IRF-1 Inhibits Angiogenic Activity of HPV16 E6 Oncoprotein in Cervical Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
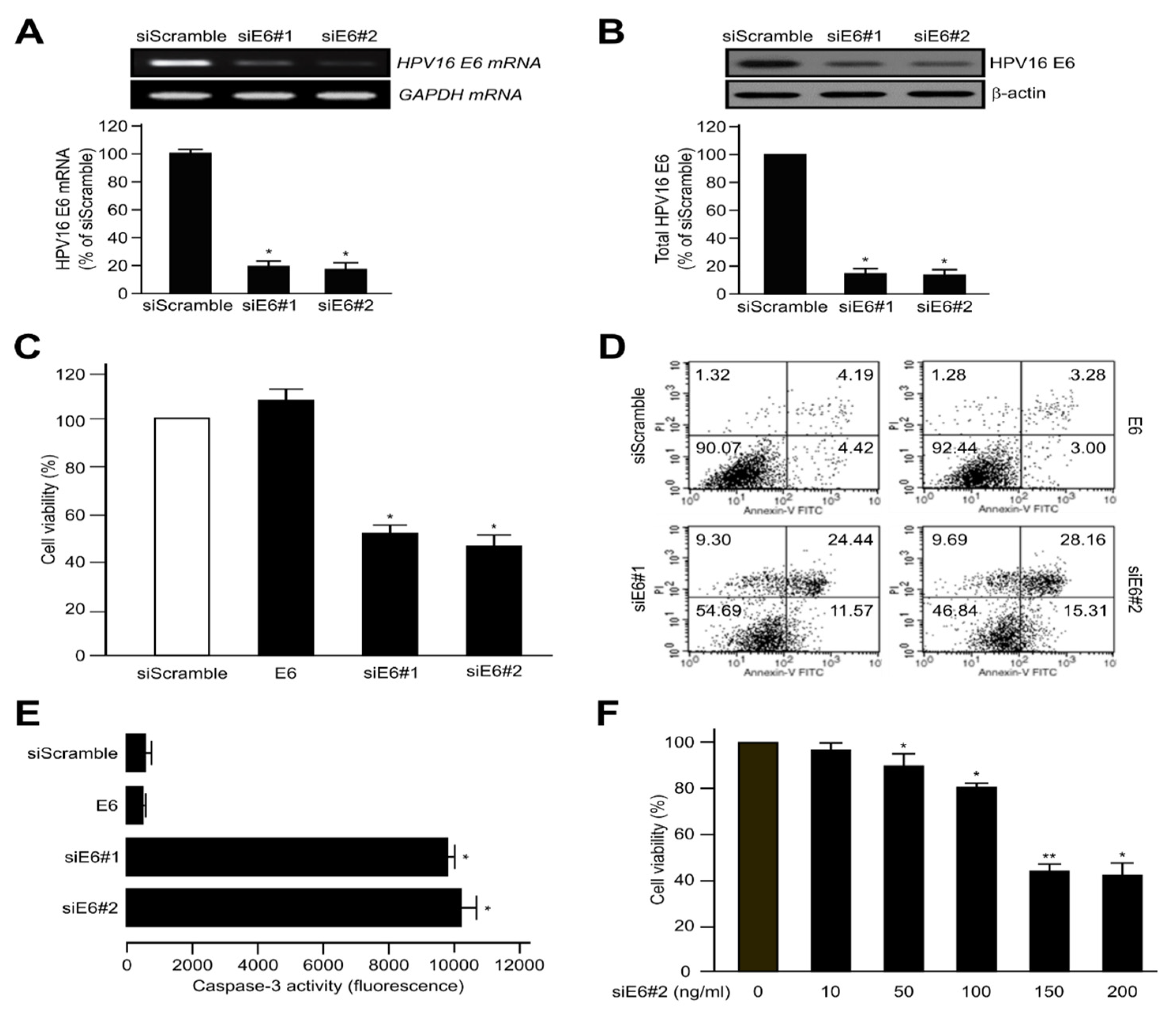
2.1. HPV16 E6 Oncoprotein Was Strongly Expressed by Inhibiting Apoptosis and Inactivating Caspase-3 in Human Cervical Cancer Cells
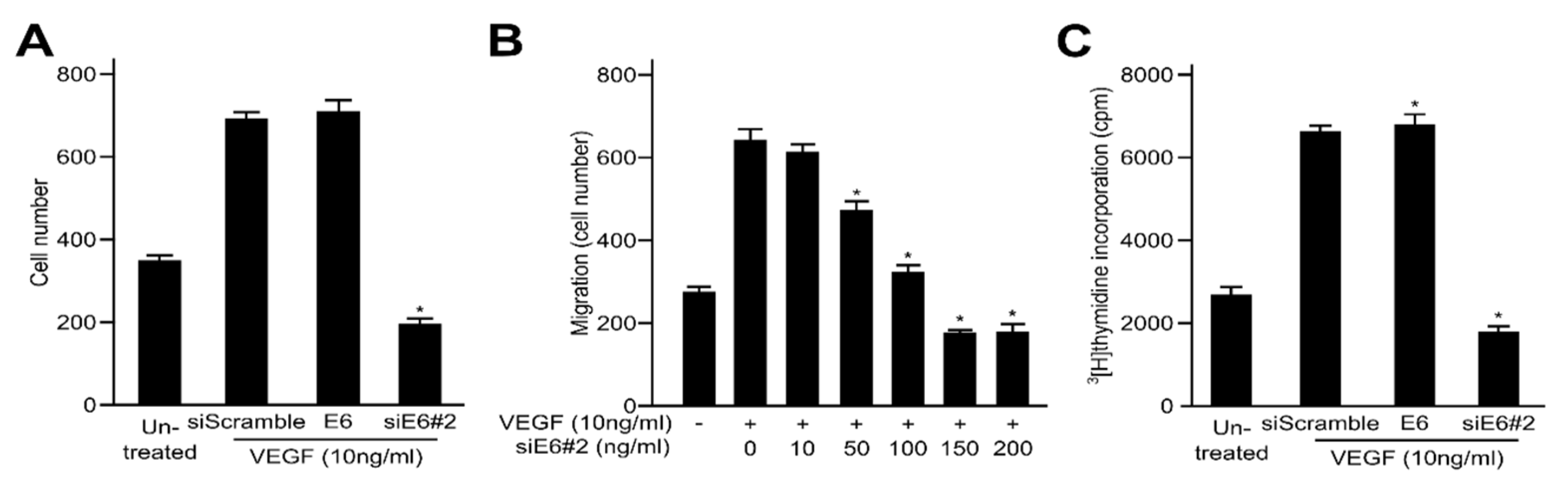
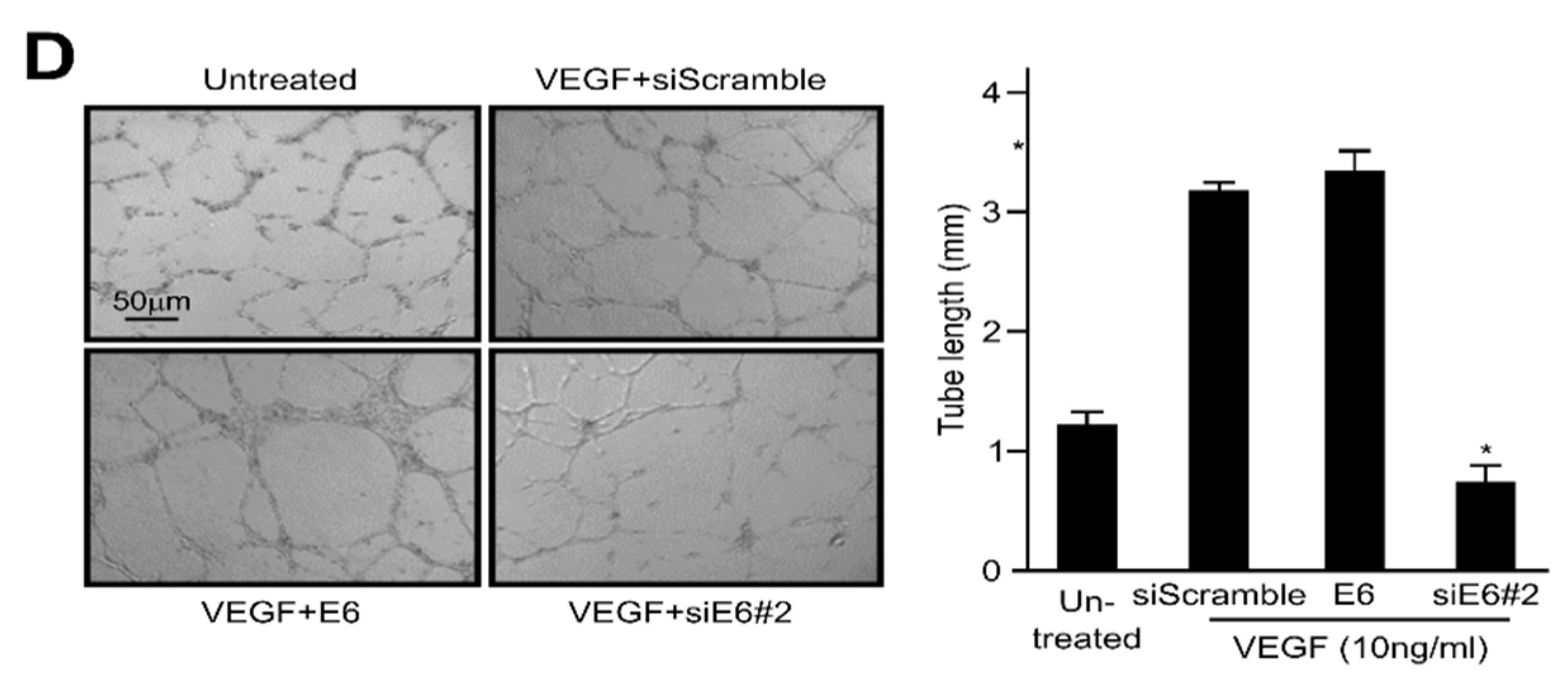
2.2. HPV16 E6 Facilitates VEGF-Induced Endothelial Cell Migration, Proliferation, and Tube Formation In Vitro
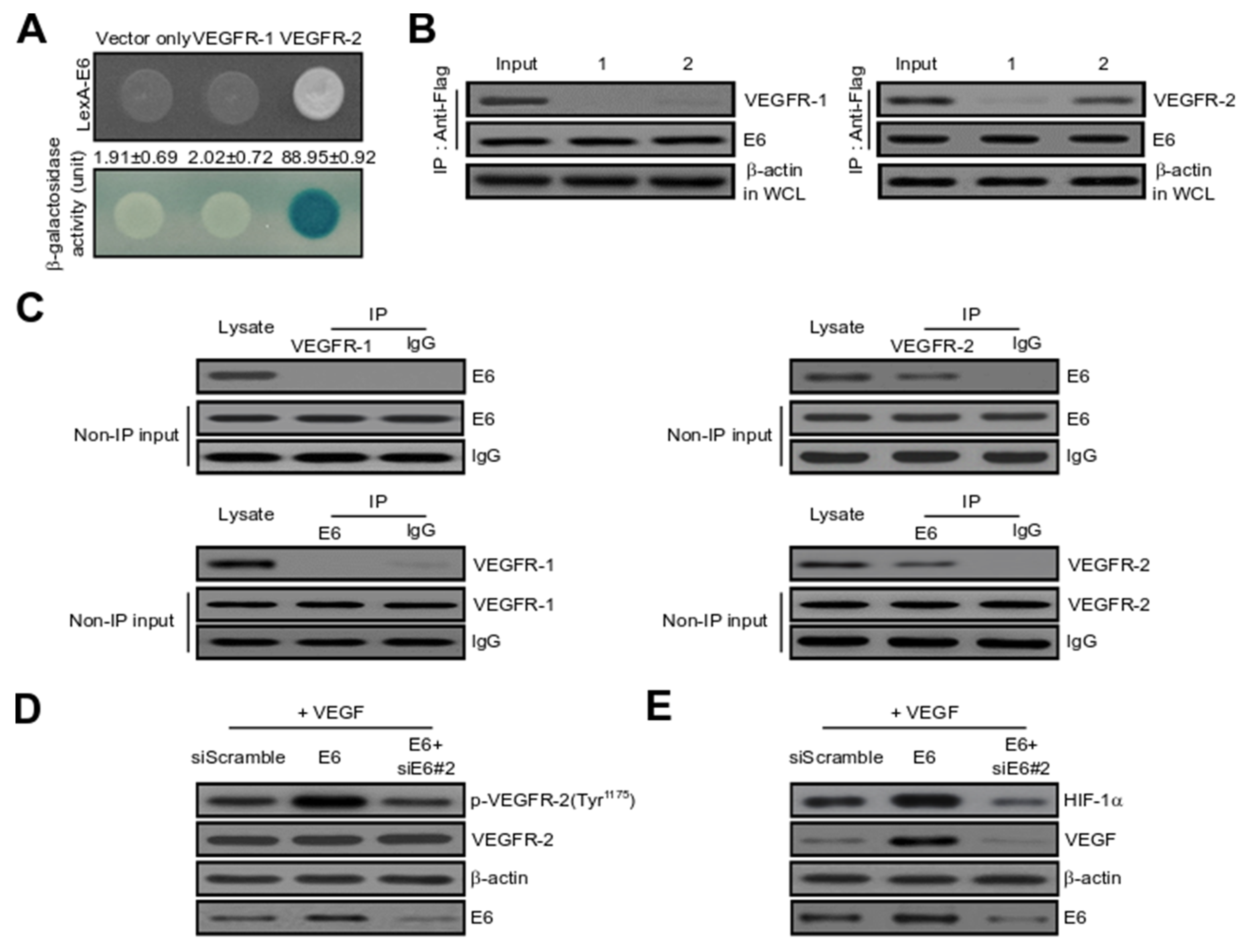
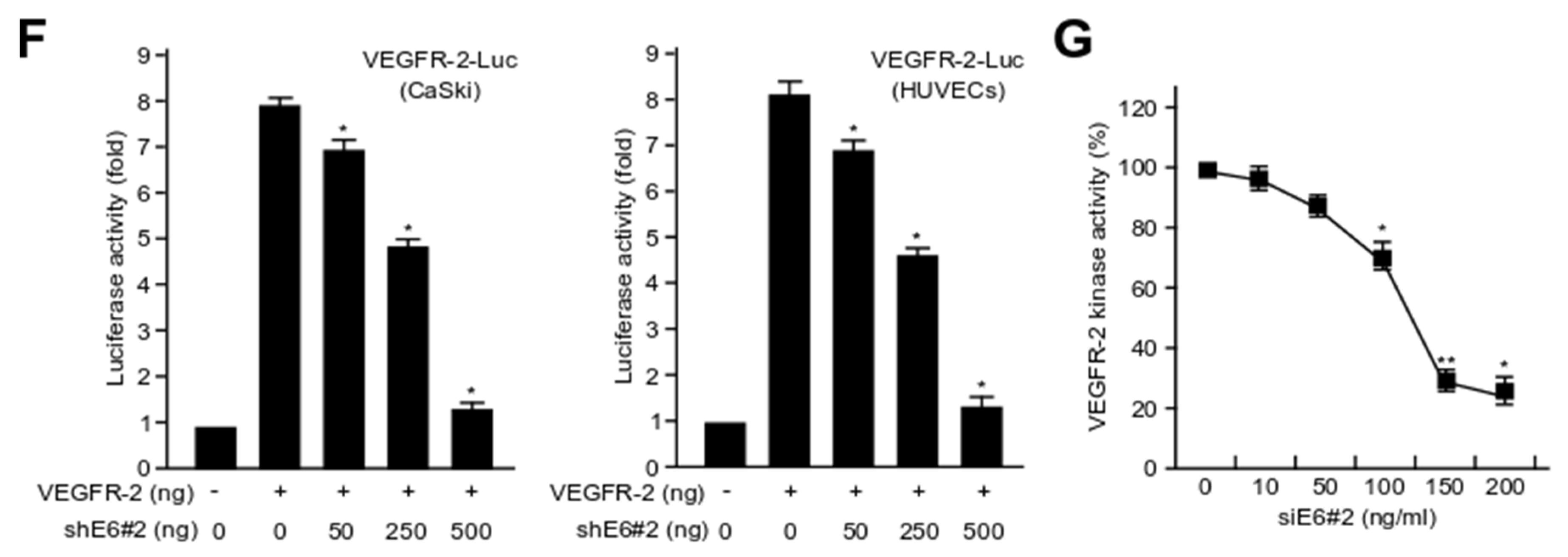
2.3. HPV16 E6 Directly Binds with VEGFR-2 but Not VEGFR-1
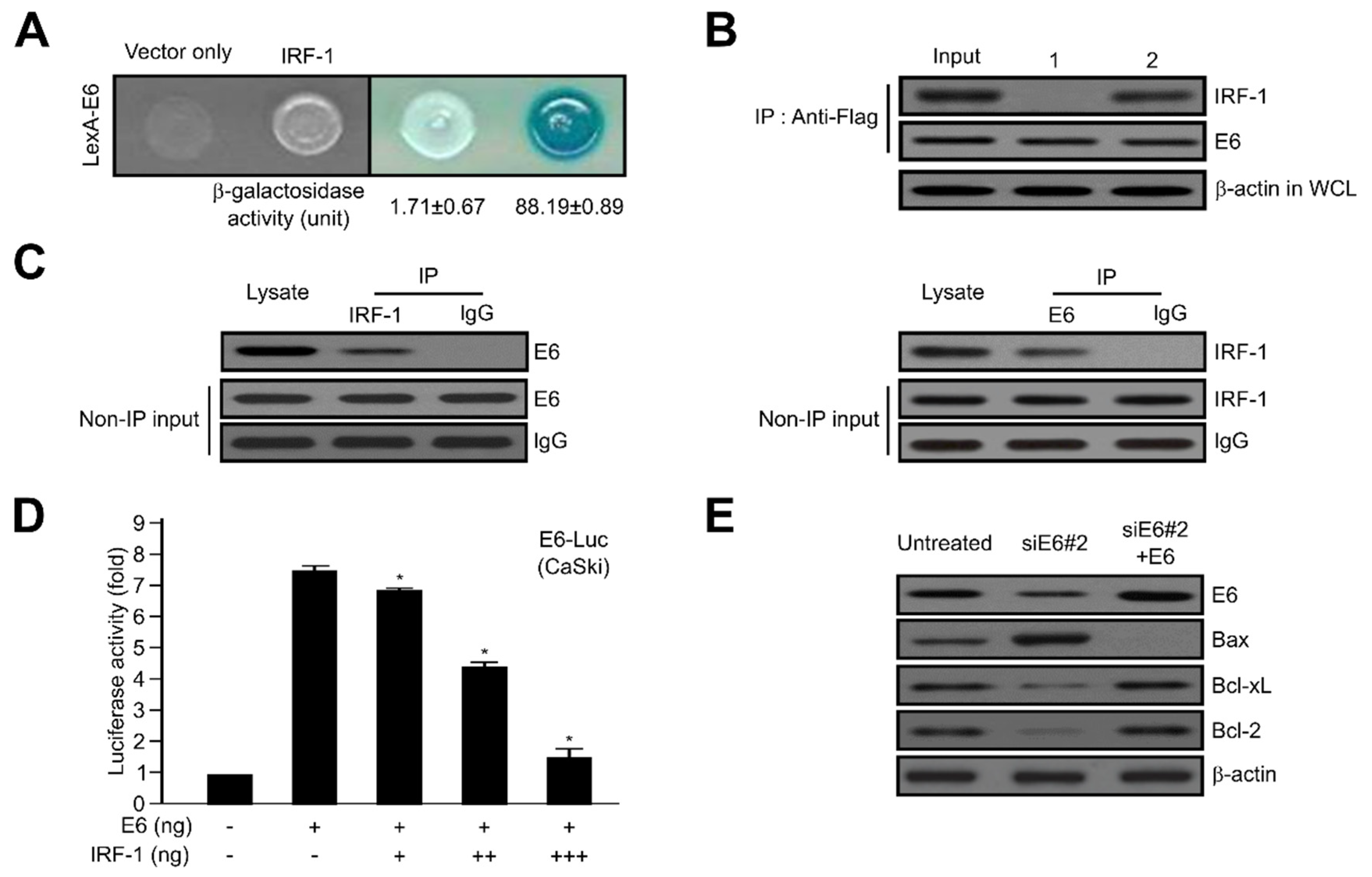
2.4. HPV16 E6 Directly Interacts with IRF-1
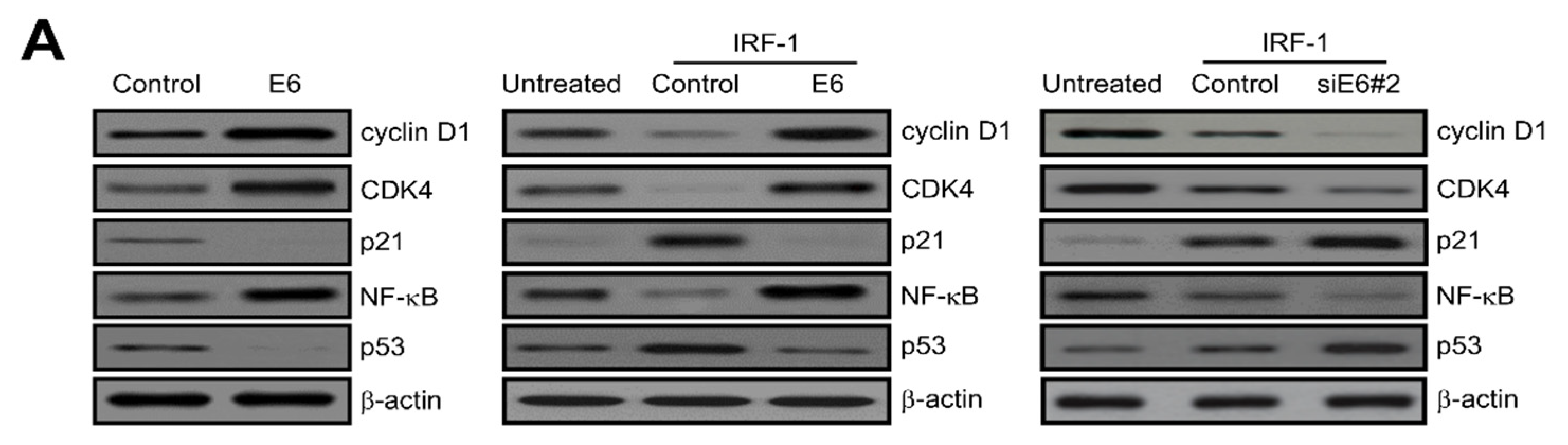
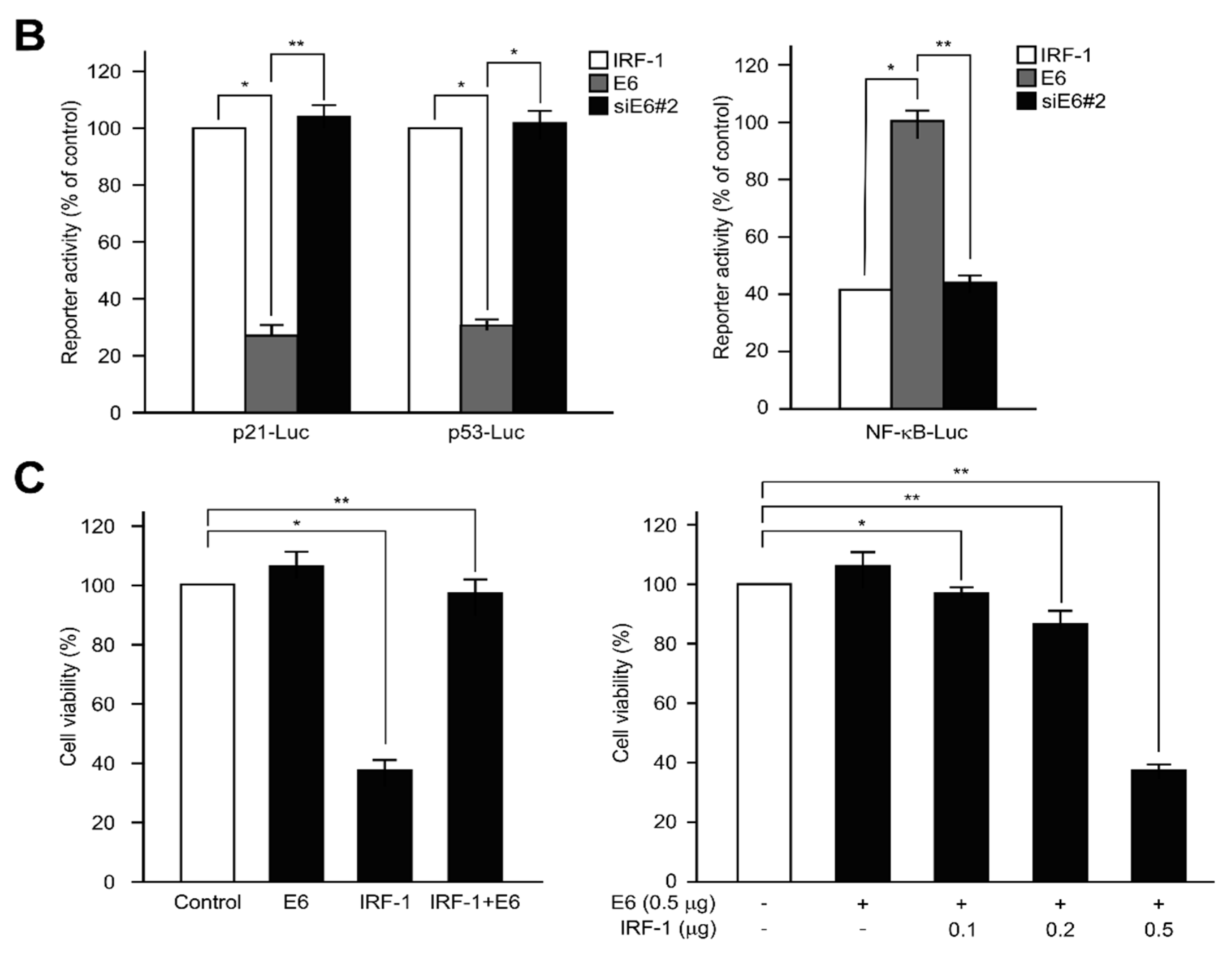
2.5. IRF-1 Controls the Expression of Cell Cycle- and Apoptosis-Regulatory Proteins
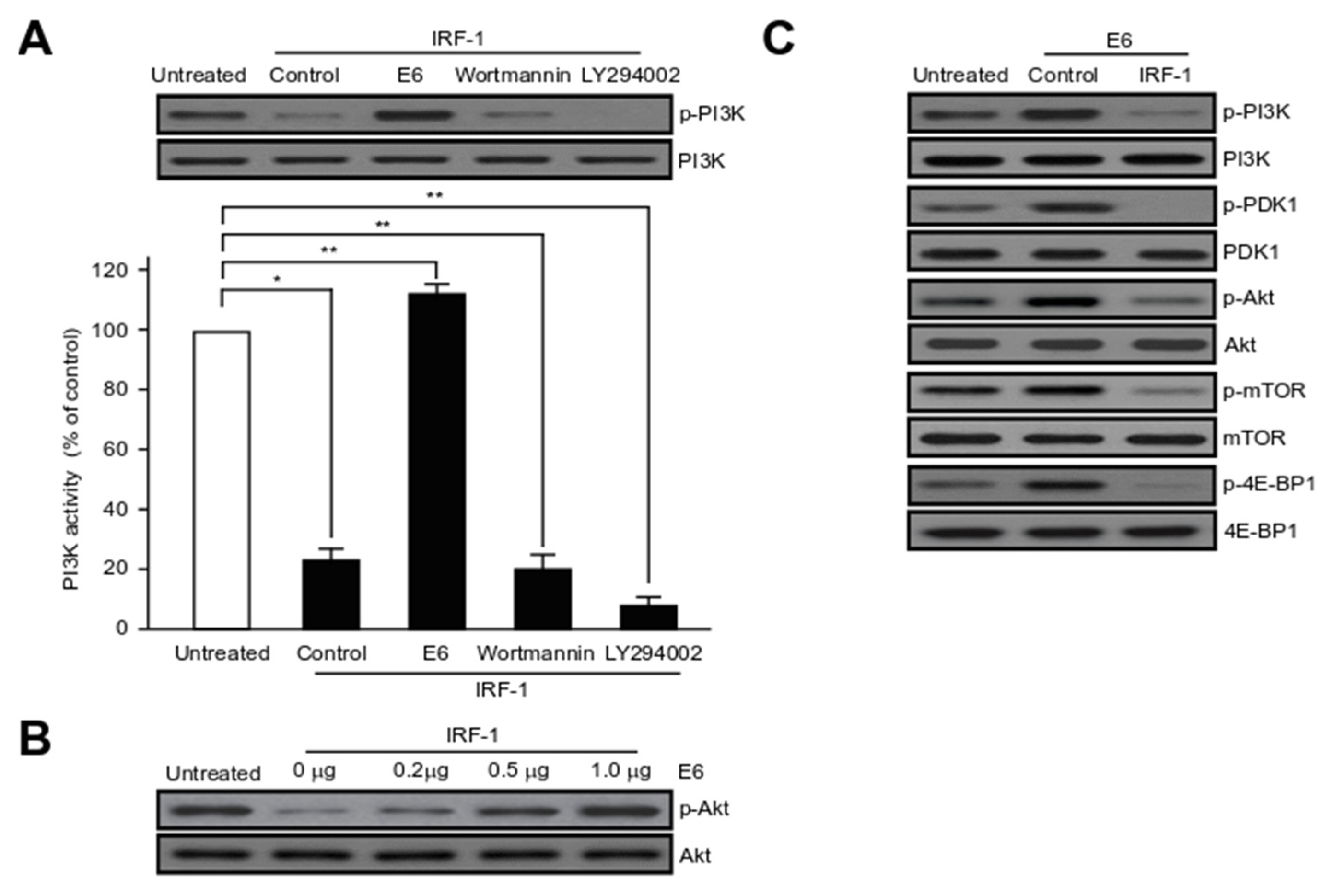
2.6. HPV16 E6 Activates Major Component Phosphorylation in PI3K/mTOR Signalling Pathway
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Reagents, and Antibodies
4.2. Flow Cytometric Analysis of Apoptosis and Cell Viability
4.3. Construction of Small Interfering RNA (siRNA) and Caspase-3 Activity Assay
4.4. Yeast Two-Hybrid (Y2H) Screening and Quantification of Binding Interaction
4.5. Co-Immunoprecipitation (Co-IP) and Western Blot Analysis
4.6. Luciferase, VEGFR-2 Kinase, and PI3K Activity Assays
4.7. Endothelial Cell Migration and Capillary-Like Tubular Structure Formation
4.8. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HPV | Human papillomavirus |
| IRF-1 | Interferon regulatory factor-1 |
| Rb | Retinoblastoma |
| E6AP | E6-associated protein |
| TRADD | Tumour necrosis factor receptor type 1-associated death domain protein |
| TNFR-1 | Tumour necrosis factor receptor 1 |
| FADD | Fas-associated death domain |
| DISC | Death-inducing signalling complex |
| LOH | Loss of heterozygosity |
| AML | Acute myelogenous leukaemia |
| DBD | DNA-binding domain |
| NLS | Nuclear location signal |
| TA | Transactivation domain |
| VEGFR-2 | Vascular endothelial growth factor receptor 2 |
| VEGF | Vascular endothelial growth factor |
| HIF1-α | Hypoxia inducible factor-1α |
| HUVECs | Human umbilical vein endothelial cells |
| siRNA | Small interfering RNA |
| Y2H | Yeast two-hybrid |
| Co-IP | Co-immunoprecipitation |
References
- Snijders, P.J.; Steenbergen, R.D.; Heideman, D.A.; Meijer, C.J. HPV-mediated cervical carcinogenesis: Concepts and clinical implications. J. Pathol. 2006, 208, 152–164. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- De Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; Hausen, H.Z. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef]
- McLaughlin-Drubin, M.E.; Munger, K. Oncogenic activities of human papillomaviruses. Virus Res. 2009, 143, 195–208. [Google Scholar] [CrossRef]
- Tomaic, V. Functional Roles of E6 and E7 Oncoproteins in HPV-Induced Malignancies at Diverse Anatomical Sites. Cancers 2016, 8, 95. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef]
- Balasubramaniam, S.D.; Balakrishnan, V.; Oon, C.E.; Kaur, G. Key Molecular Events in Cervical Cancer Development. Medicina 2019, 55, 384. [Google Scholar] [CrossRef]
- Howie, H.L.; Katzenellenbogen, R.A.; Galloway, D.A. Papillomavirus E6 proteins. Virology 2009, 384, 324–334. [Google Scholar] [CrossRef]
- Mittal, S.; Banks, L. Molecular mechanisms underlying human papillomavirus E6 and E7 oncoprotein-induced cell transformation. Mutat. Res. Rev. Mutat. Res. 2017, 772, 23–35. [Google Scholar] [CrossRef]
- Vande Pol, S.B.; Klingelhutz, A.J. Papillomavirus E6 oncoproteins. Virology 2013, 445, 115–137. [Google Scholar] [CrossRef]
- Ganti, K.; Broniarczyk, J.; Manoubi, W.; Massimi, P.; Mittal, S.; Pim, D.; Szalmas, A.; Thatte, J.; Thomas, M.; Tomaic, V.; et al. The Human Papillomavirus E6 PDZ Binding Motif: From Life Cycle to Malignancy. Viruses 2015, 7, 3530–3551. [Google Scholar] [CrossRef]
- Filippova, M.; Song, H.; Connolly, J.L.; Dermody, T.S.; Duerksen-Hughes, P.J. The human papillomavirus 16 E6 protein binds to tumor necrosis factor (TNF) R1 and protects cells from TNF-induced apoptosis. J. Biol. Chem. 2002, 277, 21730–21739. [Google Scholar] [CrossRef]
- Filippova, M.; Parkhurst, L.; Duerksen-Hughes, P.J. The human papillomavirus 16 E6 protein binds to Fas-associated death domain and protects cells from Fas-triggered apoptosis. J. Biol. Chem. 2004, 279, 25729–25744. [Google Scholar] [CrossRef]
- Garnett, T.O.; Filippova, M.; Duerksen-Hughes, P.J. Accelerated degradation of FADD and procaspase 8 in cells expressing human papilloma virus 16 E6 impairs TRAIL-mediated apoptosis. Cell Death Differ. 2006, 13, 1915–1926. [Google Scholar] [CrossRef]
- Filippova, M.; Johnson, M.M.; Bautista, M.; Filippov, V.; Fodor, N.; Tungteakkhun, S.S.; Williams, K.; Duerksen-Hughes, P.J. The large and small isoforms of human papillomavirus type 16 E6 bind to and differentially affect procaspase 8 stability and activity. J. Virol. 2007, 81, 4116–4129. [Google Scholar] [CrossRef]
- Tungteakkhun, S.S.; Filippova, M.; Fodor, N.; Duerksen-Hughes, P.J. The full-length isoform of human papillomavirus 16 E6 and its splice variant E6* bind to different sites on the procaspase 8 death effector domain. J. Virol. 2010, 84, 1453–1463. [Google Scholar] [CrossRef]
- Rataj, O.; Haedicke-Jarboui, J.; Stubenrauch, F.; Iftner, T. Brd4 inhibition suppresses HPV16 E6 expression and enhances chemoresponse: A potential new target in cervical cancer therapy. Int. J. Cancer 2019, 144, 2330–2338. [Google Scholar] [CrossRef]
- Miyamoto, M.; Fujita, T.; Kimura, Y.; Maruyama, M.; Harada, H.; Sudo, Y.; Miyata, T.; Taniguchi, T. Regulated expression of a gene encoding a nuclear factor, IRF-1, that specifically binds to IFN-beta gene regulatory elements. Cell 1988, 54, 903–913. [Google Scholar] [CrossRef]
- Fujita, T.; Kimura, Y.; Miyamoto, M.; Barsoumian, E.L.; Taniguchi, T. Induction of endogenous IFN-alpha and IFN-beta genes by a regulatory transcription factor, IRF-1. Nature 1989, 337, 270–272. [Google Scholar] [CrossRef]
- Harada, H.; Fujita, T.; Miyamoto, M.; Kimura, Y.; Maruyama, M.; Furia, A.; Miyata, T.; Taniguchi, T. Structurally similar but functionally distinct factors, IRF-1 and IRF-2, bind to the same regulatory elements of IFN and IFN-inducible genes. Cell 1989, 58, 729–739. [Google Scholar] [CrossRef]
- Taniguchi, T.; Ogasawara, K.; Takaoka, A.; Tanaka, N. IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol. 2001, 19, 623–655. [Google Scholar] [CrossRef]
- Park, J.; Kim, K.; Lee, E.J.; Seo, Y.J.; Lim, S.N.; Park, K.; Rho, S.B.; Lee, S.H.; Lee, J.H. Elevated level of SUMOylated IRF-1 in tumor cells interferes with IRF-1-mediated apoptosis. Proc. Natl. Acad. Sci. USA 2007, 104, 17028–17033. [Google Scholar] [CrossRef]
- Willman, C.L.; Sever, C.E.; Pallavicini, M.G.; Harada, H.; Tanaka, N.; Slovak, M.L.; Yamamoto, H.; Harada, K.; Meeker, T.C.; List, A.F.; et al. Deletion of IRF-1, mapping to chromosome 5q31.1, in human leukemia and preleukemic myelodysplasia. Science 1993, 259, 968–971. [Google Scholar] [CrossRef]
- Harada, H.; Kondo, T.; Ogawa, S.; Tamura, T.; Kitagawa, M.; Tanaka, N.; Lamphier, M.S.; Hirai, H.; Taniguchi, T. Accelerated exon skipping of IRF-1 mRNA in human myelodysplasia/leukemia; a possible mechanism of tumor suppressor inactivation. Oncogene 1994, 9, 3313–3320. [Google Scholar]
- Sugimura, J.; Tamura, G.; Suzuki, Y.; Fujioka, T. Allelic loss on chromosomes 3p, 5q and 17p in renal cell carcinomas. Pathol. Int. 1997, 47, 79–83. [Google Scholar] [CrossRef]
- Nozawa, H.; Oda, E.; Ueda, S.; Tamura, G.; Maesawa, C.; Muto, T.; Taniguchi, T.; Tanaka, N. Functionally inactivating point mutation in the tumor-suppressor IRF-1 gene identified in human gastric cancer. Int. J. Cancer 1998, 77, 522–527. [Google Scholar] [CrossRef]
- Green, W.B.; Slovak, M.L.; Chen, I.M.; Pallavicini, M.; Hecht, J.L.; Willman, C.L. Lack of IRF-1 expression in acute promyelocytic leukemia and in a subset of acute myeloid leukemias with del(5)(q31). Leukemia 1999, 13, 1960–1971. [Google Scholar] [CrossRef][Green Version]
- Moriyama, Y.; Nishiguchi, S.; Tamori, A.; Koh, N.; Yano, Y.; Kubo, S.; Hirohashi, K.; Otani, S. Tumor-suppressor effect of interferon regulatory factor-1 in human hepatocellular carcinoma. Clin. Cancer Res. 2001, 7, 1293–1298. [Google Scholar]
- Lee, E.J.; Jo, M.; Park, J.; Zhang, W.; Lee, J.H. Alternative splicing variants of IRF-1 lacking exons 7, 8, and 9 in cervical cancer. Biochem. Biophys. Res. Commun. 2006, 347, 882–888. [Google Scholar] [CrossRef]
- Cavalli, L.R.; Riggins, R.B.; Wang, A.; Clarke, R.; Haddad, B.R. Frequent loss of heterozygosity at the interferon regulatory factor-1 gene locus in breast cancer. Breast Cancer Res. Treat. 2010, 121, 227–231. [Google Scholar] [CrossRef]
- Drew, P.D.; Franzoso, G.; Becker, K.G.; Bours, V.; Carlson, L.M.; Siebenlist, U.; Ozato, K. NFκB and interferon regulatory factor 1 physically interact and synergistically induce major histocompatibility class I gene expression. J. Interf. Cytokine Res. 1995, 15, 1037–1045. [Google Scholar] [CrossRef]
- Gao, J.; Senthil, M.; Ren, B.; Yan, J.; Xing, Q.; Yu, J.; Zhang, L.; Yim, J. IRF-1 transcriptionally upregulates PUMA, which mediates the mitochondrial apoptotic pathway in IRF-1-induced apoptosis in cancer cells. Cell Death Differ. 2010, 17, 699–709. [Google Scholar] [CrossRef]
- Marotte, H.; Tsou, P.S.; Rabquer, B.J.; Pinney, A.J.; Fedorova, T.; Lalwani, N.; Koch, A.E. Blocking of interferon regulatory factor 1 reduces tumor necrosis factor α-induced interleukin-18 bioactivity in rheumatoid arthritis synovial fibroblasts by induction of interleukin-18 binding protein a: Role of the nuclear interferon regulatory factor 1–NF-κB–c-jun complex. Arthritis Rheum. 2011, 63, 3253–3262. [Google Scholar]
- Jeong, S.-I.; Kim, J.-W.; Ko, K.-P.; Ryu, B.-K.; Lee, M.-G.; Kim, H.-J.; Chi, S.-G. XAF1 forms a positive feedback loop with IRF-1 to drive apoptotic stress response and suppress tumorigenesis. Cell Death Dis. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Nakagawa, K.; Yokosawa, H. Degradation of transcription factor IRF-1 by the ubiquitin–proteasome pathway: The C-terminal region governs the protein stability. Eur. J. Biochem. 2000, 267, 1680–1686. [Google Scholar] [CrossRef]
- Nakagawa, K.; Yokosawa, H. PIAS3 induces SUMO-1 modification and transcriptional repression of IRF-1. FEBS Lett. 2002, 530, 204–208. [Google Scholar] [CrossRef]
- Pion, E.; Narayan, V.; Eckert, M.; Ball, K.L. Role of the IRF-1 enhancer domain in signalling polyubiquitination and degradation. Cell. Signal. 2009, 21, 1479–1487. [Google Scholar] [CrossRef]
- Escalante, C.R.; Yie, J.; Thanos, D.; Aggarwal, A.K. Structure of IRF-1 with bound DNA reveals determinants of interferon regulation. Nature 1998, 391, 103–106. [Google Scholar] [CrossRef]
- Tamura, T.; Yanai, H.; Savitsky, D.; Taniguchi, T. The IRF family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol. 2008, 26, 535–584. [Google Scholar] [CrossRef]
- Yanai, H.; Negishi, H.; Taniguchi, T. The IRF family of transcription factors: Inception, impact and implications in oncogenesis. Oncoimmunology 2012, 1, 1376–1386. [Google Scholar] [CrossRef]
- Schaper, F.; Kirchhoff, S.; Posern, G.; Köster, M.; Oumard, A.; Sharf, R.; Levi, B.-Z.; Hauser, H. Functional domains of interferon regulatory factor I (IRF-1). Biochem. J. 1998, 335, 147–157. [Google Scholar] [CrossRef]
- Harada, H.; Willison, K.; Sakakibara, J.; Miyamoto, M.; Fujita, T.; Taniguchi, T. Absence of the type I IFN system in EC cells: Transcriptional activator (IRF-1) and repressor (IRF-2) genes are developmentally regulated. Cell 1990, 63, 303–312. [Google Scholar] [CrossRef]
- Harada, H.; Kitagawa, M.; Tanaka, N.; Yamamoto, H.; Harada, K.; Ishihara, M.; Taniguchi, T. Anti-oncogenic and oncogenic potentials of interferon regulatory factors-1 and-2. Science 1993, 259, 971–974. [Google Scholar] [CrossRef]
- Lee, J.H.; Chun, T.; Park, S.Y.; Rho, S.B. Interferon regulatory factor-1 (IRF-1) regulates VEGF-induced angiogenesis in HUVECs. Biochim. Biophys. Acta 2008, 1783, 1654–1662. [Google Scholar] [CrossRef][Green Version]
- Nigg, E.A. Cyclin-dependent protein kinases: Key regulators of the eukaryotic cell cycle. Bioessays 1995, 17, 471–480. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, J.; Ling, M.T.; Zhao, L.; Zhao, K.-N. The role of the PI3K/Akt/mTOR signalling pathway in human cancers induced by infection with human papillomaviruses. Mol. Cancer 2015, 14, 1–13. [Google Scholar] [CrossRef]
- Barillari, G.; Monini, P.; Sgadari, C.; Ensoli, B. The impact of human papilloma viruses, matrix metallo-proteinases and HIV protease inhibitors on the onset and progression of uterine cervix epithelial tumors: A review of preclinical and clinical studies. Int. J. Mol. Sci. 2018, 19, 1418. [Google Scholar] [CrossRef]
- Keysar, S.B.; Astling, D.P.; Anderson, R.T.; Vogler, B.W.; Bowles, D.W.; Morton, J.J.; Paylor, J.J.; Glogowska, M.J.; Le, P.N.; Eagles-Soukup, J.R.; et al. A patient tumor transplant model of squamous cell cancer identifies PI3K inhibitors as candidate therapeutics in defined molecular bins. Mol. Oncol. 2013, 7, 776–790. [Google Scholar] [CrossRef]
- Ma, Y.Y.; Wei, S.J.; Lin, Y.C.; Lung, J.C.; Chang, T.C.; Whang-Peng, J.; Liu, J.M.; Yang, D.M.; Yang, W.K.; Shen, C.Y. PIK3CA as an oncogene in cervical cancer. Oncogene 2000, 19, 2739–2744. [Google Scholar] [CrossRef]
- Bertelsen, B.I.; Steine, S.J.; Sandvei, R.; Molven, A.; Laerum, O.D. Molecular analysis of the PI3K-AKT pathway in uterine cervical neoplasia: Frequent PIK3CA amplification and AKT phosphorylation. Int. J. Cancer 2006, 118, 1877–1883. [Google Scholar] [CrossRef]
- Chiosea, S.I.; Grandis, J.R.; Lui, V.W.; Diergaarde, B.; Maxwell, J.H.; Ferris, R.L.; Kim, S.W.; Luvison, A.; Miller, M.; Nikiforova, M.N. PIK3CA, HRAS and PTEN in human papillomavirus positive oropharyngeal squamous cell carcinoma. BMC Cancer 2013, 13, 602. [Google Scholar] [CrossRef]
- Lui, V.W.; Hedberg, M.L.; Li, H.; Vangara, B.S.; Pendleton, K.; Zeng, Y.; Lu, Y.; Zhang, Q.; Du, Y.; Gilbert, B.R.; et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013, 3, 761–769. [Google Scholar] [CrossRef]
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Muñoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef]
- Zuna, R.E.; Allen, R.A.; Moore, W.E.; Mattu, R.; Dunn, S.T. Comparison of human papillomavirus genotypes in high-grade squamous intraepithelial lesions and invasive cervical carcinoma: Evidence for differences in biologic potential of precursor lesions. Mod. Pathol. 2004, 17, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Meng, X.; Wang, Q.; Wang, Y.; Shang, H. The ING4 binding with p53 and induced p53 acetylation were attenuated by human papillomavirus 16 E6. PLoS ONE 2013, 8, e71453. [Google Scholar] [CrossRef]
- Liu, L.; Qin, Z.Y.; Liu, Q.; Wen, L.Z.; Liu, K.W.; Guo, Y.; Zhou, Y.B.; Wang, B.; Chen, D.F.; Wang, T. Norrin maintains malignancy of gastric cancer cells in part through activating AKT signaling. Biochem. Biophys. Res. Commun. 2019, 512, 405–411. [Google Scholar] [CrossRef]
- Bousarghin, L.; Touze, A.; Gaud, G.; Iochmann, S.; Alvarez, E.; Reverdiau, P.; Gaitan, J.; Jourdan, M.L.; Sizaret, P.Y.; Coursaget, P.L. Inhibition of cervical cancer cell growth by human papillomavirus virus-like particles packaged with human papillomavirus oncoprotein short hairpin RNAs. Mol. Cancer Ther. 2009, 8, 357–365. [Google Scholar] [CrossRef]
- Gu, W.; Putral, L.; Hengst, K.; Minto, K.; Saunders, N.A.; Leggatt, G.; McMillan, N.A. Inhibition of cervical cancer cell growth in vitro and in vivo with lentiviral-vector delivered short hairpin RNA targeting human papillomavirus E6 and E7 oncogenes. Cancer Gene Ther. 2006, 13, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Waltenberger, J.; Claesson-Welsh, L.; Siegbahn, A.; Shibuya, M.; Heldin, C.-H. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J. Biol. Chem. 1994, 269, 26988–26995. [Google Scholar]
- Ferrara, N.; Davis-Smyth, T. The biology of vascular endothelial growth factor. Endocr. Rev. 1997, 18, 4–25. [Google Scholar] [CrossRef] [PubMed]
- Gille, H.; Kowalski, J.; Li, B.; LeCouter, J.; Moffat, B.; Zioncheck, T.F.; Pelletier, N.; Ferrara, N. Analysis of biological effects and signaling properties of Flt-1 (VEGFR-1) and KDR (VEGFR-2) A reassessment using novel receptor-specific vascular endothelial growth factor mutants. J. Biol. Chem. 2001, 276, 3222–3230. [Google Scholar] [CrossRef] [PubMed]
- Ohno-Matsui, K.; Morita, I.; Tombran-Tink, J.; Mrazek, D.; Onodera, M.; Uetama, T.; Hayano, M.; Murota, S.i.; Mochizuki, M. Novel mechanism for age-related macular degeneration: An equilibrium shift between the angiogenesis factors VEGF and PEDF. J. Cell. Physiol. 2001, 189, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ye, D. Progress in anti-angiogenesis targeted drugs. China Oncol. 2009, 19, 401–405. [Google Scholar]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, A.; Tarnawski, A.S. Critical role of hypoxia sensor-HIF-1α in VEGF gene activation. Implications for angiogenesis and tissue injury healing. Curr. Med. Chem. 2012, 19, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Guo, X.; Hu, Z.; Shi, H. Interfering Human Papillomavirus E6/E7 Oncogenes in Cervical Cancer Cells Inhibits the Angiogenesis of Vascular Endothelial Cells via Increasing miR-377 in Cervical Cancer Cell-Derived Microvesicles. Onco Targets Ther. 2020, 13, 4145–4155. [Google Scholar] [CrossRef]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Sekaric, P.; Cherry, J.J.; Androphy, E.J. Binding of human papillomavirus type 16 E6 to E6AP is not required for activation of hTERT. J. Virol. 2008, 82, 71–76. [Google Scholar] [CrossRef]
- Park, J.S.; Kim, E.J.; Kwon, H.J.; Hwang, E.S.; Namkoong, S.E.; Um, S.J. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J. Biol. Chem. 2000, 275, 6764–6769. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Franklin, R.A.; Montalto, G.; Cervello, M.; Libra, M.; Candido, S.; Malaponte, G. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors: How mutations can result in therapy resistance and how to overcome resistance. Oncotarget 2012, 3, 1068. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.-F.; Wang, J.; Shao, W.; Wu, C.-P.; Chen, Z.-P.; To, S.-S.T.; Li, W.-P. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: Current preclinical and clinical development. Mol. Cancer 2017, 16, 100. [Google Scholar] [CrossRef] [PubMed]
- Bouker, K.B.; Skaar, T.C.; Riggins, R.B.; Harburger, D.S.; Fernandez, D.R.; Zwart, A.; Wang, A.; Clarke, R. Interferon regulatory factor-1 (IRF-1) exhibits tumor suppressor activities in breast cancer associated with caspase activation and induction of apoptosis. Carcinogenesis 2005, 26, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Cheng, X.; Lu, B.; Yang, G. Activation of interleukin-6/signal transducer and activator of transcription 3 by human papillomavirus early proteins 6 induces fibroblast senescence to promote cervical tumourigenesis through autocrine and paracrine pathways in tumour microenvironment. Eur. J. Cancer 2013, 49, 3889–3899. [Google Scholar] [CrossRef]
- Walch-Rückheim, B.; Pahne-Zeppenfeld, J.; Fischbach, J.; Wickenhauser, C.; Horn, L.C.; Tharun, L.; Büttner, R.; Mallmann, P.; Stern, P.; Kim, Y.-J. STAT3/IRF1 pathway activation sensitizes cervical cancer cells to chemotherapeutic drugs. Cancer Res. 2016, 76, 3872–3883. [Google Scholar] [CrossRef] [PubMed]
- Byun, H.-J.; Kim, B.-R.; Yoo, R.; Park, S.-Y.; Rho, S.B. sMEK1 enhances gemcitabine anti-cancer activity through inhibition of phosphorylation of Akt/mTOR. Apoptosis 2012, 17, 1095–1103. [Google Scholar] [CrossRef]
- Lee, J.H.; Rho, S.B.; Chun, T. Programmed cell death 6 (PDCD6) protein interacts with death-associated protein kinase 1 (DAPk1): Additive effect on apoptosis via caspase-3 dependent pathway. Biotechnol. Lett. 2005, 27, 1011–1015. [Google Scholar] [CrossRef]
- Rho, S.B.; Lee, K.H.; Kim, J.W.; Shiba, K.; Jo, Y.J.; Kim, S. Interaction between human tRNA synthetases involves repeated sequence elements. Proc. Natl. Acad. Sci. USA 1996, 93, 10128–10133. [Google Scholar] [CrossRef]
- Rho, S.B.; Kim, M.J.; Lee, J.S.; Seol, W.; Motegi, H.; Kim, S.; Shiba, K. Genetic dissection of protein–protein interactions in multi-tRNA synthetase complex. Proc. Natl. Acad. Sci. USA 1999, 96, 4488–4493. [Google Scholar] [CrossRef]
- Kim, B.-R.; Seo, S.H.; Park, M.S.; Lee, S.-H.; Kwon, Y.; Rho, S.B. sMEK1 inhibits endothelial cell proliferation by attenuating VEGFR-2-dependent-Akt/eNOS/HIF-1α signaling pathways. Oncotarget 2015, 6, 31830. [Google Scholar] [CrossRef] [PubMed]
- Rho, S.B.; Song, Y.J.; Lim, M.C.; Lee, S.H.; Kim, B.R.; Park, S.Y. Programmed cell death 6 (PDCD6) inhibits angiogenesis through PI3K/mTOR/p70S6K pathway by interacting of VEGFR-2. Cell. Signal. 2012, 24, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Dong, S.M.; Kim, B.R.; Park, M.S.; Trink, B.; Byun, H.J.; Rho, S.B. Thioridazine induces apoptosis by targeting the PI3K/Akt/mTOR pathway in cervical and endometrial cancer cells. Apoptosis 2012, 17, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Mauvais-Jarvis, F.; Pollard, D.A.; Yballe, C.M.; Brazil, D.; Bronson, R.T.; Kahn, C.R.; Cantley, L.C. Hypoglycaemia, liver necrosis and perinatal death in mice lacking all isoforms of phosphoinositide 3-kinase p85 alpha. Nat. Genet. 2000, 26, 379–382. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rho, S.B.; Lee, S.-H.; Byun, H.-J.; Kim, B.-R.; Lee, C.H. IRF-1 Inhibits Angiogenic Activity of HPV16 E6 Oncoprotein in Cervical Cancer. Int. J. Mol. Sci. 2020, 21, 7622. https://doi.org/10.3390/ijms21207622
Rho SB, Lee S-H, Byun H-J, Kim B-R, Lee CH. IRF-1 Inhibits Angiogenic Activity of HPV16 E6 Oncoprotein in Cervical Cancer. International Journal of Molecular Sciences. 2020; 21(20):7622. https://doi.org/10.3390/ijms21207622
Chicago/Turabian StyleRho, Seung Bae, Seung-Hoon Lee, Hyun-Jung Byun, Boh-Ram Kim, and Chang Hoon Lee. 2020. "IRF-1 Inhibits Angiogenic Activity of HPV16 E6 Oncoprotein in Cervical Cancer" International Journal of Molecular Sciences 21, no. 20: 7622. https://doi.org/10.3390/ijms21207622
APA StyleRho, S. B., Lee, S.-H., Byun, H.-J., Kim, B.-R., & Lee, C. H. (2020). IRF-1 Inhibits Angiogenic Activity of HPV16 E6 Oncoprotein in Cervical Cancer. International Journal of Molecular Sciences, 21(20), 7622. https://doi.org/10.3390/ijms21207622