genetic testing in the epilepsies—developments and dilemmas. Nat. Rev. Neurol. 2014, 10, 293–299. [Google Scholar] [CrossRef]
- Kearney, H.; Byrne, S.; Cavalleri, G.L.; Delanty, N. Tackling Epilepsy with High-definition Precision Medicine. JAMA Neurol. 2019, 76, 1109. [Google Scholar] [CrossRef]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. Q. Publ. Hell. Soc. Gastroenterol. 2015, 28, 203. [Google Scholar]
- Sharp, A.J.; Mefford, H.C.; Li, K.; Baker, C.; Skinner, C.; Stevenson, R.E.; Schroer, R.J.; Novara, F.; De Gregori, M.; Ciccone, R.; et al. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat. Genet. 2008, 40, 322–328. [Google Scholar] [CrossRef]
- Helbig, I.; Mefford, H.C.; Sharp, A.J.; Guipponi, M.; Fichera, M.; Franke, A.; Muhle, H.; De Kovel, C.; Baker, C.; Von Spiczak, S.; et al. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat. Genet. 2009, 41, 160–162. [Google Scholar] [CrossRef]
- Dibbens, L.M.; Mullen, S.; Helbig, I.; Mefford, H.C.; Bayly, M.A.; Bellows, S.; Leu, C.; Trucks, H.; Obermeier, T.; Wittig, M.; et al. Familial and sporadic 15q13.3 microdeletions in idiopathic generalized epilepsy: Precedent for disorders with complex inheritance. Hum. Mol. Genet. 2009, 18, 3626–3631. [Google Scholar] [CrossRef]
- De Kovel, C.G.F.; Trucks, H.; Helbig, I.; Mefford, H.C.; Baker, C.; Leu, C.; Kluck, C.; Muhle, H.; Von Spiczak, S.; Ostertag, P.; et al. Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain 2009, 133, 23–32. [Google Scholar] [CrossRef]
- Mefford, H.C.; Yendle, S.C.; Hsu, C.; Cook, J.; Ba, E.G.; Bsc, J.M.M.; Eeg-Olofsson, O.; Sadleir, L.G.; Gill, D.; Ben-Zeev, B.; et al. Rare copy number variants are an important cause of epileptic encephalopathies. Ann. Neurol. 2011, 70, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Møller, R.S.; Weber, Y.G.; Klitten, L.L.; Trucks, H.; Muhle, H.; Kunz, W.S.; Mefford, H.C.; Franke, A.; Kautza, M.; Wolf, P.; et al. Exon-disrupting deletions ofNRXN1in idiopathic generalized epilepsy. Epilepsia 2013, 54, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Lal, D.; Trucks, H.; Møller, R.S.; Hjalgrim, H.; Koeleman, B.P.; De Kovel, C.G.; Visscher, F.; Weber, Y.G.; Lerche, H.; Becker, F.; et al. Rare exonic deletions of theRBFOX1gene increase risk of idiopathic generalized epilepsy. Epilepsia 2013, 54, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Lionel, A.C.; Vaags, A.K.; Sato, D.; Gazzellone, M.J.; Mitchell, E.B.; Chen, H.Y.; Costain, G.; Walker, S.; Egger, G.; Thiruvahindrapuram, B.; et al. Rare exonic deletions implicate the synaptic organizer Gephyrin (GPHN) in risk for autism, schizophrenia and seizures. Hum. Mol. Genet. 2013, 22, 2055–2066. [Google Scholar] [CrossRef]
- Harrison, V.; Connell, L.; Hayesmoore, J.; McParland, J.; Pike, M.G.; Blair, E. Compound heterozygous deletion of NRXN1 causing severe developmental delay with early onset epilepsy in two sisters. Am. J. Med Genet. Part A 2011, 155, 2826–2831. [Google Scholar] [CrossRef]
- Krepischi, A.C.V.; Knijnenburg, J.; Bertola, D.R.; Kim, C.A.; Pearson, P.L.; Bijlsma, E.; Szuhai, K.; Kok, F.; Vianna-Morgante, A.M.; Rosenberg, C. Two distinct regions in 2q24.2-q24.3 associated with idiopathic epilepsy. Epilepsia 2010, 51, 2457–2460. [Google Scholar] [CrossRef]
- Naseer, M.I.; Faheem, M.; Chaudhary, A.G.; Kumosani, T.A.; AlQuaiti, M.; Jan, M.M.; Jamal, H.S.; Al-Qahtani, M. Genome wide analysis of novel copy number variations duplications/deletions of different epileptic patients in Saudi Arabia. Bmc Genom. 2015, 16, S10. [Google Scholar] [CrossRef]
- Mefford, H.C.; Muhle, H.; Ostertag, P.; Von Spiczak, S.; Buysse, K.; Baker, C.; Franke, A.; Malafosse, A.; Genton, P.; Thomas, P.; et al. Genome-Wide Copy Number Variation in Epilepsy: Novel Susceptibility Loci in Idiopathic Generalized and Focal Epilepsies. PLoS Genet. 2010, 6, e1000962. [Google Scholar] [CrossRef]
- Liu, J.Y.W.; Kasperaviciute, D.; Martinian, L.; Thom, M.; Sisodiya, S. Neuropathology of 16p13.11 Deletion in Epilepsy. PLoS ONE 2012, 7, e34813. [Google Scholar] [CrossRef] [PubMed]
- Dimassi, S.; Labalme, A.; Lesca, G.; Rudolf, G.; Bruneau, N.; Hirsch, E.; Arzimanoglou, A.; Motte, J.; De Saint-Martin, A.; Boutry-Kryza, N.; et al. A subset of genomic alterations detected in rolandic epilepsies contains candidate or known epilepsy genes includingGRIN2AandPRRT2. Epilepsia 2013, 55, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Strehlow, V.; Swinkels, M.E.; Thomas, R.H.; Rapps, N.; Syrbe, S.; Dorn, T.; Lemke, J.R. Generalized Epilepsy and Myoclonic Seizures in 22q11.2 Deletion Syndrome. Mol. Syndr. 2016, 7, 239–246. [Google Scholar] [CrossRef]
- Biervert, C.; Schroeder, B.C.; Kubisch, C.; Berkovic, S.F.; Propping, P.; Jentsch, T.J.; Steinlein, O.K. A Potassium Channel Mutation in Neonatal Human Epilepsy. Science 1998, 279, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.A.; Charlier, C.; Stauffer, D.; Dupont, B.R.; Leach, R.J.; Melis, R.; Ronen, G.M.; Bjerre, I.; Quattlebaum, T.; Murphy, J.V.; et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat. Genet. 1998, 18, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Maljevic, S.; Wuttke, T.; Seebohm, G.; Lerche, H. K V 7 channelopathies. Pflügers Archiv-Eur. J. Physiol. 2010, 460, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Heron, S.E.; Crossland, K.M.; Andermann, E.; Phillips, H.A.; Hall, A.J.; Bleasel, A.; Shevell, M.; Mercho, S.; Seni, M.-H.; Guiot, M.-C.; et al. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet 2002, 360, 851–852. [Google Scholar] [CrossRef]
- Scalmani, P.; Rusconi, R.; Armatura, E.; Zara, F.; Avanzini, G.; Franceschetti, S.; Mantegazza, M. Effects in Neocortical Neurons of Mutations of the Nav1.2 Na+ Channel causing Benign Familial Neonatal-Infantile Seizures. J. Neurosci. 2006, 26, 10100–10109. [Google Scholar] [CrossRef]
- Liao, Y.; Deprez, L.; Maljevic, S.; Pitsch, J.; Claes, L.; Hristova, D.; Jordanova, A.; Ala-Mello, S.; Bellan-Koch, A.; Blazevic, D.; et al. Molecular correlates of age-dependent seizures in an inherited neonatal-infantile epilepsy. Brain 2010, 133, 1403–1414. [Google Scholar] [CrossRef]
- Berkovic, S.F. Genetics of Epilepsy in Clinical Practice: Genetics of Epilepsy in Clinical Practice. Epilepsy Curr. 2015, 15, 192–196. [Google Scholar] [CrossRef]
- Lü, J.-J.; Zhang, Y.; Chen, Y.-C.; Pan, H.; Wang, J.-L.; Zhang, L.; Wu, H.-S.; Xu, K.-M.; Liu, X.-Y.; Tao, L.-D.; et al. T-type calcium channel gene-CACNA1H is a susceptibility gene to childhood absence epilepsy. Zhonghua Chin. J. Pediatr. 2005, 43, 133–136. [Google Scholar]
- Koeleman, B.P.; De Kovel, C.G.; Trenité, D.G.K.-N. Photoparoxysmal EEG response and genetic dissection of juvenile myoclonic epilepsy. Epilepsy Behav. 2013, 28, S69–S71. [Google Scholar] [CrossRef] [PubMed]
- Caciagli, L.; Wandschneider, B.; Ecenteno, M.; Vollmar, C.; Vos, S.B.; Trimmel, K.; Long, L.; Xiao, F.; Lowe, A.J.; Sidhu, M.K.; et al. Motor hyperactivation during cognitive tasks: An endophenotype of juvenile myoclonic epilepsy. Epilepsia 2020, 61, 1438–1452. [Google Scholar] [CrossRef]
- Alhusaini, S.; Whelan, C.D.; Dohertya, C.P.; Delanty, N.; Fitzsimons, M.; Cavalleri, G.L. Temporal Cortex Morphology in Mesial Temporal Lobe Epilepsy Patients and Their Asymptomatic Siblings. Cereb. Cortex 2015, 26, 1234–1241. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, B.; Goldenholz, D.M. Machine learning applications in epilepsy. Epilepsia 2019, 60, 2037–2047. [Google Scholar] [CrossRef]
- Zhang, J.; Cheng, W.; Wang, Z.; Zhang, Z.; Lu, W.; Lu, G.-M.; Feng, J. Pattern Classification of Large-Scale Functional Brain Networks: Identification of Informative Neuroimaging Markers for Epilepsy. PLoS ONE 2012, 7, e36733. [Google Scholar] [CrossRef]
- Stewart, J.D.; Horvath, R.; Baruffini, E.; Ferrero, I.; Bulst, S.; Watkins, P.B.; Fontana, R.J.; Day, C.P.; Chinnery, P.F. Polymerase γ gene POLG determines the risk of sodium valproate-induced liver toxicity. Hepatology 2010, 52, 1791–1796. [Google Scholar] [CrossRef]
- Ihtisham, K.; Ramanujam, B.; Srivastava, S.; Mehra, N.K.; Kaur, G.; Khanna, N.; Jain, S.; Kumar, S.; Kaul, B.; Samudrala, R.; et al. Association of cutaneous adverse drug reactions due to antiepileptic drugs with HLA alleles in a North Indian population. Seizure 2019, 66, 99–103. [Google Scholar] [CrossRef]
- McCormack, M.; Alfirevic, A.; Bourgeois, S.; Farrell, J.J.; Kasperavičiūtė, D.; Carrington, M.; Sills, G.J.; Marson, T.; Jia, X.; de Bakker, P.I.; et al. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N. Engl. J. Med. 2011, 364, 1134–1143. [Google Scholar] [CrossRef]
- Registry, N.-G. Carbamazepine Response. NCBI Genetic Testing Registry. 2014. Available online: https://www.ncbi.nlm.nih.gov/gtr/conditions/CN077964/ (accessed on 16 October 2020).
- Silvado, C.E.; Terra, V.C.; Twardowschy, C.A. CYP2C9 polymorphisms in epilepsy: Influence on phenytoin treatment. Pharm. Pers. Med. 2018, 11, 51–58. [Google Scholar] [CrossRef]
- Chaudhary, N.; Kabra, M.; Gulati, S.; Gupta, Y.K.; Pandey, R.M.; Bhatia, B.D. Frequencies of CYP2C9 polymorphisms in North Indian population and their association with drug levels in children on phenytoin monotherapy. BMC Pediatr. 2016, 16, 66. [Google Scholar] [CrossRef] [PubMed]
- Caudle, K.E.; Rettie, A.E.; Whirl-Carrillo, M.; Smith, L.H.; Mintzer, S.; Lee, M.T.M.; Klein, T.E.; Callaghan, J.T. Clinical Pharmacogenetics Implementation Consortium Guidelines for CYP2C9 and HLA-B Genotypes and Phenytoin Dosing. Clin. Pharm. 2014, 96, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, E.S. Psychiatric issues in epilepsy. Curr Opin Neurol. 2001, 14, 217–224. [Google Scholar] [CrossRef] [PubMed]




| Sl. No. | Locus | Size | CNVs | Gene | Phenotype | Consequences | Country | Year | References | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2p16.3 | ~287 and ~79 kb | Exon-disrupting deletion | Nrxn1 | Severe early onset epilepsy | Alteration in the calcium-dependent release of neurotransmitters | UK | 2011 (case report) | [251] | |
| 2 | 2q24.2-q24.3 | 11Mb | Duplication/deletion | SCN1A, SCN2A, SLC4A10 | Idiopathic epilepsy | Imbalance in sodium channel | Brazil | 2010 (case report) | [252] | |
| 3 | 6p12.1 | 99.9kb | Micro-duplication | BMP5 | Epilepsy | Increased cell death in ventral forebrain | Saudi Arabia | 2015 | [253] | |
| 4 | 7q11.22 | 78.7kb | Deletion | AUTS2 | JME | Role in neurodevelopment | Switzerland, Germany, USA | 2010 | [254] | |
| 5 | 7q32.3 | 63.9kb | Microdeletion | PODXL | Epilepsy | Malignant progression of astrocytic tumors | Saudi Arabia | 2015 | [253] | |
| 6 | 7q35 | 785.8kb | Deletion, hemizygous deletions | CNTNAP2 | GTCS | Affects cell–cell interaction in nervous system | Switzerland, Germany, USA | 2010 | [254] | |
| 7 | 15q11.2 | Microdeletion | NIPA2, CYFIP1 | GGE | Role in neuronal growth and differentiation | Austria, Belgium, Denmark, Germany, the Netherlands | 2010 | [246] | ||
| 8 | 15q13.3 | 1.4Mb | Microdeletion | CHRNA7 | GGE (specifically JME) | Modulates thalamo-cortical pathways | Switzerland, Germany, USA | 2009, 2010 | [244,254] | |
| 9 | 16p13.11 | 800Kb | Deletion | NDE1 | MTLE | Brain structural alterations | UK | 2012 | [255] | |
| 10 | 16p13.2 | 47Kb | Microdeletion | GRIN2A | RE (childhood epilepsy) | Disrupt GRIN2A | France | 2014 | [256] | |
| 11 | 22q11.2 | 3Mb | Microdeletion | DGCR6, DGCR6L | GGE (specifically JME) | Haplo-insufficiency of DGCR6 in 22q11 could disturb interaction with GABABR | The Netherlands | 2016 | [257] | |
| Sl. No. | Gene | Genomic Loci | Putative Markers | Gene Function | Phenotype Prediction | Company | Major Ethnic Group |
|---|---|---|---|---|---|---|---|
| 1 | SLC2A1 | 1p34.2 | - | Solute carrier transporter | Genetic generalized epilepsy | Centogene AG the Rare Disease Company and Blueprint Genetics | European |
| 2 | CACNB4 | 2q23.3 | - | Voltage-gated calcium channel | |||
| 3 | GABRG2 | 5q34 | - | GABA receptor | Generalized epilepsy with febrile seizures plus | Invitae, GeneDx, Fulgent Genetics, LifeLabs Genetics, Laboratoria de Genetica Clinica SL, Institute of Human Genetics and Cologne University | American and European |
| 4 | STX1B | 16p11.2 | - | Synaptic vesicle | |||
| 5 | SCN1A | 2q24.3 | rs8191987, rs16851381, rs2298771 | Voltage-dependent sodium channel | |||
| 6 | SCN1B | 19q13.11 | Voltage-gated sodium channel | ||||
| 7 | SCN9A | 2q24.3 | Voltage-gated sodium channel | ||||
| 8 | GABRA1 | 5q34 | rs1581220270 | GABA receptor | Childhood absence epilepsy | Invitae, LifeLabs Genetics and Clinical Molecular Genetics Laboratory | American |
| 9 | GABRB3 | 15q12 | rs25409 | Ligand-gated ionic channel | |||
| 10 | GABRG2 | 5q34 | rs1561645243 | GABA receptor | |||
| 11 | JRK | 8q24.3 | T456M | DNA-binding protein | |||
| 12 | CACNA1H | 16p13.3 | rs9934839, rs2745150, rs8044363, rs8043905, rs9934839, rs3751664, c.937A>G, rs119454947, rs119454949 | Voltage-dependent calcium channel | |||
| 13 | CACNB4 | 2q23.3 | R482X | Voltage-dependent calcium channel | Juvenile myoclonic epilepsy | Athena Diagnostics Inc, GeneDx, Invitae, MedGen and Illumina Clinical Services Laboratory | American & European |
| 14 | EFHC1 | 6p12.2 | rs137852778, rs137852776, rs137852777, rs149055334, rs79761183 | Calcium-binding protein | |||
| 15 | GABRA1 | 5q34 | rs121434579 | GABA receptor | |||
| 16 | CILK1 | 6p12.1 | rs376111440, rs55932059, rs1554169267, rs765078446 | Protein kinases | |||
| 17 | GAL | 11q13.2 | C116A | Galanin and GMAP prepropeptide | Familial temporal lobe epilepsy | Invitae, GeneDx, Fulgent Genetics, LifeLabs Genetics, Laboratoria de Genetica Clinica SL, Institute of Human Genetics, Cologne University, Athena Diagnostics Inc, CGC Genetics, MNG Laboratories (Medical Neurogenetics, LLC), PreventionGenetics, Amplexa Genetics | American & European |
| 18 | CPA6 | 8q13.2 | rs114402678, rs61738009 | Metallocarboxypeptidases | |||
| 19 | RELN | 7q22.1 | - | Cell–cell interaction | |||
| 20 | LGI1 | 10q23.33 | - | - | |||
| 21 | MICAL1 | 6q21 | - | Depolymerization of actin filaments |
| Sl No | Gene | Variant | Location | Risk Allele | GMAF | Phenotype | Sample Size (P/C) | Country | Study (PMID) | Sensitivity | Specificity | PPV | OR |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1. | Cx36 | rs3743123 | Coding | T | 0.401 | JME | 169/123 | UK, Denmark, France, Greece, Portugal, and Sweden | 15235036 | 37.8 | 70.7 | 37.4 | 1.4 |
| 247/621 | Germany | 16876983 | |||||||||||
| 2. | SCN1A | rs3812718 | Intronic | A | 0.581 | Epilepsy | 76/701 | Australia | 19949041 | 56.6 | 47.6 | 39.4 | 1.2 |
| 90/701 | 19289736 | ||||||||||||
| 97/837 | China | 20477842 | |||||||||||
| 362/86 | India | 20602612 | |||||||||||
| 62/199 | Switzerland | 21762453 | |||||||||||
| 138/282 | India | 22578703 | |||||||||||
| 234/189 | Taiwan | 22188362 | |||||||||||
| 485/298 | India | 23466530 | |||||||||||
| 133/209 | Austria | 24014518 | |||||||||||
| 212/344 | German | ||||||||||||
| 282/470 | Malaysian Chinese | 25668517 | |||||||||||
| 151/244 | India | ||||||||||||
| 243/358 | Malaysia | ||||||||||||
| 200/200 | Greece | 28144265 | |||||||||||
| 3. | GABRG2 | rs211037 | Coding | T | 0.222 | Epilepsy | 135/154 | Germany | 12117362 | 32.6 | 45.1 | 38.5 | 0.6 |
| 53/96 | Italy | 12694927 | |||||||||||
| 104/83 | Taiwan | 12672902 | |||||||||||
| 94/106 | Japan | 12759178 | |||||||||||
| 569/330 | British | 16806831 | |||||||||||
| 684/284 | Ireland | ||||||||||||
| 74/118 | America | 16256272 | |||||||||||
| 77/83 | Taiwan | 17162195 | |||||||||||
| 100/120 | Egypt | 21983990 | |||||||||||
| 441/267 | India | 24061200 | |||||||||||
| 60/153 | Roman | 29379546 | |||||||||||
| 100/100 | Brazil | 23287319 | |||||||||||
| 1719/4672 | Malaysia, Hong Kong, Korea, India | 26452361 | |||||||||||
| 4 | BRD2 | rs3918149 | 5′ UTR | A | 0.246 | JME | 531/1390 | Britain, Iran, Germany, Australia, India | 17437413 | 17 | 13 | 27.4 | 1.14 |
| 116/470 | Germany | 30719712 | |||||||||||
| 5 | CACNA1H | rs9934839 | Coding | G | 0.340 | CAE | 100/191 | China | 16905256 | 9.17 | 5.49 | 65.6 | 1.73 |
| 218/191 | China | 17156077 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thakran, S.; Guin, D.; Singh, P.; Singh, P.; Kukal, S.; Rawat, C.; Yadav, S.; Kushwaha, S.S.; Srivastava, A.K.; Hasija, Y.; et al. Genetic Landscape of Common Epilepsies: Advancing towards Precision in Treatment. Int. J. Mol. Sci. 2020, 21, 7784. https://doi.org/10.3390/ijms21207784
Thakran S, Guin D, Singh P, Singh P, Kukal S, Rawat C, Yadav S, Kushwaha SS, Srivastava AK, Hasija Y, et al. Genetic Landscape of Common Epilepsies: Advancing towards Precision in Treatment. International Journal of Molecular Sciences. 2020; 21(20):7784. https://doi.org/10.3390/ijms21207784
Chicago/Turabian StyleThakran, Sarita, Debleena Guin, Pooja Singh, Priyanka Singh, Samiksha Kukal, Chitra Rawat, Saroj Yadav, Suman S. Kushwaha, Achal K. Srivastava, Yasha Hasija, and et al. 2020. "Genetic Landscape of Common Epilepsies: Advancing towards Precision in Treatment" International Journal of Molecular Sciences 21, no. 20: 7784. https://doi.org/10.3390/ijms21207784
APA StyleThakran, S., Guin, D., Singh, P., Singh, P., Kukal, S., Rawat, C., Yadav, S., Kushwaha, S. S., Srivastava, A. K., Hasija, Y., Saso, L., Ramachandran, S., & Kukreti, R. (2020). Genetic Landscape of Common Epilepsies: Advancing towards Precision in Treatment. International Journal of Molecular Sciences, 21(20), 7784. https://doi.org/10.3390/ijms21207784