Atorvastatin Augments Gemcitabine-Mediated Anti-Cancer Effects by Inhibiting Yes-Associated Protein in Human Cholangiocarcinoma Cells

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Results
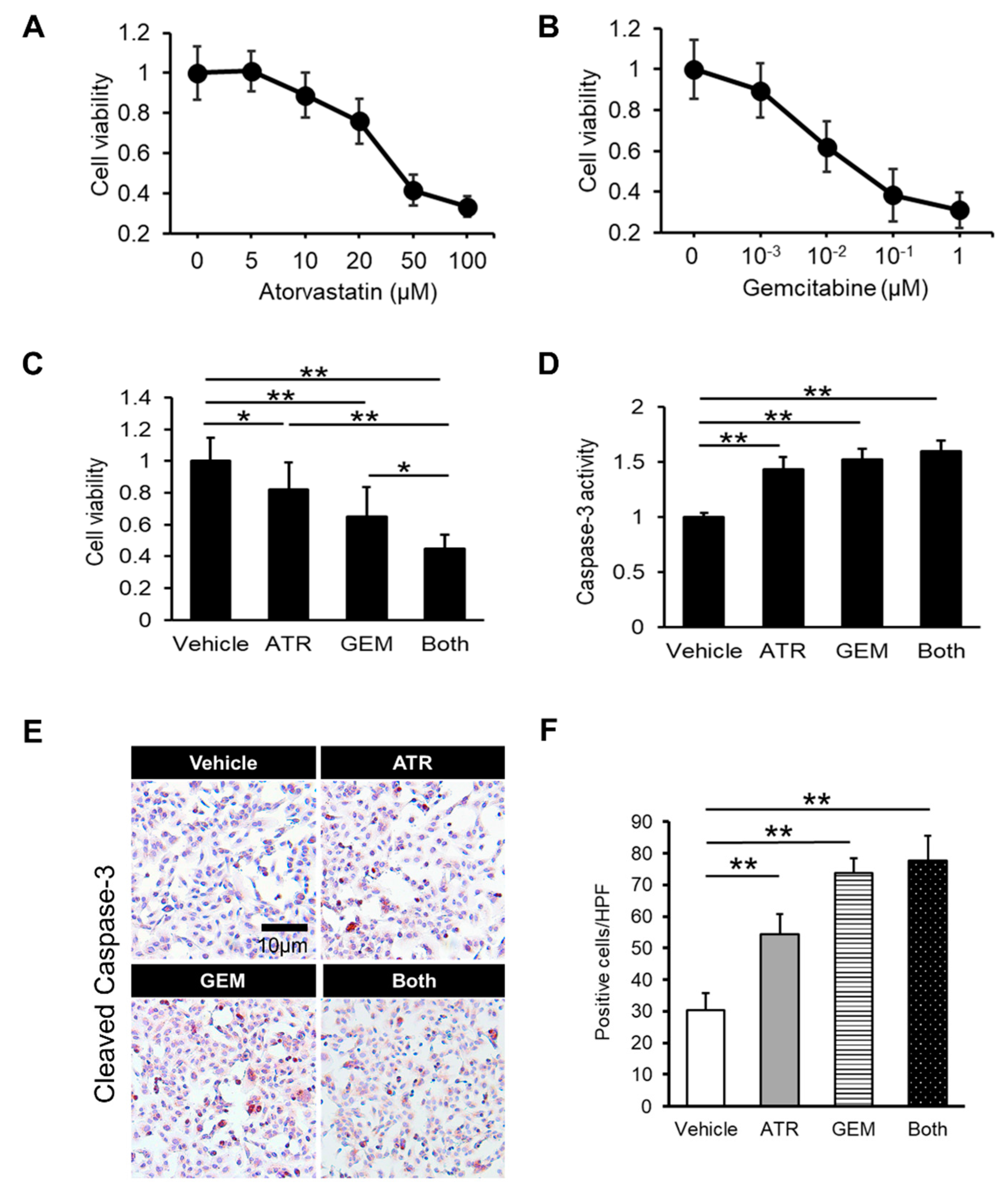
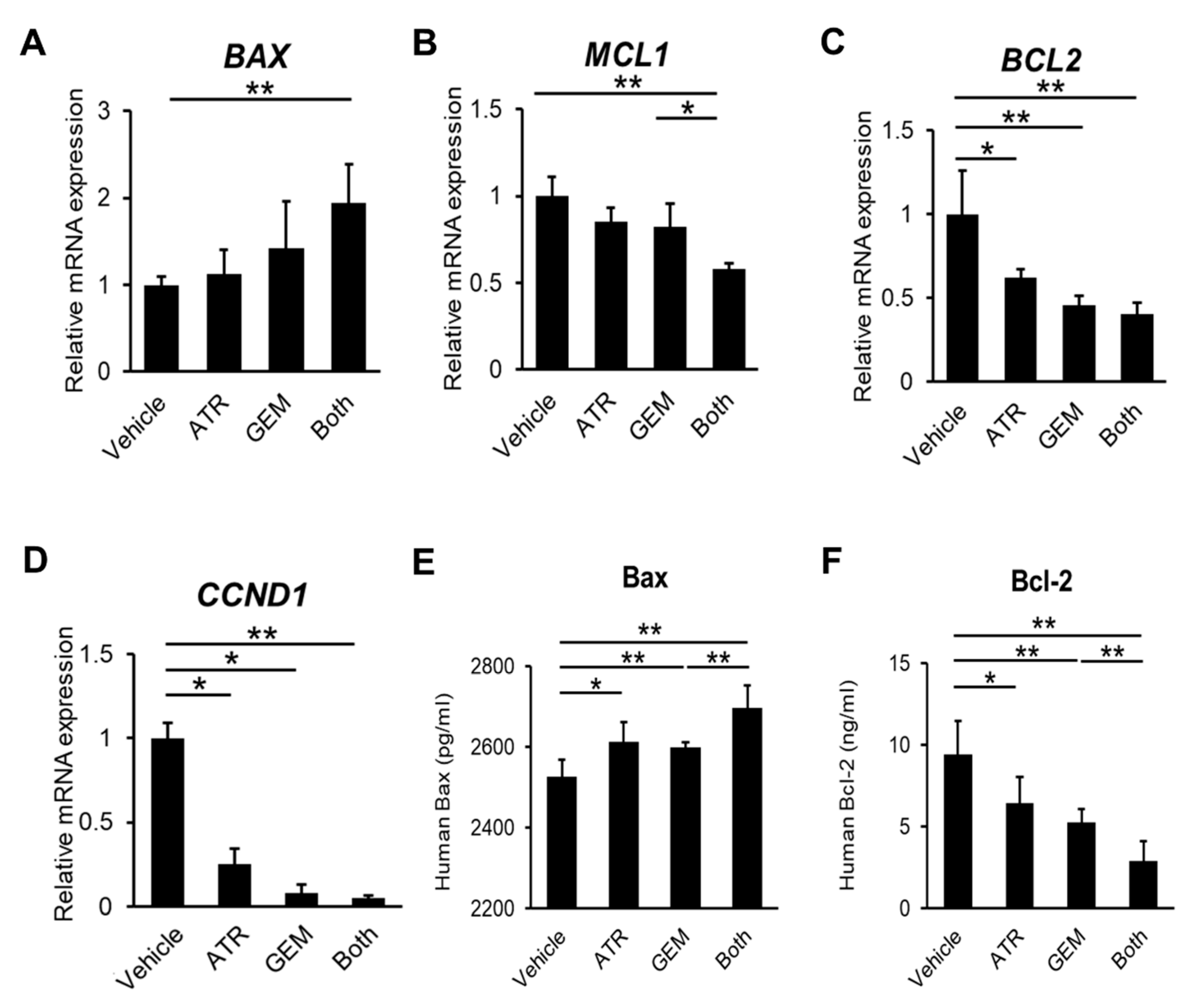
2.1. Gemcitabine Plus Atorvastatin Efficiently Suppressed Human Cholangiocarcinoma Cell Growth
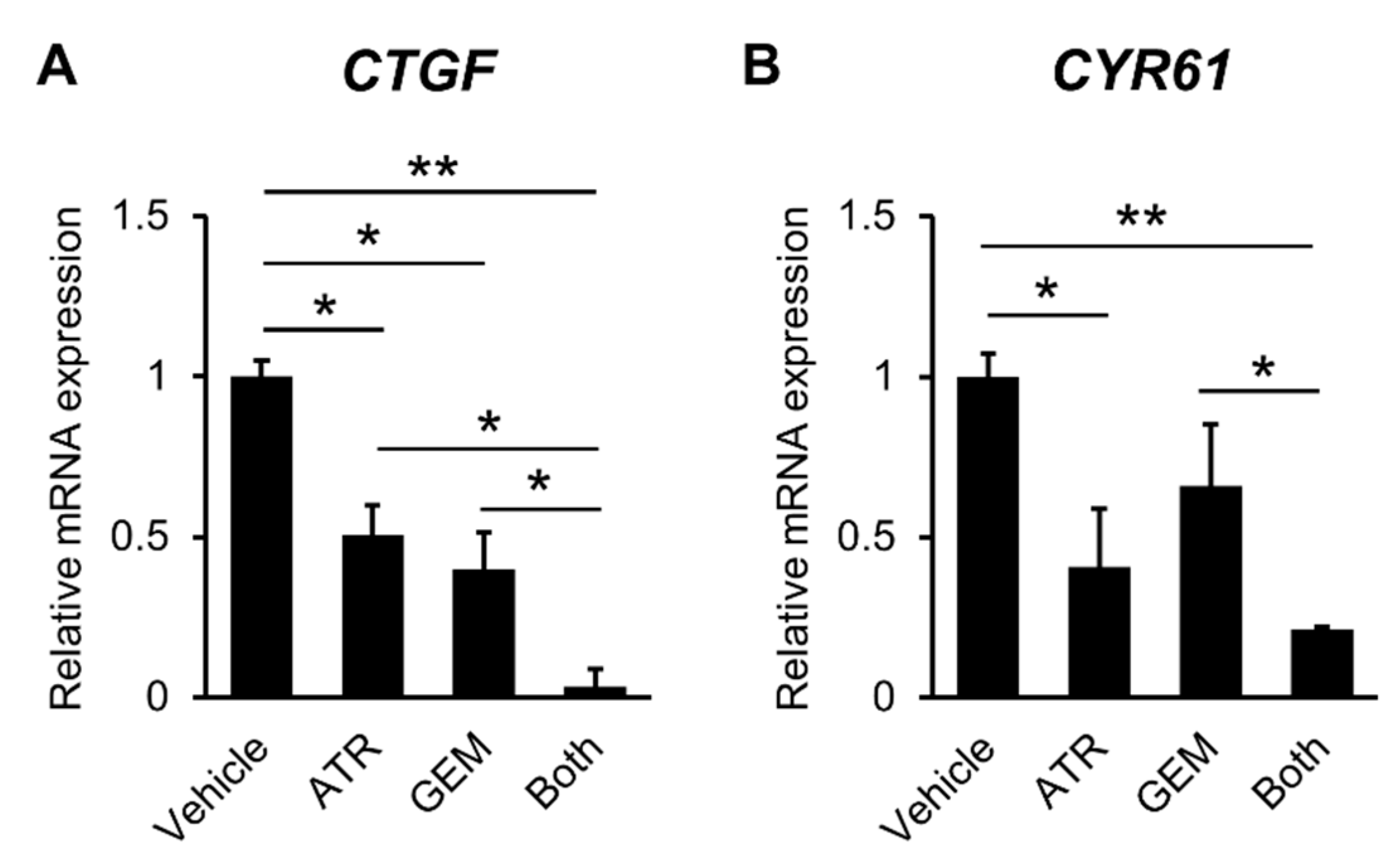
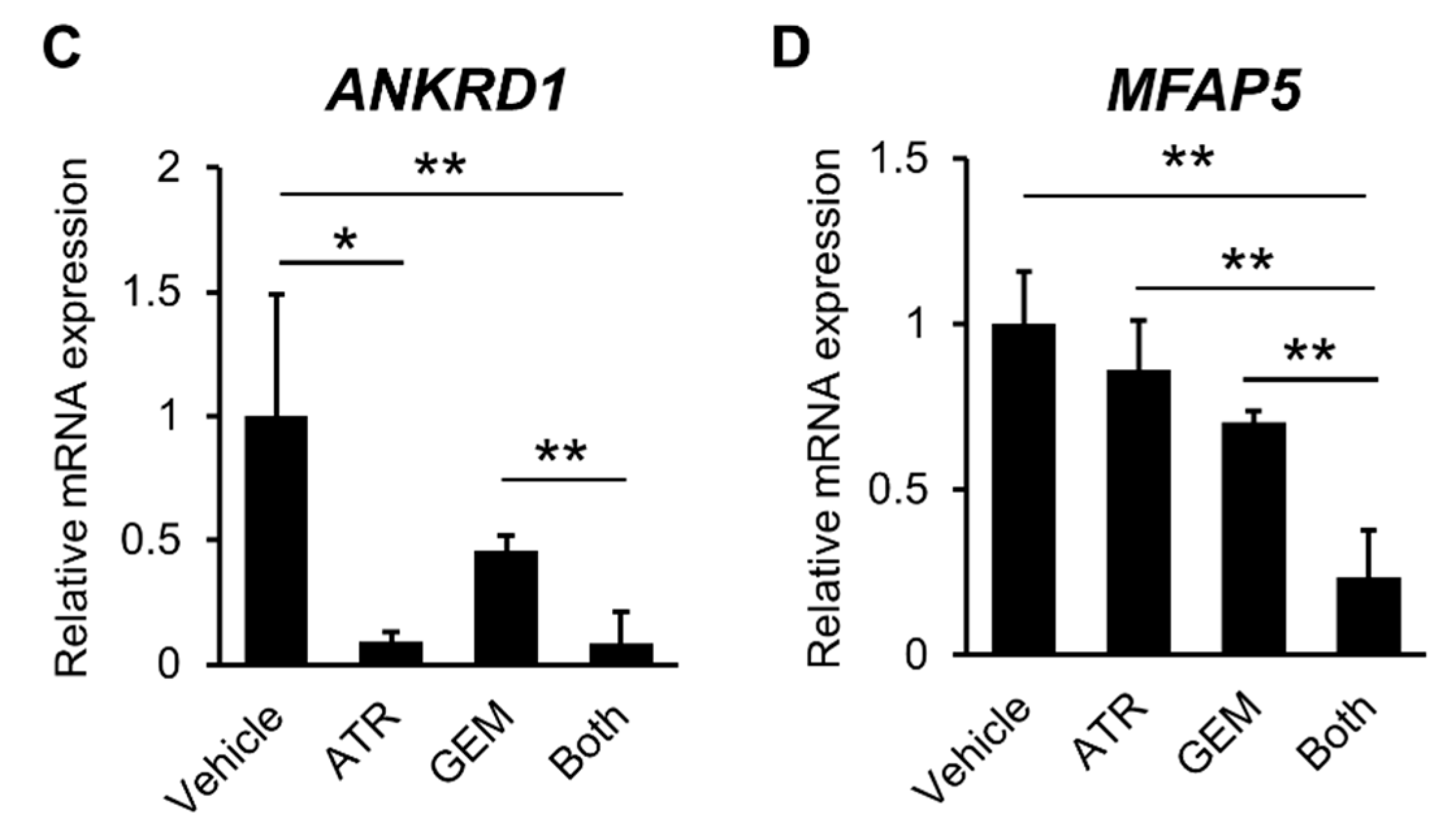
2.2. Gemcitabine Plus Atorvastatin Inhibited YAP/TAZ-TEAD Activation in Human CCA Cells
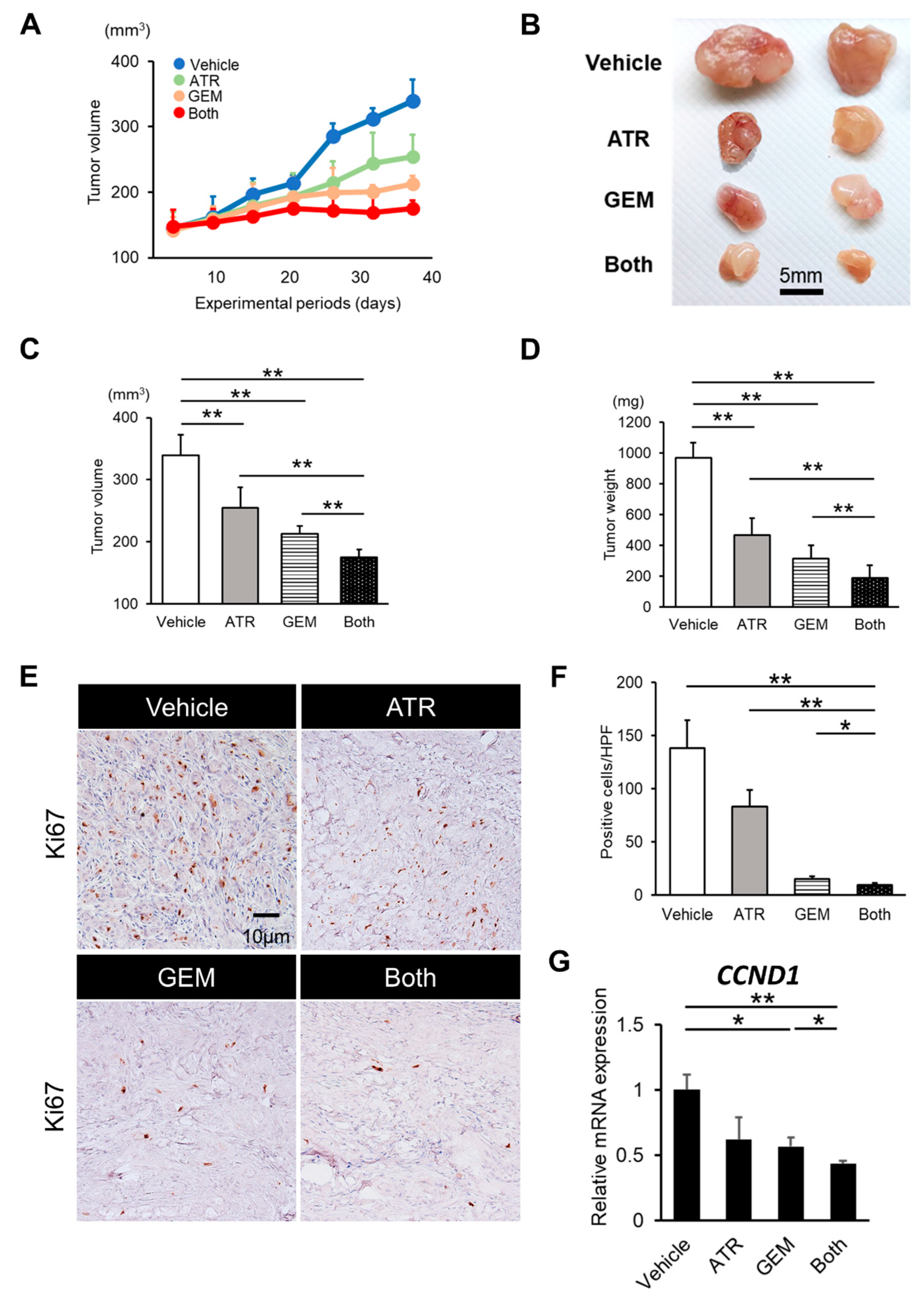
2.3. Gemcitabine Plus Atorvastatin Reduced the Human CCA Cell-Derived Xenograft Tumor Burden
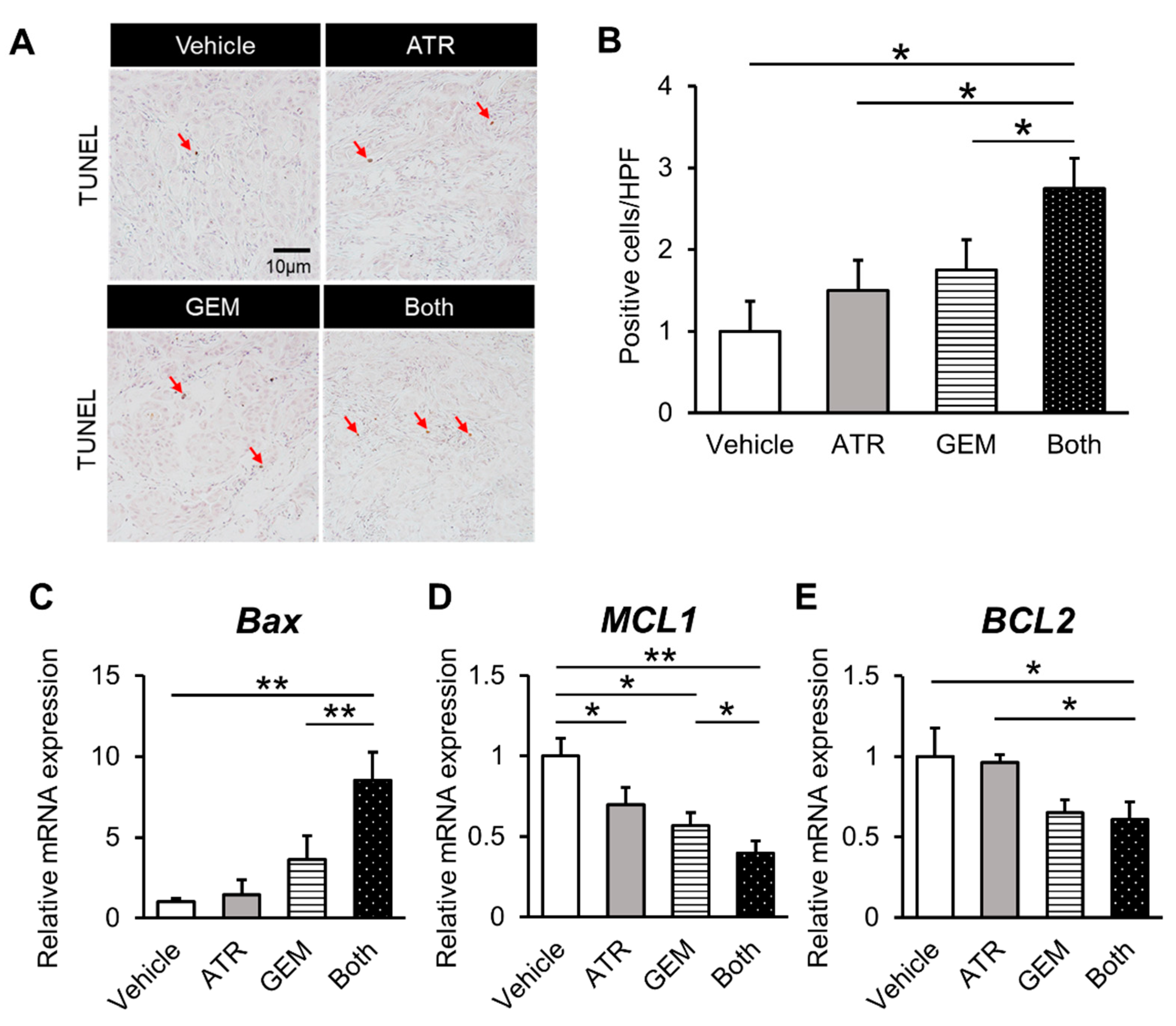
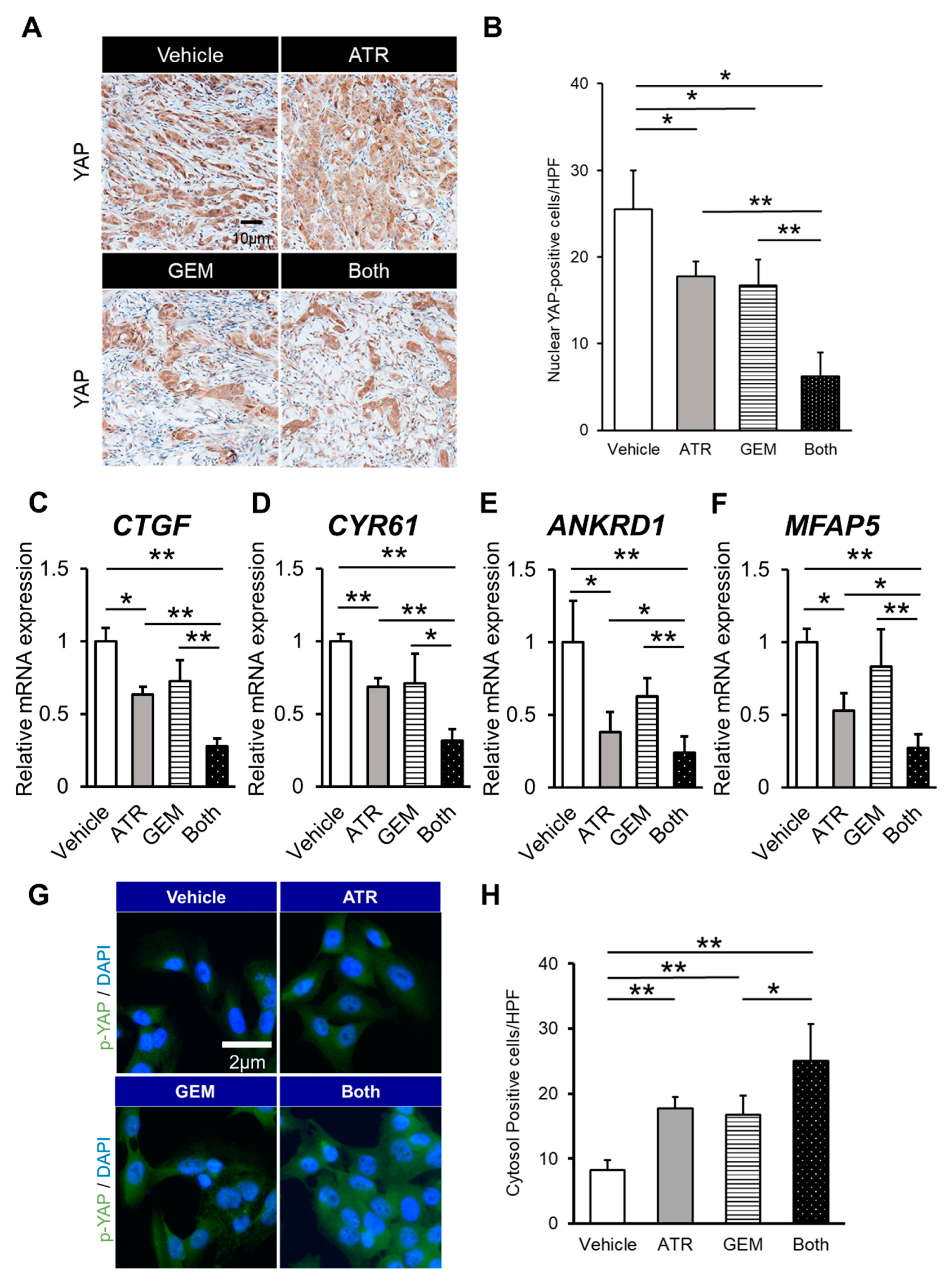
2.4. Gemcitabine Plus Atorvastatin Suppressed YAP/TAZ-TEAD Activation in Human CCA Cell-Derived Xenograft Tumors
3. Discussion
4. Materials and Methods
4.1. Compounds and Cell Culture
4.2. Cell Proliferation Assay
4.3. Measurement of Caspase-3 Activity
4.4. Human CCA Cell Xenografts
4.5. RNA Extraction and Quantitative Real-Time PCR
4.6. Immunohistochemical Analyses
4.7. Immunocytochemical Analyses
4.8. Immunofluorescence
4.9. Enzyme-Linked Immunosorbent Assay (Elisa) for Pro- and Anti-Apoptotic Markers
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Razumilava, N.; Gores, G.J. Cholangiocarcinoma. Lancet 2014, 383, 2168–2179. [Google Scholar] [CrossRef]
- Rizvi, S.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma-evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef]
- Morise, Z.; Sugioka, A.; Tokoro, T.; Tanahashi, Y.; Okabe, Y.; Kagawa, T.; Takeura, C. Surgery and chemotherapy for intrahepatic cholangiocarcinoma. World J. Hepatol. 2010, 2, 58–64. [Google Scholar] [CrossRef]
- Tarchi, P.; Tabrizian, P.; Prigoff, J.; Schwartz, M. Outcomes of resection for solitary ≤5 cm intrahepatic cholangiocarcinoma. Surgery 2018, 163, 698–702. [Google Scholar] [CrossRef]
- Bridgewater, J.; Galle, P.R.; Khan, S.A.; Llovet, J.M.; Park, J.W.; Patel, T.; Pawlik, T.M.; Gores, G.J. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J. Hepatol. 2014, 60, 1268–1289. [Google Scholar] [CrossRef]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef]
- Lee, J.; Park, S.H.; Chang, H.-M.; Kim, J.S.; Choi, H.J.; Lee, M.A.; Chang, J.S.; Jeung, H.C.; Kang, J.H.; Lee, H.W.; et al. Gemcitabine and oxaliplatin with or without erlotinib in advanced biliary-tract cancer: A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2012, 13, 181–188. [Google Scholar] [CrossRef]
- Malka, D.; Cervera, P.; Foulon, S.; Trarbach, T.; De La Fouchardière, C.; Boucher, E.; Fartoux, L.; Faivre, S.; Blanc, J.-F.; Viret, F.; et al. Gemcitabine and oxaliplatin with or without cetuximab in advanced biliary-tract cancer (BINGO): A randomised, open-label, non-comparative phase 2 trial. Lancet Oncol. 2014, 15, 819–828. [Google Scholar] [CrossRef]
- Chen, J.S.; Hsu, C.; Chiang, N.J.; Tsai, C.S.; Tsou, H.H.; Huang, S.F.; Bai, L.Y.; Chang, I.C.; Shiah, H.S.; Ho, C.L.; et al. A KRAS mutation status-stratified randomized phase II trial of gemcitabine and oxaliplatin alone or in combination with cetuximab in advanced biliary tract cancer. Ann. Oncol. 2015, 26, 943–949. [Google Scholar] [CrossRef]
- Valle, J.W.; Wasan, H.; Lopes, A.; Backen, A.C.; Palmer, D.H.; Morris, K.; Duggan, M.; Cunningham, D.; Anthoney, D.A.; Corrie, P.; et al. Cediranib or placebo in combination with cisplatin and gemcitabine chemotherapy for patients with advanced biliary tract cancer (ABC-03): A randomised phase 2 trial. Lancet Oncol. 2015, 16, 967–978. [Google Scholar] [CrossRef]
- Leone, F.; Marino, D.; Cereda, S.; Filippi, R.; Belli, C.; Spadi, R.; Nasti, G.; Montano, M.; Amatu, A.; Aprile, G.; et al. Panitumumab in combination with gemcitabine and oxaliplatin does not prolong survival in wild-type KRAS advanced biliary tract cancer: A randomized phase 2 trial (Vecti-BIL study). Cancer 2016, 122, 574–581. [Google Scholar] [CrossRef]
- Phelip, J.-M.; Vendrely, V.; Rostain, F.; Subtil, F.; Jouve, J.-L.; Gasmi, M.; Michel, P.; Le Malicot, K.; Smith, D.; Seitz, J.-F.; et al. Gemcitabine plus cisplatin versus chemoradiotherapy in locally advanced biliary tract cancer: Fédération Francophone de Cancérologie Digestive 9902 phase II randomised study. Eur. J. Cancer 2014, 50, 2975–2982. [Google Scholar] [CrossRef]
- Yu, F.; Guan, K. The Hippo pathway: Regulators and regulations. Genes Dev. 2013, 1, 1–17. [Google Scholar] [CrossRef]
- Cottini, F.; Hideshima, T.; Xu, C.; Sattler, M.; Dori, M.; Agnelli, L.; Hacken, E.T.; Bertilaccio, M.T.; Antonini, E.; Neri, A.; et al. Rescue of Hippo coactivator YAP1 triggers DNA damage-induced apoptosis in hematological cancers. Nat. Med. 2014, 20, 599–606. [Google Scholar] [CrossRef]
- Mo, J.; Park, H.W.; Guan, K.-L. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep. 2014, 15, 642–656. [Google Scholar] [CrossRef]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.-J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef]
- Zhao, B.; Tumaneng, K.; Guan, K.L. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat. Cell Biol. 2011, 13, 877–883. [Google Scholar] [CrossRef]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a Universal Size-Control Mechanism in Drosophila and Mammals Jixin. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef]
- Zeng, Q.; Hong, W. The Emerging Role of the Hippo Pathway in Cell Contact Inhibition, Organ Size Control, and Cancer Development in Mammals. Cancer Cell 2008, 13, 188–192. [Google Scholar] [CrossRef]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer Access options Rent or Buy Subscription info for Japanese customers. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef]
- Gomez, M.; Gomez, V.; Hergovich, A. The Hippo pathway in disease and therapy: Cancer and beyond. Clin. Transl. Med. 2014, 3, 1–12. [Google Scholar] [CrossRef]
- Zhou, T.Y.; Zhuang, L.H.; Hu, Y.; Zhou, Y.L.; Lin, W.K.; Wang, D.D.; Wan, Z.Q.; Chang, L.L.; Chen, Y.; Ying, M.D.; et al. Inactivation of hypoxia-induced YAP by statins overcomes hypoxic resistance tosorafenib in hepatocellular carcinoma cells article. Sci. Rep. 2016, 6, 1–12. [Google Scholar]
- Tschaharganeh, D.F.; Chen, X.; Latzko, P.; Malz, M.; Gaida, M.M.; Felix, K.; Ladu, S.; Singer, S.; Pinna, F.; Gretz, N.; et al. Yes-associated protein up-regulates jagged-1 and activates the NOTCH pathway in human hepatocellular carcinoma. Gastroenterology 2013, 144, 1530–1542. [Google Scholar] [CrossRef]
- Schlegelmilch, K.; Mohseni, M.; Kirak, O.; Pruszak, J.; Rodriguez, J.R.; Zhou, D.; Kreger, B.T.; Vasioukhin, V.; Avruch, J.; Brummelkamp, T.R.; et al. Yap1 acts downstream of α-catenin to control epidermal proliferation. Cell 2011, 144, 782–795. [Google Scholar] [CrossRef]
- Piccolo, S.; Cordenonsi, M.; Dupont, S. Molecular pathways: YAP and TAZ take center stage in organ growth and tumorigenesis. Clin. Cancer Res. 2013, 19, 4925–4930. [Google Scholar] [CrossRef]
- Pei, T.; Li, Y.; Wang, J.; Wang, H.; Liang, Y.; Shi, H.; Sun, B.; Yin, D.; Sun, J.; Song, R.; et al. YAP is a critical oncogene in human cholangiocarcinoma. Oncotarget 2015, 6, 17206–17220. [Google Scholar] [CrossRef]
- Morvaridi, S.; Dhall, D.; Greene, M.I.; Pandol, S.J.; Wang, Q. Role of YAP and TAZ in pancreatic ductal adenocarcinoma and in stellate cells associated with cancer and chronic pancreatitis. Sci. Rep. 2015, 5, 1–14. [Google Scholar] [CrossRef]
- Marti, P.; Stein, C.; Blumer, T.; Abraham, Y.; Dill, M.T.; Pikiolek, M.; Orsini, V.; Jurisic, G.; Megel, P.; Makowska, Z.; et al. YAP promotes proliferation, chemoresistance, and angiogenesis in human cholangiocarcinoma through TEAD transcription factors. Hepatology 2015, 62, 1497–1510. [Google Scholar] [CrossRef]
- Kowalik, M.A.; Saliba, C.; Pibiri, M.; Perra, A.; Ledda-Columbano, G.M.; Sarotto, I.; Ghiso, E.; Giordano, S.; Columbano, A. Yes-associated protein regulation of adaptive liver enlargement and hepatocellular carcinoma development in mice. Hepatology 2011, 53, 2086–2096. [Google Scholar] [CrossRef]
- Tanaka, K.; Osada, H.; Murakami-Tonami, Y.; Horio, Y.; Hida, T.; Sekido, Y. Statin suppresses Hippo pathway- inactivated malignant mesothelioma cells and blocks the YAP/CD44 growth stimulatory axis. Cancer Lett. 2017, 385, 215–224. [Google Scholar] [CrossRef]
- Saikawa, S.; Kaji, K.; Nishimura, N.; Seki, K.; Sato, S.; Nakanishi, K.; Kitagawa, K.; Kawaratani, H.; Kitade, M.; Moriya, K.; et al. Angiotensin receptor blockade attenuates cholangiocarcinoma cell growth by inhibiting the oncogenic activity of Yes-associated protein. Cancer Lett. 2018, 434, 120–129. [Google Scholar] [CrossRef]
- Peng, Y.-C.; Lin, C.-L.; Hsu, W.-Y.; Chang, C.-S.; Yeh, H.-Z.; Tung, C.-F.; Wu, Y.-L.; Sung, F.-C.; Kao, C.-H. Statins are associated with a reduced risk of cholangiocarcinoma: A population-based case-control study. Br. J. Clin. Pharmacol. 2015, 80, 755–761. [Google Scholar] [CrossRef]
- Oates, J.A.; Wood, A.J.; Grundy, S.M. HMG-CoA Reductase Inhibitors for Treatment of Hypercholesterolemia. N. Engl. J. Med. 1988, 319, 24–33. [Google Scholar] [CrossRef]
- Liu, Z.; AlSaggaf, R.; McGlynn, K.A.; Anderson, L.A.; Tsai, H.-T.; Zhu, B.; Zhu, Y.; Mbulaiteye, S.M.; Gadalla, S.M.; Koshiol, J. Statin use and reduced risk of biliary tract cancers in the UK Clinical Practice Research Datalink. Gut 2019, 68, 1458–1464. [Google Scholar] [CrossRef]
- Kamigaki, M.; Sasaki, T.; Serikawa, M.; Inoue, M.; Kobayashi, K.; Itsuki, H.; Minami, T.; Yukutake, M.; Okazaki, A.; Ishigaki, T.; et al. Statins induce apoptosis and inhibit proliferation in cholangiocarcinoma cells. Int. J. Oncol. 2011, 39, 561–568. [Google Scholar] [CrossRef][Green Version]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef]
- Jiang, Z.; Chen, X.; Chen, K.; Sun, L.; Gao, L.; Zhou, C.; Lei, M.; Duan, W.; Wang, Z.; Ma, Q.; et al. YAP inhibition by resveratrol via activation of AMPK enhances the sensitivity of pancreatic cancer cells to gemcitabine. Nutrients 2016, 8, 546. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Chen, Y.-C.; Lin, C.-C.; Hsieh, Y.-P.; Hsu, C.-S.; Hsieh, M.-C. Synergistic Anticancer Effects of Gemcitabine with Pitavastatin on Pancreatic Cancer Cell Line MIA PaCa-2 in vitro and in vivo. Cancer Manag. Res. 2020, 12, 4645–4665. [Google Scholar] [CrossRef]
- Buranrat, B.; Senggunprai, L.; Prawan, A.; Kukongviriyapan, V. Simvastatin and atorvastatin as inhibitors of proliferation and inducers of apoptosis in human cholangiocarcinoma cells. Life Sci. 2016, 153, 41–49. [Google Scholar] [CrossRef]
- Yang, S.-H.; Lin, H.-Y.; Changou, C.A.; Chen, C.-H.; Liu, Y.-R.; Wang, J.; Jiang, X.; Luh, F.; Yen, Y. Integrin β3 and LKB1 are independently involved in the inhibition of proliferation by lovastatin in human intrahepatic cholangiocarcinoma. Oncotarget 2016, 7, 362–373. [Google Scholar] [CrossRef]
- Seeree, P.; Janvilisri, T.; Kangsamaksin, T.; Tohtong, R.; Kumkate, S. Downregulation of abca1 and abcg1 transporters by simvastatin in cholangiocarcinoma cells. Oncol. Lett. 2019, 18, 5173–5184. [Google Scholar] [CrossRef]
- Pan, Z.; Tian, Y.; Cao, C.; Niu, G. The emerging role of YAP/TAZ in tumor immunity. Mol. Cancer Res. 2019, 17, 1777–1786. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sense (5′–3′) | Antisense (5′–3′) |
|---|---|---|
| CTGF | TGCTTTGAACGATCAGACAA | CTTGTGGCAAGTGAATTTCC |
| CYR61 | AAGAAACCCGGATTTGTGAG | GCTGCATTTCTTGCCCTTT |
| ANKRD1 | GCCTACGTTTCTGAAGGCTG | GTGGATTCAAGCATATCACGGAA |
| MFAP5 | GTGACTCAAGCGACTCCAGAA | AGTCATCTGTGGAAGGTGCAAT |
| BAX | TCTGACGGCAACTTCAACTG | GGAGGAAGTCCAATGTCCAG |
| MCL1 | GAGGGCGACTTTTGGCTAC | GTACCCGTCCAGCTCCTCTT |
| BCL2 | CATGTGTGTGGAGAGCGTCAA | GCCGGTTCAGGTACTCAGTCA |
| CCND1 | CCCTCGGTGTCCTACTTCAA | CTTAGAGGCCACGAACATGC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitagawa, K.; Moriya, K.; Kaji, K.; Saikawa, S.; Sato, S.; Nishimura, N.; Namisaki, T.; Akahane, T.; Mitoro, A.; Yoshiji, H. Atorvastatin Augments Gemcitabine-Mediated Anti-Cancer Effects by Inhibiting Yes-Associated Protein in Human Cholangiocarcinoma Cells. Int. J. Mol. Sci. 2020, 21, 7588. https://doi.org/10.3390/ijms21207588
Kitagawa K, Moriya K, Kaji K, Saikawa S, Sato S, Nishimura N, Namisaki T, Akahane T, Mitoro A, Yoshiji H. Atorvastatin Augments Gemcitabine-Mediated Anti-Cancer Effects by Inhibiting Yes-Associated Protein in Human Cholangiocarcinoma Cells. International Journal of Molecular Sciences. 2020; 21(20):7588. https://doi.org/10.3390/ijms21207588
Chicago/Turabian StyleKitagawa, Koh, Kei Moriya, Kosuke Kaji, Soichiro Saikawa, Shinya Sato, Norihisa Nishimura, Tadashi Namisaki, Takemi Akahane, Akira Mitoro, and Hitoshi Yoshiji. 2020. "Atorvastatin Augments Gemcitabine-Mediated Anti-Cancer Effects by Inhibiting Yes-Associated Protein in Human Cholangiocarcinoma Cells" International Journal of Molecular Sciences 21, no. 20: 7588. https://doi.org/10.3390/ijms21207588
APA StyleKitagawa, K., Moriya, K., Kaji, K., Saikawa, S., Sato, S., Nishimura, N., Namisaki, T., Akahane, T., Mitoro, A., & Yoshiji, H. (2020). Atorvastatin Augments Gemcitabine-Mediated Anti-Cancer Effects by Inhibiting Yes-Associated Protein in Human Cholangiocarcinoma Cells. International Journal of Molecular Sciences, 21(20), 7588. https://doi.org/10.3390/ijms21207588