STAT3 Differentially Regulates TLR4-Mediated Inflammatory Responses in Early or Late Phases



Abstract
1. Introduction
2. Results
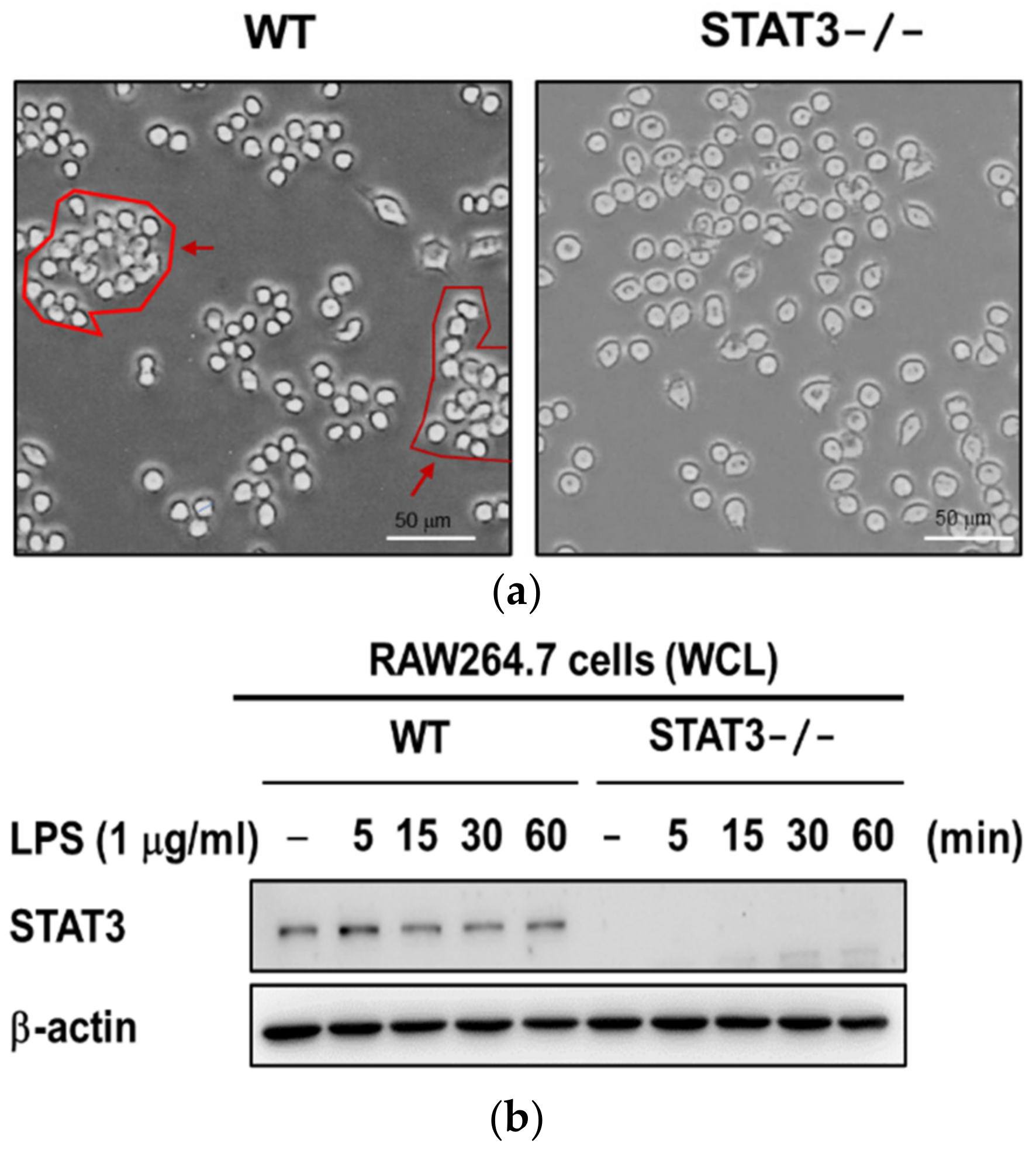
2.1. Establishment of STAT3 Knockout RAW264.7 Macrophage Cell Line
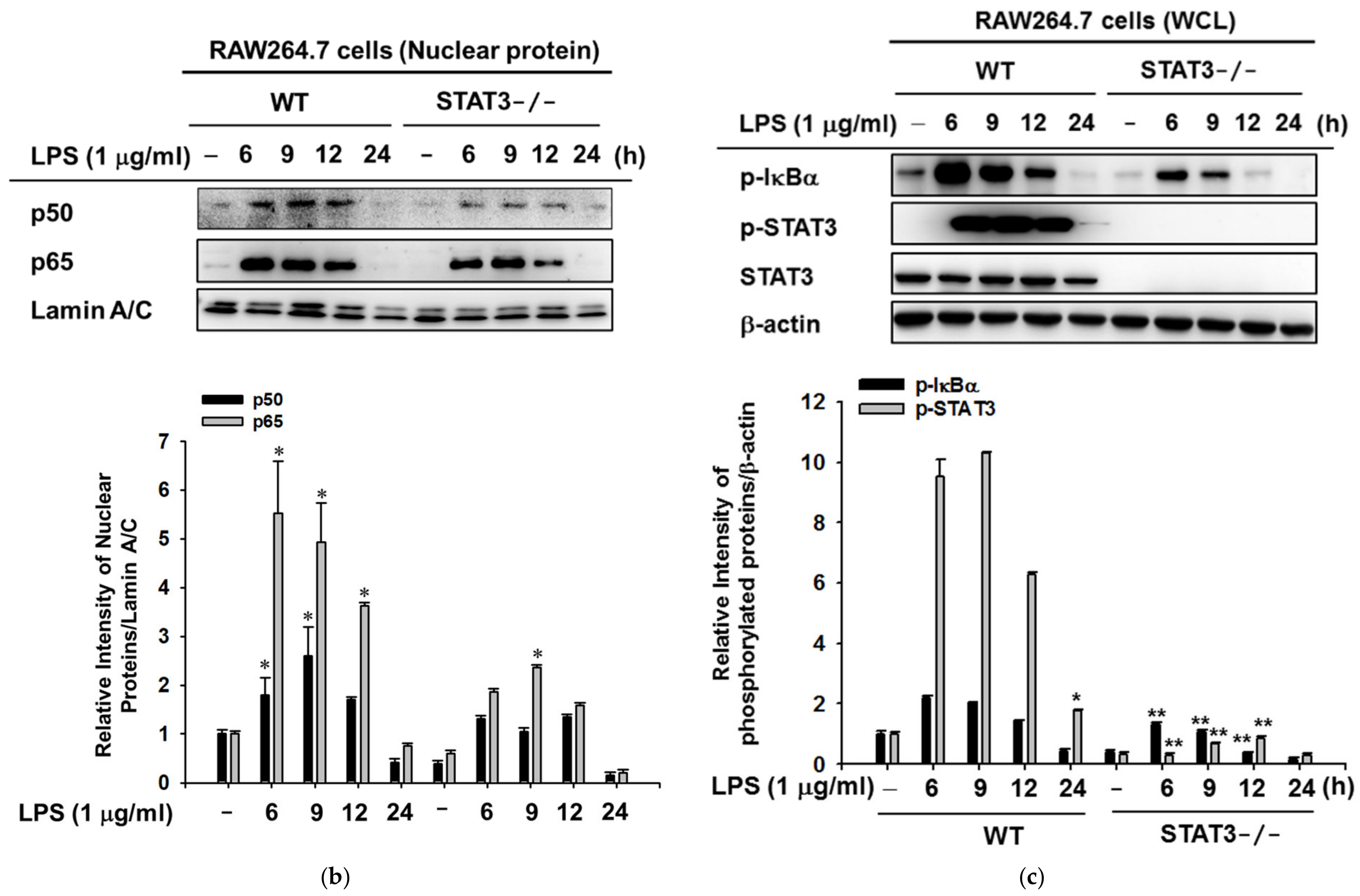
2.2. STAT3 Deletion in Macrophages Leads to Enhanced Lyn/AKT/NF-κB-Dependent Inflammatory Signaling Pathways in Early Stages of Inflammation
2.3. STAT3 Deletion in Macrophages Display Reduced Gene Expressions Profiles of iNOS and IL-10, and NF-κB Signaling in Macrophages at Later Stages of LPS Stimulation
2.4. Effects of STAT3 on NO Production, Invasion, and Colony Formation
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. STAT3 Knockout Cell Preparation
4.3. Cell Culture
4.4. Migration Assay
4.5. Transwell Migration and Invasion Assays
4.6. Nitric Oxide Assay
4.7. Cell Viability Assay
4.8. mRNA Isolation and Real-Time PCR
4.9. Preparation of Total and Nuclear Cell Lysate
4.10. Immunoblotting Analysis
4.11. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ERK | Extracellular signal-regulated kinase |
| iNOS | Inducible nitric oxide synthase |
| cNOS | Constitutive nitric oxide synthase |
| BCR | B-cell receptor |
| JNK | c-Jun N-terminal kinase |
| KO | Knockout |
| IBD | Inflammatory bowel disease |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| NF-κB | Nuclear factor-κB |
| NO | Nitric oxide |
| PCR | Polymerase chain reaction |
| STAT | Signal transducers and activators of transcription |
| TLR | Toll-like receptor |
| TNF | Tumor necrosis factor |
| JAK | Janus kinase |
| CRISPR | Clustered regulatory interspaced short palindromic repeats |
References
- Torrado, E.; Cooper, A.M. Cytokines in the balance of protection and pathology during mycobacterial infections. Adv. Exp. Med. Biol. 2013, 783, 121–140. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.; Jiang, Z.; Georgel, P.; Crozat, K.; Croker, B.; Rutschmann, S.; Du, X.; Hoebe, K. Genetic analysis of host resistance: Toll-like receptor signaling and immunity at large. Annu. Rev. Immunol. 2006, 24, 353–389. [Google Scholar] [CrossRef]
- Rescigno, M.; Granucci, F.; Citterio, S.; Foti, M.; Ricciardi-Castagnoli, P. Coordinated events during bacteria-induced DC maturation. Immunol. Today 1999, 20, 200–203. [Google Scholar] [CrossRef]
- Suresh, R.; Mosser, D.M. Pattern recognition receptors in innate immunity, host defense, and immunopathology. Adv. Physiol. Educ. 2013, 37, 284–291. [Google Scholar] [CrossRef]
- Shin, J.S.; Noh, Y.S.; Lee, Y.S.; Cho, Y.W.; Baek, N.I.; Choi, M.S.; Jeong, T.S.; Kang, E.; Chung, H.G.; Lee, K.T. Arvelexin from Brassica rapa suppresses NF-kappaB-regulated pro-inflammatory gene expression by inhibiting activation of IkappaB kinase. Br. J. Pharmacol. 2011, 164, 145–158. [Google Scholar] [CrossRef]
- Koh, Y.C.; Yang, G.; Lai, C.S.; Weerawatanakorn, M.; Pan, M.H. Chemopreventive effects of phytochemicals and medicines on M1/M2 polarized macrophage role in inflammation-related diseases. Int. J. Mol. Sci. 2018, 19, 2208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, F.; Liu, L.; Feng, L.; Wu, X.; Shen, Y.; Sun, Y.; Wu, X.; Xu, Q. (+)-Borneol improves the efficacy of edaravone against DSS-induced colitis by promoting M2 macrophages polarization via JAK2-STAT3 signaling pathway. Int. Immunopharmacol. 2017, 53, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.O.; Lim, J.W.; Kim, H. Red ginseng extract inhibits the expression of MCP-1 and iNOS in Helicobacter pylori-infected gastric epithelial cells by suppressing the activation of NADPH oxidase and Jak2/Stat3. J. Ethnopharmacol. 2013, 150, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Li, J.R.; Wang, Y.; Lei, L.S.; Yu, C.L.; Chen, N.N. Cyclovirobuxinum D suppresses lipopolysaccharide-induced inflammatory responses in murine macrophages in vitro by blocking JAK-STAT signaling pathway. Acta Pharmacol. Sin. 2014, 35, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Verstrepen, L.; Bekaert, T.; Chau, T.L.; Tavernier, J.; Chariot, A.; Beyaert, R. TLR-4, IL-1R and TNF-R signaling to NF-kappaB: Variations on a common theme. Cell. Mol. Life Sci. 2008, 65, 2964–2978. [Google Scholar] [CrossRef] [PubMed]
- Tabarsa, M.; Karnjanapratum, S.; Cho, M.; Kim, J.K.; You, S. Molecular characteristics and biological activities of anionic macromolecules from Codium fragile. Int. J. Biol. Macromol. 2013, 59, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Kwon, M.J.; Kim, I.H.; Kim, Y.M.; Lee, M.K.; Nam, T.J. Pyropia yezoensis glycoprotein promotes the M1 to M2 macrophage phenotypic switch via the STAT3 and STAT6 transcription factors. Int. J. Mol. Med. 2016, 38, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Park, S.Y.; Joe, E.H.; Jou, I. 15d-PGJ2 and rosiglitazone suppress Janus kinase-STAT inflammatory signaling through induction of suppressor of cytokine signaling 1 (SOCS1) and SOCS3 in glia. J. Biol. Chem. 2003, 278, 14747–14752. [Google Scholar] [CrossRef]
- Lee, J.O.; Choi, E.; Shin, K.K.; Hong, Y.H.; Kim, H.G.; Jeong, D.; Hossain, M.A.; Kim, H.S.; Yi, Y.S.; Kim, D.; et al. Compound K, a ginsenoside metabolite, plays an antiinflammatory role in macrophages by targeting the AKT1-mediated signaling pathway. J. Ginseng. Res. 2019, 43, 154–160. [Google Scholar] [CrossRef]
- Kim, E.; Yi, Y.S.; Son, Y.J.; Han, S.Y.; Kim, D.H.; Nam, G.; Hossain, M.A.; Kim, J.H.; Park, J.; Cho, J.Y. BIOGF1K, a compound K-rich fraction of ginseng, plays an antiinflammatory role by targeting an activator protein-1 signaling pathway in RAW264.7 macrophage-like cells. J. Ginseng. Res. 2018, 42, 233–237. [Google Scholar] [CrossRef]
- Han, S.Y.; Kim, J.; Kim, E.; Kim, S.H.; Seo, D.B.; Kim, J.H.; Shin, S.S.; Cho, J.Y. AKT-targeted anti-inflammatory activity of Panax ginseng calyx ethanolic extract. J. Ginseng. Res. 2018, 42, 496–503. [Google Scholar] [CrossRef]
- Subbian, S.; Bandyopadhyay, N.; Tsenova, L.; O’Brien, P.; Khetani, V.; Kushner, N.L.; Peixoto, B.; Soteropoulos, P.; Bader, J.S.; Karakousis, P.C.; et al. Early innate immunity determines outcome of Mycobacterium tuberculosis pulmonary infection in rabbits. Cell Commun. Signal. 2013, 11, 60. [Google Scholar] [CrossRef]
- Koo, M.S.; Subbian, S.; Kaplan, G. Strain specific transcriptional response in Mycobacterium tuberculosis infected macrophages. Cell Commun. Signal. 2012, 10, 2–10. [Google Scholar] [CrossRef]
- Rottenberg, M.E.; Carow, B. SOCS3 and STAT3, major controllers of the outcome of infection with Mycobacterium tuberculosis. Semin. Immunol. 2014, 26, 518–532. [Google Scholar] [CrossRef]
- Akira, S. Roles of STAT3 defined by tissue-specific gene targeting. Oncogene 2000, 19, 2607–2611. [Google Scholar] [CrossRef]
- Lovato, P.; Brender, C.; Agnholt, J.; Kelsen, J.; Kaltoft, K.; Svejgaard, A.; Eriksen, K.W.; Woetmann, A.; Odum, N. Constitutive STAT3 activation in intestinal T cells from patients with Crohn’s disease. J. Biol. Chem. 2003, 278, 16777–16781. [Google Scholar] [CrossRef] [PubMed]
- Atreya, R.; Mudter, J.; Finotto, S.; Mullberg, J.; Jostock, T.; Wirtz, S.; Schutz, M.; Bartsch, B.; Holtmann, M.; Becker, C.; et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: Evidence in crohn disease and experimental colitis in vivo. Nat. Med. 2000, 6, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Queval, C.J.; Song, O.R.; Deboosere, N.; Delorme, V.; Debrie, A.S.; Iantomasi, R.; Veyron-Churlet, R.; Jouny, S.; Redhage, K.; Deloison, G.; et al. STAT3 Represses nitric oxide synthesis in human macrophages upon Mycobacterium tuberculosis infection. Sci. Rep. 2016, 6, 29297. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Ihle, J.N. The Stat family in cytokine signaling. Curr. Opin. Cell Biol. 2001, 13, 211–217. [Google Scholar] [CrossRef]
- Welte, T.; Zhang, S.S.; Wang, T.; Zhang, Z.; Hesslein, D.G.; Yin, Z.; Kano, A.; Iwamoto, Y.; Li, E.; Craft, J.E.; et al. STAT3 deletion during hematopoiesis causes Crohn’s disease-like pathogenesis and lethality: A critical role of STAT3 in innate immunity. Proc. Natl. Acad. Sci. USA 2003, 100, 1879–1884. [Google Scholar] [CrossRef]
- Liu, X.; Lee, Y.S.; Yu, C.R.; Egwuagu, C.E. Loss of STAT3 in CD4+ T cells prevents development of experimental autoimmune diseases. J. Immunol. 2008, 180, 6070–6076. [Google Scholar] [CrossRef]
- Prada, C.E.; Jousma, E.; Rizvi, T.A.; Wu, J.; Dunn, R.S.; Mayes, D.A.; Cancelas, J.A.; Dombi, E.; Kim, M.O.; West, B.L.; et al. Neurofibroma-associated macrophages play roles in tumor growth and response to pharmacological inhibition. Acta Neuropathol. 2013, 125, 159–168. [Google Scholar] [CrossRef]
- Gavino, A.C.; Nahmod, K.; Bharadwaj, U.; Makedonas, G.; Tweardy, D.J. STAT3 inhibition prevents lung inflammation, remodeling, and accumulation of Th2 and Th17 cells in a murine asthma model. Allergy 2016, 71, 1684–1692. [Google Scholar] [CrossRef]
- Santana, F.P.R.; da Silva, R.C.; Grecco, S.D.S.; Pinheiro, A.; Caperuto, L.C.; Arantes-Costa, F.M.; Claudio, S.R.; Yoshizaki, K.; Macchione, M.; Ribeiro, D.A.; et al. Inhibition of MAPK and STAT3-SOCS3 by sakuranetin attenuated chronic allergic airway inflammation in mice. Mediators Inflamm. 2019, 2019, 1356356. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, A.; Jeong, D.; Kim, M.Y.; Cho, J.Y. Trichosanthes tricuspidata Lour. methanol extract exhibits anti-inflammatory activity by targeting Syk, Src, and IRAK1 kinase activity. Evid. Based Complement. Alternat. Med. 2019, 2019, 6879346. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.S.; Springer, M.G.; Choi, K.; Jousma, E.; Rizvi, T.A.; Dombi, E.; Kim, M.O.; Wu, J.; Ratner, N. STAT3 inhibition reduces macrophage number and tumor growth in neurofibroma. Oncogene 2019, 38, 2876–2884. [Google Scholar] [CrossRef] [PubMed]
- Furtek, S.L.; Backos, D.S.; Matheson, C.J.; Reigan, P. Strategies and approaches of targeting STAT3 for cancer treatment. ACS Chem. Biol. 2016, 11, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Greenhill, C.J.; Rose-John, S.; Lissilaa, R.; Ferlin, W.; Ernst, M.; Hertzog, P.J.; Mansell, A.; Jenkins, B.J. IL-6 trans-signaling modulates TLR4-dependent inflammatory responses via STAT3. J. Immunol. 2011, 186, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guan, J.; Wang, W.; Hou, C.; Zhou, L.; Ma, J.; Cheng, Y.; Jiao, S.; Zhou, Z. TRAF3-interacting JNK-activating modulator promotes inflammation by stimulating translocation of Toll-like receptor 4 to lipid rafts. J. Biol. Chem. 2019, 294, 2744–2756. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Suzuki, H. Toll-like receptor 4 as a possible therapeutic target for delayed brain injuries after aneurysmal subarachnoid hemorrhage. Neural Regen. Res. 2017, 12, 193. [Google Scholar]
- Kim, E.; Yoon, J.Y.; Lee, J.; Jeong, D.; Park, J.G.; Hong, Y.H.; Kim, J.H.; Aravinthan, A.; Kim, J.H.; Cho, J.Y. TANK-binding kinase 1 and Janus kinase 2 play important roles in the regulation of mitogen-activated protein kinase phosphatase-1 expression after toll-like receptor 4 activation. J Cell. Physiol. 2018, 233, 8790–8801. [Google Scholar] [CrossRef]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef]
- Buchert, M.; Burns, C.J.; Ernst, M. Targeting JAK kinase in solid tumors: Emerging opportunities and challenges. Oncogene 2016, 35, 939–951. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Terzic, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and colon cancer. Gastroenterology 2010, 138, 2101–2114. [Google Scholar] [CrossRef] [PubMed]
- West, N.R.; McCuaig, S.; Franchini, F.; Powrie, F. Emerging cytokine networks in colorectal cancer. Nat. Rev. Immunol. 2015, 15, 615–629. [Google Scholar] [CrossRef]
- Zhuang, Z.; Zhao, X.; Wu, Y.; Huang, R.; Zhu, L.; Zhang, Y.; Shi, J. The anti-apoptotic effect of PI3K-Akt signaling pathway after subarachnoid hemorrhage in rats. Ann. Clin. Lab. Sci. 2011, 41, 364–372. [Google Scholar]
- Ozes, O.N.; Mayo, L.D.; Gustin, J.A.; Pfeffer, S.R.; Pfeffer, L.M.; Donner, D.B. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 1999, 401, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Madrid, L.V.; Wang, C.Y.; Guttridge, D.C.; Schottelius, A.J.; Baldwin, A.S., Jr.; Mayo, M.W. Akt suppresses apoptosis by stimulating the transactivation potential of the RelA/p65 subunit of NF-kappaB. Mol. Cell. Biol. 2000, 20, 1626–1638. [Google Scholar] [CrossRef]
- Lee, H.S.; Moon, C.; Lee, H.W.; Park, E.-M.; Cho, M.-S.; Kang, J.L. Src tyrosine kinases mediate activations of NF-κB and integrin signal during lipopolysaccharide-induced acute lung injury. J. Immunol. 2007, 179, 7001–7011. [Google Scholar] [CrossRef]
- Toubiana, J.; Rossi, A.-L.; Belaidouni, N.; Grimaldi, D.; Pene, F.; Chafey, P.; Comba, B.; Camoin, L.; Bismuth, G.; Claessens, Y.-E. Src-family-tyrosine kinase Lyn is critical for TLR2-mediated NF-κB activation through the PI 3-kinase signaling pathway. Innate Immun. 2015, 21, 685–697. [Google Scholar] [CrossRef]
- Takada, Y.; Mukhopadhyay, A.; Kundu, G.C.; Mahabeleshwar, G.H.; Singh, S.; Aggarwal, B.B. Hydrogen peroxide activates NF-κB through tyrosine phosphorylation of IκBα and serine phosphorylation of p65 evidence for the involvement of IκBα kinase and Syk protein-tyrosine kinase. J. Biol. Chem. 2003, 278, 24233–24241. [Google Scholar] [CrossRef]
- Takada, Y.; Aggarwal, B.B. TNF activates Syk protein tyrosine kinase leading to TNF-induced MAPK activation, NF-κB activation, and apoptosis. J. Immunol. 2004, 173, 1066–1077. [Google Scholar] [CrossRef]
- Song, C.; Hong, Y.H.; Park, J.G.; Kim, H.G.; Jeong, D.; Oh, J.; Sung, G.-H.; Hossain, M.A.; Taamalli, A.; Kim, J.H. Suppression of Src and Syk in the NF-κB signaling pathway by Olea europaea methanol extract is leading to its anti-inflammatory effects. J. Ethnopharmacol. 2019, 235, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Kovarova, M.; Tolar, P.; Arudchandran, R.; Draberova, L.; Rivera, J.; Draber, P. Structure-function analysis of Lyn kinase association with lipid rafts and initiation of early signaling events after Fcepsilon receptor I aggregation. Mol. Cell. Biol. 2001, 21, 8318–8328. [Google Scholar] [CrossRef]
- Takata, M.; Sabe, H.; Hata, A.; Inazu, T.; Homma, Y.; Nukada, T.; Yamamura, H.; Kurosaki, T. Tyrosine kinases Lyn and Syk regulate B cell receptor-coupled Ca2+ mobilization through distinct pathways. EMBO J. 1994, 13, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xiong, L.; Gong, J. Lyn kinase enhanced hepatic fibrosis by modulating the activation of hepatic stellate cells. Am. J. Transl. Res. 2017, 9, 2865–2877. [Google Scholar]
- Avila, M.; Martinez-Juarez, A.; Ibarra-Sanchez, A.; Gonzalez-Espinosa, C. Lyn kinase controls TLR4-dependent IKK and MAPK activation modulating the activity of TRAF-6/TAK-1 protein complex in mast cells. Innate Immun. 2012, 18, 648–660. [Google Scholar] [CrossRef]
- Lee, J.; Rhee, M.H.; Kim, E.; Cho, J.Y. BAY 11-7082 is a broad-spectrum inhibitor with anti-inflammatory activity against multiple targets. Mediators Inflamm. 2012, 2012, 416036. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.G.; Yang, W.S.; Hong, Y.H.; Kweon, D.H.; Lee, J.; Kim, S.; Cho, J.Y. Anti-inflammatory functions of the CDC25 phosphatase inhibitor BN82002 via targeting AKT2. Biochem. Pharmacol. 2019, 164, 216–227. [Google Scholar] [CrossRef]
- Gharbi, S.I.; Zvelebil, M.J.; Shuttleworth, S.J.; Hancox, T.; Saghir, N.; Timms, J.F.; Waterfield, M.D. Exploring the specificity of the PI3K family inhibitor LY294002. Biochem. J. 2007, 404, 15–21. [Google Scholar] [CrossRef]
- Wang, W.B.; Levy, D.E.; Lee, C.K. STAT3 negatively regulates type I IFN-mediated antiviral response. J. Immunol. 2011, 187, 2578–2585. [Google Scholar] [CrossRef]
- Stark, G.R.; Darnell, J.E., Jr. The JAK-STAT pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef]
- McFarland, B.C.; Gray, G.K.; Nozell, S.E.; Hong, S.W.; Benveniste, E.N. Activation of the NF-kappaB pathway by the STAT3 inhibitor JSI-124 in human glioblastoma cells. Mol. Cancer Res. 2013, 11, 494–505. [Google Scholar] [CrossRef]
- Akira, S. Functional roles of STAT family proteins: Lessons from knockout mice. Stem Cells 1999, 17, 138–146. [Google Scholar] [CrossRef]
- Xin, G.; Qin, S.; Wang, S.; Wang, X.; Zhang, Y.; Wang, J. Sex hormone affects the severity of non-alcoholic steatohepatitis through the MyD88-dependent IL-6 signaling pathway. Exp. Biol. Med. 2015, 240, 1279–1286. [Google Scholar] [CrossRef]
- Dalwadi, H.; Krysan, K.; Heuze-Vourc’h, N.; Dohadwala, M.; Elashoff, D.; Sharma, S.; Cacalano, N.; Lichtenstein, A.; Dubinett, S. Cyclooxygenase-2-dependent activation of signal transducer and activator of transcription 3 by interleukin-6 in non-small cell lung cancer. Clin. Cancer Res. 2005, 11, 7674–7682. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-kappaB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- O’Dea, E.L.; Barken, D.; Peralta, R.Q.; Tran, K.T.; Werner, S.L.; Kearns, J.D.; Levchenko, A.; Hoffmann, A. A homeostatic model of IκB metabolism to control constitutive NF-κB activity. Mol. Syst. Biol. 2007, 3, 111. [Google Scholar] [CrossRef]
- Tergaonkar, V.; Bottero, V.; Ikawa, M.; Li, Q.; Verma, I.M. IκB kinase-independent IκBα degradation pathway: Functional NF-κB activity and implications for cancer therapy. Mol. Cell. Biol. 2003, 23, 8070–8083. [Google Scholar] [CrossRef]
- Han, S.S.; Yun, H.; Son, D.J.; Tompkins, V.S.; Peng, L.; Chung, S.T.; Kim, J.S.; Park, E.S.; Janz, S. NF-kappaB/STAT3/PI3K signaling crosstalk in iMyc E mu B lymphoma. Mol. Cancer 2010, 9, 97. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Sethi, G.; Vadhan-Raj, S.; Bueso-Ramos, C.; Takada, Y.; Gaur, U.; Nair, A.S.; Shishodia, S.; Aggarwal, B.B. Resveratrol inhibits proliferation, induces apoptosis, and overcomes chemoresistance through down-regulation of STAT3 and nuclear factor-kappaB-regulated antiapoptotic and cell survival gene products in human multiple myeloma cells. Blood 2007, 109, 2293–2302. [Google Scholar] [CrossRef]
- Lee, H.; Herrmann, A.; Deng, J.H.; Kujawski, M.; Niu, G.; Li, Z.; Forman, S.; Jove, R.; Pardoll, D.M.; Yu, H. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell 2009, 15, 283–293. [Google Scholar] [CrossRef]
- Duan, W.; Li, H. Combination of NF-kB targeted siRNA and methotrexate in a hybrid nanocarrier towards the effective treatment in rheumatoid arthritis. J. Nanobiotechnol. 2018, 16, 58. [Google Scholar] [CrossRef]
- Fuchs, O. Targeting of NF-kappaB signaling pathway, other signaling pathways and epigenetics in therapy of multiple myeloma. Cardiovasc. Hematol. Disord. Drug Targets 2013, 13, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Jiang, H.; Li, Y.; Chen, W.; Li, H.; Peng, K.; Zhang, Z.; Sun, X. Targeting NF-kB signaling with polymeric hybrid micelles that co-deliver siRNA and dexamethasone for arthritis therapy. Biomaterials 2017, 122, 10–22. [Google Scholar] [CrossRef]
- Senis, Y.A.; Mazharian, A.; Mori, J. Src family kinases: At the forefront of platelet activation. Blood 2014, 124, 2013–2024. [Google Scholar] [CrossRef]
- Gong, P.; Angelini, D.J.; Yang, S.; Xia, G.; Cross, A.S.; Mann, D.; Bannerman, D.D.; Vogel, S.N.; Goldblum, S.E. TLR4 signaling is coupled to SRC family kinase activation, tyrosine phosphorylation of zonula adherens proteins, and opening of the paracellular pathway in human lung microvascular endothelia. J. Biol. Chem. 2008, 283, 13437–13449. [Google Scholar] [CrossRef]
- Zhang, Q.; Meng, X.; Qin, G.; Xue, X.; Dang, N. Lyn kinase promotes the proliferation of malignant melanoma cells through inhibition of apoptosis and autophagy via the PI3K/Akt signaling pathway. J. Cancer 2019, 10, 1197–1208. [Google Scholar] [CrossRef]
- Tsuda, M.; Tozaki-Saitoh, H.; Masuda, T.; Toyomitsu, E.; Tezuka, T.; Yamamoto, T.; Inoue, K. Lyn tyrosine kinase is required for P2X(4) receptor upregulation and neuropathic pain after peripheral nerve injury. Glia 2008, 56, 50–58. [Google Scholar] [CrossRef]
- Park, Y.H.; Kim, D.K.; Kim, H.S.; Lee, D.; Lee, M.B.; Min, K.Y.; Jo, M.G.; Lee, J.E.; Kim, Y.M.; Choi, W.S. WZ3146 inhibits mast cell Lyn and Fyn to reduce IgE-mediated allergic responses in vitro and in vivo. Toxicol. Appl. Pharmacol. 2019, 383, 114763. [Google Scholar] [CrossRef]
- Park, Y.H.; Kim, D.K.; Kim, H.W.; Kim, H.S.; Lee, D.; Lee, M.B.; Min, K.Y.; Koo, J.; Kim, S.J.; Kang, C.; et al. Repositioning of anti-cancer drug candidate, AZD7762, to an anti-allergic drug suppressing IgE-mediated mast cells and allergic responses via the inhibition of Lyn and Fyn. Biochem. Pharmacol. 2018, 154, 270–277. [Google Scholar] [CrossRef]
- Hong, J.J.; Yankee, T.M.; Harrison, M.L.; Geahlen, R.L. Regulation of signaling in B cells through the phosphorylation of Syk on linker region tyrosines. A mechanism for negative signaling by the Lyn tyrosine kinase. J. Biol. Chem. 2002, 277, 31703–31714. [Google Scholar] [CrossRef]
- Ruiz-Medina, B.E.; Lerma, D.; Hwang, M.; Ross, J.A.; Skouta, R.; Aguilera, R.J.; Kirken, R.A.; Varela-Ramirez, A.; Robles-Escajeda, E. Green barley mitigates cytotoxicity in human lymphocytes undergoing aggressive oxidative stress, via activation of both the Lyn/PI3K/Akt and MAPK/ERK pathways. Sci. Rep. 2019, 9, 6005. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liu, R.; Liu, Y.; Zhang, R.; He, L. Sinomenine potentiates P815 cell degranulation via upregulation of Ca2+ mobilization through the Lyn/PLCgamma/IP3R pathway. Int. J. Immunopathol. Pharmacol. 2016, 29, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Fang, L.; Pu, Q.; Lin, P.; Hoggarth, A.; Huang, H.; Li, X.; Li, G.; Wu, M. Lyn prevents aberrant inflammatory responses to Pseudomonas infection in mammalian systems by repressing a SHIP-1-associated signaling cluster. Signal Transduct. Target Ther. 2016, 1, 16032. [Google Scholar] [CrossRef] [PubMed]
- Shivakrupa, R.; Linnekin, D. Lyn contributes to regulation of multiple Kit-dependent signaling pathways in murine bone marrow mast cells. Cell. Signal. 2005, 17, 103–109. [Google Scholar] [CrossRef]
- Wang, L.; Kurosaki, T.; Corey, S. Engagement of the B-cell antigen receptor activates STAT through Lyn in a Jak-independent pathway. Oncogene 2007, 26, 2851. [Google Scholar] [CrossRef]
- Steer, S.A.; Moran, J.M.; Maggi, L.B., Jr.; Buller, R.M.; Perlman, H.; Corbett, J.A. Regulation of cyclooxygenase-2 expression by macrophages in response to double-stranded RNA and viral infection. J. Immunol. 2003, 170, 1070–1076. [Google Scholar] [CrossRef]
- Chiou, W.F.; Don, M.J.; Liao, J.F.; Wei, B.L. Psoralidin inhibits LPS-induced iNOS expression via repressing Syk-mediated activation of PI3K-IKK-IkappaB signaling pathways. Eur. J. Pharmacol. 2011, 650, 102–109. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, L.; Kouadir, M.; Tan, R.; Lu, Y.; Chang, J.; Xu, B.; Yin, X.; Zhou, X.; Zhao, D. PP2 and piceatannol inhibit PrP106-126-induced iNOS activation mediated by CD36 in BV2 microglia. Acta. Biochim. Biophys. Sin. 2013, 45, 763–772. [Google Scholar] [CrossRef]
- Shaheen, Z.R.; Naatz, A.; Corbett, J.A. CCR5-dependent activation of mTORC1 regulates translation of inducible NO synthase and COX-2 during encephalomyocarditis virus infection. J. Immunol. 2015, 195, 4406–4414. [Google Scholar] [CrossRef]
- Xu, N.; Yuan, H.; Liu, W.; Li, S.; Liu, Y.; Wan, J.; Li, X.; Zhang, R.; Chang, Y. Activation of RAW264.7 mouse macrophage cells in vitro through treatment with recombinant ricin toxin-binding subunit B: Involvement of protein tyrosine, NF-kappaB and JAK-STAT kinase signaling pathways. Int. J. Mol. Med. 2013, 32, 729–735. [Google Scholar] [CrossRef]
- Zhou, P.; Iadecola, C. iNOS and COX-2 in ischemic stroke. In Handbook of Neurochemistry and Molecular Neurobiology: Acute Ischemic Injury and Repair in the Nervous System; Springer: Berlin, Germany, 2007; pp. 33–45. [Google Scholar]
- Tripathi, P.; Tripathi, P.; Kashyap, L.; Singh, V. The role of nitric oxide in inflammatory reactions. FEMS Immunol. Med. Microbiol. 2007, 51, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Salvemini, D.; Wang, Z.Q.; Wyatt, P.S.; Bourdon, D.M.; Marino, M.H.; Manning, P.T.; Currie, M.G. Nitric oxide: A key mediator in the early and late phase of carrageenan-induced rat paw inflammation. Br. J. Pharmacol. 1996, 118, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Naka, T.; Kubo, M. SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 2007, 7, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, H.; Ohishi, M.; Mori, H.; Murakami, M.; Chinen, T.; Aki, D.; Hanada, T.; Takeda, K.; Akira, S.; Hoshijima, M.; et al. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat. Immunol. 2003, 4, 551–556. [Google Scholar] [CrossRef]
- Pengal, R.A.; Ganesan, L.P.; Wei, G.; Fang, H.; Ostrowski, M.C.; Tridandapani, S. Lipopolysaccharide-induced production of interleukin-10 is promoted by the serine/threonine kinase Akt. Mol. Immunol. 2006, 43, 1557–1564. [Google Scholar] [CrossRef]
- Murray, M.Y.; Birkland, T.P.; Howe, J.D.; Rowan, A.D.; Fidock, M.; Parks, W.C.; Gavrilovic, J. Macrophage migration and invasion is regulated by MMP10 expression. PLoS ONE 2013, 8, e63555. [Google Scholar] [CrossRef]
- Kharraz, Y.; Guerra, J.; Mann, C.J.; Serrano, A.L.; Muñoz-Cánoves, P. Macrophage plasticity and the role of inflammation in skeletal muscle repair. Mediators Inflamm. 2013, 2013, 1–9. [Google Scholar] [CrossRef]
- Grabner, B.; Schramek, D.; Mueller, K.M.; Moll, H.P.; Svinka, J.; Hoffmann, T.; Bauer, E.; Blaas, L.; Hruschka, N.; Zboray, K. Disruption of STAT3 signalling promotes KRAS-induced lung tumorigenesis. Nat. Commun. 2015, 6, 6285. [Google Scholar] [CrossRef]
- Jenkins, S.J.; Ruckerl, D.; Cook, P.C.; Jones, L.H.; Finkelman, F.D.; van Rooijen, N.; MacDonald, A.S.; Allen, J.E. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science 2011, 332, 1284–1288. [Google Scholar] [CrossRef]
- Gu, Z.; Li, Y.; Lee, P.; Liu, T.; Wan, C.; Wang, Z. Protein arginine methyltransferase 5 functions in opposite ways in the cytoplasm and nucleus of prostate cancer cells. PLoS ONE 2012, 7, e44033. [Google Scholar] [CrossRef]
- Wang, L.; Jiang, Z.; Huang, D.; Duan, J.; Huang, C.; Sullivan, S.; Vali, K.; Yin, Y.; Zhang, M.; Wegrzyn, J. Jak/Stat3 regulated global gene expression dynamics during late-stage reprogramming process. BMC Genom. 2018, 19, 183. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.; Kim, E.; Kim, J.H.; Yoon, K.; Kim, S.; Lee, J.; Cho, J.Y. AKT1-targeted proapoptotic activity of compound K in human breast cancer cells. J. Ginseng. Res. 2019, 43, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.O.; Kim, E.; Kim, J.H.; Hong, Y.H.; Kim, H.G.; Jeong, D.; Kim, J.; Kim, S.H.; Park, C.; Seo, D.B.; et al. Antimelanogenesis and skin-protective activities of Panax ginseng calyx ethanol extract. J. Ginseng. Res. 2018, 42, 389–399. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Direction | Primer Sequence |
|---|---|---|
| iNOS | Forward | AGGAGGAGAGAGATCCGATTTAG |
| Reverse | CTACTGAGACAGGGAAGTCTGA | |
| TNF-α | Forward | CTGGAGGACAGAGAAGAAATGG |
| Reverse | AGATTGCCACAGAATCCTGG | |
| IL-6 | Forward | ACCCTTCCAGATGGCAATATC |
| Reverse | CTACTCTATGCTGGGCAGTTT | |
| COX-2 | Forward | CACTACATCCTGACCCACTT |
| Reverse | ATGCTCCTGCTTGAGTATGT | |
| IL-10 | Forward | TCCTGCCATCACCTGAAATATG |
| Reverse | CTTTCTCCTCCTCTGCTTTCTC | |
| SOCS-3 | Forward | TGTGAAGAGGCAGTAGCATTTA |
| Reverse | CAGATCAACAGATGAGCCATCT | |
| IRF-3 | Forward | CAACCAACAAGTGATATTCTCCATG |
| Reverse | CAGGCCATCAGCAACAACAT | |
| GAPDH | Forward | CAATGAATACGGCTACAGCA |
| Reverse | AGGGAGATGCTCAGTGTTGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahuja, A.; Kim, E.; Sung, G.-H.; Cho, J.Y. STAT3 Differentially Regulates TLR4-Mediated Inflammatory Responses in Early or Late Phases. Int. J. Mol. Sci. 2020, 21, 7675. https://doi.org/10.3390/ijms21207675
Ahuja A, Kim E, Sung G-H, Cho JY. STAT3 Differentially Regulates TLR4-Mediated Inflammatory Responses in Early or Late Phases. International Journal of Molecular Sciences. 2020; 21(20):7675. https://doi.org/10.3390/ijms21207675
Chicago/Turabian StyleAhuja, Akash, Eunji Kim, Gi-Ho Sung, and Jae Youl Cho. 2020. "STAT3 Differentially Regulates TLR4-Mediated Inflammatory Responses in Early or Late Phases" International Journal of Molecular Sciences 21, no. 20: 7675. https://doi.org/10.3390/ijms21207675
APA StyleAhuja, A., Kim, E., Sung, G.-H., & Cho, J. Y. (2020). STAT3 Differentially Regulates TLR4-Mediated Inflammatory Responses in Early or Late Phases. International Journal of Molecular Sciences, 21(20), 7675. https://doi.org/10.3390/ijms21207675