Oncolytic Virotherapy in Glioma Tumors

 , and
, and
Abstract
1. Introduction
2. DNA Viruses Proposed as Glioma Oncolytic Agents
2.1. Herpes Simplex Virus Type I
2.1.1. Herpes Simplex Virus-1 Pre-Clinical Research
2.1.2. Herpes Simplex Virus-1 Clinical Studies
2.2. Adenovirus
2.2.1. Adenovirus Pre-Clinical Research
2.2.2. Adenovirus Clinical Studies
2.3. Vaccinia Virus (VV)
2.3.1. Vaccinia Virus Pre-Clinical Research
2.3.2. VV Clinical Studies
2.4. Myxoma
Myxoma Virus Pre-Clinical Research
2.5. Parvovirus
2.5.1. Parvoviridae Pre-Clinical Research
2.5.2. Parvoviridae Clinical Studies
3. RNA Viruses Proposed as Glioma Oncolytic Agents
3.1. Reovirus
3.1.1. Reovirus Pre-Clinical Research
3.1.2. Reovirus Clinical Studies
3.2. Measles
3.2.1. Measles Pre-Clinical Research
3.2.2. Measles Clinical Studies
3.3. Vesicular Stomatitis
Vesicular Stomatitis Virus Pre-Clinical Research
3.4. Newcastle Disease Virus (NDV)
3.4.1. Newcastle Disease Virus Pre-Clinical Research
3.4.2. Newcastle Disease Virus Clinical Studies
3.5. Seneca Valley
Seneca Valley Virus Pre-Clinical Research
3.6. Poliovirus
3.6.1. Poliovirus Pre-Clinical Research
3.6.2. Poliovirus Clinical Studies
3.7. Sindbis
Sindbis Pre-Clinical Research
3.8. Rift Valley Fever Virus (RVFV)
4. Current OV Challenges for Malignant Glioma
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| WHO | World Health Organization |
| TMZ | Temozolomide |
| DNA | Deoxyribonucleic acid |
| RNA | Ribonucleic acid |
| GBM | Glioblastoma |
| OV | Oncolytic virotherapy |
| CNS | Central nervous system |
| DAMPs | Danger-Associated molecular patterns |
| OS | Overall survival |
| ICD | Immunogenic cell death |
| PRRs | Pattern recognition receptors |
| HSV-1 | Herpes simplex virus type 1 |
| LAMR | Laminin receptor |
| VV | Vaccinia virus |
| MYXV | Myxoma virus |
| NDV | Newcastle disease virus |
| TVEC | Talimogene laherparepvec |
| TTFields | Tumor treating fields |
References
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2012–2016. Neuro-Oncology 2019, 21, v1–v100. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.G.; Smith, T.R.; Gittleman, H.; Ostrom, Q.T.; Kruchko, C.; Barnholtz-Sloan, J.S. Glioblastoma incidence rate trends in Canada and the United States compared with England, 1995–2015. Neuro-Oncology 2019, 22, 301–302. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of tumor-treating fields plus maintenance temozolomide vs. maintenance temozolomide alone on survival in patients with glioblastoma. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed]
- Shergalis, A.; Bankhead, A.; Luesakul, U.; Muangsin, N.; Neamati, N. Current challenges and opportunities in treating glioblastoma. Pharmacol. Rev. 2018, 70, 412–445. [Google Scholar] [CrossRef]
- Velasquez, C.; Mansouri, S.; Mora, C.; Nassiri, F.; Suppiah, S.; Martino, J.; Zadeh, G.; Fernandez-Luna, J.L. Molecular and clinical insights into the invasive capacity of glioblastoma Cells. J. Oncol. 2019, 2019, 1740763. [Google Scholar] [CrossRef]
- Sundar, S.J.; Hsieh, J.K.; Manjila, S.; Lathia, J.D.; Sloan, A. The role of cancer stem cells in glioblastoma. Neurosurg. Focus 2014, 37, E6. [Google Scholar] [CrossRef]
- Mallick, S.; Benson, R.; Hakim, A.; Rath, G.K. Management of glioblastoma after recurrence: A changing paradigm. J. Egypt. Natl. Cancer Inst. 2016, 28, 199–210. [Google Scholar] [CrossRef]
- Chinot, O.L.; Rouge, T.D.L.M.; Moore, N.; Zeaiter, A.; Das, A.; Phillips, H.; Modrusan, Z.; Cloughesy, T. AVAglio: Phase 3 trial of bevacizumab plus temozolomide and radiotherapy in newly diagnosed glioblastoma multiforme. Adv. Ther. 2011, 28, 334–340. [Google Scholar] [CrossRef]
- McGranahan, T.; Therkelsen, K.E.; Ahmad, S.; Nagpal, S. Current state of immunotherapy for treatment of glioblastoma. Curr. Treat. Options Oncol. 2019, 20, 24. [Google Scholar] [CrossRef] [PubMed]
- Wollmann, G.; Özduman, K.; Pol, A.N.V.D. Oncolytic virus therapy for glioblastoma multiforme. Cancer J. 2012, 18, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.J.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.-C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.; Russell, S.J. History of oncolytic viruses: Genesis to genetic engineering. Mol. Ther. 2007, 15, 651–659. [Google Scholar] [CrossRef]
- Paun, A.; Pitha, P.M. The innate antiviral response: New insights into a continuing story. Intern. Rev. Cytol. 2006, 69, 1–66. [Google Scholar] [CrossRef]
- De Matos, A.L.; Franco, L.S.; McFadden, G. Oncolytic viruses and the immune system: The dynamic duo. Mol. Ther.-Methods Clin. Dev. 2020, 17, 349–358. [Google Scholar] [CrossRef]
- Martikainen, M.; Essand, M. Virus-based immunotherapy of glioblastoma. Cancers 2019, 11, 186. [Google Scholar] [CrossRef]
- Zheng, M.; Huang, J.; Tong, A.; Yang, H. Oncolytic viruses for cancer therapy: Barriers and recent advances. Mol. Ther.-Oncolytics 2019, 15, 234–247. [Google Scholar] [CrossRef]
- Krummenacher, C.; Nicola, A.V.; Whitbeck, J.C.; Lou, H.; Hou, W.; Lambris, J.D.; Geraghty, R.J.; Spear, P.G.; Cohen, G.H.; Eisenberg, R.J. Herpes simplex virus glycoprotein D can bind to poliovirus receptor-Related protein 1 or herpesvirus entry mediator, two structurally unrelated mediators of virus entry. J. Virol. 1998, 72, 7064–7074. [Google Scholar] [CrossRef]
- Friedman, G.K.; Langford, C.P.; Coleman, J.M.; Cassady, K.A.; Parker, J.N.; Markert, J.M.; Gillespie, G.Y. Engineered herpes simplex viruses efficiently infect and kill CD133 + human glioma xenograft cells that express CD111. J. Neuro-Oncol. 2009, 95, 199–209. [Google Scholar] [CrossRef]
- Andreansky, S.; He, B.; Van Cott, J.; McGhee, J.; Markert, J.M.; Gillespie, G.Y.; Roizman, B.; Whitley, R.J. Treatment of intracranial gliomas in immunocompetent mice using herpes simplex viruses that express murine interleukins. Gene Ther. 1998, 5, 121–130. [Google Scholar] [CrossRef]
- Martuza, R.L.; Malick, A.; Markert, J.M.; Ruffner, K.L.; Coen, D.M. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 1991, 252, 854–856. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.; Roizman, B. Herpes simplex virus 1 gamma (1) 34.5 gene function, which blocks the host response to infection, maps in the homologous domain of the genes expressed during growth arrest and DNA damage. Proc. Natl. Acad. Sci. USA 1994, 91, 5247–5251. [Google Scholar] [CrossRef] [PubMed]
- McKie, E.; MacLean, A.; Lewis, A.; Cruickshank, G.; Rampling, R.; Barnett, S.; Kennedy, P.; Brown, S. Selective in vitro replication of herpes simplex virus type 1 (HSV-1) ICP34.5 null mutants in primary human CNS tumours-evaluation of a potentially effective clinical therapy. Br. J. Cancer 1996, 74, 745–752. [Google Scholar] [CrossRef]
- Todo, T. “Armed” oncolytic herpes simplex viruses for brain tumor therapy. Cell Adhes. Migr. 2008, 2, 208–213. [Google Scholar] [CrossRef]
- Mineta, T.; Rabkin, S.D.; Yazaki, T.; Hunter, W.D.; Martuza, R.L. Attenuated multi–mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat. Med. 1995, 1, 938–943. [Google Scholar] [CrossRef] [PubMed]
- Chung, R.Y.; Saeki, Y.; Chiocca, E.A. B-myb promoter retargeting of herpes simplex virus γ34.5 gene-mediated virulence toward tumor and cycling cells. J. Virol. 1999, 73, 7556–7564. [Google Scholar] [CrossRef] [PubMed]
- Kambara, H.; Saeki, Y.; Okano, H.; Chiocca, E.A. An oncolytic HSV-1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res. 2005, 65, 2832–2839. [Google Scholar] [CrossRef]
- Fulci, G.; Breymann, L.; Gianni, D.; Kurozomi, K.; Rhee, S.S.; Yu, J.; Kaur, B.; Louis, D.N.; Weissleder, R.; Caligiuri, M.A.; et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12873–12878. [Google Scholar] [CrossRef]
- Nakashima, H.; Nguyen, T.; Kasai, K.; Passaro, C.; Ito, H.; Goins, W.; Shaikh, I.; Erdelyi, R.; Nishihara, R.; Nakano, I.; et al. Toxicity and efficacy of a novel GADD34-expressing oncolytic HSV-1 for the treatment of experimental glioblastoma. Clin. Cancer Res. 2018, 24, 2574–2584. [Google Scholar] [CrossRef]
- Passaro, C.; Alayo, Q.; De Laura, I.; McNulty, J.; Grauwet, K.; Ito, H.; Bhaskaran, V.; Mineo, M.; Lawler, S.E.; Shah, K.; et al. Arming an oncolytic herpes simplex virus type 1 with a single-chain fragment variable antibody against PD-1 for experimental glioblastoma therapy. Clin. Cancer Res. 2018, 25, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Ghonime, M.G.; Jackson, J.; Shah, A.; Roth, J.; Li, M.; Saunders, U.; Coleman, J.; Gillespie, G.Y.; Markert, J.M.; Cassady, K.A. Chimeric HCMV/HSV-1 and Δγ 1 34.5 oncolytic herpes simplex virus elicit immune mediated antigliomal effect and antitumor memory. Transl. Oncol. 2018, 11, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.C.; Parker, J.N.; Gillespie, G.Y.; Lakeman, F.D.; Meleth, S.; Markert, J.M.; Cassady, K.A. Enhanced antiglioma activity of chimeric HCMV/HSV-1 oncolytic viruses. Gene Ther. 2007, 14, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Todo, T.; Martuza, R.L.; Rabkin, S.D.; Johnson, P.A. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. USA 2001, 98, 6396–6401. [Google Scholar] [CrossRef]
- Gambini, E.; Reisoli, E.; Appolloni, I.; Gatta, V.; Campadelli-Fiume, G.; Menotti, L.; Malatesta, P. Replication-competent herpes simplex virus retargeted to HER2 as therapy for high-grade glioma. Mol. Ther. 2012, 20, 994–1001. [Google Scholar] [CrossRef]
- Hardcastle, J.; Kurozumi, K.; Dmitrieva, N.; Sayers, M.P.; Ahmad, S.; Waterman, P.; Weissleder, R.; Chiocca, E.A.; Kaur, B. Enhanced antitumor efficacy of vasculostatin (Vstat120) expressing oncolytic HSV-1. Mol. Ther. 2010, 18, 285–294. [Google Scholar] [CrossRef]
- Hellums, E.K.; Markert, J.M.; Parker, J.N.; He, B.; Perbal, B.; Roizman, B.; Whitley, R.J.; Langford, C.P.; Bharara, S.; Gillespie, G.Y. Increased efficacy of an interleukin-12-secreting herpes simplex virus in a syngeneic intracranial murine glioma model. Neuro-Oncology 2005, 7, 213–224. [Google Scholar] [CrossRef]
- Rampling, R.; Cruickshank, G.; Papanastassiou, V.; Nicoll, J.; Hadley, D.; Brennan, D.; Petty, R.; MacLean, A.; Harland, J.; McKie, E.; et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 2000, 7, 859–866. [Google Scholar] [CrossRef]
- Harrow, S.; Papanastassiou, V.; Harland, J.; Mabbs, R.; Petty, R.; Fraser, M.; Hadley, D.; Patterson, J.; Brown, S.M.; Rampling, R. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: Safety data and long-term survival. Gene Ther. 2004, 11, 1648–1658. [Google Scholar] [CrossRef]
- Markert, J.M.; Medlock, M.D.; Rabkin, S.D.; Gillespie, G.Y.; Todo, T.; Hunter, W.D.; Palmer, C.A.; Feigenbaum, F.; Tornatore, C.; Tufaro, F.; et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: Results of a phase I trial. Gene Ther. 2000, 7, 867–874. [Google Scholar] [CrossRef]
- Markert, J.M.; Liechty, P.G.; Wang, W.; Gaston, S.; Braz, E.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Lakeman, A.D.; Palmer, C.A.; et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol. Ther. 2009, 17, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Markert, J.M.; Razdan, S.N.; Kuo, H.-C.; Cantor, A.; Knoll, A.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Agee, B.S.; Coleman, J.M.; et al. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol. Ther. 2014, 22, 1048–1055. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Nakashima, H.; Kasai, K.; Fernandez, S.A.; Oglesbee, M. Preclinical toxicology of rQNestin34.5v.2: An oncolytic herpes virus with transcriptional regulation of the ICP34.5 neurovirulence gene. Mol. Ther.-Methods Clin. Dev. 2020, 17, 871–893. [Google Scholar] [CrossRef] [PubMed]
- Summary for policymakers. Phys. Sci. Basis 2014, 53, 1–30. [CrossRef]
- Sharma, A.; Li, X.; Bangari, D.S.; Mittal, S.K. Adenovirus receptors and their implications in gene delivery. Virus Res. 2009, 143, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Kiyokawa, J.; Wakimoto, H. Preclinical and clinical development of oncolytic adenovirus for the treatment of malignant glioma. Oncolytic Virother. 2019, 8, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. China approves world’s first oncolytic virus therapy for cancer treatment. J. Natl. Cancer Inst. 2006, 98, 298–300. [Google Scholar] [CrossRef]
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-Johannes, A.; Fattaey, A.; et al. An adenovirus mutant that replicates selectively in p53- deficient human tumor cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef]
- Geoerger, B.; Grill, J.; Opolon, P.; Morizet, J.; Aubert, G.; Terrier-Lacombe, M.-J.; Paillerets, B.B.-D.; Barrois, M.; Feunteun, J.; Kirn, D.H.; et al. Oncolytic activity of the E1B-55 kDa-deleted adenovirus ONYX-015 is independent of cellular p53 status in human malignant glioma xenografts. Cancer Res. 2002, 62, 764–772. [Google Scholar]
- Pasqualini, R.; Koivunen, E.; Ruoslahti, E. αv Integrins as receptors for tumor targeting by circulating ligands. Nat. Biotechnol. 1997, 15, 542–546. [Google Scholar] [CrossRef]
- Gladson, C.L.; Cheresh, D.A. Glioblastoma expression of vitronectin and the alpha v beta 3 integrin. Adhesion mechanism for transformed glial cells. J. Clin. Investig. 1991, 88, 1924–1932. [Google Scholar] [CrossRef]
- Suzuki, K.; Fueyo, J.; Krasnykh, V.; Reynolds, P.N.; Curiel, D.T.; Alemany, R. A conditionally replicative adenovirus with enhanced infectivity shows improved oncolytic potency. Clin. Cancer Res. 2001, 7, 120–126. [Google Scholar]
- Fueyo, J.; Gomez-Manzano, C.; Alemany, R.; Lee, P.S.; McDonnell, T.J.; Mitlianga, P.; Shi, Y.-X.; Levin, V.A.; Yung, W.K.A.; Kyritsis, A.P. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene 2000, 19, 2–12. [Google Scholar] [CrossRef]
- Alonso, M.M.; Cascallo, M.; Gomez-Manzano, C.; Jiang, H.; Bekele, B.N.; Pérez-Giménez, A.; Lang, F.F.; Piao, Y.; Alemany, R.; Fueyo, J.; et al. ICOVIR-5 Shows E2F1 addiction and potent antiglioma effect in vivo. Cancer Res. 2007, 67, 8255–8263. [Google Scholar] [CrossRef]
- Rojas, J.J.; Cascallo, M.; Guedan, S.; Gros, A.; Martinez-Quintanilla, J.; Hemminki, A.; Alemany, R. A modified E2F-1 promoter improves the efficacy to toxicity ratio of oncolytic adenoviruses. Gene Ther. 2009, 16, 1441–1451. [Google Scholar] [CrossRef]
- Rojas, J.J.; Guedan, S.; Searle, P.F.; Martinez-Quintanilla, J.; Gil-Hoyos, R.; Alcayaga-Miranda, F.; Cascallo, M.; Alemany, R. Minimal RB-responsive E1A promoter modification to attain potency, selectivity, and transgene-arming capacity in oncolytic adenoviruses. Mol. Ther. 2010, 18, 1960–1971. [Google Scholar] [CrossRef]
- Guedan, S.; Rojas, J.J.; Gros, A.; Mercade, E.; Cascallo, M.; Alemany, R. Hyaluronidase expression by an oncolytic adenovirus enhances its intratumoral spread and suppresses tumor growth. Mol. Ther. 2010, 18, 1275–1283. [Google Scholar] [CrossRef]
- Toole, B.P. Hyaluronan: From extracellular glue to pericellular cue. Nat. Rev. Cancer 2004, 4, 528–539. [Google Scholar] [CrossRef]
- Desoize, B. Multicellular resistance: A paradigm for clinical resistance? Crit. Rev. Oncol. 2000, 36, 193–207. [Google Scholar] [CrossRef]
- Martinez-Quintanilla, J.; He, D.; Wakimoto, H.; Alemany, R.; Shah, K. Encapsulated stem cells loaded with hyaluronidase-expressing oncolytic virus for brain tumor therapy. Mol. Ther. 2015, 23, 108–118. [Google Scholar] [CrossRef]
- Vera, B.; Martínez-Vélez, N.; Xipell, E.; De La Rocha, A.A.; Patiño-García, A.; Saez-Castresana, J.; Gonzalez-Huarriz, M.; Cascallo, M.; Alemany, R.; Alonso, M.M. Characterization of the antiglioma effect of the oncolytic adenovirus VCN-01. PLoS ONE 2016, 11, e0147211. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Rivera-Molina, Y.; Gomez-Manzano, C.; Clise-Dwyer, K.; Bover, L.; Vence, L.M.; Yuan, Y.; Lang, F.F.; Toniatti, C.; Hossain, M.B.; et al. Oncolytic adenovirus and tumor-targeting immune modulatory therapy improve autologous cancer vaccination. Cancer Res. 2017, 77, 3894–3907. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Molina, Y.; Jiang, H.; Fueyo, J.; Nguyen, T.; Shin, D.H.; Youssef, G.; Fan, X.; Gumin, J.; Alonso, M.M.; Phadnis, S.; et al. GITRL-armed Delta-24-RGD oncolytic adenovirus prolongs survival and induces anti-glioma immune memory. Neuro-Oncol. Adv. 2019, 1. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.A.; Cai, H.; Miao, J.; Khare, P.D.; Gonzalez, P.; Dalsing-Hernandez, J.; Sharma, G.; Chan, T.; Cooper, L.J.; Lebel, F. Regulated intratumoral expression of IL-12 using a RheoSwitch therapeutic system® (RTS®) gene switch as gene therapy for the treatment of glioma. Cancer Gene Ther. 2018, 25, 106–116. [Google Scholar] [CrossRef]
- Ricobaraza, A.; Gonzalez-Aparicio, M.; Mora-Jimenez, L.; Lumbreras, S.; Hernandez-Alcoceba, R. High-capacity adenoviral vectors: Expanding the scope of gene therapy. Int. J. Mol. Sci. 2020, 21, 3643. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Abbed, K.M.; Tatter, S.; Louis, D.N.; Hochberg, F.H.; Barker, F.; Kracher, J.; Grossman, S.A.; Fisher, J.D.; Carson, K.; et al. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol. Ther. 2004, 10, 958–966. [Google Scholar] [CrossRef]
- Thorne, S.H.; Bartlett, D.L.; Kirn, D.H. The use of oncolytic vaccinia viruses in the treatment of cancer: A new role for an old ally? Curr. Gene Ther. 2005, 5, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Al Yaghchi, C.; Zhang, Z.; Alusi, G.; Lemoine, N.R.; Wang, Y. Vaccinia virus, a promising new therapeutic agent for pancreatic cancer. Immunotherapy 2015, 7, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Timiryasova, T.M.; Li, J.; Chen, B.; Chong, D.; Langridge, W.H.; Gridley, D.S.; Fodor, I. Antitumor effect of vaccinia virus in glioma model. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 1999, 11, 133–144. [Google Scholar]
- Chen, B.; Timiryasova, T.M.; Haghighat, P.; Andres, M.L.; Kajioka, E.H.; Dutta-Roy, R.; Gridley, D.S.; Fodor, I. Low-dose vaccinia virus-mediated cytokine gene therapy of glioma. J. Immunother. 2001, 24, 46–57. [Google Scholar] [CrossRef]
- Chen, B.; Timiryasova, T.M.; Andres, M.L.; Kajioka, E.H.; Dutta-roy, R.; Gridley, D.S.; Fodor, I. Evaluation of combined vaccinia virus ± mediated. Cancer Gene Ther. 2000, 7, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- McCart, J.A.; Ward, J.M.; Lee, J.; Hu, Y.; Alexander, H.R.; Libutti, S.K.; Moss, B.; Bartlett, D.L. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001, 61, 8751–8757. [Google Scholar] [PubMed]
- Naik, A.M.; Chalikonda, S.; McCart, J.A.; Xu, H.; Guo, Z.S.; Langham, G.; Gardner, N.; Mocellin, S.; Lokshin, A.E.; Moss, B.; et al. Intravenous and isolated limb perfusion delivery of wild type and a tumor-selective replicating mutant vaccinia virus in nonhuman primates. Hum. Gene Ther. 2006, 17, 31–45. [Google Scholar] [CrossRef]
- Lun, X.Q.; Jang, J.-H.; Tang, N.; Deng, H.; Head, R.; Bell, J.C.; Stojdl, D.F.; Nutt, C.L.; Senger, D.L.; Forsyth, P.A.; et al. Efficacy of systemically administered oncolytic vaccinia virotherapy for malignant gliomas is enhanced by combination therapy with rapamycin or cyclophosphamide. Clin. Cancer Res. 2009, 15, 2777–2788. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Guo, Z.S.; Bartlett, D.L.; Yan, D.Z.; Schane, C.P.; Thomas, D.L.; Liu, J.; McFadden, G.; Shisler, J.L.; Roy, E.J. Synergistic combination of oncolytic virotherapy and immunotherapy for glioma. Clin. Cancer Res. 2020, 26, 2216–2230. [Google Scholar] [CrossRef]
- Idbaih, A.; Erbs, P.; Foloppe, J.; Chneiweiss, H.; Kempf, J.; Homerin, M.; Schmitt, C.; Them, L.N.; Delattre, J.-Y. TG6002: A novel oncolytic and vectorized gene pro-drug therapy approach to treat glioblastoma. J. Clin. Oncol. 2017, 35, e13510. [Google Scholar] [CrossRef]
- Kerr, P.J.; McFadden, G. Immune responses to Myxoma virus. Viral Immunol. 2002, 15, 229–246. [Google Scholar] [CrossRef]
- Wang, F.; Ma, Y.; Barrett, J.W.; Gao, X.; Loh, J.; Barton, E.; Iv, H.W.V.; McFadden, G. Disruption of Erk-dependent type I interferon induction breaks the myxoma virus species barrier. Nat. Immunol. 2004, 5, 1266–1274. [Google Scholar] [CrossRef]
- Lun, X.; Yang, W.; Alain, T.; Shi, Z.-Q.; Muzik, H.; Barrett, J.W.; McFadden, G.; Bell, J.; Hamilton, M.G.; Senger, D.L.; et al. Myxoma virus is a novel oncolytic virus with significant antitumor activity against experimental human gliomas. Cancer Res. 2005, 65, 9982–9990. [Google Scholar] [CrossRef]
- Ogbomo, H.; Zemp, F.J.; Lun, X.; Zhang, J.; Stack, D.; Rahman, M.M.; McFadden, G.; Mody, C.H.; Forsyth, P.A. Myxoma virus infection promotes NK lysis of malignant gliomas in vitro and in vivo. PLoS ONE 2013, 8, e66825. [Google Scholar] [CrossRef]
- Burton, C.; Das, A.; McDonald, D.; Vandergrift, W.A.; Patel, S.J.; Cachia, D.; Bartee, E. Oncolytic myxoma virus synergizes with standard of care for treatment of glioblastoma multiforme. Oncolytic Virother. 2018, 7, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Josiah, D.T.; Zhu, D.; Dreher, F.; Olson, J.; McFadden, G.; Caldas, H. Adipose-derived stem cells as therapeutic delivery vehicles of an oncolytic virus for glioblastoma. Mol. Ther. 2010, 18, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Pisklakova, A.; McKenzie, B.; Zemp, F.; Lun, X.; Kenchappa, R.S.; Etame, A.B.; Rahman, M.M.; Reilly, K.; Pilon-Thomas, S.; McFadden, G.; et al. M011L-deficient oncolytic myxoma virus induces apoptosis in brain tumor-initiating cells and enhances survival in a novel immunocompetent mouse model of glioblastoma. Neuro-Oncology 2016, 18, 1088–1098. [Google Scholar] [CrossRef]
- Marchini, A.; Bonifati, S.; Scott, E.M.; Angelova, A.L.; Rommelaere, J. Oncolytic parvoviruses: From basic virology to clinical applications. Virol. J. 2015, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Vihinen-Ranta, M.; Suikkanen, S.; Parrish, C.R. Pathways of cell infection by parvoviruses and adeno-associated viruses. J. Virol. 2004, 78, 6709–6714. [Google Scholar] [CrossRef]
- Cotmore, S.F.; Tattersall, P. Parvoviral host range and cell entry mechanisms. Int. Rev. Cytol. 2007, 70, 183–232. [Google Scholar] [CrossRef]
- Di Piazza, M.; Mader, C.; Geletneky, K.; Calle, M.H.Y.; Weber, E.; Schlehofer, J.; Deleu, L.; Rommelaere, J. Cytosolic activation of cathepsins mediates parvovirus H-1-induced killing of cisplatin and TRAIL-resistant glioma cells. J. Virol. 2007, 81, 4186–4198. [Google Scholar] [CrossRef]
- Geletneky, K.; Hartkopf, A.D.; Krempien, R.; Rommelaere, J.; Schlehofer, J.R. Improved killing of human high-grade glioma cells by combining ionizing radiation with oncolytic parvovirus H-1 infection. J. Biomed. Biotechnol. 2010, 2010, 1–9. [Google Scholar] [CrossRef]
- Geletneky, K.; Kiprianova, I.; Ayache, A.; Koch, R.; Calle, M.H.Y.; Deleu, L.; Sommer, C.; Thomas, N.; Rommelaere, J.; Schlehofer, J.R. Regression of advanced rat and human gliomas by local or systemic treatment with oncolytic parvovirus H-1 in rat models. Neuro-oncology 2010, 12, 804–814. [Google Scholar] [CrossRef]
- Paglino, J.C.; Ozduman, K.; Van den Pol, A.N. Lu III parvovirus selectively and efficiently targets, replicates in, and kills human glioma cells. J. Virol. 2012, 86, 7280–7291. [Google Scholar] [CrossRef]
- Rubio, M.-P.; Guerra, S.; Almendral, J.M. Genome replication and postencapsidation functions mapping to the nonstructural gene restrict the host range of a murine parvovirus in human cells. J. Virol. 2001, 75, 11573–11582. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wollmann, G.; Tattersall, P.; Van den Pol, A.N. Targeting human glioblastoma cells: Comparison of nine viruses with oncolytic potential. J. Virol. 2005, 79, 6005–6022. [Google Scholar] [CrossRef] [PubMed]
- Abschuetz, A.; Kehl, T.; Geibig, R.; Leuchs, B.; Rommelaere, J.; Régnier-Vigouroux, A. Oncolytic murine autonomous parvovirus, a candidate vector for glioma gene therapy, is innocuous to normal and immunocompetent mouse glial cells. Cell Tissue Res. 2006, 325, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Hüsing, J.; Rommelaere, J.; Schlehofer, J.; Leuchs, B.; Dahm, M.; Krebs, O.; Doeberitz, M.V.K.; Huber, B.; Hajda, J. Phase I/IIa study of intratumoral/intracerebral or intravenous/intracerebral administration of Parvovirus H-1 (ParvOryx) in patients with progressive primary or recurrent glioblastoma multiforme: ParvOryx01 protocol. BMC Cancer 2012, 12, 99. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Hajda, J.; Angelova, A.L.; Leuchs, B.; Capper, D.; Bartsch, A.J.; Neumann, J.-O.; Schöning, T.; Hüsing, J.; Beelte, B.; et al. Oncolytic H-1 Parvovirus shows safety and signs of immunogenic activity in a first phase I/IIa glioblastoma trial. Mol. Ther. 2017, 25, 2620–2634. [Google Scholar] [CrossRef]
- Angelova, A.; Barf, M.; Geletneky, K.; Unterberg, A.W.; Rommelaere, J. Immunotherapeutic potential of oncolytic H-1 parvovirus: Hints of glioblastoma microenvironment conversion towards immunogenicity. Viruses 2017, 9, 382. [Google Scholar] [CrossRef]
- Strong, J.E.; Tang, D.; Lee, P.W. Evidence that the epidermal growth factor receptor on host cells confers reovirus infection efficiency. Virology 1993, 197, 405–411. [Google Scholar] [CrossRef]
- Strong, J.E.; Coffey, M.C.; Tang, D.; Sabinin, P.; Lee, P.W. The molecular basis of viral oncolysis: Usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998, 17, 3351–3362. [Google Scholar] [CrossRef]
- Wilcox, M.E.; Yang, W.; Senger, D.; Rewcastle, N.B.; Morris, D.G.; Brasher, P.M.A.; Shi, Z.Q.; Johnston, R.N.; Nishikawa, S.; Lee, P.W.K.; et al. Reovirus as an oncolytic agent against experimental human malignant gliomas. J. Natl. Cancer Inst. 2001, 93, 903–912. [Google Scholar] [CrossRef]
- Samson, A.; Scott, K.J.; Taggart, D.; West, E.J.; Wilson, E.; Nuovo, G.J.; Thomson, S.; Corns, R.; Mathew, R.K.; Fuller, M.J.; et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci. Transl. Med. 2018, 10, eaam7577. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, P.A.; Roldán, G.; George, D.; Wallace, C.; Palmer, C.A.; Morris, D.; Cairncross, G.; Matthews, M.V.; Markert, J.; Gillespie, Y.; et al. A phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Mol. Ther. 2008, 16, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Kicielinski, K.P.; Chiocca, E.A.; Yu, J.S.; Gill, G.M.; Coffey, M.; Markert, J.M. Phase 1 clinical trial of intratumoral reovirus infusion for the treatment of recurrent malignant gliomas in adults. Mol. Ther. 2014, 22, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Bateman, A.; Johnson, K.; Diaz, R.M.; James, C.D.; Vile, R.; Russell, S.J. Use of viral fusogenic membrane glycoproteins as novel therapeutic transgenes in gliomas. Hum. Gene Ther. 2001, 12, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.D.; Nakamura, T.; Russell, S.J.; Peng, K.-W. High CD46 Receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res. 2004, 64, 4919–4926. [Google Scholar] [CrossRef] [PubMed]
- Phuong, L.K.; Allen, C.; Peng, K.-W.; Giannini, C.; Greiner, S.; TenEyck, C.J.; Mishra, P.; Macura, S.I.; Russell, S.J.; Galanis, E. Use of a vaccine strain of measles virus genetically engineered to produce carcinoembryonic antigen as a novel therapeutic agent against glioblastoma multiforme. Cancer Res. 2003, 63, 2462–2469. [Google Scholar]
- Opyrchal, M.; Allen, C.; Iankov, I.; Aderca, I.; Schroeder, M.; Sarkaria, J.; Galanis, E. Effective radiovirotherapy for malignant gliomas by using oncolytic measles virus strains encoding the sodium iodide symporter (MV-NIS). Hum. Gene Ther. 2012, 23, 419–427. [Google Scholar] [CrossRef]
- Paraskevakou, G.; Allen, C.; Nakamura, T.; Zollman, P.; James, C.D.; Peng, K.W.; Schroeder, M.; Russell, S.J.; Galanis, E. Epidermal growth factor receptor (EGFR)-retargeted measles virus strains effectively target EGFR- or EGFRvIII expressing gliomas. Mol. Ther. 2007, 15, 677–686. [Google Scholar] [CrossRef]
- Bach, P.; Abel, T.; Hoffmann, C.; Gal, Z.; Braun, G.; Voelker, I.; Ball, C.R.; Johnston, I.C.D.; Lauer, U.M.; Herold-Mende, C.; et al. Specific elimination of CD133 + tumor cells with targeted oncolytic measles virus. Cancer Res. 2013, 73, 865–874. [Google Scholar] [CrossRef]
- Studebaker, A.W.; Raffel, C.; Hutzen, B. Advances in the design and development of oncolytic measles viruses. Oncolytic Virotherapy 2015, 4, 109–118. [Google Scholar] [CrossRef][Green Version]
- Nikolic, J.; Belot, L.; Raux, H.; Legrand, P.; Gaudin, Y.; Albertini, A.A. Structural basis for the recognition of LDL-receptor family members by VSV glycoprotein. Nat. Commun. 2018, 9, 1029. [Google Scholar] [CrossRef]
- Duntsch, C.D.; Zhou, Q.; Jayakar, H.R.; Weimar, J.D.; Robertson, J.H.; Pfeffer, L.M.; Wang, L.; Xiang, Z.; Whitt, M.A. Recombinant vesicular stomatitis virus vectors as oncolytic agents in the treatment of high-grade gliomas in an organotypic brain tissue slice-glioma coculture model. J. Neurosurg. 2004, 100, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, S.; Barber, G.N. Vesicular Stomatitis Virus (VSV) therapy of tumors. IUBMB Life 2000, 50, 135–138. [Google Scholar] [CrossRef]
- Stojdl, D.F.; Lichty, B.D.; Tenoever, B.R.; Paterson, J.M.; Power, A.T.; Knowles, S.; Marius, R.; Reynard, J.; Poliquin, L.; Atkins, H.; et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 2003, 4, 263–275. [Google Scholar] [CrossRef]
- Wollmann, G.; Rogulin, V.; Simon, I.; Rose, J.K.; Van den Pol, A.N. Some attenuated variants of vesicular stomatitis virus show enhanced oncolytic activity against human glioblastoma cells relative to normal brain cells. J. Virol. 2009, 84, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Lun, X.; Senger, D.L.; Alain, T.; Oprea, A.; Parato, K.; Stojdl, D.; Lichty, B.; Power, A.; Johnston, R.N.; Hamilton, M.; et al. Effects of intravenously administered recombinant vesicular stomatitis virus (VSV ΔM51) on multifocal and invasive gliomas. J. Natl. Cancer Inst. 2006, 98, 1546–1557. [Google Scholar] [CrossRef] [PubMed]
- Özduman, K.; Wollmann, G.; Piepmeier, J.M.; Van den Pol, A.N. Systemic vesicular stomatitis virus selectively destroys multifocal glioma and metastatic carcinoma in brain. J. Neurosci. 2008, 28, 1882–1893. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Kretzschmar, E.; Perkins, A.S.; Forman, J.; Price, R.; Buonocore, L.; Kawaoka, Y.; Rose, J.K. Vaccination with a recombinant vesicular stomatitis virus expressing an influenza virus hemagglutinin provides complete protection from influenza virus challenge. J. Virol. 1998, 72, 4704–4711. [Google Scholar] [CrossRef] [PubMed]
- Publicover, J.; Ramsburg, E.; Rose, J.K. Characterization of nonpathogenic, live, viral vaccine vectors inducing potent cellular immune responses. J. Virol. 2004, 78, 9317–9324. [Google Scholar] [CrossRef]
- Özduman, K.; Wollmann, G.; Ahmadi, S.A.; Van den Pol, A.N. Peripheral immunization blocks lethal actions of vesicular stomatitis virus within the brain. J. Virol. 2009, 83, 11540–11549. [Google Scholar] [CrossRef]
- Flanagan, E.B.; Zamparo, J.M.; Ball, L.A.; Rodriguez, L.L.; Wertz, G.W. Rearrangement of the genes of vesicular stomatitis virus eliminates clinical disease in the natural host: New strategy for vaccine development. J. Virol. 2001, 75, 6107–6114. [Google Scholar] [CrossRef]
- Cooper, D.; Wright, K.J.; Calderon, P.C.; Guo, M.; Nasar, F.; Johnson, J.E.; Coleman, J.W.; Lee, M.; Kotash, C.; Yurgelonis, I.; et al. Attenuation of recombinant vesicular stomatitis virus-human immunodeficiency virus type 1 vaccine vectors by gene translocations and G gene truncation reduces neurovirulence and enhances immunogenicity in mice. J. Virol. 2007, 82, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Wollmann, G.; Paglino, J.C.; Maloney, P.R.; Ahmadi, S.A.; Van den Pol, A.N. Attenuation of vesicular stomatitis virus infection of brain using antiviral drugs and an adeno-associated virus-interferon vector. Virology 2015, 475, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ganar, K.; Das, M.; Sinha, S.; Kumar, S. Newcastle disease virus: Current status and our understanding. Virus Res. 2014, 184, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, J.M.; Mustafa, M.Z.; Ideris, A. New Castle disease virus interaction in targeted therapy against proliferation and invasion pathways of glioblastoma multiforme. BioMed Res. Int. 2014, 2014, 1–11. [Google Scholar] [CrossRef]
- Schirrmacher, V. Signaling through RIG-I and type I interferon receptor: Immune activation by Newcastle disease virus in man versus immune evasion by Ebola virus (review). Int. J. Mol. Med. 2015, 36, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.-J.; Tsai, J.-C.; Tseng, Y.-T.; Wu, M.-S.; Liu, W.-S.; Lam, H.-I.; Yu, J.-H.; Nozell, S.E.; Benveniste, E.N. Small G protein Rac GTPases regulate the maintenance of glioblastoma stem-like cells in vitro and in vivo. Oncotarget 2017, 8, 18031–18049. [Google Scholar] [CrossRef]
- Puhlmann, J.; Puehler, F.; Mumberg, D.; Boukamp, P.; Beier, R. Rac1 is required for oncolytic NDV replication in human cancer cells and establishes a link between tumorigenesis and sensitivity to oncolytic virus. Oncogene 2010, 29, 2205–2216. [Google Scholar] [CrossRef]
- Krishnamurthy, S.; Takimoto, T.; Scroggs, R.A.; Portner, A. Differentially regulated interferon response determines the outcome of Newcastle disease virus infection in normal and tumor cell lines. J. Virol. 2006, 80, 5145–5155. [Google Scholar] [CrossRef]
- García-Romero, N.; Palacín-Aliana, I.; Esteban-Rubio, S.; Madurga, R.; Rius-Rocabert, S.; Carrión-Navarro, J.; Presa, J.; Cuadrado-Castano, S.; Sánchez-Gómez, P.; García-Sastre, A.; et al. Newcastle disease virus (NDV) oncolytic activity in human glioma tumors is dependent on CDKN2A-type I IFN gene cluster codeletion. Cells 2020, 9, 1405. [Google Scholar] [CrossRef]
- Zulkifli, M.M.; Ibrahim, R.; Ali, A.M.; Aini, I.; Jaafar, H.; Hilda, S.S.; Alitheen, N.B.; Abdullah, J.M. Newcastle diseases virus strain V4UPM displayed oncolytic ability against experimental human malignant glioma. Neurol. Res. 2009, 31, 3–10. [Google Scholar] [CrossRef]
- Kazimirsky, G.; Jiang, W.; Slavin, S.; Ziv-Av, A.; Brodie, C. Mesenchymal stem cells enhance the oncolytic effect of Newcastle disease virus in glioma cells and glioma stem cells via the secretion of TRAIL. Stem Cell Res. Ther. 2016, 7, 149. [Google Scholar] [CrossRef] [PubMed]
- Koks, C.A.; Garg, A.D.; Ehrhardt, M.; Riva, M.; Vandenberk, L.; Boon, L.; De Vleeschouwer, S.; Agostinis, P.; Graf, N.; Van Gool, S.W. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int. J. Cancer 2014, 136, E313–E325. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Chen, Y.; Hong, X.; Liu, X.; Su, X.; Li, S.; Dong, X.; Zhao, G.; Li, Y. Newcastle disease virus enhances the growth-inhibiting and proapoptotic effects of temozolomide on glioblastoma cells in vitro and in vivo. Sci. Rep. 2018, 8, 11470. [Google Scholar] [CrossRef] [PubMed]
- Csatary, L.; Gosztonyi, G.; Szeberenyi, J.; Fabian, Z.; Liszka, V.; Bodey, B.; Csatary, C. MTH-68/H oncolytic viral treatment in human high-grade gliomas. J. Neuro-Oncol. 2004, 67, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V.; Lorenzen, D.; Van Gool, S.W.; Stuecker, W. A New strategy of cancer immunotherapy combining hyperthermia/oncolytic virus pretreatment with specific autologous anti-tumor vaccination-A review. Austin Oncol. Case Rep. 2017, 2, 1–8. [Google Scholar] [CrossRef]
- Freeman, A.I.; Zakay-Rones, Z.; Gomori, J.M.; Linetsky, E.; Rasooly, L.; Greenbaum, E.; Rozenman-Yair, S.; Panet, A.; Libson, E.; Irving, C.S.; et al. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol. Ther. 2006, 13, 221–228. [Google Scholar] [CrossRef]
- Reddy, P.S.; Burroughs, K.D.; Hales, L.M.; Ganesh, S.; Jones, B.H.; Idamakanti, N.; Hay, C.; Li, S.S.; Skele, K.L.; Vasko, A.-J.; et al. Seneca Valley Virus, a systemically deliverable oncolytic picornavirus, and the treatment of neuroendocrine cancers. J. Natl. Cancer Inst. 2007, 99, 1623–1633. [Google Scholar] [CrossRef]
- Bs, C.L.M.; Houghton, P.J.; Kolb, E.A.; Gorlick, R.G.; Reynolds, C.P.; Kang, M.H.; Maris, J.M.; Keir, S.T.; Wu, J.; Smith, M.A.; et al. Initial testing of the replication competent Seneca Valley virus (NTX-010) by the pediatric preclinical testing program. Pediatr. Blood Cancer 2010, 55, 295–303. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, X.; Mao, H.; Baxter, P.A.; Huang, Y.; Yu, L.; Wadhwa, L.; Su, J.M.; Adesina, A.; Perlaky, L.; et al. Intravenous injection of oncolytic picornavirus SVV-001 prolongs animal survival in a panel of primary tumor–based orthotopic xenograft mouse models of pediatric glioma. Neuro-Oncology 2013, 15, 1173–1185. [Google Scholar] [CrossRef]
- Mehndiratta, M.M.; Mehndiratta, P.; Pande, R. Poliomyelitis. Neurohospitalist 2014, 4, 223–229. [Google Scholar] [CrossRef]
- Gromeier, M.; Alexander, L.; Wimmer, E. Internal ribosomal entry site substitution eliminates neurovirulence in intergeneric poliovirus recombinants. Proc. Natl. Acad. Sci. USA 1996, 93, 2370–2375. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, E.; Jiang, H.; Muszynski, K.; Harris, R.D.; Giardina, S.L.; Gromeier, M.; Mitra, G.; Soman, G. Evaluation of IRES-mediated, cell-type-specific cytotoxicity of poliovirus using a colorimetric cell proliferation assay. J. Virol. Methods 2009, 155, 44–54. [Google Scholar] [CrossRef]
- Merrill, M.K.; Bernhardt, G.; Sampson, J.H.; Wikstrand, C.J.; Bigner, D.D.; Gromeier, M. Poliovirus receptor CD155-targeted oncolysis of glioma. Neuro-Oncology 2004, 6, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Dobrikova, E.Y.; Broadt, T.; Poiley-Nelson, J.; Yang, X.; Soman, G.; Giardina, S.; Harris, R.; Gromeier, M. Recombinant oncolytic poliovirus eliminates glioma in vivo without genetic adaptation to a pathogenic phenotype. Mol. Ther. 2008, 16, 1865–1872. [Google Scholar] [CrossRef] [PubMed]
- Gromeier, M.; Lachmann, S.; Rosenfeld, M.R.; Gutin, P.H.; Wimmer, E. Intergeneric poliovirus recombinants for the treatment of malignant glioma. Proc. Natl. Acad. Sci. USA 2000, 97, 6803–6808. [Google Scholar] [CrossRef]
- Goetz, C.; Dobrikova, E.; Shveygert, M.; Dobrikov, M.; Gromeier, M. Oncolytic poliovirus against malignant glioma. Futur. Virol. 2011, 6, 1045–1058. [Google Scholar] [CrossRef]
- Desjardins, A.; Gromeier, M.; Ii, J.E.H.; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.; Nair, S.; et al. Recurrent glioblastoma treated with recombinant poliovirus. N. Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef]
- Wang, K.S.; Kuhn, R.J.; Strauss, E.G.; Ou, S.; Strauss, J.H. High-affinity laminin receptor is a receptor for Sindbis virus in mammalian cells. J. Virol. 1992, 66, 4992–5001. [Google Scholar] [CrossRef]
- Tseng, J.-C.; Levin, B.; Hirano, T.; Yee, H.; Pampeno, C.; Meruelo, D. In vivo antitumor activity of Sindbis viral vectors. J. Natl. Cancer Inst. 2002, 94, 1790–1802. [Google Scholar] [CrossRef]
- Zhang, J.; Frolov, I.; Russell, S.J. Gene therapy for malignant glioma using Sindbis vectors expressing a fusogenic membrane glycoprotein. J. Gene Med. 2004, 6, 1082–1091. [Google Scholar] [CrossRef]
- Tseng, J.-C.; Granot, T.; Digiacomo, V.; Levin, B.; Meruelo, D. Enhanced specific delivery and targeting of oncolytic Sindbis viral vectors by modulating vascular leakiness in tumor. Cancer Gene Ther. 2009, 17, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Tseng, J.-C.; Levin, B.; Hurtado, A.; Yee, H.; De Castro, I.P.; Jimenez, M.; Shamamian, P.; Jin, R.; Novick, R.P.; Pellicer, A.; et al. Systemic tumor targeting and killing by Sindbis viral vectors. Nat. Biotechnol. 2003, 22, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Harmon, B.; Schudel, B.R.; Maar, D.; Kozina, C.; Ikegami, T.; Tseng, C.-T.K.; Negrete, O.A. Rift Valley fever virus strain MP-12 enters mammalian host cells via caveola-mediated endocytosis. J. Virol. 2012, 86, 12954–12970. [Google Scholar] [CrossRef] [PubMed]
- Ritter, M.; Bouloy, M.; Vialat, P.; Janzen, C.; Haller, O.; Frese, M. Resistance to Rift Valley fever virus in Rattus norvegicus: Genetic variability within certain ‘inbred’ strains. J. Gen. Virol. 2000, 81, 2683–2688. [Google Scholar] [CrossRef]
- Filone, C.M.; Heise, M.; Doms, R.W.; Bertolotti-Ciarlet, A. Development and characterization of a Rift Valley fever virus cell–cell fusion assay using alphavirus replicon vectors. Virology 2006, 356, 155–164. [Google Scholar] [CrossRef]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus: Replication and immunotherapeutic effects in recurrent malignant glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef]
- Steiner, H.-H.; Bonsanto, M.M.; Beckhove, P.; Brysch, M.; Geletneky, K.; Ahmadi, R.; Schuele-Freyer, R.; Kremer, P.; Ranaie, G.; Matejic, D.; et al. Antitumor vaccination of patients with glioblastoma multiforme: A pilot study to assess feasibility, safety, and clinical benefit. J. Clin. Oncol. 2004, 22, 4272–4281. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug and gene delivery to the brain. Neuron 2002, 36, 555–558. [Google Scholar] [CrossRef]
- Li, L.; Liu, S.; Han, D.; Tang, B.; Ma, J. Delivery and biosafety of oncolytic virotherapy. Front. Oncol. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Howells, A.; Marelli, G.; Lemoine, N.R.; Wang, Y. Oncolytic viruses-interaction of virus and tumor cells in the battle to eliminate cancer. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef]
- Nguyen, A.; Ho, L.; Wan, Y. Chemotherapy and oncolytic virotherapy: Advanced tactics in the war against cancer. Front. Oncol. 2014, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mulder, W.J.M.; Ochando, J.; Joosten, L.A.; Fayad, Z.A.; Netea, M. Therapeutic targeting of trained immunity. Nat. Rev. Drug Discov. 2019, 18, 553–566. [Google Scholar] [CrossRef] [PubMed]


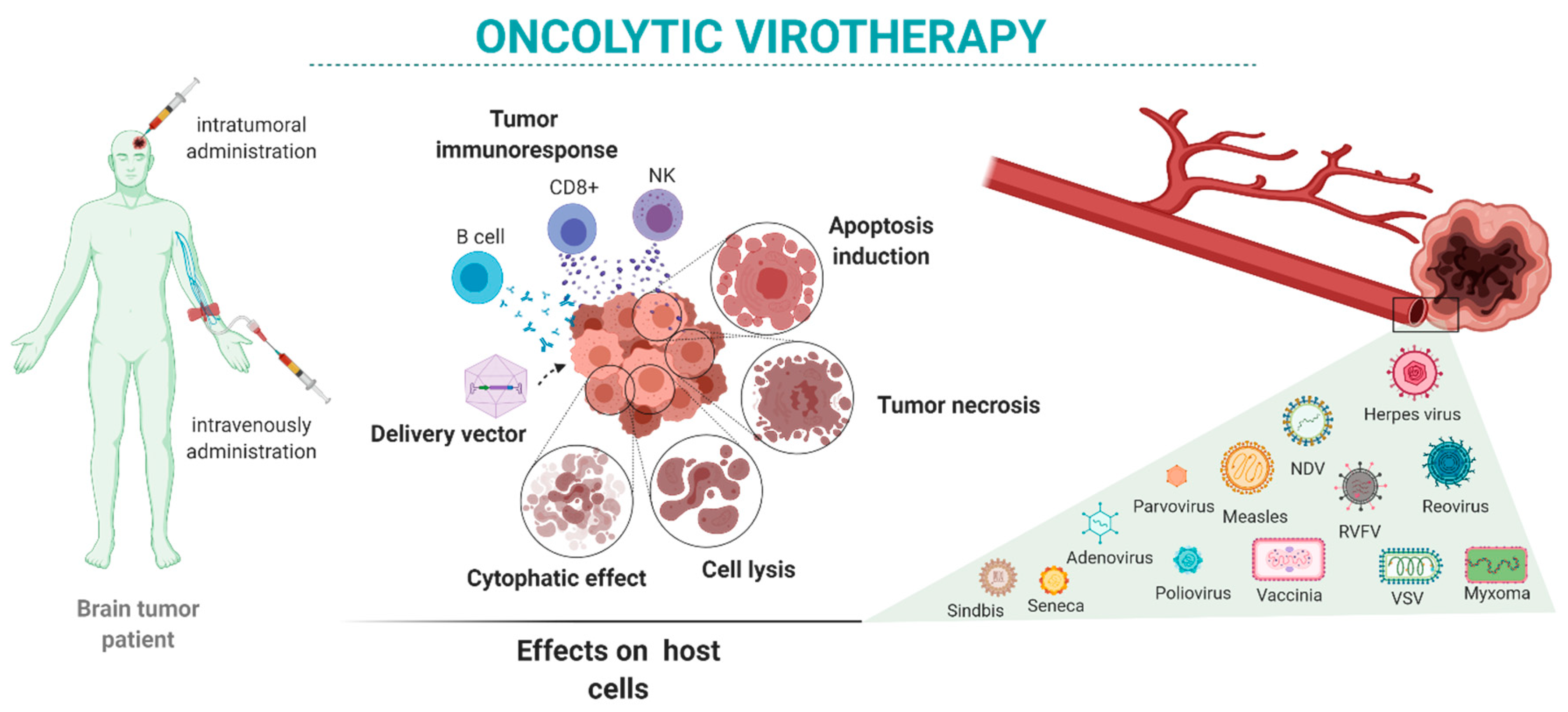{kind=link}
| Virus | Modifications | Cell Lines | In Vivo Models | Results |
|---|---|---|---|---|
| Herpes | dlsptk: TK deletion. | Human: U87 and T98G [22] | U87 i.c. and s.c. nude mice | Tumor cell infection and death. In vivo tumor reduction and increased surveillance. |
| HSV-1716: γ134.5 loci partial deletion. | Human: U87, T98G, SB18, U373 and U251 [24] | - | Tumor cell infection and death. | |
| G207: γ134.5 loci deletion. ICP6 truncation. | Human: U87, U373, U138 and T98G [26] | U87 i.c. and s.c. nude mice, i.c. owl monkeys | Elimination of tumor cells, necrosis and no toxicity. | |
| rQNestin34.5: ICP6 deletion. γ134.5 expression under Nestin promoter. | Human: U251, U87dEGFR, T98G, Gli36d5, U138, and MGH238 [28] | U87dEGFR i.c. and s.c. nude mice | Increase of oncolytic activity at in vitro and in vivo models | |
| NG34: γ134.5 loci deletion. ICP6 deletion. GADD34 expression under Nestin promoter. | Human: U251, U87ΔEGFR and primary glioma cells Murine: GL261 [30] | U87ΔEGRF-RliFluc and G35 i.c. nude mice, BALB/c mice | Similar oncolytic activity as rQNestin34.5 with lower neurotoxicity. | |
| NG34scFvPD-1: γ134.5 loci deletion. ICP6 deletion. GADD34 expression under Nestin promoter. scFvPD-1 expression under CMV’s IE promoter. | Human: U87ΔEGFR and U251 Murine: GL261N4 and CT2A [31] | GL261N4 and CT2A i.c. C57Bl/6J mice, GL261N4 and U87ΔEGFR i.c. nude mice | Increased oncolytic activity in comparison to NG34 in immunocompetent mice. Development of specific immunity and memory. | |
| G47Δ: γ134.5 loci deletion. ICP6 truncation. α47 deletion. US11 expression under α47 promoter. | Human: U87 and U373 [34] | U87 s.c. in nude mice | Increased survival, higher number of cured mice than G207. | |
| C134: γ134.5 loci deletion. HCMV’s IRS1 protein expression. | Human: D54, U87 and U251 Murine: N2A [32] | U87 i.c. in SCID mice | Reduced tumor volume and increased surveillance. | |
| Human and murine: 12 established GBM [33] | N2A orthotopic in A/J and BALB/c mice | Improved replication and longer survival in vivo | ||
| HSV-1 R-LM113: insertion of scFvHER2 in gD protein. | Murine: established GBM [35] | PDGFB/DsRed-induced gliomas in nude mice | No toxicity in nude mice and oncolytic effect in HER2 overexpressing and established tumors in vivo. | |
| RAMBO: γ134.5 loci deletion. ICP6 truncation. Vstat120 expression under IE4/5 HSV promoter. | Human: U343, U87, U87ΔEGFR, LN229, Gli36ΔEGFR-H2B-RFP, U251-T2, U87ΔEGFR-Luc [36] | U87ΔEGFR-Luc and Gli36ΔEGFR-H2B-RFP i.c. and U87ΔEGFR-Luc s.c. nude mice | Increased survival in vivo and inhibition of tumor vascularization. | |
| M002: γ134.5 loci deletion. IL-12 expression. | Murine: 4C8 [37] | 4C8 i.c. gliomas in B6D2F1 mice | Increase mice survival, infiltration of CD4+, CD8+ and NK cells. Longer viral persistence in tumors | |
| HSV-IL4: γ134.5 loci deletion. IL-4 expression. | Human: U251 and D54 [21] | GL-261 i.c. in C57BL/6 | Infiltration of macrophages, CD4+ and CD8+. Longer survival. | |
| Adenovirus | ONIX-15: E1B-55kD deletion. | Human: 4 primary GBM [49] | S.c. xenograft in nude mice | Tumor regression. |
| Delta-24-RGD: E1A partial deletion. RGD tripeptide incorporation. | Human: U251, U373, U87 and D54 [53] | D54 s.c. in nude mice | Cell death with low doses, single injection inhibits tumor growth, several injections resulted in 36% of animals with tumor regression. | |
| ICOVIR-5: E1A expression under E2F-1 promoter. E1A partial deletion. RGD tripeptide incorporation. | Human: U251 and U87 [54] | U87 i.c. xenograft in nude mice | Tumor cytotoxic effect in vitro high tumor selectivity and increase of survival in vivo. | |
| Adenovirus | ICOVIR-17: E1A expression under a promoter including four palindromic E2F-1 sites and a Sp-1-binding site. E1A partial deletion. RGD tripeptide incorporation. PH20 expression under MLP promoter. | Human: U87, U138, LN308, Gli36, U373, LN229 and 6 primary GBM [60] | U87 and CSCs i.c. in nude mice | Better distribution in HA tumors. Longer mice survival. |
| VCN-01: E1A expression under a promoter including four palindromic E2F-1 sites and a Sp-1-binding site. E1A partial deletion. RGD relocated in fiver shaft protein. PH20 expression under MLP promoter. | Human: U87, A172, T98G, U251, U373, SNB19 and 2 GBM CSC [61] | U87 and GBM CSC i.c. xenografts in nude mice | Control of tumor growth One single injection improves survival in aggressive infiltrative tumor. | |
| Delta-24-RGDOX: E1A partial deletion. RGD tripeptide incorporation. OX40L expression. | Human: U87 Murine: GL261 [62] | GL261 i.c. in C57BL/6 mice | Proliferation of tumor specific T cells. Sinergy with anti PD-L1. | |
| Delta-24-GREAT: E1A partial deletion. RGD tripeptide incorporation. GITRL expression. | Human: U87 and U251 Murine: GL261 [63] | GL261 i.c. in C57BL/6 mice | Extended survival and development of antiviral and antitumor specific response and memory. | |
| Ad-RTS-IL-12: No replicative. Expression of IL-12 under RTS® system with veledimex as a co-treatment. | Murine: GL261 [64] | GL261 i.c. in C57BL/6 mice | Tumor infiltration with CD8, extended survival and immune memory development. | |
| Vaccinia | rVV-p53: p53 expression. | Rat: C6 [69] | C6 s.c. in nude mice | Moderate cell apoptosis. Tumor growth control. |
| rVV-mIL12/mIL2: IL12 expression. IL2 expression. | Rat: C6 [71] | C6 s.c. in nude mice | Cytokine toxicity at high dose Antitumor NK dependent effect. | |
| rVV-p53 and rVV-mL12: p53 expression. IL12 expression. | Rat: C6 [74] | C6 s.c. in nude mice | Better tumor growth control. Higher NK and macrophage infiltration. | |
| vvDD: TK deletion. VGF deletion. | Human: A172, U87MG and U118 Rat: RG2, F98 and C6 [73] | U87, U118 and C6 s.c. and RG2, F98 i.c. in nude mice | Control of tumor growth. Sinergy with rapamycin or cyclophosphamide. | |
| Rhesus macaques [75] | No adverse effects. | |||
| vvDD-IL15Rα: TK deletion. VGF deletion. IL15Rα expression. | Murine: GL261 [75] | GL261 i.c. in C57BL/6J | Increase of NK and CD8+ in tumor. Prolonged survival. | |
| TG6002: TK deletion. ribonucleotide reductase genes deletion. FCU1 expression. | Human: U87 and patient derived GBM [76] | U87 i.c. and s.c. in nude mice | Prolonged survival in s.c. and i.c. Synergic effect with 5FC in i.c. model. | |
| Myxoma | MYXV WT | Human: U87, U251, U373, U343, A172 and U118 Rat: RG2 and 9L [79] | U87 and U251 i.c. in nude mice | Regression and longer survival in both models. |
| Human: U87, U251, and U118 [80] | U87 orthotopic in CB-17 SCID mice | Inhibition of MHC-I tumor expression and promotes NK mediated death. | ||
| Human: U118 and 3 patient samples Murine: GL261 Rat: T9 [81] | - | SOC co-treatment increases results of MYXV. | ||
| MYXV WT: administered in ADSCs | Human: U87 and U251 [82] | U87 orthotopic in nude mice | Increase the tumor infection rate | |
| MYXV-M011L-KO: M11L deletion | Human: Brain tumor initiating cells (BTIC) [80] | mBITCs i.c. in C57Bl/6J mice | Prolonged survival. TMZ increases oncolysis | |
| Parvovirus | Human: U87 Rat: RG-2 [89] | U87 i-deficient rats and RG-2 i-competent | Complete remission of the tumors | |
| H-1PV WT | Human: U373, U138 and 5 CSCs [87] Human: U87 Rat: RG-2 [89] | RGD orthotopic ratsU87 i-deficient rats and RG-2 i-competent | Cathepsin B activation induces cell death in H-1PVComplete remission of the tumors | |
| Human: U87, U373, U118, MO59J and A172 Murine: GL261 [90] Human: U373, U138 and 5 CSCs [87] | U87 and U373 s.c. U87 orthotopic CB17-SCID miceRGD orthotopic rats | Selective infection, no toxicity, reduce tumor volume in vivo Cathepsin B activation induces cell death in H-1PV | ||
| MVMp WT | Human: U373, U87, SW1088, SK-N-SH Rat: C6 [91] Human: U87, U373, U118, MO59J and A172 Murine: GL261 [90] | -U87 and U373 s.c. U87 orthotopic CB17-SCID mice | MVM p strain cytotoxic only in U373 and C6 (MVM) selective infection, no toxicity, reduce tumor volume in vivo | |
| Human: U87 and MO59J [92] Human: U373, U87, SW1088, SK-N-SH Rat: C6 [91] | - | Selective infection MVM p strain cytotoxic only in U373 and C6 (MVM) | ||
| Murine: Fibroblast L929 and A9. Astrocytoma MT539MG [93], Human: U87 and MO59J [92] | - | Safe for microglia (MVMp) selective GBM infection (MVM) | ||
| Murine: Fibroblast L929 and A9. Astrocytoma MT539MG [93], | - | Safe for microglia (MVMp) |
| Virus | Modifications | Cell Lines | In Vivo Models | Results |
|---|---|---|---|---|
| Reovirus | Reovirus | Human: 24 GBM cell lines [99] | U87 and U251 intracranial and subcutaneous in SCID mice | Death in 20 out of 24 GBM lines Regression in both in vivo models Toxicity in nude mice. |
| Human: U87 and 2 patient-derived lines [100] | GL261 intracranial in C57/BL6 | i.v. administration reaches brain tumors. T cell tumor recruitment and cytotoxicity. Synergy with anti PD-L1. | ||
| Measles | MV-CEA: CEA expression. | Human: U87, U251, and U118 [105] | U87 intracranial and subcutaneous in nude mice | Regression in s.c. tumor after intravenous and intratumor administration. Regression in intracranial tumor after intratumor administration. |
| MV-NIS: NIS expression. | Human: U87, U251 and 6 patient derived GBM [106] | U251 subcutaneous and GBM intracranial in nude mice | Synergic effect of virotherapy and radiotherapy. | |
| MV-GFP-HAA-scEGFR: H protein partial deletion. scEGFR insertion in H protein. | Human: 5 patient derived GBM [107] | GBM intracranial in nude mice Mouse model Ifnarko CD46 Ge | Tumor regression after intratumor administration. No toxicity in CNS. | |
| MV-141.4: scFvCD133 insertion in H. | Human: primary GBM [111] | GBM intracranial in NOD/SCID | Better survival rate in comparison with MV-Edm. | |
| VSV | VSV WT | - | C6 subcutaneous nude mice [112] | Inhibition of tumor growth. |
| VSV-ΔG: G protein deletion. | Human: U87 Rat: C6 [111] | - | Infection of cell lines. Rapid lysis. | |
| VSVΔM51: M51 single nucleotide deletion. | Human: 14 glioma cell lines and 15 primary gliomas [115] | U87 and U118 subcutaneous in nude mice | Infection and elimination of all cell lines. Tumor regression and prolonged survival. | |
| VSV-rp30: unknown viral glioma adaptation | Human: U87, U118, U373 and A172 [92] | U87 subcutaneous in nude mice | Increased selectivity and lytic capacity in glioblastoma cells. Tumor selectivity and cytopathic effect. | |
| Human: U87 and U118 [116] | U87 orthotopic in nude mice | Infection and lysis of brain and peripheral tumors. | ||
| VSV-CT1/CT2: G protein partial deletion | Human: U87, U118, U373 and A172 [114] | U87 orthotopic in nude mice | Elimination of tumor cells. Normal cell toxicity can be eliminated with IFN co-treatment VSV-1p-GFP: infection and potent apoptosis over tumor. | |
| VSV-1p-GFP: GFP at the first position in the genome. VSV-CT9-M51: G protein partial deletion. M51 single nucleotide deletion. | Human: U87, U118, U373 and A172 Rat: 9L [119] | U87 orthotopic in CB17-SCID mice | VSV-CT1 and VSV-CT9-M51 have less toxicity than wt VSV. VSV-CT9-M51 is able to infect and kill tumors in brain. | |
| VSV-CT9-M51: G protein partial deletion. M51 single nucleotide deletion. | Human: primary GBM [122] | Orthotopic CB17 SCID | Coinfection with AAV-mIFN-β or with ribavirin enhances oncolytic properties. | |
| Seneca Valley | SVV-001 WT | Human: primary GBM [138] | 4 orthotopic models in nude mice | Partial response against glioma cells Effectivity in 2 of 4 in vivo tumors |
| Human: GBM CSCs [139] | 6 GBM CSC orthotopic nude mice | 4 of 6 prolonged survival, tumor infection and cell lysis. Susceptibility dependent of sialic acid presence. | ||
| NDV | NDV WT | Human: 6 GBM CSCs [129] | Orthotopic nude mice | NDV replication is dependent on IFN deletion. |
| Human: U87 and DBTRG.05MG [130] | Subcutaneous nude mice | Induce apoptosis. Decrease tumor volume. | ||
| Human: A172 and U87 and 2 CSCs [131] | - | Induce apoptosis. | ||
| Murine: GL261 [132] | GL261 orthotopic mice | NDV induces ICD. | ||
| Human: T98G, LN18, U251, U87. Rat: C6 [133] | C6 in rats | Synergistic effects with TMZ. Decrease tumor volumes and increase OS. | ||
| Poliovirus | PVS-RIPO: IRES replaced with HRV2 | Human: U87 [142] | - | Reduce viability. |
| Human: CSCs and established cell lines [145] | HTB14 orthotopic and HTB15 flanks | Tumor regression. | ||
| Human: 6 CSCs [143] | - | Cytolysis. | ||
| Human: U87, HTB14 and HTB15 [144] | HTB15 in athymic Balb/c mice | Tumor regression. | ||
| Sindbis | Sindbis WT | Human: U87, U-118, U373, M059J, A172 [92] | U87 in flanks CB17-SCID mice | Effective replication and selective kill U87. |
| Sindbis Gal.fu | Human: U87 [150] | U87 orthotopic in nude mice | Cytopathic activity. | |
| RVFV | RVFV MP-12 and ZH548: attenuated strains | Rat: C6 [154] Human: U87 [155] | - | Infection occurs. |
| Virus | Phase and Reference | n Patients | Results |
|---|---|---|---|
| Herpes | Phase I: HSV-1716 [38] | 9 | Two 24 moth survivors |
| Phase Ib: HSV-1716 [39] | 12 | Evidence of tumor infection Three patients clinically stable for two years | |
| Phase II: HSV-1716 NCT02031965 | 2 | No results available | |
| Phase I: G207 [41] | 21 | No toxicities | |
| Phase Ib: G207 [41] | 6 | No toxicity Evidence of tumor infection | |
| Phase I: G207 [42] | 9 | No toxicities in combination with 5 Gy | |
| Phase I: rQNestin34.5v2 NCT03152318 | 108 | Recruiting | |
| Phase I: C134 NCT03657576 | 24 | Recruiting | |
| Adenovirus | Phase I: ONYX-015 [66] | 24 | No toxicity One patient without progression and some with regression |
| Phase I: Delta-24-RGD NCT03896568 | 36 | Recruiting | |
| Phase I: Delta-24-RGD NCT03178032 | 12 | No results available | |
| Phase II: Delta-24-RGD NCT02798406 | 49 | Active | |
| Phase I: Delta-24-RGD NCT02197169 | 37 | No toxicities | |
| Phase I: Delta-24-RGD NCT01956734 | 31 | No results available | |
| Phase I and II: Delta-24-RGD NCT01582516 [156] | 20 | Virus spread in tumor, oncolytic effect and immunostimulation | |
| Phase I: Delta-24-RGD NCT00805376 | 37 | 20% of >3 year survivors 12% of >95% tumor regression Evidence of immunostimulation | |
| Phase II Delta-24-RGD (2016-001600-40) | - | Discontinued | |
| Phase I: Delta-24-RGD NCT03714334 | 24 | Recruiting | |
| Phase I: Delta-24-RGD NCT03072134 | 36 | No results available | |
| Phase I: DNX-2440 NCT03714334 | 24 | Recruiting | |
| Phase I/II: Ad-RTS-IL-12 NCT03330197 | 45 | Recruiting | |
| Reovirus | Phase I: Reovirus [101] | 12 | No toxicities |
| Phase I: Reovirus NCT00528684 [102] | 15 | One 2 year survivor One 3 year survivor | |
| Phase Ib: Reovirus [100] | 9 | Evidence of T cell tumor infiltration and upregulation of IFN and PD-1/PD-L1 axis | |
| Phase I: Reovirus/Sargramostim NCT02444546 | 6 | Active | |
| Vaccinia | Phase I and II: TG6002 NCT03294486 | 78 | Recruiting |
| Measles | Phase I: MV-CEA NCT00390299 | 23 | No toxicities |
| NDV | Phase I/II: NDV-HUJ NCT01174537 [136] | 14 | No toxicities Complete regression in 1 patient |
| Phase 0: MTH-68/H [134] | 4 | OS 5–9 years | |
| VOL-DC vaccine [135] | 10 | Increased OS | |
| Phase II: ATV-NDV vaccine [157] | 23 | PFS 40 weeks vs. 26 weeks | |
| Parvovirus | H-1PV [94] | 18 | Enhanced immunogenicity |
| Poliovirus | Phase I: NCT01491893 [147] | 61 | No neurovirulence and increased survival rate |
| Phase II: NCT02986178 | 122 | Active | |
| Phase Ib: NCT03043391 | 12 | Recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rius-Rocabert, S.; García-Romero, N.; García, A.; Ayuso-Sacido, A.; Nistal-Villan, E. Oncolytic Virotherapy in Glioma Tumors. Int. J. Mol. Sci. 2020, 21, 7604. https://doi.org/10.3390/ijms21207604
Rius-Rocabert S, García-Romero N, García A, Ayuso-Sacido A, Nistal-Villan E. Oncolytic Virotherapy in Glioma Tumors. International Journal of Molecular Sciences. 2020; 21(20):7604. https://doi.org/10.3390/ijms21207604
Chicago/Turabian StyleRius-Rocabert, Sergio, Noemí García-Romero, Antonia García, Angel Ayuso-Sacido, and Estanislao Nistal-Villan. 2020. "Oncolytic Virotherapy in Glioma Tumors" International Journal of Molecular Sciences 21, no. 20: 7604. https://doi.org/10.3390/ijms21207604
APA StyleRius-Rocabert, S., García-Romero, N., García, A., Ayuso-Sacido, A., & Nistal-Villan, E. (2020). Oncolytic Virotherapy in Glioma Tumors. International Journal of Molecular Sciences, 21(20), 7604. https://doi.org/10.3390/ijms21207604