Development of Conformational Antibodies to Detect Bcl-xL’s Amyloid Aggregates in Metal-Induced Apoptotic Neuroblastoma Cells

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
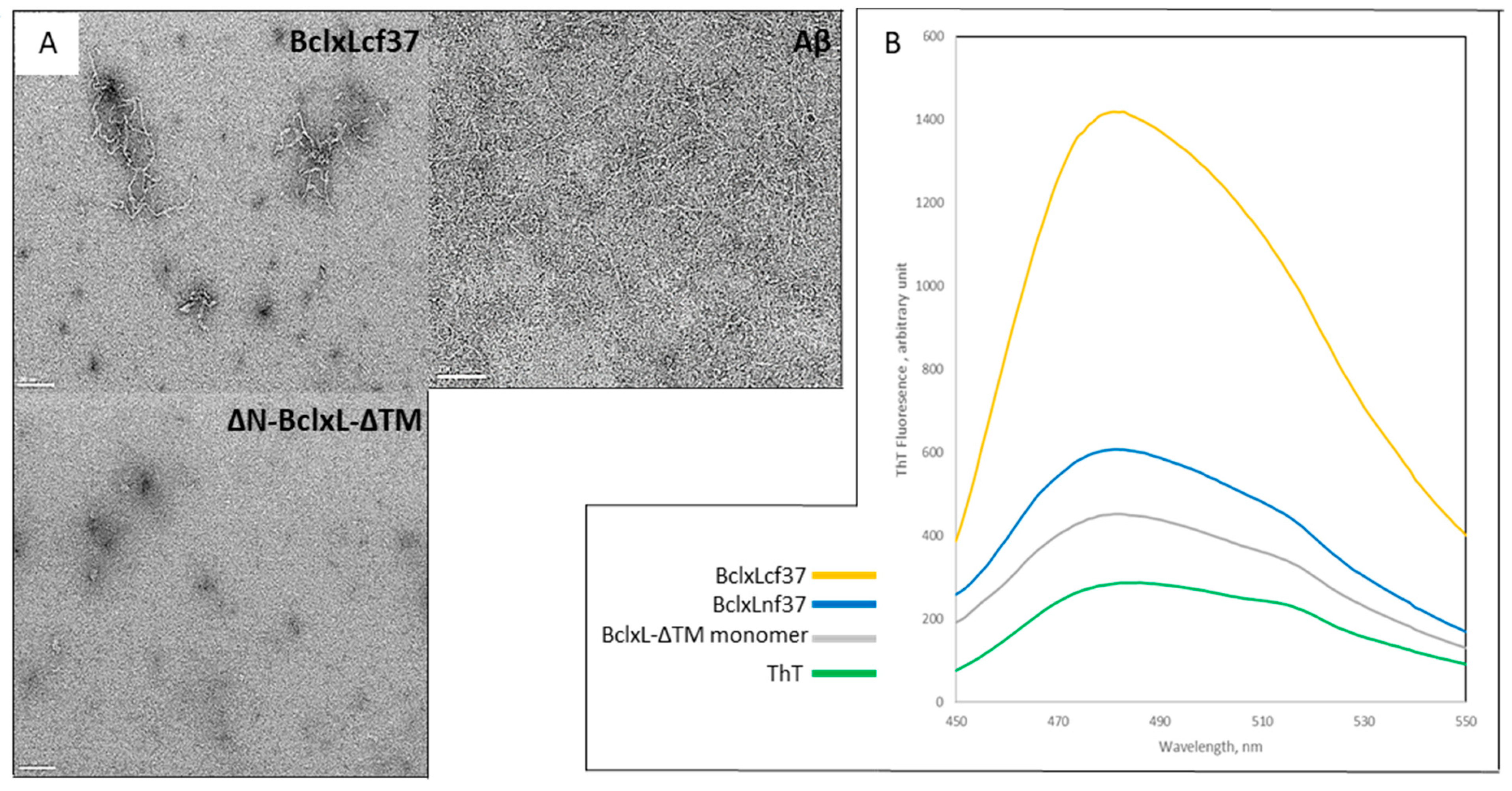
2.1. Trypsin Proteolysis of Bcl-xL-ΔTM Monomer Induces Its Conversion into Amyloid Fibrils
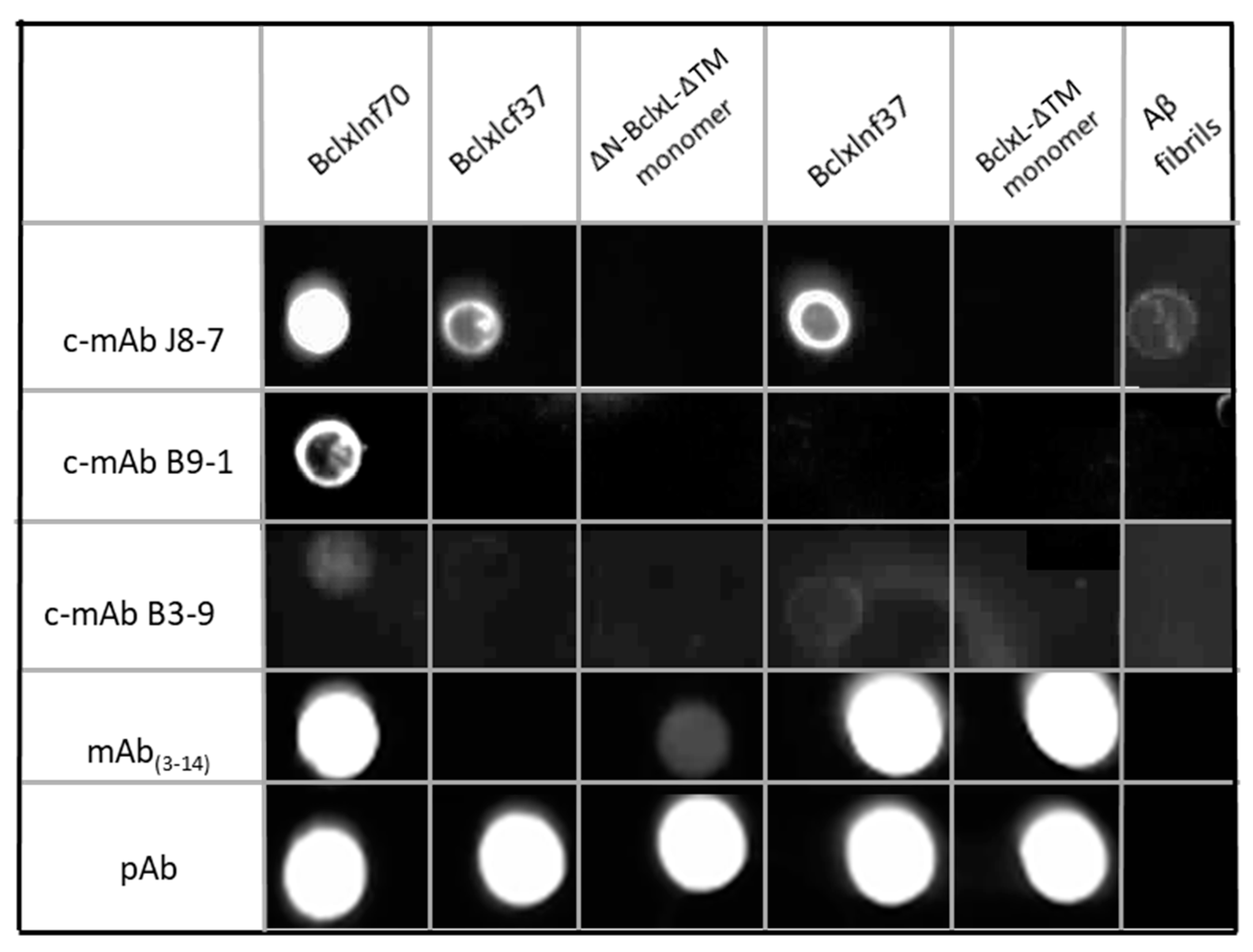
2.2. Production, Selection, and Characterization of Conformational Antibodies Directed against BclxLcf37 (c-mAbs)
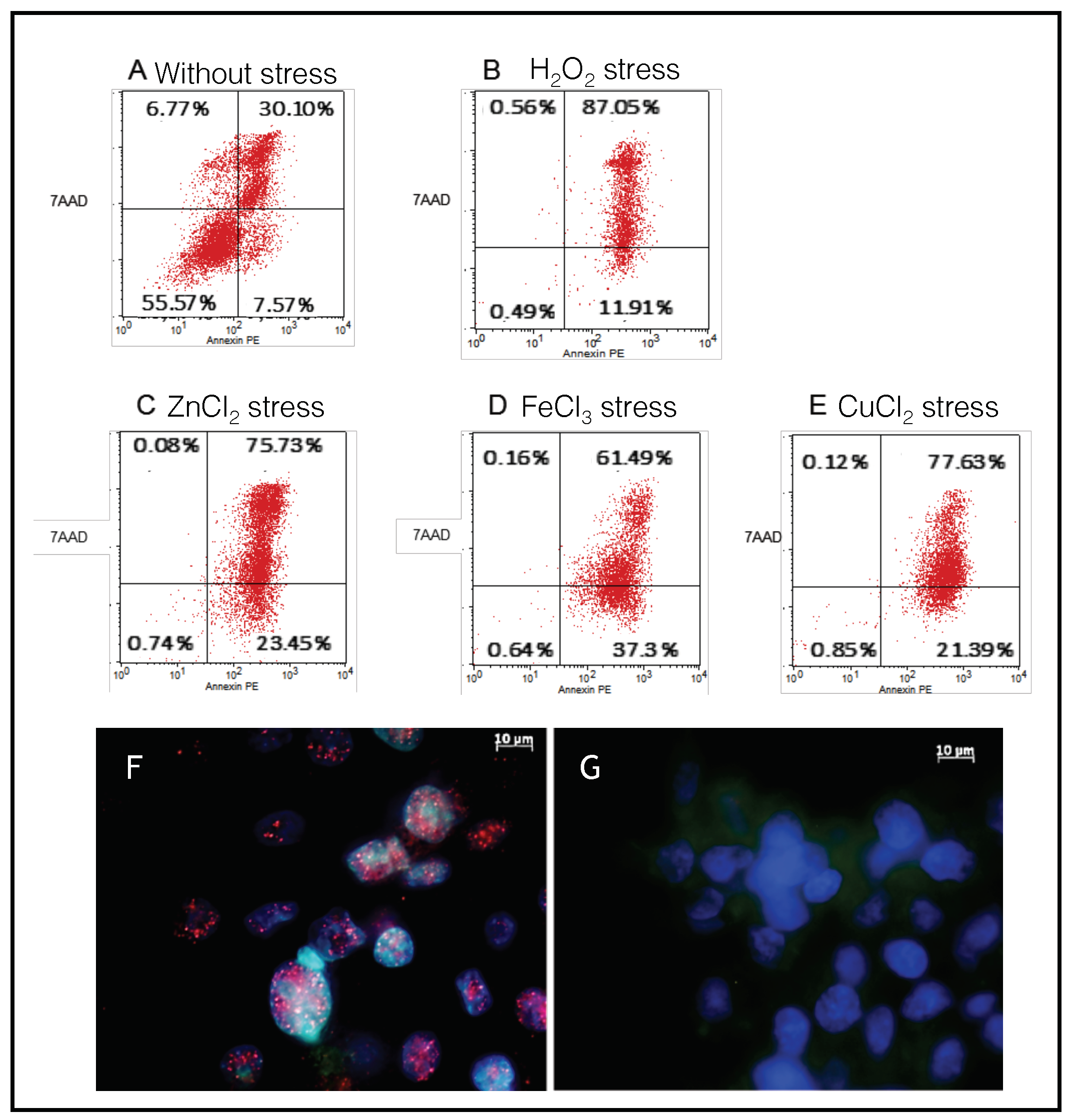
2.3. SH-SY5Y Cells as a Model to Monitor the Apoptosis Induced by Metal Stress
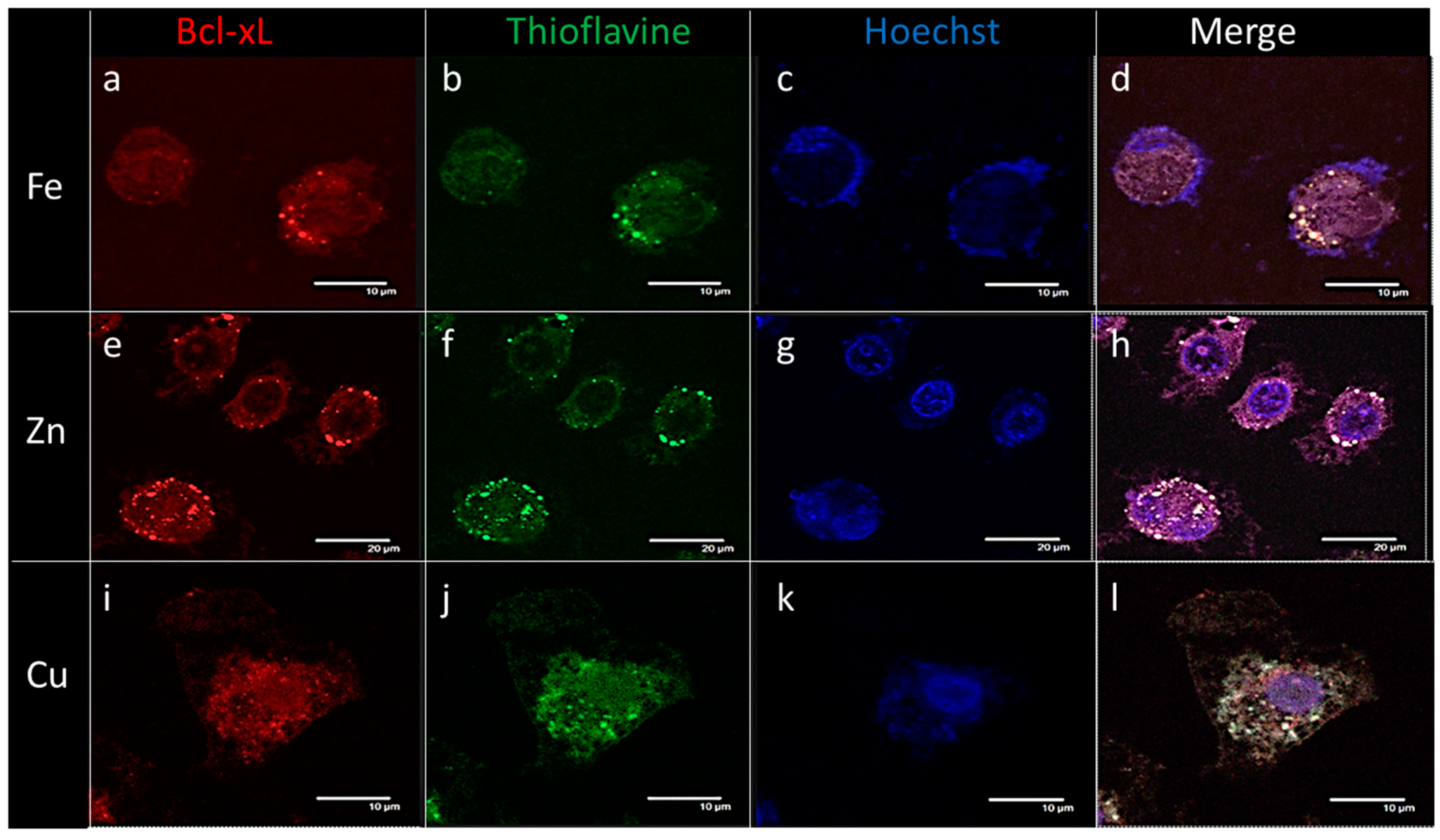
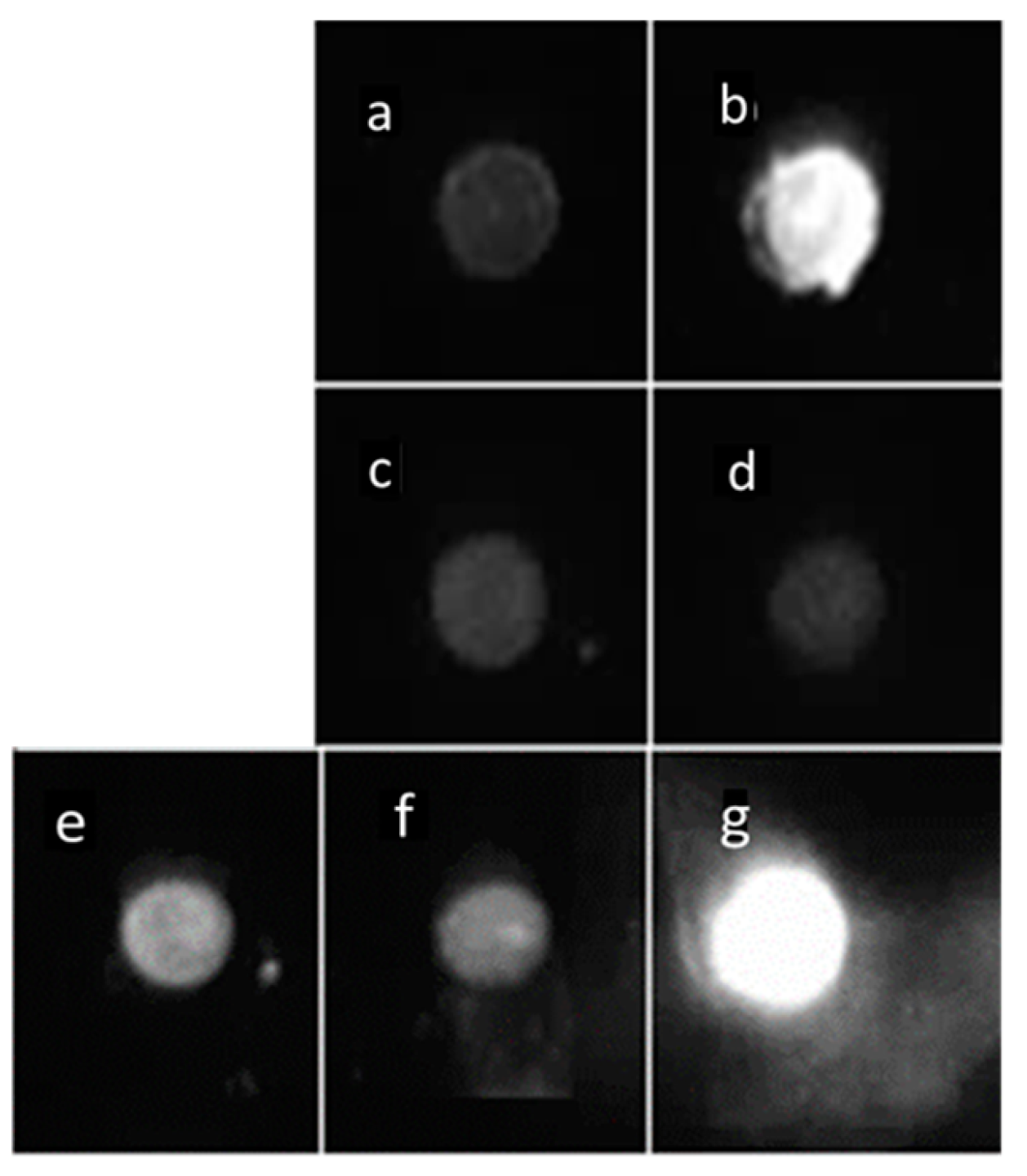
2.4. Endogenous Bcl-xL Is Converted into Amyloid Aggregates Following Apoptosis Induced by Metal Stress in SH-SY5Y Cells
2.5. The B3-9 c-mAb Specifically Immunoprecipitates Bcl-xL from Cells Subjected to Metal-Induced Apoptosis
3. Discussion
4. Materials and Methods
4.1. Generation of Fibrils
4.2. Electron Microscopy
4.3. Fluorescence Spectroscopy Experiments
4.4. Production and Purification of Specific Conformational Monoclonal Antibodies (mAbs) Against Amyloid Bcl-xL Fibers
4.5. Dot Blotting
4.6. Cell Culture
4.7. Metal Oxidative Stress
4.8. Flow Cytometry
4.9. Immunofluorescence
4.10. Immuno-Precipitation (IP) Experiments
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
References
- Tamano, H.; Takeda, A. Dynamic action of neurometals at the synapse. Metallomics 2011, 3, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Cicero, C.E.; Mostile, G.; Vasta, R.; Rapisarda, V.; Signorelli, S.S.; Ferrante, M.; Zappia, M.; Nicoletti, A. Metals and neurodegenerative diseases. A systematic review. Environ. Res. 2017, 159, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Kato-Negishi, M.; Tanaka, K. Cross talk between neurometals and amyloidogenic proteins at the synapse and the pathogenesis of neurodegenerative diseases. Metallomics 2017, 9, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Reybier, K.; Ayala, S.; Alies, B.; Rodrigues, J.V.; Bustos Rodriguez, S.; La Penna, G.; Collin, F.; Gomes, C.M.; Hureau, C.; Faller, P. Free Superoxide is an Intermediate in the Production of H2O2 by Copper(I)-Aβ Peptide and O2. Angew. Chem. Int. Ed. 2016, 55, 1085–1089. [Google Scholar] [CrossRef] [PubMed]
- Telling, N.D.; Everett, J.; Collingwood, J.F.; Dobson, J.; van der Laan, G.; Gallagher, J.J.; Wang, J.; Hitchcock, A.P. Iron Biochemistry is Correlated with Amyloid Plaque Morphology in an Established Mouse Model of Alzheimer’s Disease. Cell Chem. Biol. 2017, 24, 1205–1215.e3. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, G.; Chakrabarti, S.; Chatterjee, U.; Saso, L. Proteinopathy, oxidative stress and mitochondrial dysfunction: Cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Des. Devel. Ther. 2017, 11, 797–810. [Google Scholar] [CrossRef]
- Leuner, K.; Müller, W.E.; Reichert, A.S. From mitochondrial dysfunction to amyloid beta formation: Novel insights into the pathogenesis of Alzheimer’s disease. Mol. Neurobiol. 2012, 46, 186–193. [Google Scholar] [CrossRef]
- Paola, D.; Domenicotti, C.; Nitti, M.; Vitali, A.; Borghi, R.; Cottalasso, D.; Zaccheo, D.; Odetti, P.; Strocchi, P.; Marinari, U.M.; et al. Oxidative stress induces increase in intracellular amyloid beta-protein production and selective activation of betaI and betaII PKCs in NT2 cells. Biochem. Biophys. Res. Commun. 2000, 268, 642–646. [Google Scholar] [CrossRef]
- Tamagno, E.; Bardini, P.; Obbili, A.; Vitali, A.; Borghi, R.; Zaccheo, D.; Pronzato, M.A.; Danni, O.; Smith, M.A.; Perry, G.; et al. Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol. Dis. 2002, 10, 279–288. [Google Scholar] [CrossRef]
- Fischer, R.; Maier, O. Interrelation of oxidative stress and inflammation in neurodegenerative disease: Role of TNF. Oxid. Med. Cell. Longev. 2015, 2015, 610813. [Google Scholar] [CrossRef]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Anilkumar, U.; Prehn, J.H.M. Anti-apoptotic BCL-2 family proteins in acute neural injury. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Wei, M.C.; Zong, W.-X.; Cheng, E.H.-Y.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A Requisite Gateway to Mitochondrial Dysfunction and Death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef]
- Hsu, Y.-T.; Wolter, K.G.; Youle, R.J. Cytosol-to-membrane redistribution of Bax and Bcl-XL during apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 3668–3672. [Google Scholar] [CrossRef]
- Happo, L.; Strasser, A.; Cory, S. BH3-only proteins in apoptosis at a glance. J. Cell Sci. 2012, 125, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Cheng, E.H. Conversion of Bcl-2 to a Bax-like Death Effector by Caspases. Science 1997, 278, 1966–1968. [Google Scholar] [CrossRef]
- Sattler, M.; Liang, H.; Nettesheim, D.; Meadows, R.P.; Harlan, J.E.; Eberstadt, M.; Yoon, H.S.; Shuker, S.B.; Chang, B.S.; Minn, A.J.; et al. Structure of Bcl-xL-Bak Peptide Complex: Recognition Between Regulators of Apoptosis. Science 1997, 275, 983–986. [Google Scholar] [CrossRef]
- Ivanovska, I.; Galonek, H.L.; Hildeman, D.A.; Hardwick, J.M. Regulation of cell death in the lymphoid system by Bcl-2 family proteins. Acta Haematol. 2004, 111, 42–55. [Google Scholar] [CrossRef]
- Soane, L.; Siegel, Z.T.; Schuh, R.A.; Fiskum, G. Postnatal Developmental Regulation of Bcl-2 Family Proteins in Brain Mitochondria. J. Neurosci. Res. 2008, 86, 1267–1276. [Google Scholar] [CrossRef]
- Beaumatin, F.; El Dhaybi, M.; Bobo, C.; Verdier, M.; Priault, M. Bcl-xL deamidation and cancer: Charting the fame trajectories of legitimate child and hidden siblings. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2017, 1864, 1734–1745. [Google Scholar] [CrossRef] [PubMed]
- Seng, N.; Megyesi, J.; Tarcsafalvi, A.; Price, P. Mimicking Cdk2 phosphorylation of Bcl-xL at Ser73 results in caspase activation and Bcl-xL cleavage. Cell Death Discov. 2016, 2, 16001. [Google Scholar] [CrossRef]
- Kharbanda, S.; Pandey, P.; Schofield, L.; Israels, S.; Roncinske, R.; Yoshida, K.; Bharti, A.; Yuan, Z.-M.; Saxena, S.; Weichselbaum, R.; et al. Role for Bcl-xL as an inhibitor of cytosolic cytochrome C accumulation in DNA damage-induced apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 6939–6942. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.N.; Wang, X.; Huang, Y.; Ibrado, A.M.; Liu, L.; Fang, G.; Bhalla, K. Overexpression of Bcl-X(L) inhibits Ara-C-induced mitochondrial loss of cytochrome c and other perturbations that activate the molecular cascade of apoptosis. Cancer Res. 1997, 57, 3115–3120. [Google Scholar] [PubMed]
- Carthy, C.M.; Yanagawa, B.; Luo, H.; Granville, D.J.; Yang, D.; Cheung, P.; Cheung, C.; Esfandiarei, M.; Rudin, C.M.; Thompson, C.B.; et al. Bcl-2 and Bcl-xL overexpression inhibits cytochrome c release, activation of multiple caspases, and virus release following coxsackievirus B3 infection. Virology 2003, 313, 147–157. [Google Scholar] [CrossRef]
- Zaidi, A.U.; D’Sa-Eipper, C.; Brenner, J.; Kuida, K.; Zheng, T.S.; Flavell, R.A.; Rakic, P.; Roth, K.A. Bcl-X (L) –Caspase-9 Interactions in the Developing Nervous System: Evidence for Multiple Death Pathways. J. Neurosci. 2001, 21, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Bruey, J.-M.; Bruey-Sedano, N.; Luciano, F.; Zhai, D.; Balpai, R.; Xu, C.; Kress, C.L.; Bailly-Maitre, B.; Li, X.; Osterman, A.; et al. Bcl-2 and Bcl-XL Regulate Proinflammatory Caspase-1 Activation by Interaction with NALP1. Cell 2007, 129, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Aon, M.A.; Hsu, Y.-T.; Soane, L.; Teng, X.; McCaffery, J.M.; Cheng, W.-C.; Qi, B.; Li, H.; Alavian, K.N.; et al. Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J. Cell Biol. 2011, 195, 263–276. [Google Scholar] [CrossRef]
- Alavian, K.N.; Li, H.; Collis, L.; Bonanni, L.; Zeng, L.; Sacchetti, S.; Lazrove, E.; Nabili, P.; Flaherty, B.; Graham, M.; et al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat. Cell Biol. 2011, 13, 1224–1233. [Google Scholar] [CrossRef]
- Li, H.; Chen, Y.; Jones, A.F.; Sanger, R.H.; Collis, L.P.; Flannery, R.; McNay, E.C.; Yu, T.; Schwarzenbacher, R.; Bossy, B.; et al. Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 2169–2174. [Google Scholar] [CrossRef]
- Park, H.-A.; Licznerski, P.; Alavian, K.N.; Shanabrough, M.; Jonas, E.A. Bcl-xL Is Necessary for Neurite Outgrowth in Hippocampal Neurons. Antioxid. Redox Signal. 2015, 22, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Hyman, B.T.; Yuan, J. Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nat. Rev. Neurosci. 2012, 13, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Nagahashi, A.; Nagashima, K.; Rokudai, S.; Tsuruo, T. Acceleration of apoptotic cell death after the cleavage of Bcl-XL protein by caspase-3-like proteases. Oncogene 1998, 17, 1295–1304. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, F.; Salomoni, P.; Cotteret, S.; Cesi, V.; Srinivasula, S.M.; Alnemri, E.S.; Calabretta, B. Caspase cleavage enhances the apoptosis-inducing effects of BAD. Mol. Cell. Biol. 2001, 21, 3025–3036. [Google Scholar] [CrossRef]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef]
- Nakagawa, T.; Yuan, J. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J. Cell Biol. 2000, 150, 887–894. [Google Scholar] [CrossRef]
- Wood, D.E.; Newcomb, E.W. Cleavage of Bax enhances its cell death function. Exp. Cell Res. 2000, 256, 375–382. [Google Scholar] [CrossRef]
- Clem, R.J.; Cheng, E.H.-Y.; Karp, C.L.; Kirsch, D.G.; Ueno, K.; Takahashi, A.; Kastan, M.B.; Griffin, D.E.; Earnshaw, W.C.; Veliuona, M.A.; et al. Modulation of cell death by Bcl-xL through caspase interaction. Proc. Natl. Acad. Sci. USA 1998, 95, 554–559. [Google Scholar] [CrossRef]
- Jonas, E.A.; Hickman, J.A.; Chachar, M.; Polster, B.M.; Brandt, T.A.; Fannjiang, Y.; Ivanovska, I.; Basañez, G.; Kinnally, K.W.; Zimmerberg, J.; et al. Proapoptotic N-truncated BCL-xL protein activates endogenous mitochondrial channels in living synaptic terminals. Proc. Natl. Acad. Sci. USA 2004, 101, 13590–13595. [Google Scholar] [CrossRef]
- Krajewska, M.; Mai, J.; Zapata, J.; Ashwell, K.; Schendel, S.; Reed, J.; Krajewski, S. Dynamics of expression of apoptosis-regulatory proteins Bid, Bcl-2, Bcl-X, Bax and Bak during development of murine nervous system. Cell Death Differ. 2002, 9, 145–157. [Google Scholar] [CrossRef]
- Hickman, J.A.; Hardwick, J.M.; Kaczmarek, L.K.; Jonas, E.A. Bcl-xL Inhibitor ABT-737 Reveals a Dual Role for Bcl-xL in Synaptic Transmission. J. Neurophysiol. 2008, 99, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- Jonas, E.A.; Hickman, J.A.; Hardwick, J.M.; Kaczmarek, L.K. Exposure to Hypoxia Rapidly Induces Mitochondrial Channel Activity within a Living Synapse. J. Biol. Chem. 2005, 280, 4491–4497. [Google Scholar] [CrossRef]
- Ofengeim, D.; Chen, Y.; Miyawaki, T.; Li, H.; Sacchetti, S.; Flannery, R.J.; Alavian, K.N.; Pontarelli, F.; Roelofs, B.A.; Hickman, J.A.; et al. N-terminally cleaved Bcl-xL mediates ischemia-induced neuronal death. Nat. Neurosci. 2012, 15, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-A.; Licznerski, P.; Mnatsakanyan, N.; Niu, Y.; Sacchetti, S.; Wu, J.; Polster, B.M.; Alavian, K.N.; Jonas, E.A. Inhibition of Bcl-xL prevents pro-death actions of ΔN-Bcl-xL at the mitochondrial inner membrane during glutamate excitotoxicity. Cell Death Differ. 2017, 24, 1963–1974. [Google Scholar] [CrossRef]
- Ruvolo, P.P.; Deng, X.; May, W.S. Phosphorylation of Bcl2 and regulation of apoptosis. Leukemia 2001, 15, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ueta, E.; Kimura, T.; Yamamoto, T.; Osaki, T. Reactive oxygen species (ROS) control the expression of Bcl-2 family proteins by regulating their phosphorylation and ubiquitination. Cancer Sci. 2004, 95, 644–650. [Google Scholar] [CrossRef]
- Boise, L.H.; Gonzalez-Garcia, M.; Postema, C.E.; Ding, L.; Lindsten, T.; Turka, L.A.; Mao, X.; Nunez, G.; Thompson, C.B. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 1993, 74, 597–608. [Google Scholar] [CrossRef]
- Urrutia, P.J.; Mena, N.P.; Nunez, M.T. The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders. Front. Pharmacol. 2014, 5. [Google Scholar] [CrossRef]
- Megyesi, J.; Tarcsafalvi, A.; Seng, N.; Hodeify, R.; Price, P.M. Cdk2 phosphorylation of Bcl-xL after stress converts it to a pro-apoptotic protein mimicking Bax/Bak. Cell Death Discov. 2016, 2, 15066. [Google Scholar] [CrossRef]
- Brutus, N.A.; Hanley, S.; Ashraf, Q.M.; Mishra, O.P.; Delivoria-Papadopoulos, M. Effect of Hyperoxia on Serine Phosphorylation of Apoptotic Proteins in Mitochondrial Membranes of the Cerebral Cortex of Newborn Piglets. Neurochem. Res. 2009, 34, 1219–1225. [Google Scholar] [CrossRef]
- Chenal, A.; Vendrely, C.; Vitrac, H.; Karst, J.C.; Gonneaud, A.; Blanchet, C.E.; Pichard, S.; Garcia, E.; Salin, B.; Catty, P.; et al. Amyloid Fibrils Formed by the Programmed Cell Death Regulator Bcl-xL. J. Mol. Biol. 2012, 415, 584–599. [Google Scholar] [CrossRef] [PubMed]
- Kutuk, O.; Basaga, H. Bcl-2 protein family: Implications in vascular apoptosis and atherosclerosis. Apoptosis 2006, 11, 1661–1675. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, D.; He, J.; Chen, L.; Li, H. Bcl-XL: A multifunctional anti-apoptotic protein. Pharmacol. Res. 2020, 151, 104547. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Guerrero, A.D.; Huang, L.; Shabier, Z.; Pan, M.; Tan, T.-H.; Wang, J. Caspase-9-induced mitochondrial disruption through cleavage of anti-apoptotic BCL-2 family members. J. Biol. Chem. 2007, 282, 33888–33895. [Google Scholar] [CrossRef]
- Chenal, A.; Dartevelle, S.; Nato, F.; Almunia, C.; Marquette, C.; Gonneaud, A.; Garcia, E.; Forge, V. Production et caractérisation d’anticorps monoclonaux spécifiques de la conformation amyloïde de la protéine Bcl-XL in vitro et in vivo. Applications à la détection de fibres amyloïdes in vivo après stress apoptotique et au criblage de molécules régulatrices de l’amyloïdogenèse in vitro. Invention deposit at the National Collection of Microorganism Culture of the Institut Pasteur. n°CNCM-30032.1110, 2012. [Google Scholar]
- Yang, T.; Zhu, Z.; Yin, E.; Wang, Y.; Zhang, C.; Yuan, H.; Zhang, H.; Jin, S.; Guo, Z.; Wang, X. Alleviation of symptoms of Alzheimer’s disease by diminishing Aβ neurotoxicity and neuroinflammation. Chem. Sci. 2019, 10, 10149–10158. [Google Scholar] [CrossRef]
- Faller, P.; Hureau, C.; La Penna, G. Metal ions and intrinsically disordered proteins and peptides: From Cu/Zn amyloid-beta to general principles. Acc. Chem. Res. 2014, 47, 2252–2259. [Google Scholar] [CrossRef]
- Bader, B.; Nubling, G.; Mehle, A.; Nobile, S.; Kretzschmar, H.; Giese, A. Single particle analysis of tau oligomer formation induced by metal ions and organic solvents. Biochem. Biophys. Res. Commun. 2011, 411, 190–196. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 2001, 276, 44284–44296. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Vardaki, I.; Sanchez, C.; Fonseca, P.; Olsson, M.; Chioureas, D.; Rassidakis, G.; Ullén, A.; Zhivotovsky, B.; Björkholm, M.; Panaretakis, T. Caspase-3–dependent cleavage of Bcl-xL in the stroma exosomes is required for their uptake by hematological malignant cells. Blood 2016, 128, 2655–2665. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonneaud, A.; Fakhir, F.-Z.; Landas, E.; Le Tallec, E.; Chartier-Garcia, E.; Almunia, C.; Chenal, A.; Forge, V.; Marquette, C. Development of Conformational Antibodies to Detect Bcl-xL’s Amyloid Aggregates in Metal-Induced Apoptotic Neuroblastoma Cells. Int. J. Mol. Sci. 2020, 21, 7625. https://doi.org/10.3390/ijms21207625
Gonneaud A, Fakhir F-Z, Landas E, Le Tallec E, Chartier-Garcia E, Almunia C, Chenal A, Forge V, Marquette C. Development of Conformational Antibodies to Detect Bcl-xL’s Amyloid Aggregates in Metal-Induced Apoptotic Neuroblastoma Cells. International Journal of Molecular Sciences. 2020; 21(20):7625. https://doi.org/10.3390/ijms21207625
Chicago/Turabian StyleGonneaud, Alexis, Fatima-Zohra Fakhir, Emeline Landas, Enora Le Tallec, Elisabeth Chartier-Garcia, Christine Almunia, Alexandre Chenal, Vincent Forge, and Christel Marquette. 2020. "Development of Conformational Antibodies to Detect Bcl-xL’s Amyloid Aggregates in Metal-Induced Apoptotic Neuroblastoma Cells" International Journal of Molecular Sciences 21, no. 20: 7625. https://doi.org/10.3390/ijms21207625
APA StyleGonneaud, A., Fakhir, F.-Z., Landas, E., Le Tallec, E., Chartier-Garcia, E., Almunia, C., Chenal, A., Forge, V., & Marquette, C. (2020). Development of Conformational Antibodies to Detect Bcl-xL’s Amyloid Aggregates in Metal-Induced Apoptotic Neuroblastoma Cells. International Journal of Molecular Sciences, 21(20), 7625. https://doi.org/10.3390/ijms21207625