Tissue-Specific Transcriptome Analysis Reveals Candidate Transcripts Associated with the Process of Programmed B Chromosome Elimination in Aegilops speltoides

 , , , and
, , , and 
Abstract
1. Introduction
2. Results and Discussion
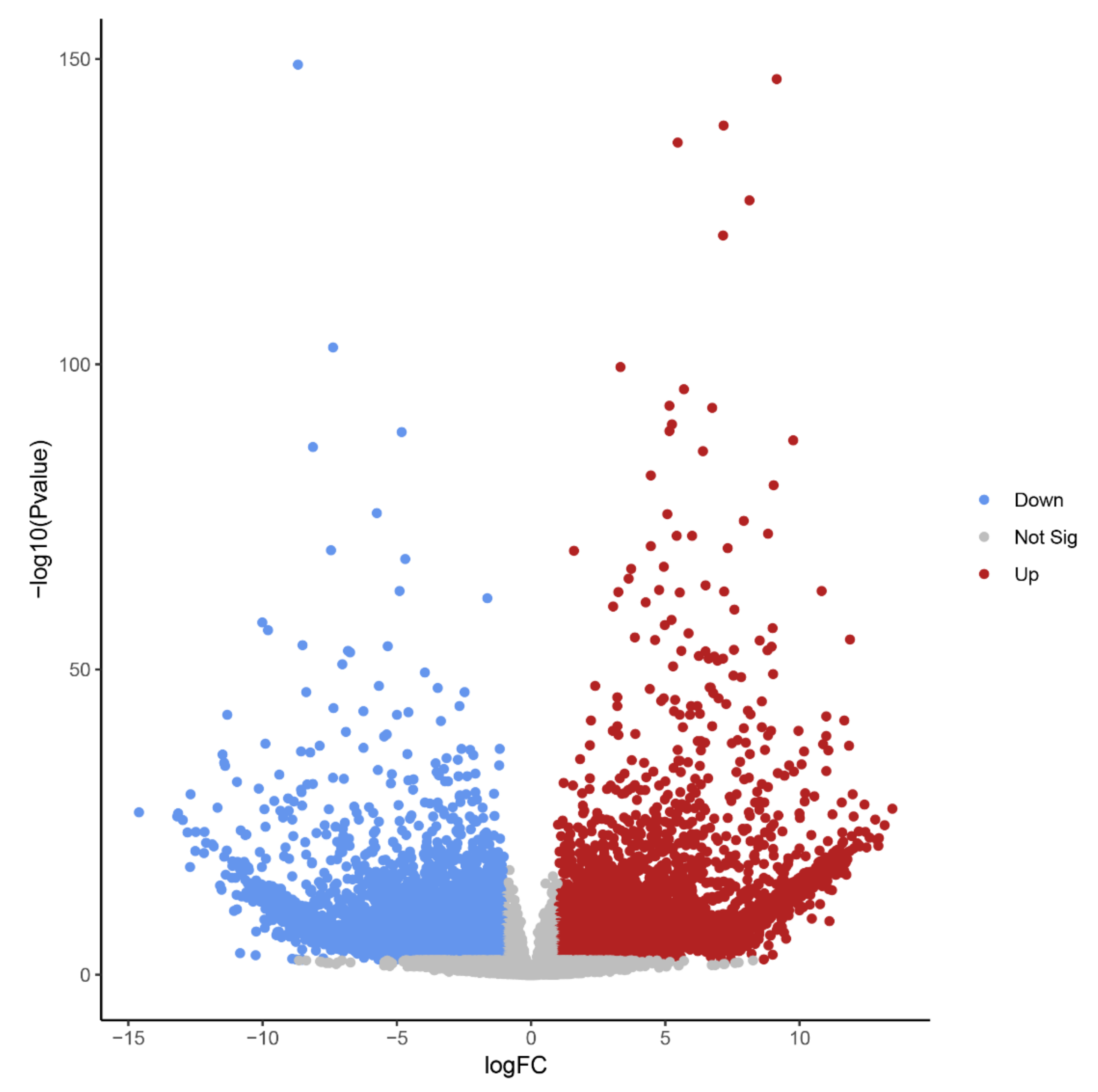
2.1. The Transcriptome of Embryos Undergoing B Chromosome Elimination Is Strongly Affected
2.2. Biological Processes Potentially Related to the Elimination of B Chromosomes
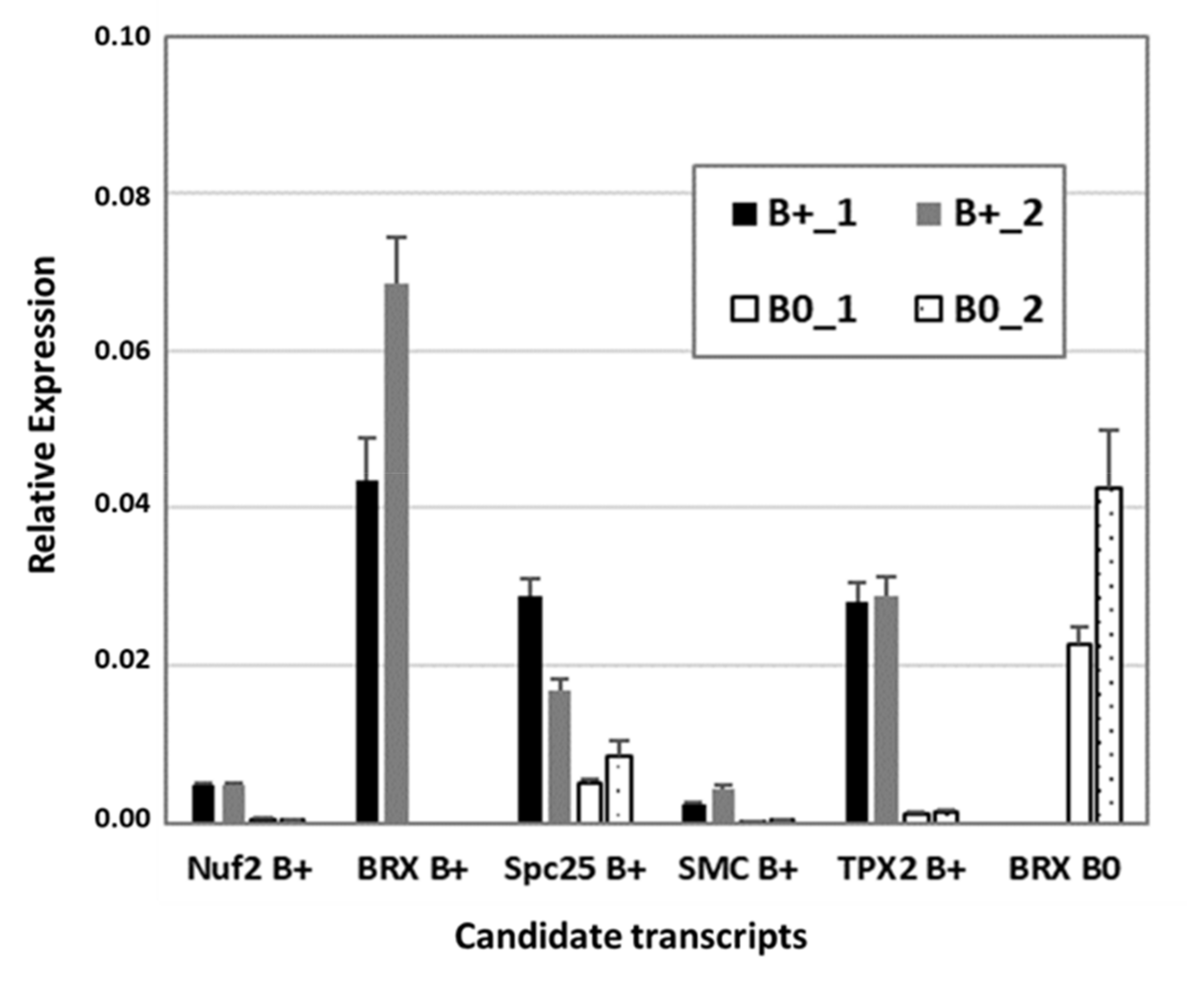
2.3. B Chromosome-Specific Transcripts
3. Materials and Methods
3.1. Plant Material
3.2. Laser Capture Microdissection (LCM) and RNA Extraction
3.3. RNA Sequencing and Data Preprocessing
3.4. Sequence Assembly and Annotation
3.5. Quality Analysis and Differential Expression Analysis
3.6. Transcriptome Annotation
3.7. GO Term Enrichment Analysis
3.8. RT-qPCR
3.9. Data Availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ENA | European Nucleotide Archive |
| DAA | Day After Anthesis |
| DEGs | Differentially Expressed Genes |
| FC | Fold Change |
| FDR | False Discovery Rate |
| GO | Gene Ontology |
| LCM | Laser Capture Microdissection |
| PCA | Principle Component Analysis |
References
- Wang, J.; Davis, R.E. Programmed DNA elimination in multicellular organisms. Curr. Opin. Genet. Dev. 2014, 27, 26–34. [Google Scholar] [CrossRef]
- Kloc, M.; Zagrodzinska, B. Chromatin elimination - an oddity or a common mechanism in differentiation and development? Differentiation 2001, 68, 84–91. [Google Scholar] [CrossRef]
- Ruban, A.; Schmutzer, T.; Wu, D.D.; Fuchs, J.; Boudichevskaia, A.; Rubtsova, M.; Pistrick, K.; Melzer, M.; Himmelbach, A.; Schubert, V.; et al. Supernumerary B chromosomes of Aegilops speltoides undergo precise elimination in roots early in embryo development. Nat. Commun. 2020, 11, 2764. [Google Scholar] [CrossRef]
- Houben, A.; Banaei-Moghaddam, A.M.; Klemme, S.; Timmis, J.N. Evolution and biology of supernumerary B chromosomes. Cell. Mol. Life Sci. 2013, 71, 467–478. [Google Scholar] [CrossRef]
- Benetta, E.D.; Akbari, O.S.; Ferree, P.M. Sequence expression of supernumerary B chromosomes: Function or fluff? Genes 2019, 10, 123. [Google Scholar] [CrossRef]
- Mendelson, D.; Zohary, D. Behavior and transmission of supernumerary chromosomes in Aegilops speltoides. Heredity 1972, 29, 329. [Google Scholar] [CrossRef]
- Berger, C.A.; McMahon, R.M.; Witkus, E.R. The cytology of Xanthisma texanum D.C. III: Differential somatic reduction. Bull. Torrey Bot. Club 1955, 82, 277–382. [Google Scholar] [CrossRef]
- Semple, J.C. Behavior of B chromosomes in Xanthisma texanum Dc-Nonrandom phenomenon. Science 1972, 175, 666. [Google Scholar] [CrossRef]
- Müntzing, A. Cytological studies of extra fragment chromosomes in rye. III. The mechanism of non-disjunction at the pollen mitosis. Hereditas 1946, 32, 97–119. [Google Scholar] [CrossRef]
- Janaki-Ammal, E.K. Chromosome diminution in a plant. Nature 1940, 146, 839–840. [Google Scholar] [CrossRef]
- Ohta, S. Mechanisms of B-chromosome accumulation in Aegilops mutica Boiss. Genes Genet. Syst. 1996, 71, 23–29. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Xiang, D.; Quilichini, T.D.; Liu, Z.; Gao, P.; Pan, Y.; Li, Q.; Nilsen, K.T.; Venglat, P.; Esteban, E.; Pasha, A.; et al. The transcriptional landscape of polyploid wheats and their diploid ancestors during embryogenesis and grain development. Plant Cell 2019, 31, 2888–2911. [Google Scholar] [CrossRef]
- Amini, H.; Naghavi, M.R.; Shen, T.; Wang, Y.; Nasiri, J.; Khan, I.A.; Fiehn, O.; Zerbe, P.; Maloof, J.N. Tissue-Specific transcriptome analysis reveals candidate genes for terpenoid and phenylpropanoid metabolism in the medicinal plant Ferula assafoetida. G3 Genes Genomes Genet. 2019, 9, 807–816. [Google Scholar] [CrossRef]
- Kushwaha, S.K.; Grimberg, A.; Carlsson, A.S.; Hofvander, P. Charting oat (Avena sativa) embryo and endosperm transcription factor expression reveals differential expression of potential importance for seed development. Mol. Genet. Genom. 2019, 294, 1183–1197. [Google Scholar] [CrossRef]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: protein domains identifier. Nucleic Acids Res. 2005, 33, W116–120. [Google Scholar] [CrossRef]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L.; et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009, 37, D211–D215. [Google Scholar] [CrossRef]
- Cock, P.J.; Chilton, J.M.; Gruning, B.; Johnson, J.E.; Soranzo, N. NCBI BLAST+ integrated into Galaxy. Gigascience 2015, 4, 39. [Google Scholar] [CrossRef]
- Ma, W.; Gabriel, T.S.; Martis, M.M.; Gursinsky, T.; Schubert, V.; Vrana, J.; Dolezel, J.; Grundlach, H.; Altschmied, L.; Scholz, U.; et al. Rye B chromosomes encode a functional Argonaute-like protein with in vitro slicer activities similar to its A chromosome paralog. New Phytol. 2017, 213, 916–928. [Google Scholar] [CrossRef]
- Luptovciak, I.; Komis, G.; Takac, T.; Ovecka, M.; Samaj, J. Katanin: A sword cutting microtubules for cellular, developmental, and physiological purposes. Front. Plant Sci. 2017, 8, 1982. [Google Scholar] [CrossRef]
- Zhang, T.; Zhou, Y.; Qi, S.T.; Wang, Z.B.; Qian, W.P.; Ouyang, Y.C.; Shen, W.; Schatten, H.; Sun, Q.Y. Nuf2 is required for chromosome segregation during mouse oocyte meiotic maturation. Cell Cycle 2015, 14, 2701–2710. [Google Scholar] [CrossRef]
- Podszywalow-Bartnicka, P.; Wolczyk, M.; Kusio-Kobialka, M.; Wolanin, K.; Skowronek, K.; Nieborowska-Skorska, M.; Dasgupta, Y.; Skorski, T.; Piwocka, K. Downregulation of BRCA1 protein in BCR-ABL1 leukemia cells depends on stress-triggered TIAR-mediated suppression of translation. Cell Cycle 2014, 13, 3727–3741. [Google Scholar] [CrossRef]
- Wang, S.; Chang, Y.; Ellis, B. Overview of OVATE FAMILY PROTEINS, a novel class of plant-specific growth regulators. Front. Plant Sci. 2016, 7, 417. [Google Scholar] [CrossRef]
- Wang, S.; Chang, Y.; Guo, J.; Chen, J.G. Arabidopsis Ovate Family Protein 1 is a transcriptional repressor that suppresses cell elongation. Plant J. 2007, 50, 858–872. [Google Scholar] [CrossRef]
- Luo, J.; Zhou, J.J.; Zhang, J.Z. Aux/IAA gene family in plants: Molecular structure, regulation, and function. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Wu, D.D.; Ruban, A.; Fuchs, J.; Macas, J.; Novák, P.; Vaio, M.; Zhou, Y.H.; Houben, A. Nondisjunction and unequal spindle organization accompany the drive of Aegilops speltoides B chromosomes. New Phytol. 2019, 223, 1340–1352. [Google Scholar] [CrossRef]
- Ruban, A.; Fuchs, J.; Marques, A.; Schubert, V.; Soloviev, A.; Raskina, O.; Badaeva, E.; Houben, A. B Chromosomes of Aegilops speltoides are enriched in organelle genome-derived sequences. PLoS ONE 2014, 9, e90214. [Google Scholar] [CrossRef]
- Brandt, R.; Mascher, M.; Thiel, J. Laser capture microdissection-based RNA-seq of barley grain tissues. Methods Mol. Biol. 2018, 1723, 397–409. [Google Scholar] [CrossRef]
- Andrews, S. Fast QC: A Quality Control Tool for High throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 13 October 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics (Oxford, England) 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Smith-Unna, R.; Boursnell, C.; Patro, R.; Hibberd, J.M.; Kelly, S. TransRate: reference-free quality assessment of de novo transcriptome assemblies. Genome Res. 2016, 26, 1134–1144. [Google Scholar] [CrossRef]
- Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate—A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Waterhouse, R.M.; Seppey, M.; Simao, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO Applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Czechowski, T.; Bari, R.P.; Stitt, M.; Scheible, W.R.; Udvardi, M.K. Real-time RT-PCR profiling of over 1400 Arabidopsis transcription factors: Unprecedented sensitivity reveals novel root- and shoot-specific genes. Plant J. 2004, 38, 366–379. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amount Studied | Differentially Expressed | Highly DE 1 | DE B0 Unique | DE Bplus Unique | ||||
|---|---|---|---|---|---|---|---|---|
| p < 0.05 | p < 0.01 | p < 0.01 | p < 0.05 | p < 0.01 | p < 0.05 | p < 0.01 | ||
| ranscr. isoforms | 41,615 | 21,197 | 16,162 | 14,578 | 390 | 387 | 245 | 240 |
| Unigenes | 45,533 | 20,276 | 15,044 | 13,519 | 561 | 524 | 597 | 519 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boudichevskaia, A.; Ruban, A.; Thiel, J.; Fiebig, A.; Houben, A. Tissue-Specific Transcriptome Analysis Reveals Candidate Transcripts Associated with the Process of Programmed B Chromosome Elimination in Aegilops speltoides. Int. J. Mol. Sci. 2020, 21, 7596. https://doi.org/10.3390/ijms21207596
Boudichevskaia A, Ruban A, Thiel J, Fiebig A, Houben A. Tissue-Specific Transcriptome Analysis Reveals Candidate Transcripts Associated with the Process of Programmed B Chromosome Elimination in Aegilops speltoides. International Journal of Molecular Sciences. 2020; 21(20):7596. https://doi.org/10.3390/ijms21207596
Chicago/Turabian StyleBoudichevskaia, Anastassia, Alevtina Ruban, Johannes Thiel, Anne Fiebig, and Andreas Houben. 2020. "Tissue-Specific Transcriptome Analysis Reveals Candidate Transcripts Associated with the Process of Programmed B Chromosome Elimination in Aegilops speltoides" International Journal of Molecular Sciences 21, no. 20: 7596. https://doi.org/10.3390/ijms21207596
APA StyleBoudichevskaia, A., Ruban, A., Thiel, J., Fiebig, A., & Houben, A. (2020). Tissue-Specific Transcriptome Analysis Reveals Candidate Transcripts Associated with the Process of Programmed B Chromosome Elimination in Aegilops speltoides. International Journal of Molecular Sciences, 21(20), 7596. https://doi.org/10.3390/ijms21207596