Cdc-Like Kinases (CLKs): Biology, Chemical Probes, and Therapeutic Potential


Abstract
1. Introduction
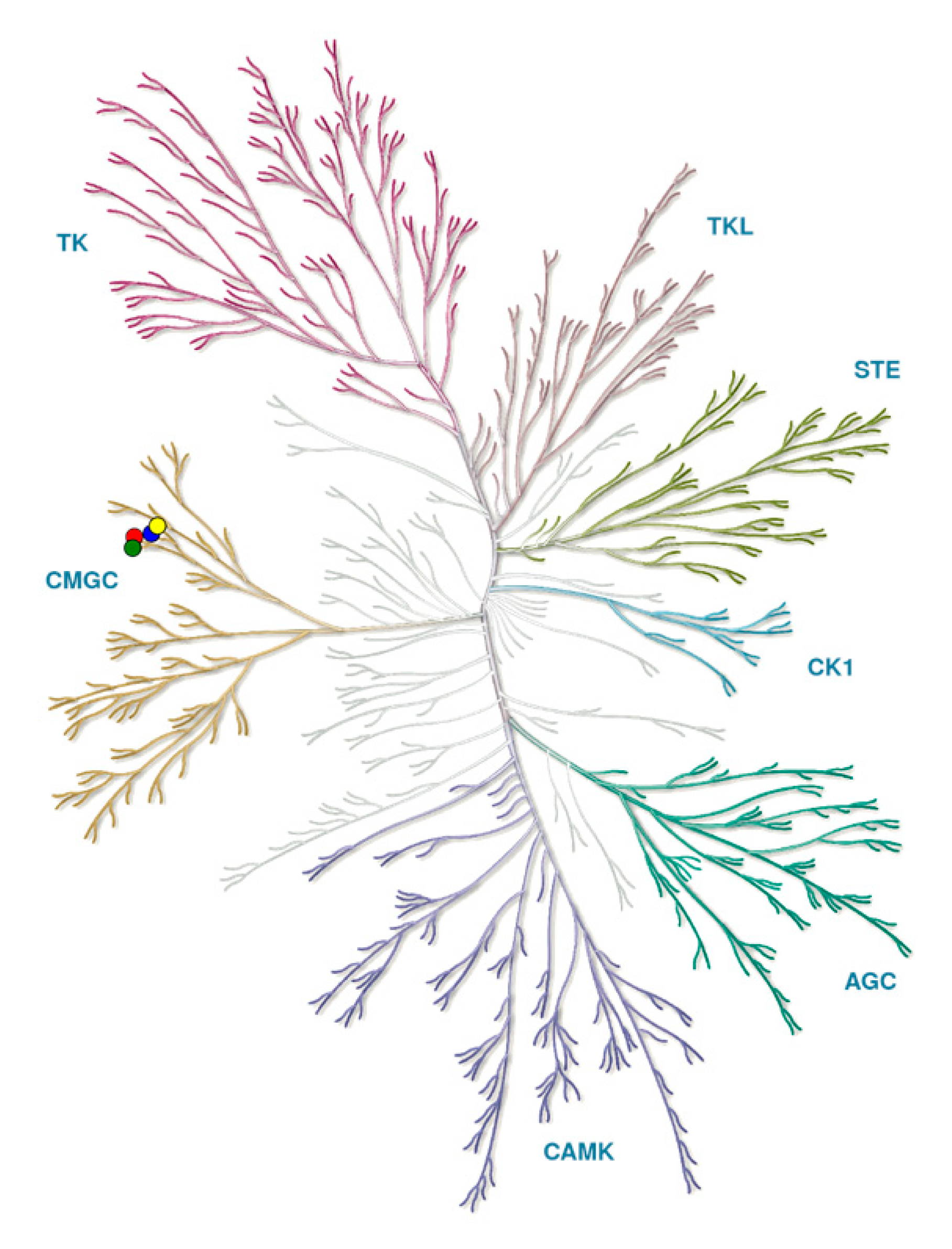
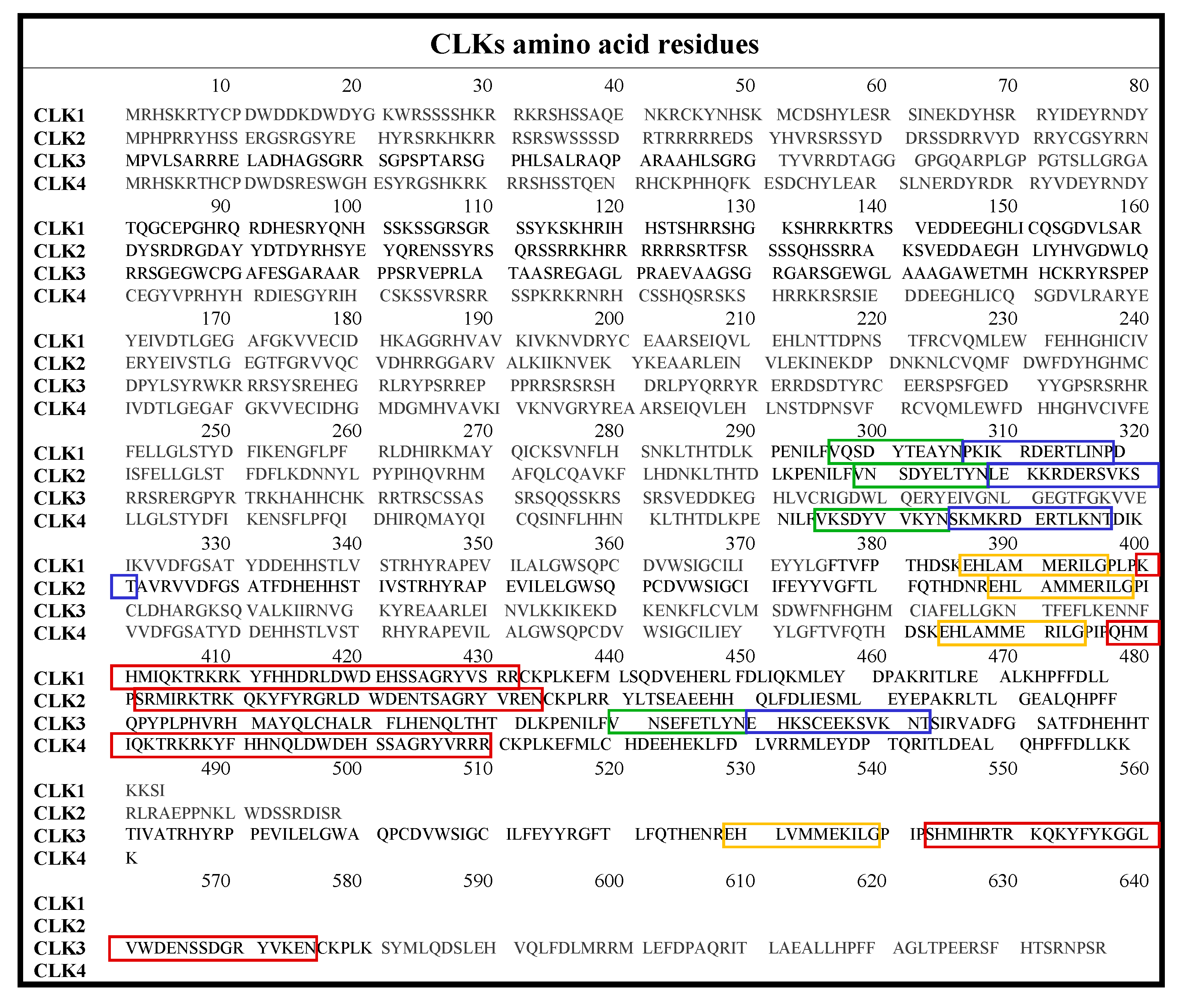
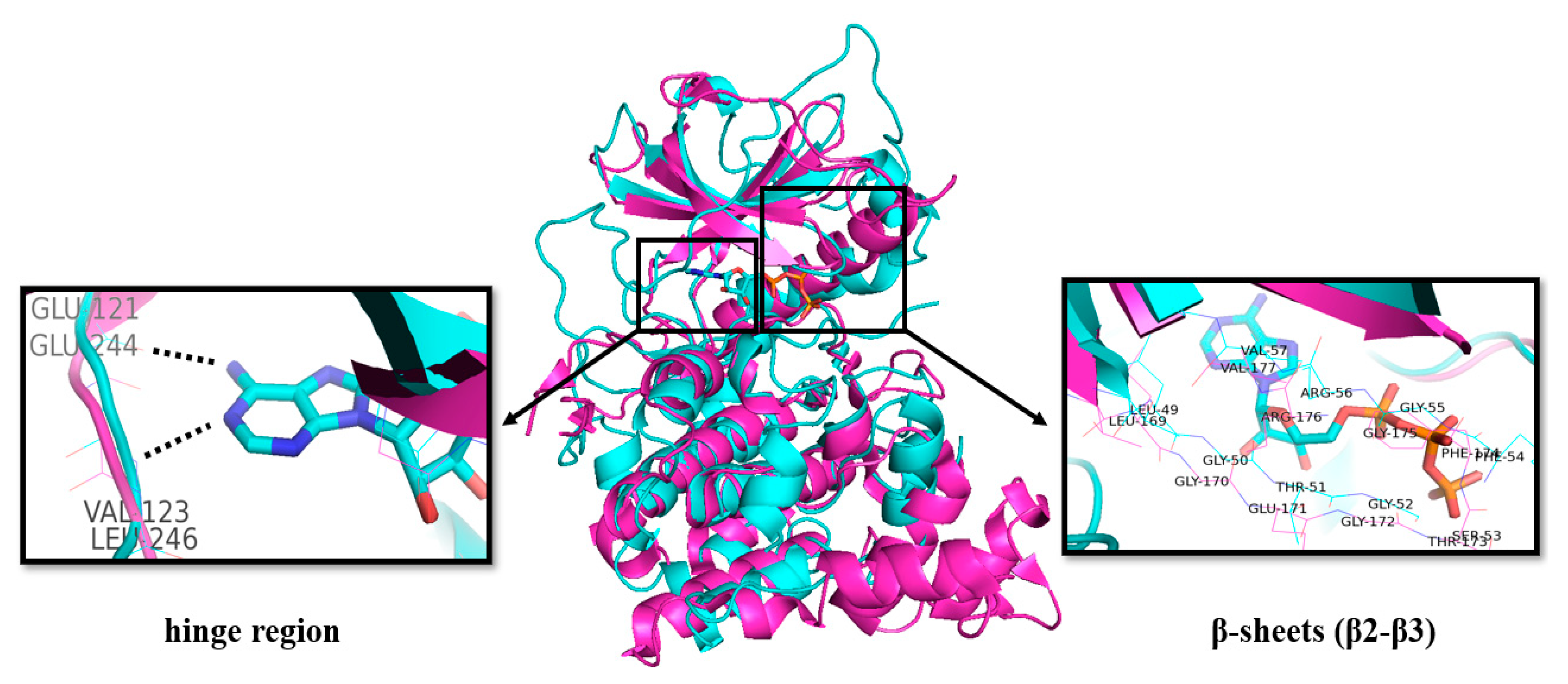
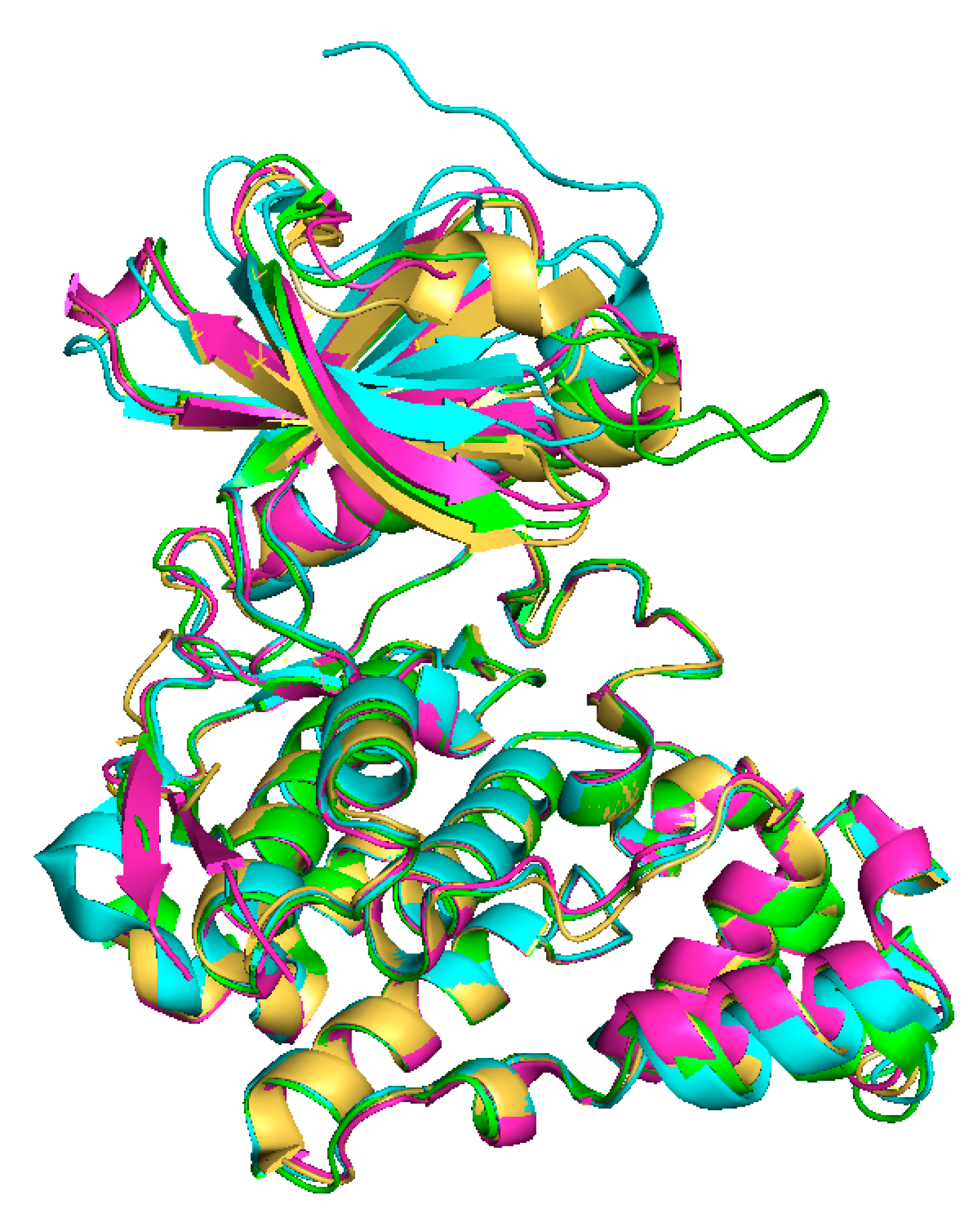
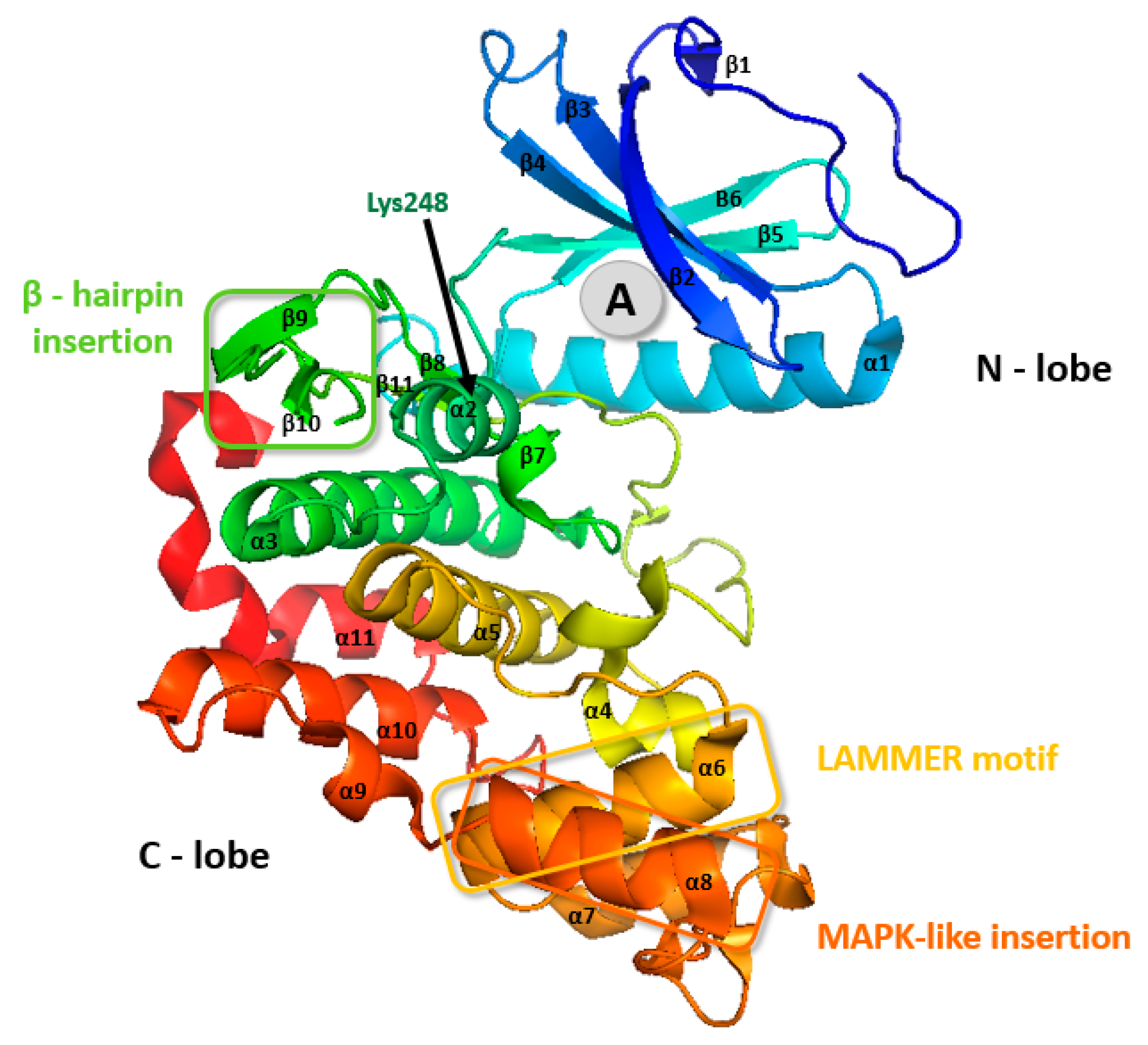
2. Classification and Structure of CLKs
3. Biology of CLKs
3.1. CLK1
3.2. CLK2
3.3. CLK3
3.4. CLK4
4. Effects of Altered CLK Expression
5. CLKs as Therapeutic Targets
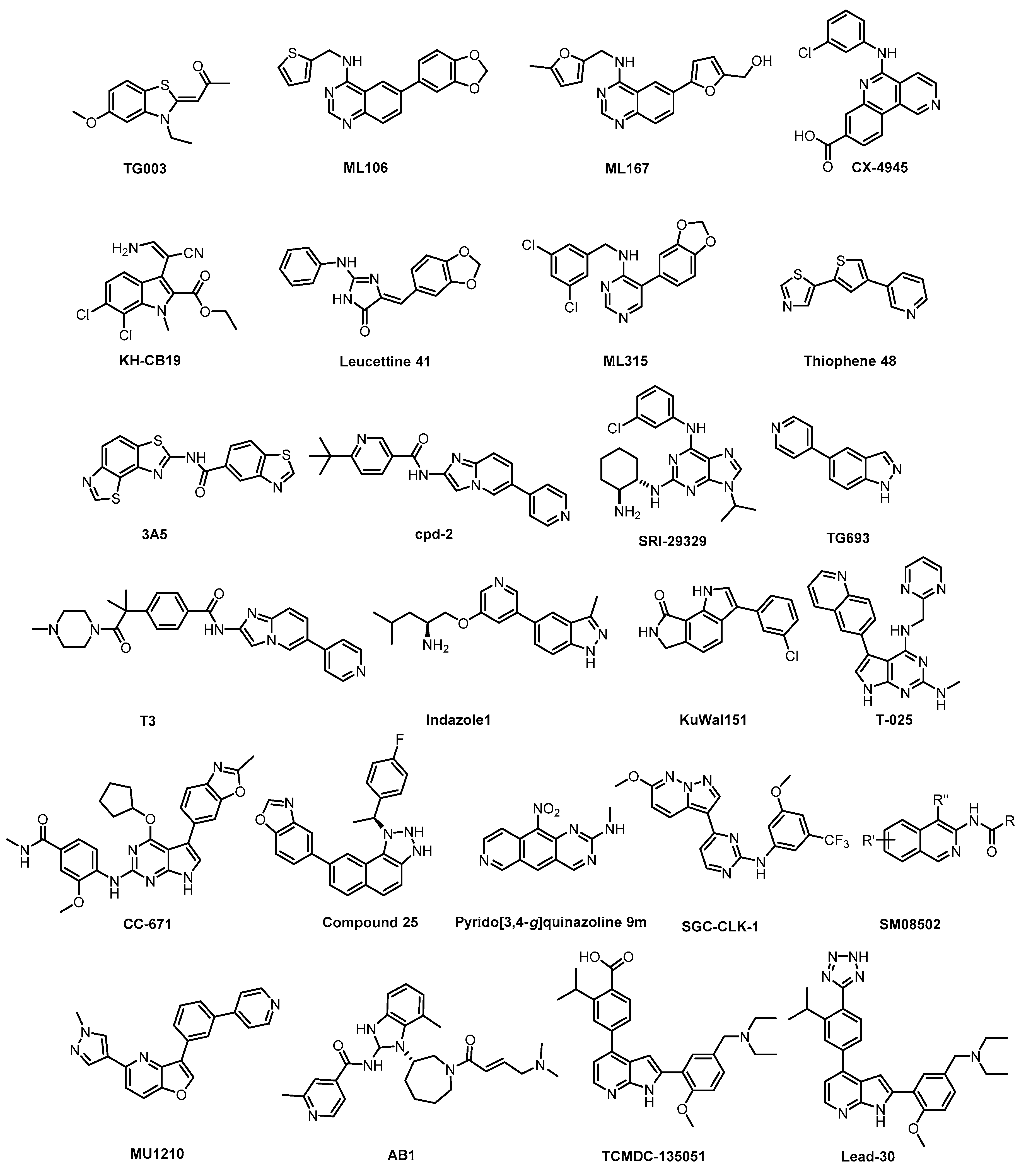
6. Small-Molecule CLK Inhibitors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CLKs | Structurally Similar Kinases | Other Significant Targets | Kinases Screened | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | CLK1 | CLK2 | CLK3 | CLK4 | DYRK1A | DYRK1B | DYRK2 | HIPK1 | HIPK2 | HIPK3 | ||
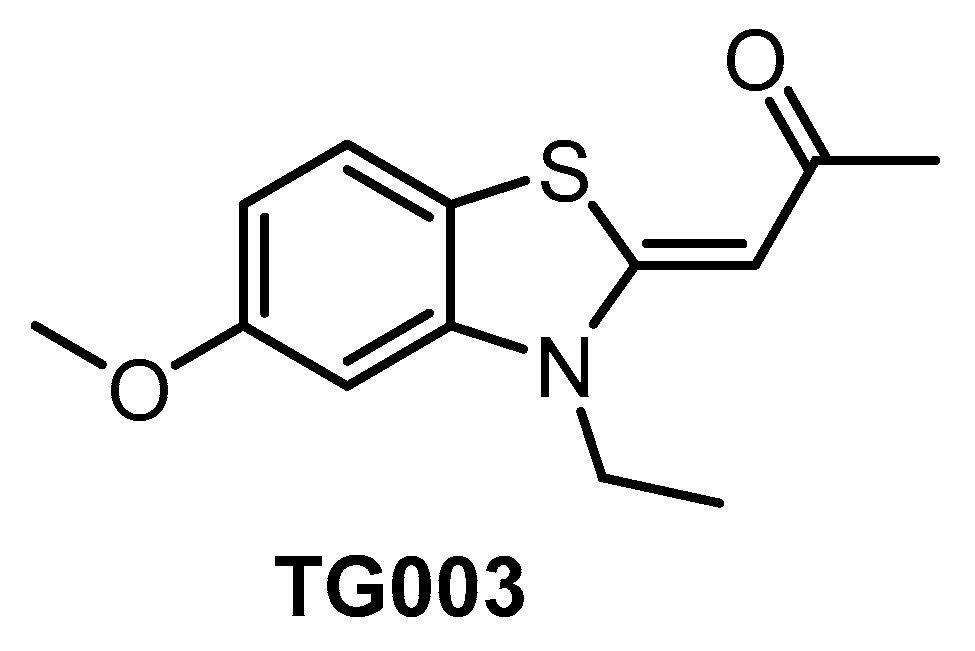
| TG003 [64,134,138] | 19 (Kd) | 95 (Kd) | inactive | 30 (Kd) | 12 (Kd) | 130 (Kd) | ND | ND | ND | ND | CK1δ = 150 CK1ε = 300 CK1γ2 = 270 CK1γ3 = 290 PIM1 = 130 PIM3 = 280 YSK4 = 290 (Kd) | ~402 kinases |
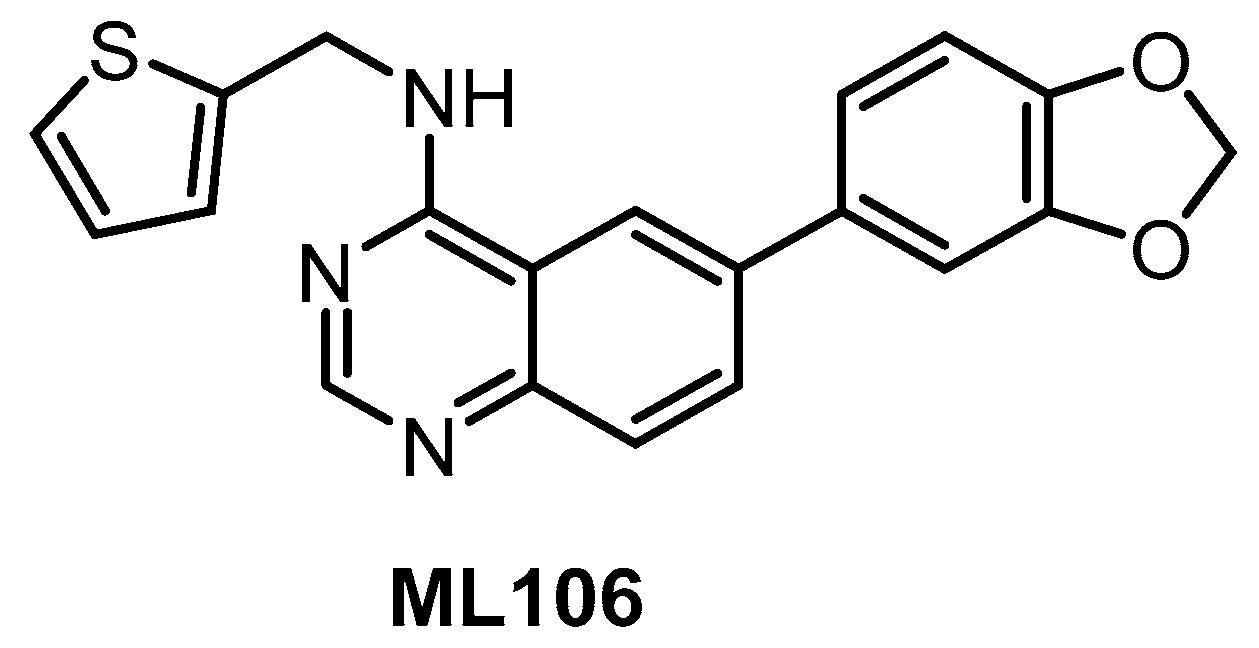
| ML106 [134,138] | 37 (Kd) | 680 (Kd) | 470 (Kd) | 50 (Kd) | 27 (Kd) | 430 (Kd) | ND | ND | ND | ND | EGFR = 230 (Kd) | 402 kinases |
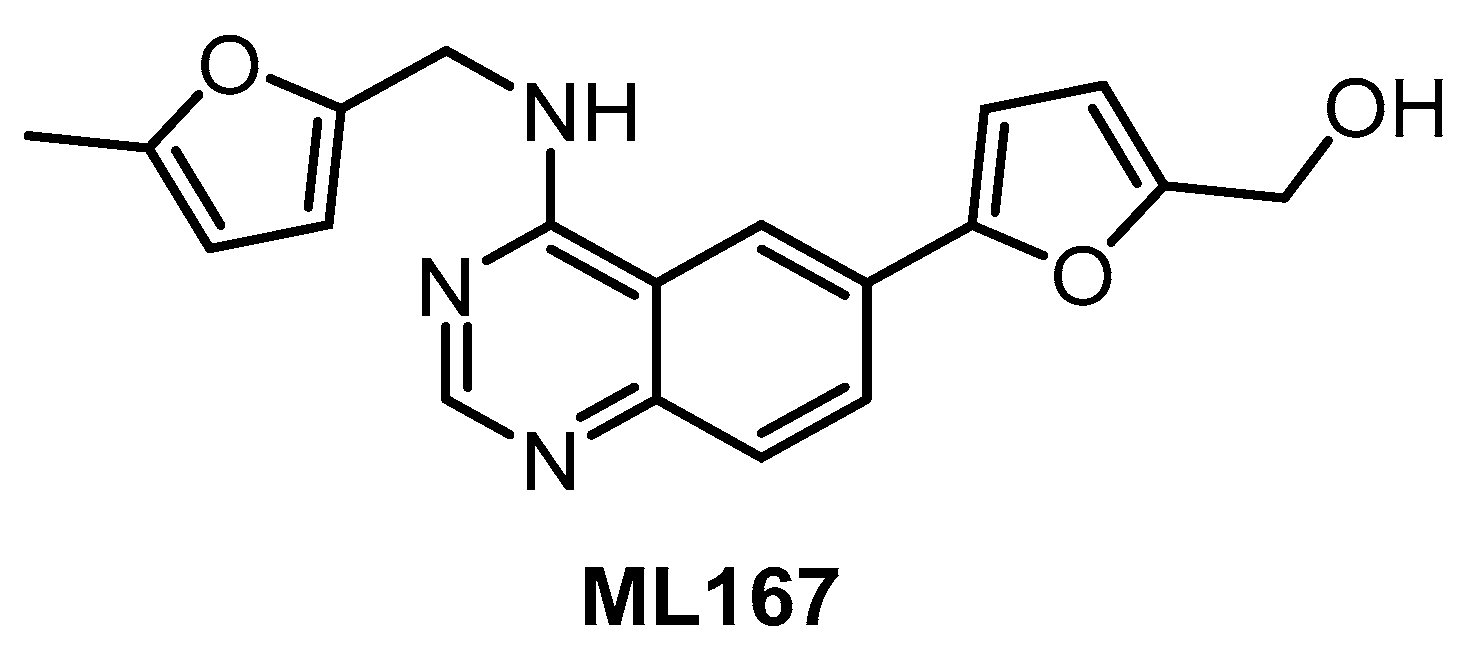
| ML167 [138] | 1522 | 1648 | inactive | 136 | inactive | 4420 | ND | ND | ND | ND | Not published | 442 kinases |
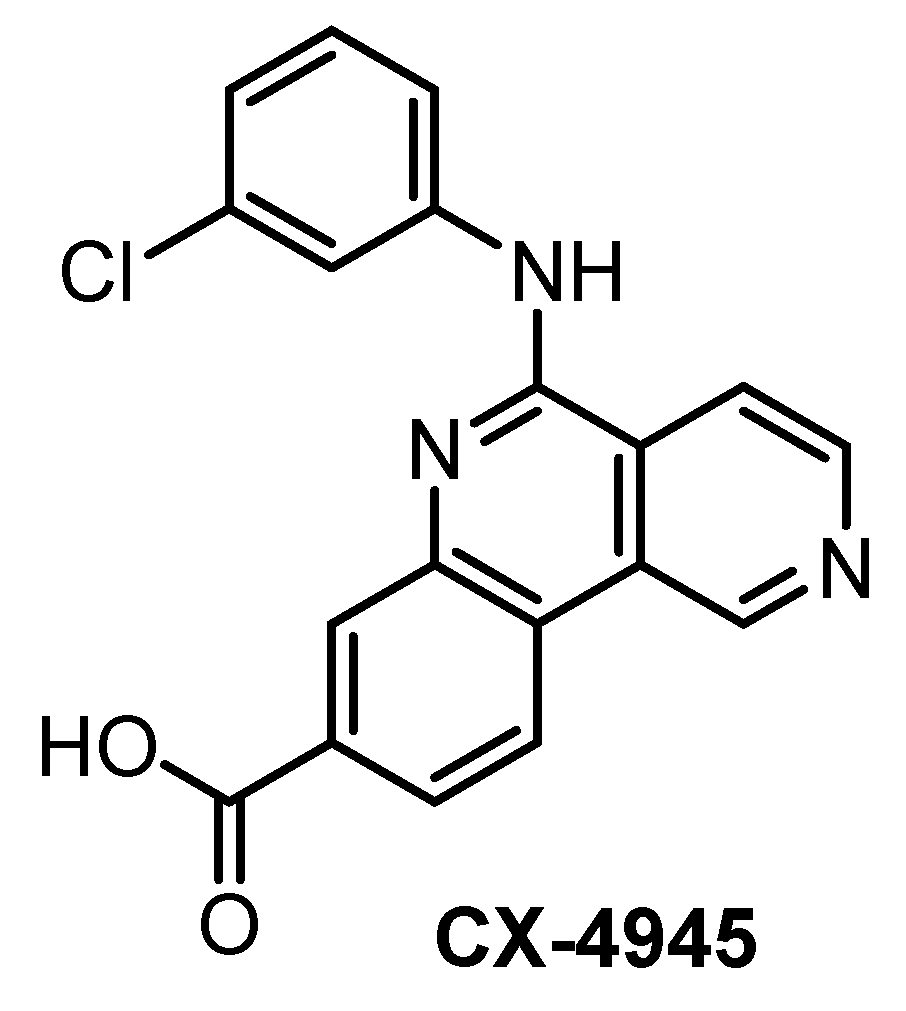
| CX-4945 [133,139,140] | 3.3 | 2.9 | 67 | 23 | 14 | ND | 5% (0.5 µM) | 11% (0.5 µM) | 15% (0.5 µM) | 7% (0.5 µM) | CK2α = 1.5 PIM1 = 216 TBK1 DAPK2 ZIPK FLT1 | ~235 kinases |
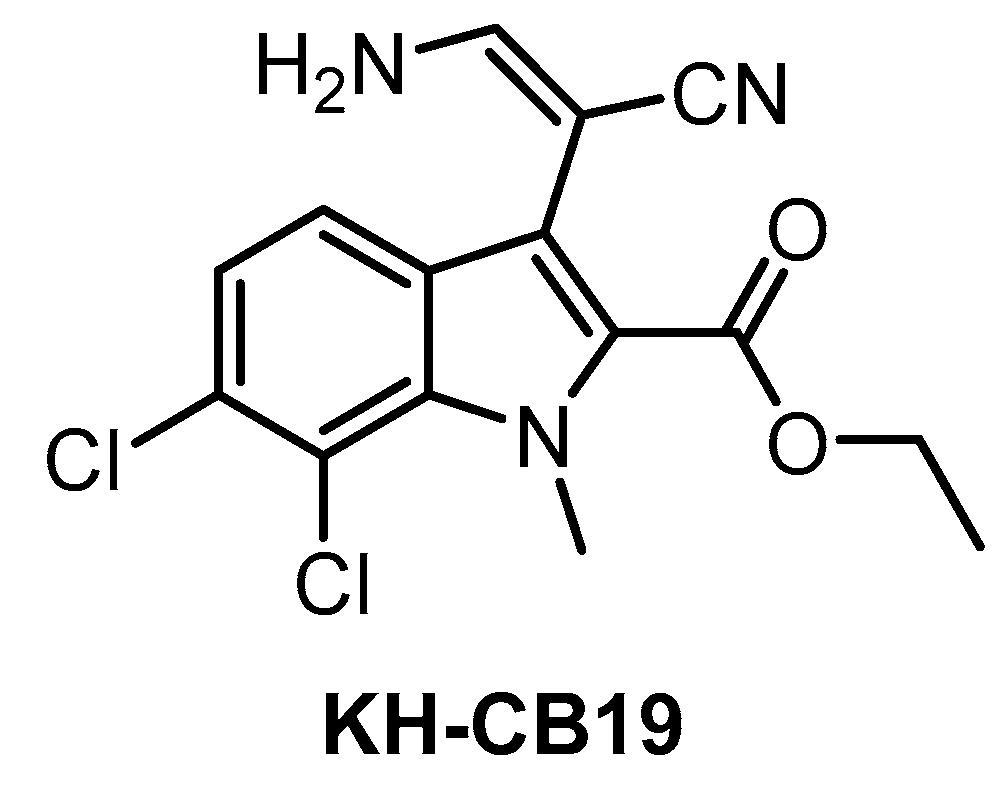
| KH-CB19 [141] | 20 | ND | 530 | ND | 55 | ND | ND | ND | ND | ND | PIM1/3 SGK085 | 106 kinases (thermal shift assay) |
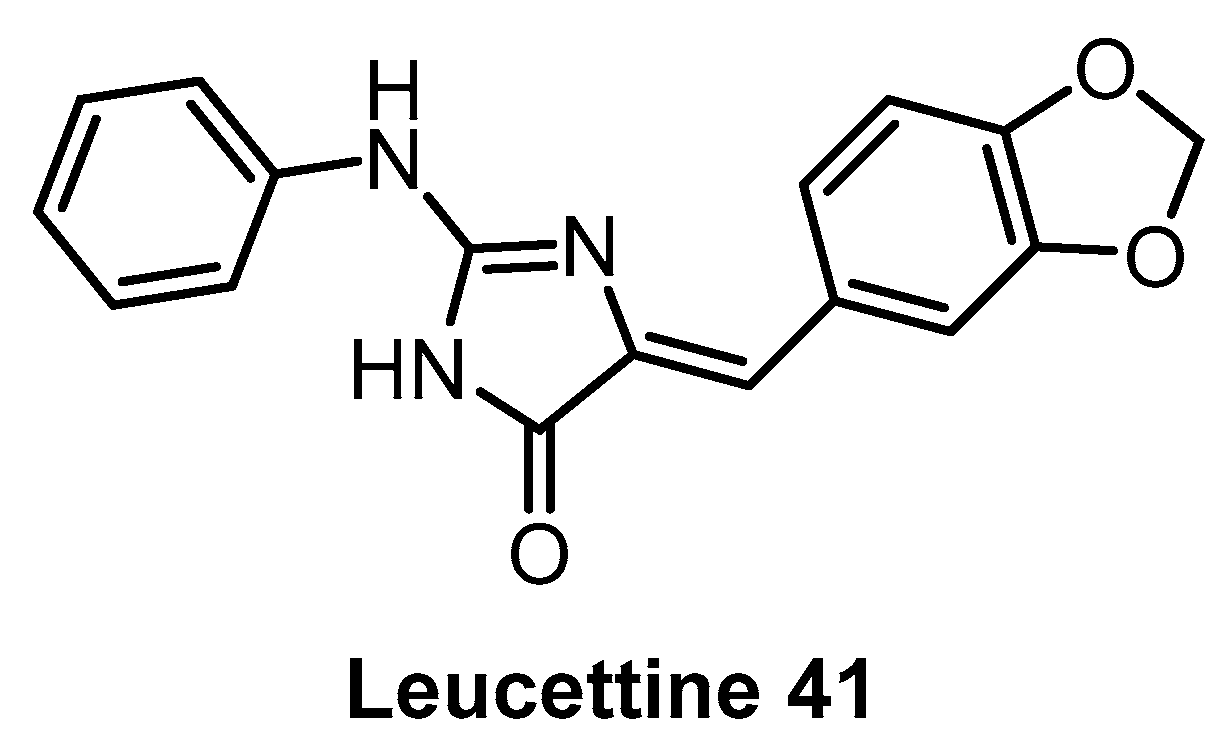
| Leucettine L41 [142] | 71 | 720 | inactive | 64 | 60 | 44 | 73 | 1% (10 µM) | 11% (10 µM) | 4% (10 µM) | GSK3α = 210 PIM1 IRAK1 TAOK1 | 402 kinases |
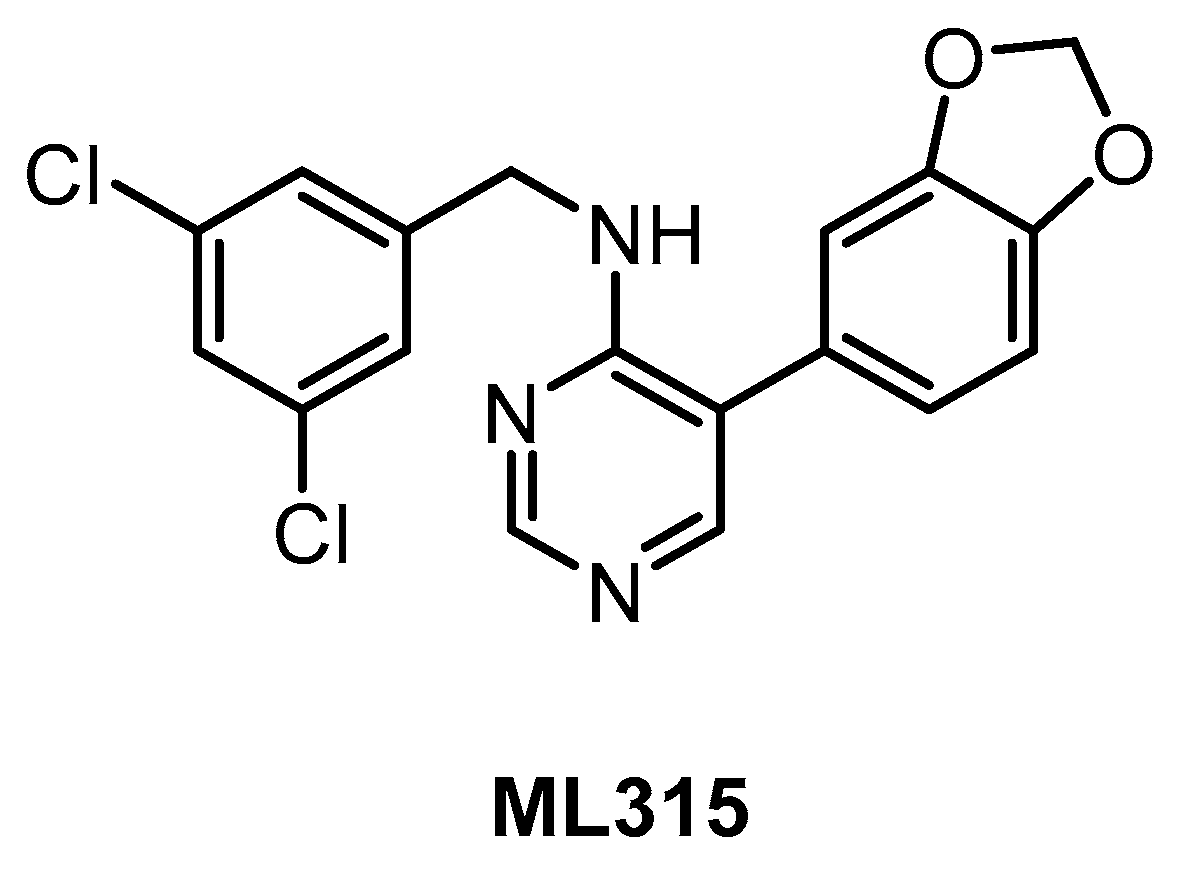
| ML315 [143] | 68 | 231 | inactive | 68 | 282 | 1156 | ND | ND | ND | ND | PRKCE MAP3K1 CK1ε | 442 kinases |
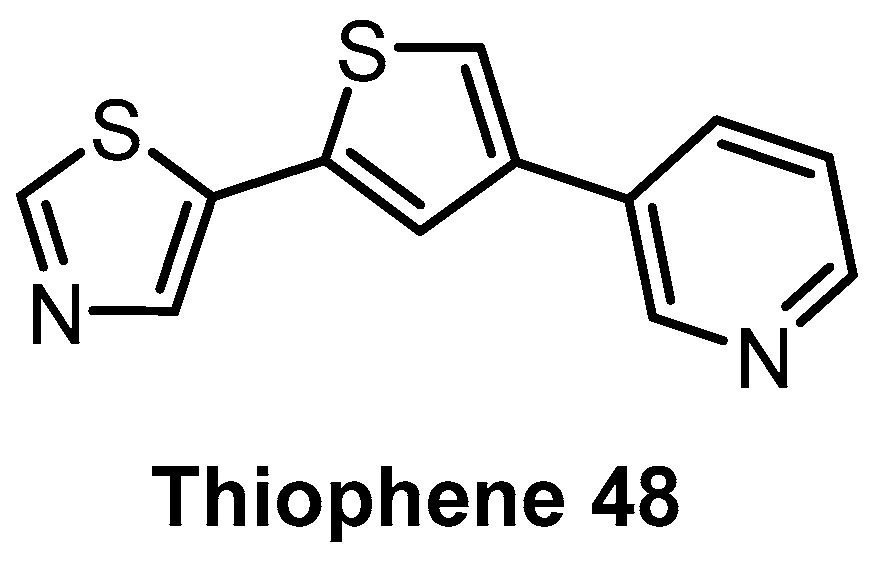
| Thiophene 48 [144] | 110 | 22% (5 µM) | 69% (5 µM) | 1% (5 µM) | 100 | 70 | 40 | 91% (5 µM) | ND | ND | - | 102 kinases |
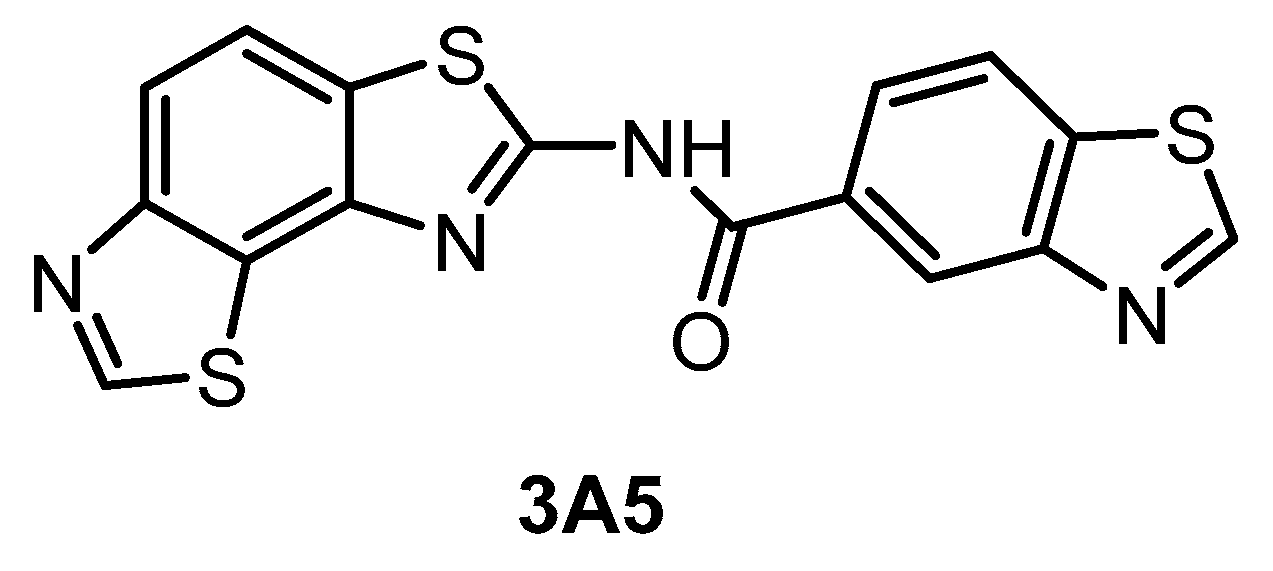
| 3A5 [145] | 51 | 68 | 346 | ND | 260 | ND | 61% (1 µM) | inactive | inactive | inactive | CK1γ2 | ~140 kinases |
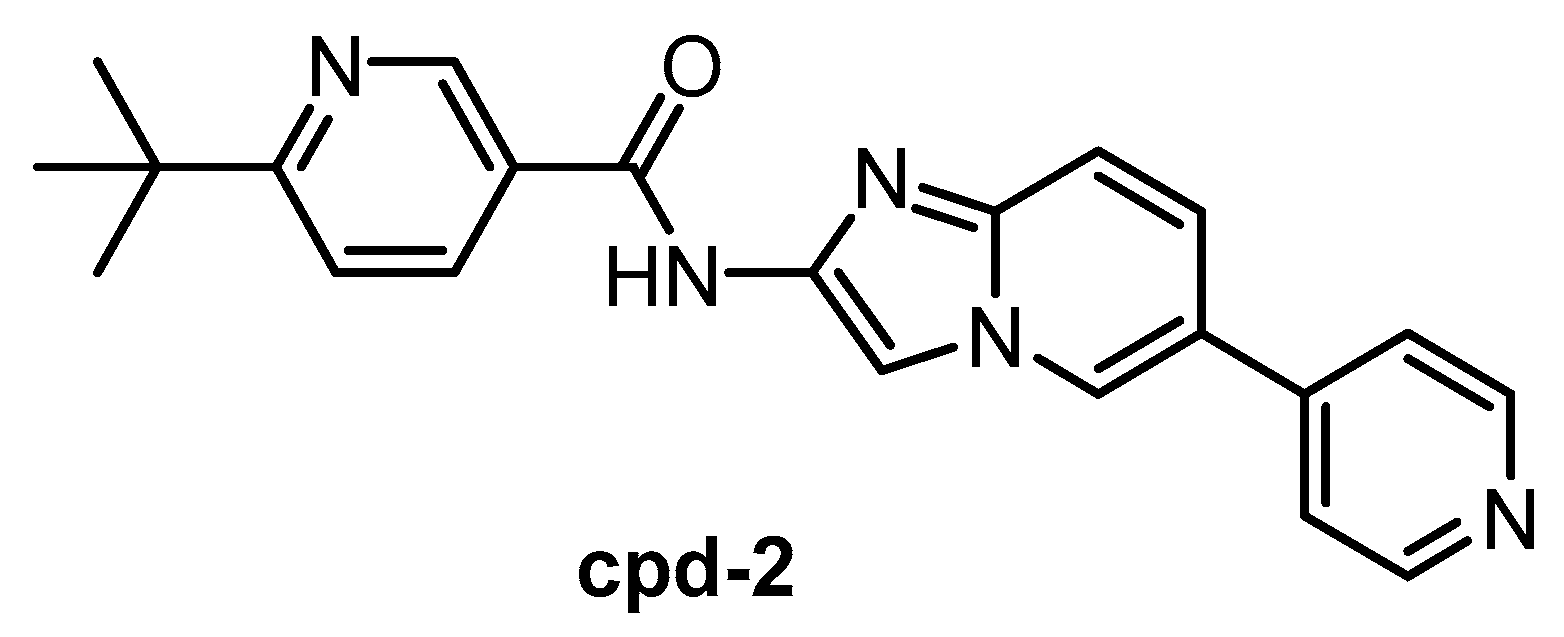
| Cpd-2 [146] | 1.1 | 2.4 | ND | ND | ND | ND | ND | ND | ND | ND | SRPK1 = 200 SRPK2 = 310 SRPK3 = 230 | 28 kinases |
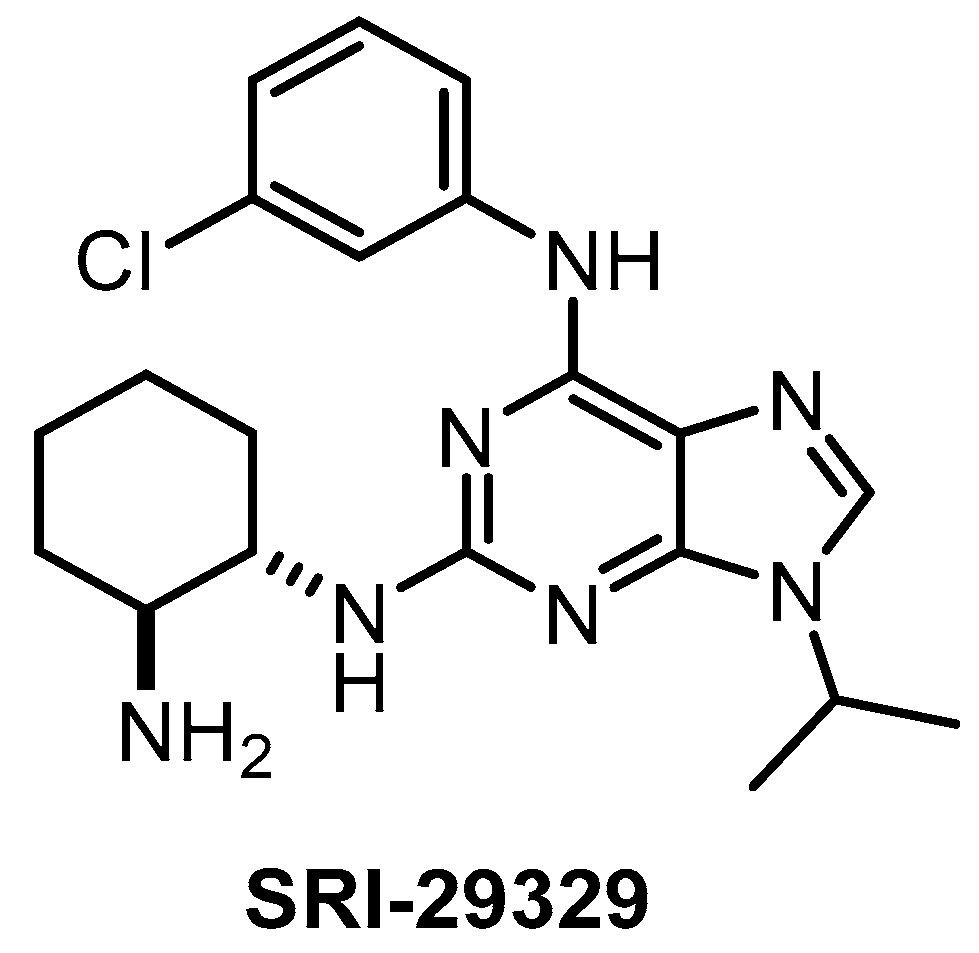
| SRI-29329 [147] | 78 | 16 | >1000 | 86 | 95% (1 µM) | ND | ND | ND | ND | ND | - | 29 kinases |
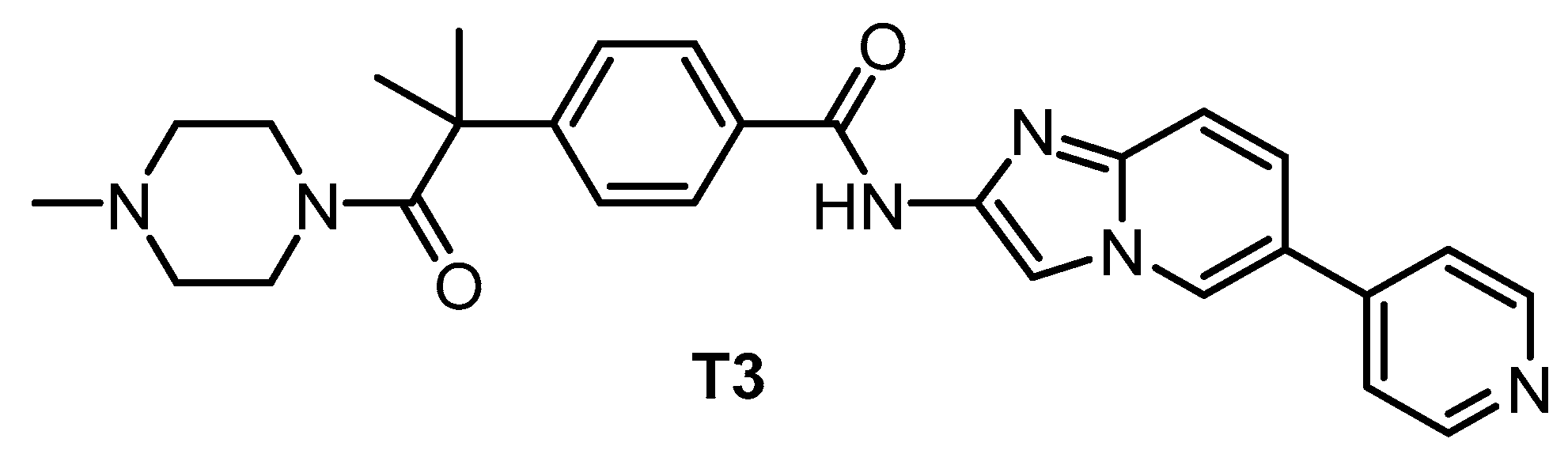
| T3 [135,148] | 0.67 | 15 | 110 | ND | 260 | 230 | ND | inactive | inactive | ND | - | 71 kinases |
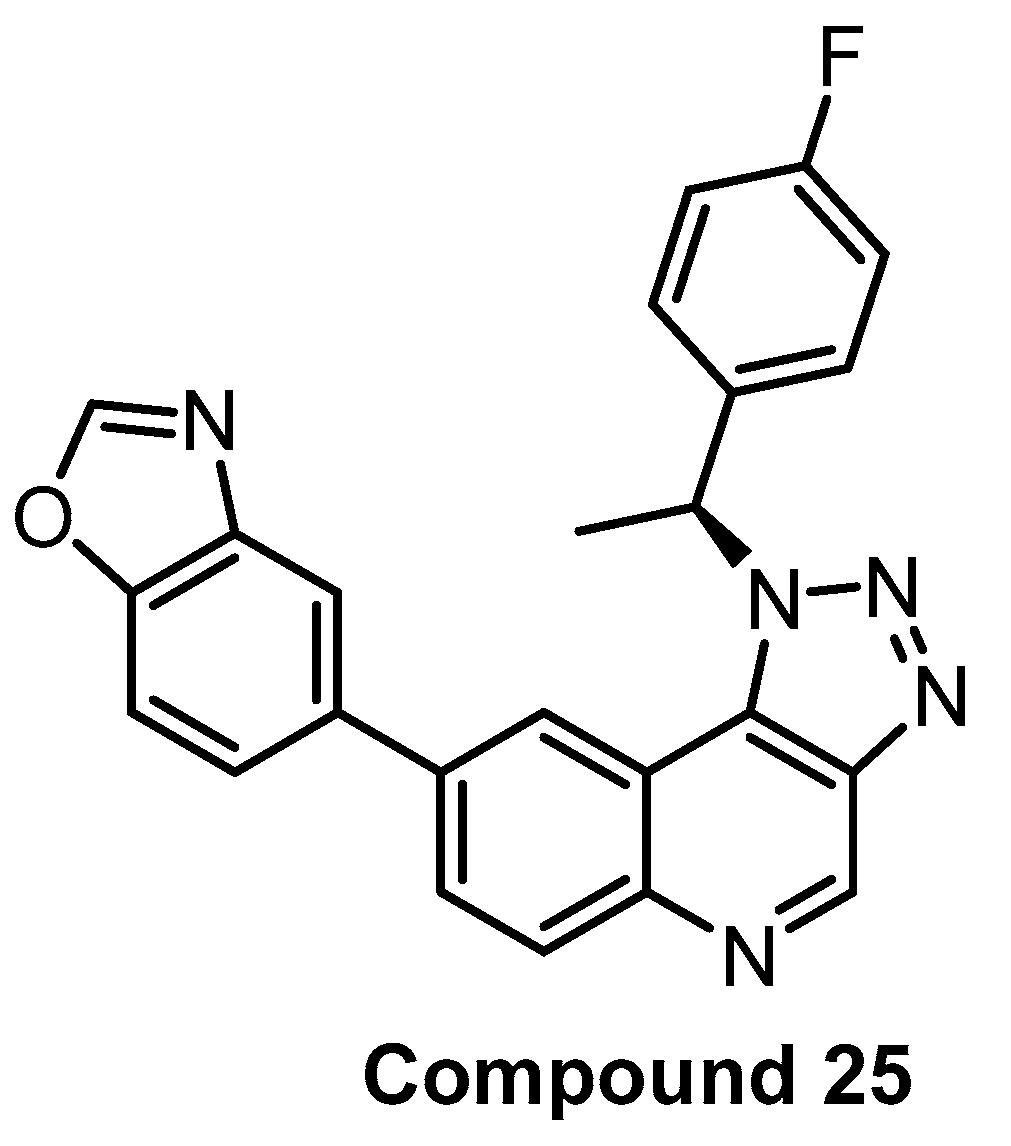
| Compound 25 [90] | 2 | 31 | inactive | 8 | 138 | 690 | inactive | inactive | inactive | inactive | - | 368 kinases |
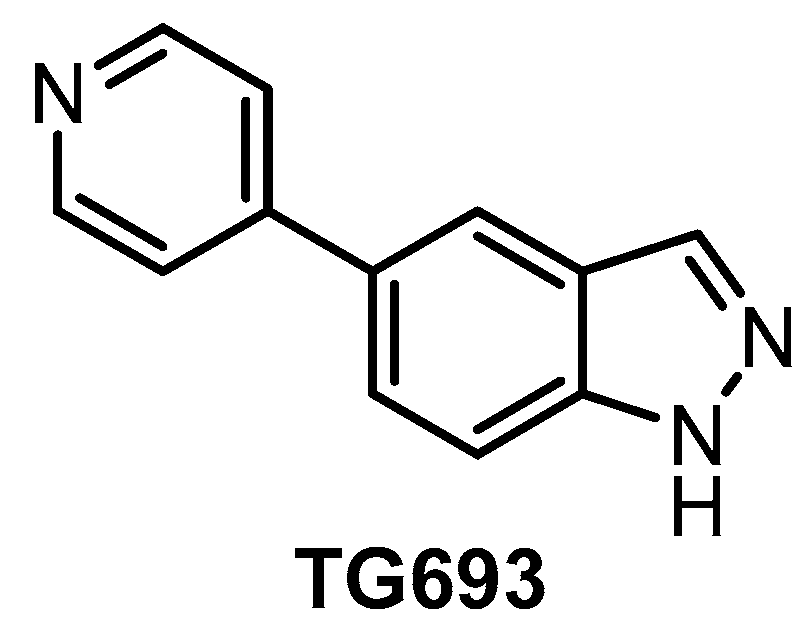
| TG693 [127] | 113 | 85% (1 µM) | 88% (1 µM) | ND | 16% (1 µM) | ND | 23% (1 µM) | inactive | ND | inactive | HASPIN | ~313 kinases |
| Indazole1 [133] | 12 | 10 | 2250 | 12 | 73 | ND | ND | ND | ND | ND | - | 34 kinases |
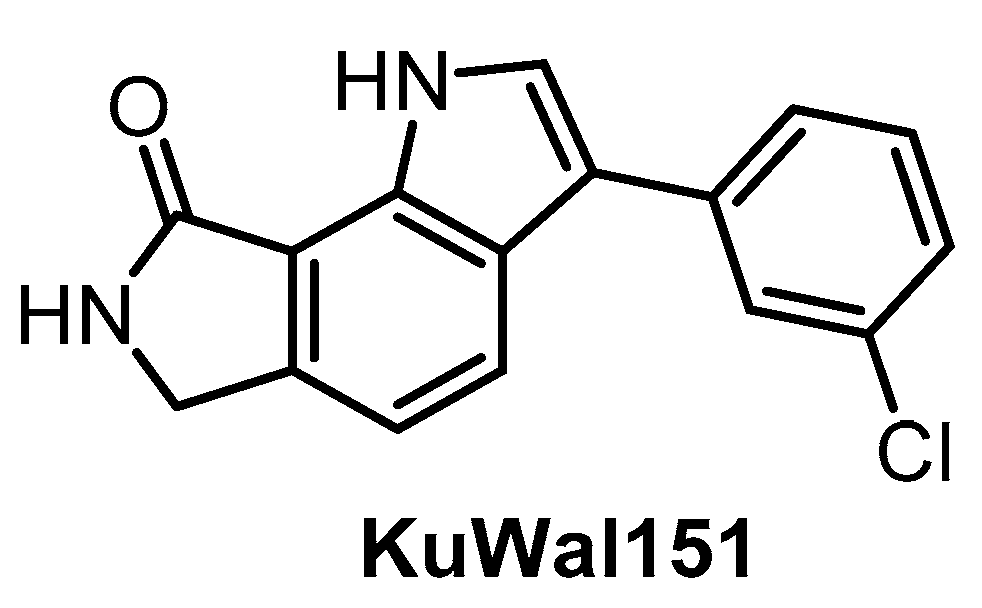
| KuWal151 [149] | 88 | 510 | inactive | 28 | inactive | inactive | inactive | ND | ND | ND | - | IC50 for 14 kinases |
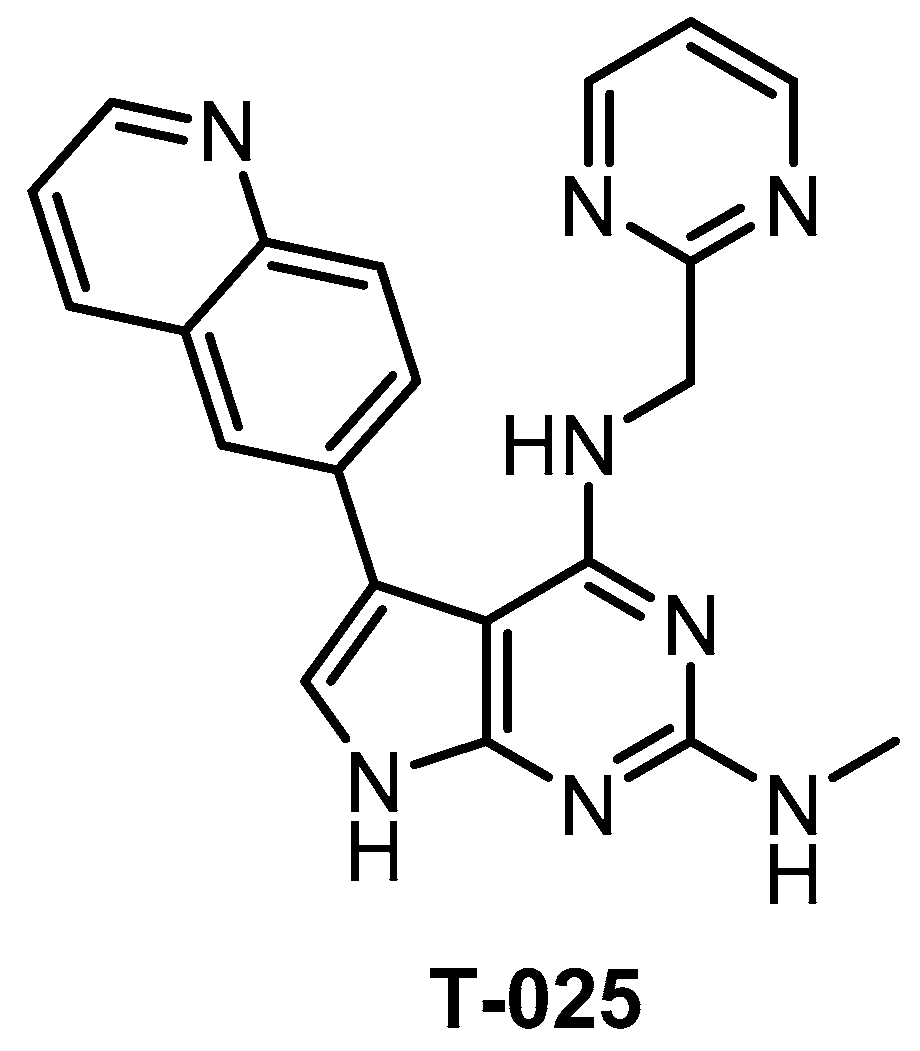
| T-025 [118] | 4.8 (Kd) | 0.096 (Kd) | 6.5 (Kd) | 0.61 (Kd) | 0.074 (Kd) | 1.5 (Kd) | 32 (Kd) | 55 (Kd) | 96 (Kd) | 5% (0.3 µM) | HIPK4 YSK4 IRAK4 FLT ERK8 | 468 kinases |
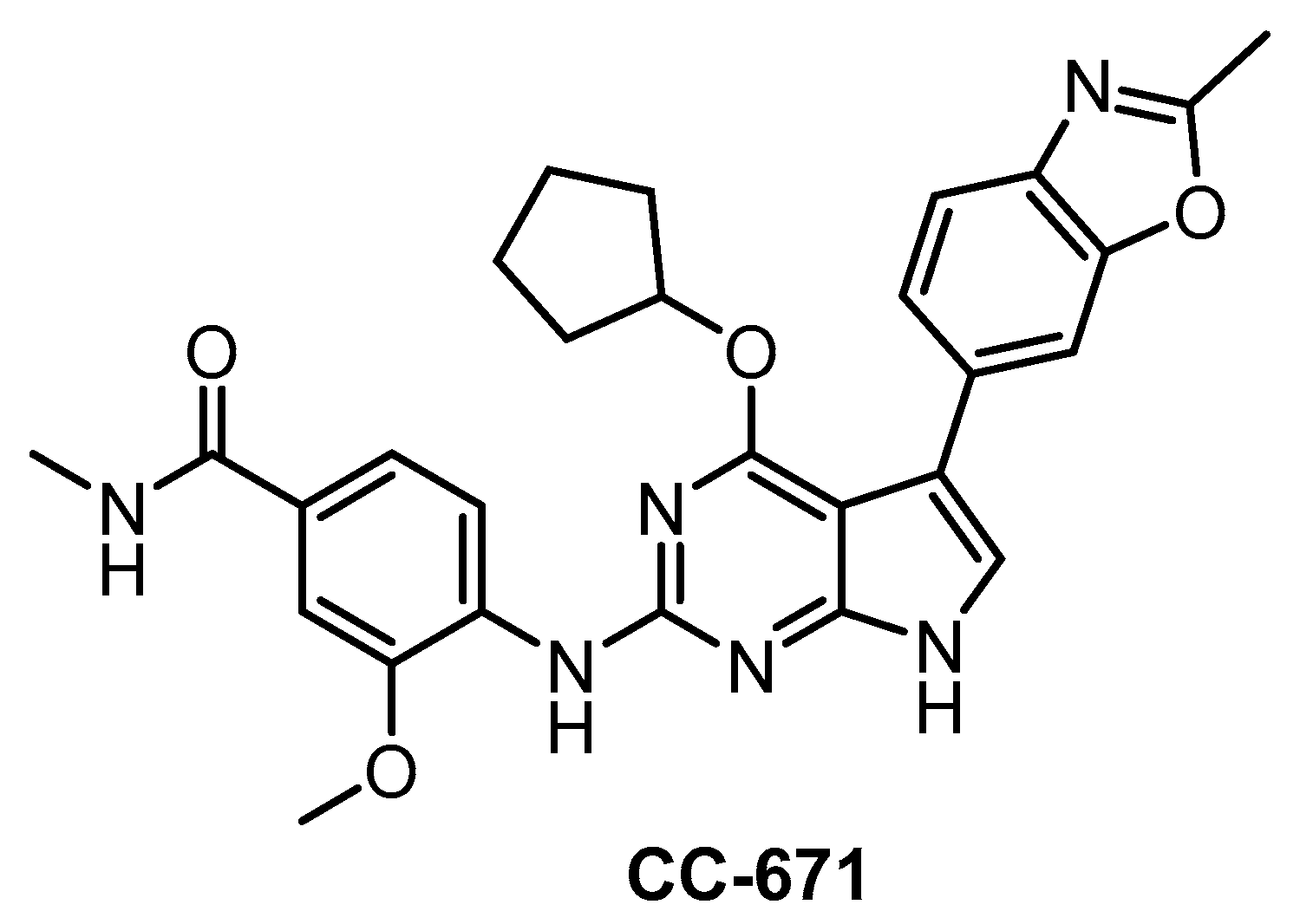
| CC-671 [137] | 300 | 6.3 | 60% (3 µM) | ND | 104 | 157 | ND | 97% (3 µM) | 96% (3 µM) | 92% (3 µM) | TTK = 5 DYRK3 = 99 PhKγ1 = 136 TSSK1 = 452 | 225 kinases |
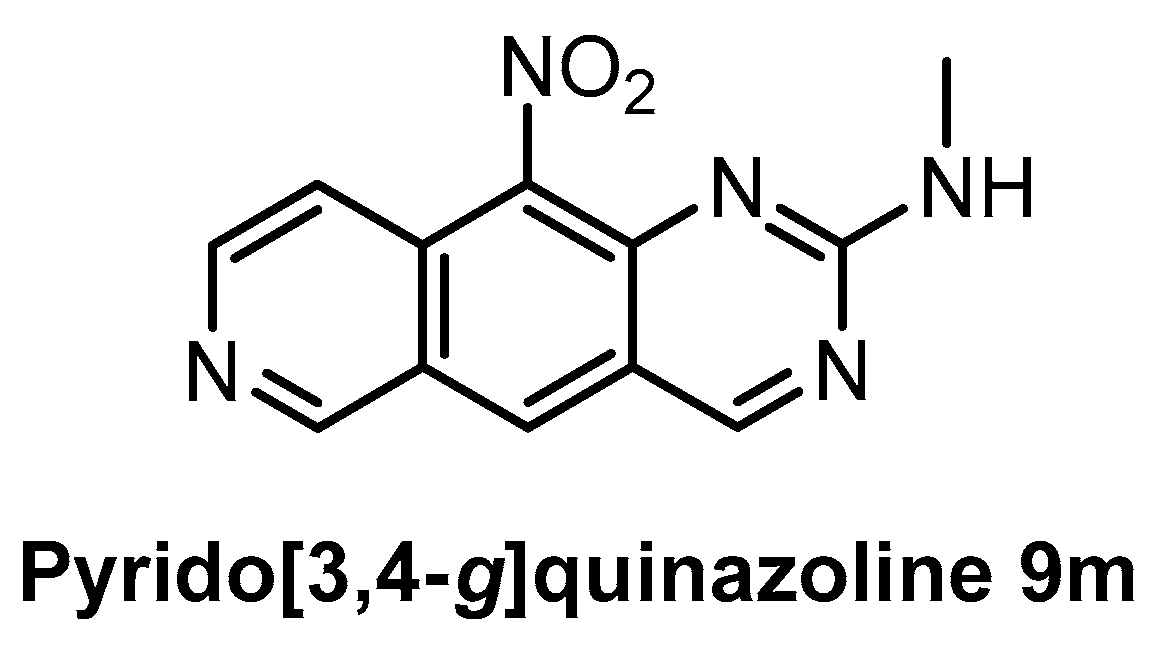
| Pyrido [3,4-g]quinazoline 9m [150] | 18 | ND | ND | ND | 39% (1 µM) | ND | ND | ND | ND | ND | CDK5 CK1 GSK3 | - |
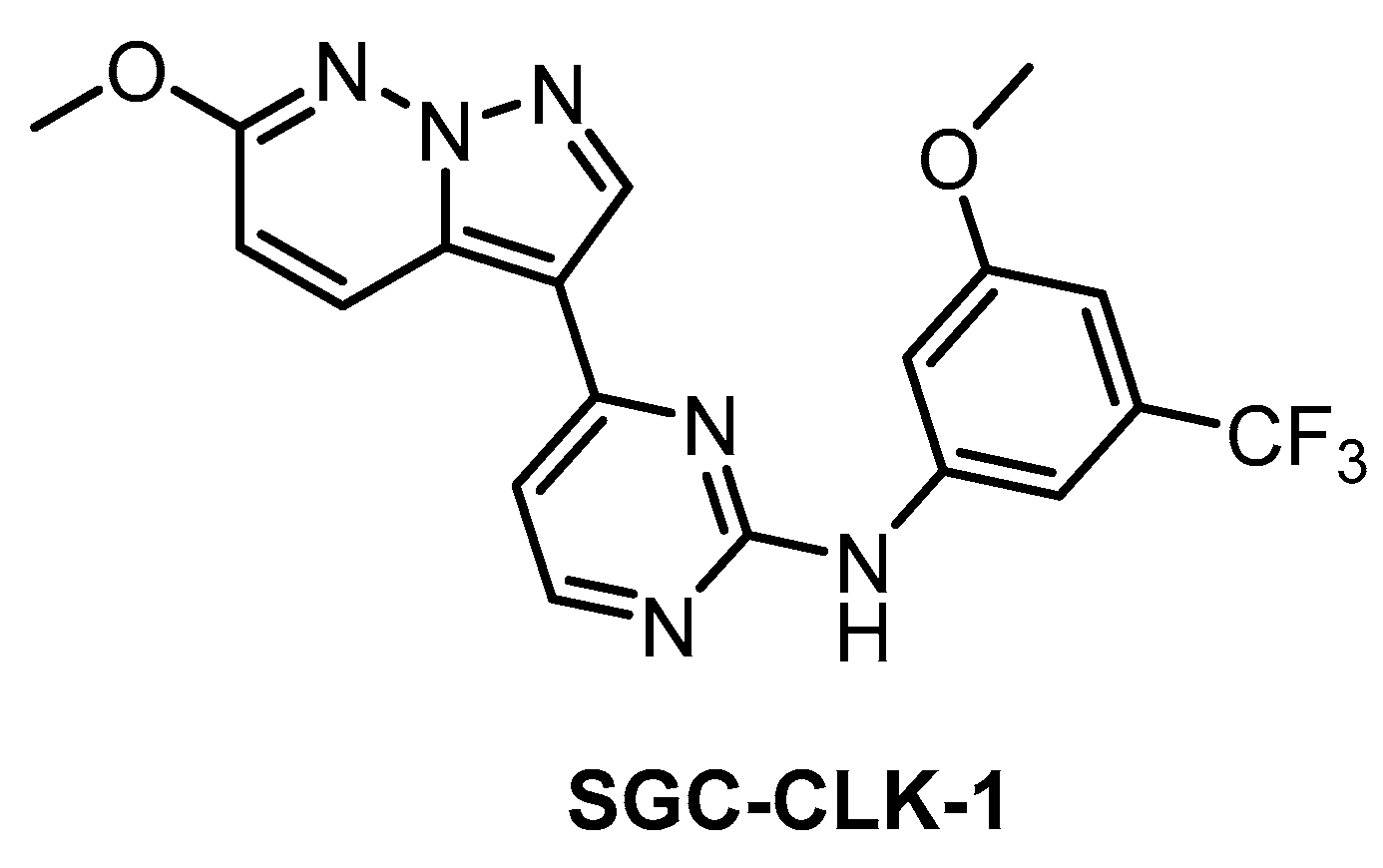
| SGC-CLK-1 [151] | 13 | 4 | 363 | 46 | ND | ND | ND | 50 | 42 | ND | ERK8 NEK7 PIP5K2B STK16 | 403 kinases |
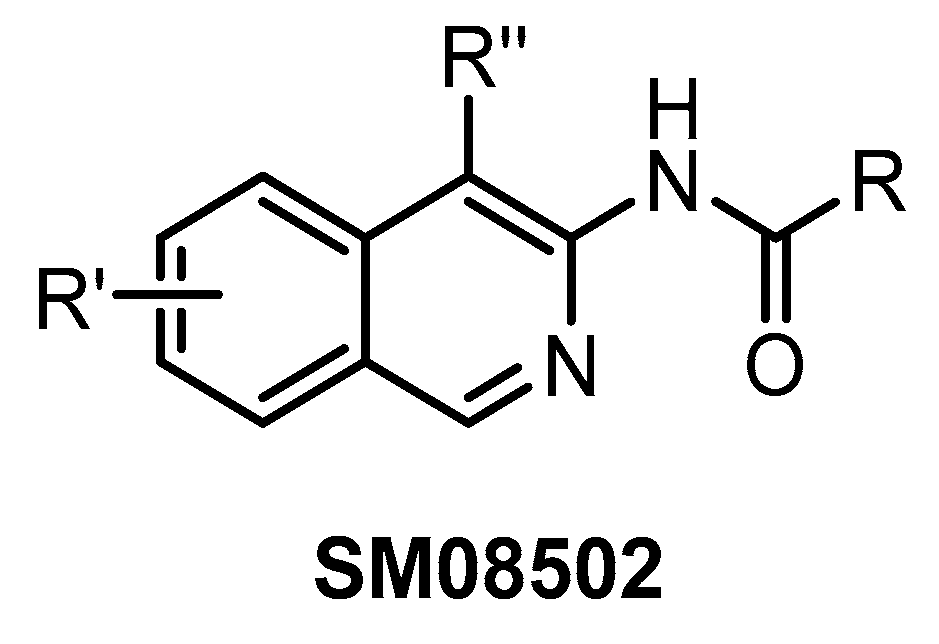
| SM08502 [76] | 8 | 1 | 22 | 1 | 1 | 1 | 3 | ND | 23 | 21 | additional 14 kinases with IC50 < 50 nM | 466 kinases |
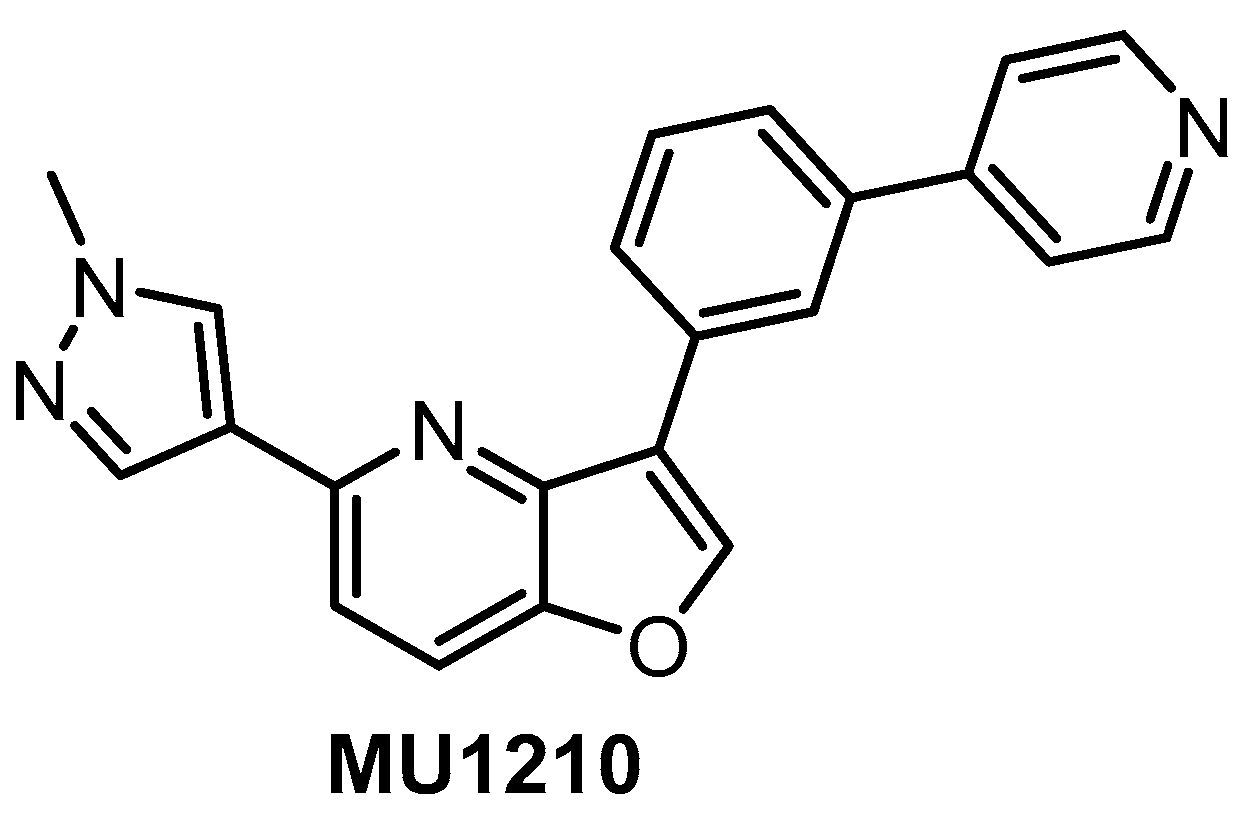
| MU1210 [47,152] | 8 | 20 | Inactive | 12 | 213 | 956 | 1309 | 187 | 29 | 159 | GSK3α PIM1/1 HASPIN | 210 kinases |
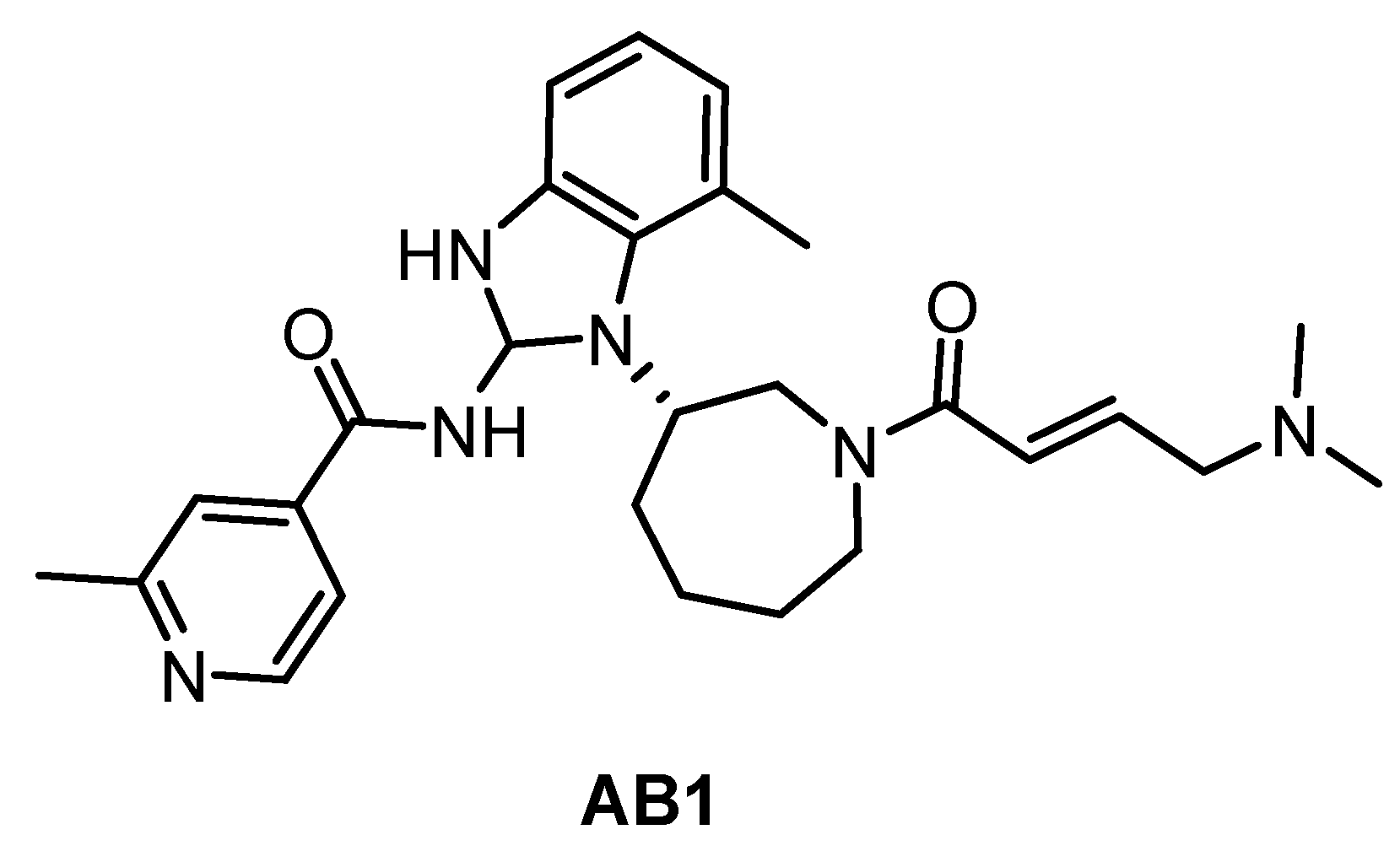
| AB1 [89] | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | BTK = 2 hERGFR = 2 | ND |
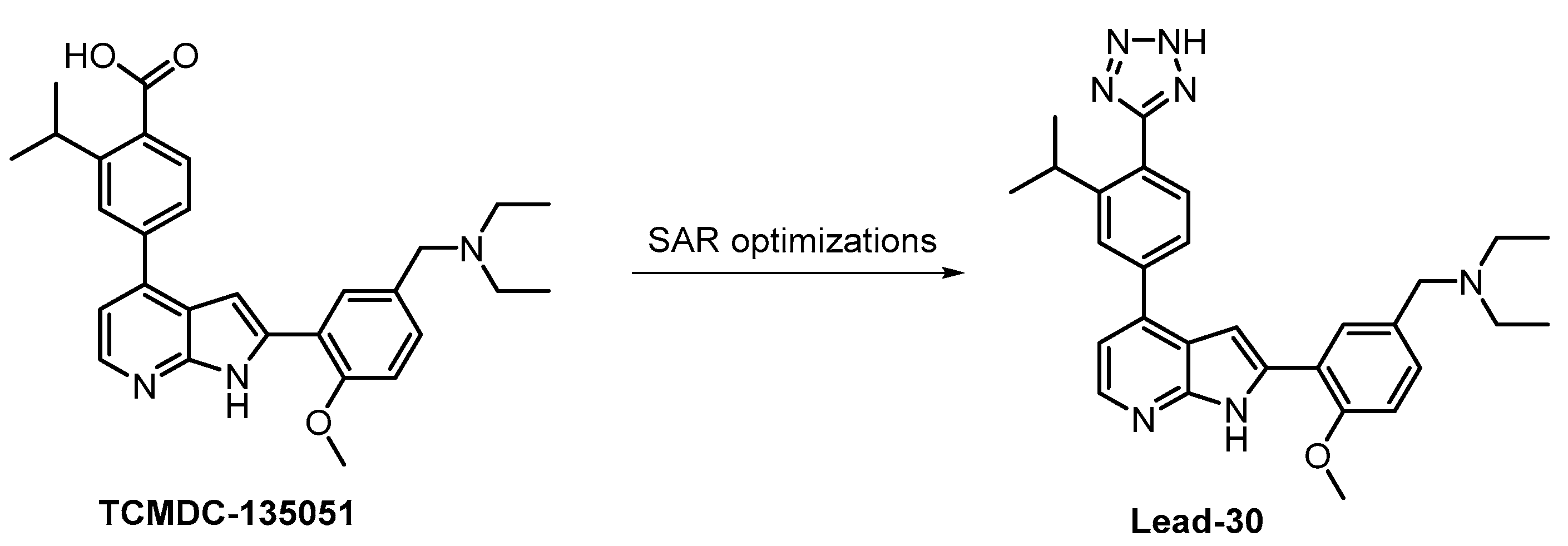
| TCMDC-135051 [130,131] | ND | 17% (1 µM) | 40 | ND | 71% (1 µM) | ND | 99% (1 µM) | 83% (1 µM) | 56% (1 µM) | 93% (1 µM) | MNK1 MAP4K3 CAMKKb CDK9 IRAK1 TGFBR1 PHK | 140 kinases |
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Roskoski, R. A Historical Overview of Protein Kinases and Their Targeted Small Molecule Inhibitors. Pharmacol. Res. 2015, 100, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Haar, E.; Walters, W.; Pazhanisamy, S.; Taslimi, P.; Pierce, A.; Bemis, G.; Salituro, F.; Harbeson, S. Kinase Chemogenomics: Targeting the Human Kinome for Target Validation and Drug Discovery. Mini-Rev. Med. Chem. 2004, 4, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Modi, V.; Dunbrack, R.L. Defining a New Nomenclature for the Structures of Active and Inactive Kinases. Proc. Natl. Acad. Sci. USA 2019, 116, 6818–6827. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. Protein Kinases—the Major Drug Targets of the Twenty-First Century? Nat. Rev. Drug Discov. 2002, 1, 309. [Google Scholar] [CrossRef]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Muzio, L.L. The Crucial Role of Protein Phosphorylation in Cell Signaling and Its Use as Targeted Therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef]
- Fleuren, E.D.G.; Zhang, L.; Wu, J.; Daly, R.J. The Kinome “at Large” in Cancer. Nat. Rev. Cancer 2016, 16, 83–98. [Google Scholar] [CrossRef]
- Roskoski, R. Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors: A 2020 Update. Pharmacol. Res. 2020, 152, 104609. [Google Scholar] [CrossRef]
- Berndt, N.; Karim, R.M.; Schönbrunn, E. Advances of Small Molecule Targeting of Kinases. Curr. Opin. Chem. Biol. 2017, 39, 126–132. [Google Scholar] [CrossRef]
- Deng, Y.N.; Bellanti, J.A.; Zheng, S.G. Essential Kinases and Transcriptional Regulators and Their Roles in Autoimmunity. Biomolecules 2019, 9, 145. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. Small-Molecule Kinase Inhibitors: An Analysis of FDA-Approved Drugs. Drug Discov. Today 2016, 21, 5–10. [Google Scholar] [CrossRef]
- Fedorov, O.; Müller, S.; Knapp, S. The (Un)Targeted Cancer Kinome. Nat. Chem. Biol. 2010, 6, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Lind, J.; Czernilofsky, F.; Vallet, S.; Podar, K. Emerging Protein Kinase Inhibitors for the Treatment of Multiple Myeloma. Expert Opin. Emerg. Drugs 2019, 24, 133–152. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, P.; Yang, H.; Zhou, J.; Li, Y.; Li, X.; Xue, W.; Yu, C.; Tian, Y.; Zhu, F. Comparison of FDA Approved Kinase Targets to Clinical Trial Ones: Insights from Their System Profiles and Drug-Target Interaction Networks. BioMed Res. Int. 2016, 2016, 2509385. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, F.M.; Gray, N.S. Kinase Inhibitors: The Road Ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S. SR Proteins: Binders, Regulators, and Connectors of RNA. Mol. Cells 2017, 40, 1. [Google Scholar] [CrossRef] [PubMed]
- Corkery, D.P.; Holly, A.C.; Lahsaee, S.; Dellaire, G. Connecting the Speckles: Splicing Kinases and Their Role in Tumorigenesis and Treatment Response. Nucleus 2015, 6, 279–288. [Google Scholar] [CrossRef]
- Chaudhary, S.; Khokhar, W.; Jabre, I.; Reddy, A.S.N.; Byrne, L.J.; Wilson, C.M.; Syed, N.H. Alternative Splicing and Protein Diversity: Plants versus Animals. Front. Plant Sci. 2019, 10, 708. [Google Scholar] [CrossRef]
- Fackenthal, J.D.; Godley, L.A. Aberrant RNA Splicing and Its Functional Consequences in Cancer Cells. Dis. Model. Mech. 2008, 1, 37–42. [Google Scholar] [CrossRef]
- Czubaty, A.; Piekiełko-Witkowska, A. Protein Kinases That Phosphorylate Splicing Factors: Roles in Cancer Development, Progression and Possible Therapeutic Options. Int. J. Biochem. Cell Biol. 2017, 91, 102–115. [Google Scholar] [CrossRef]
- Bates, D.O.; Cui, T.-G.; Doughty, J.M.; Winkler, M.; Sugiono, M.; Shields, J.D.; Peat, D.; Gillatt, D.; Harper, S.J. VEGF165b, an Inhibitory Splice Variant of Vascular Endothelial Growth Factor, Is Down-Regulated in Renal Cell Carcinoma. Cancer Res. 2003, 62, 10. [Google Scholar]
- Lu, F.; Gladden, A.B.; Diehl, J.A. An Alternatively Spliced Cyclin D1 Isoform, Cyclin D1b, Is a Nuclear Oncogene 1. Cancer Res. 2020, 63, 7056–7061. [Google Scholar]
- Naro, C.; Sette, C. Phosphorylation-Mediated Regulation of Alternative Splicing in Cancer. Int. J. Cell Biol. 2013, 2013, 151839. [Google Scholar] [CrossRef] [PubMed]
- Shepard, P.J.; Hertel, K.J. The SR Protein Family. Genome Biol. 2009, 10, 242. [Google Scholar] [CrossRef] [PubMed]
- Manley, J.L.; Tacke, R. SR Proteins and Splicing Control. Genes Dev. 1996, 10, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Cáceres, J.F.; Misteli, T.; Screaton, G.R.; Spector, D.L.; Krainer, A.R. Role of the Modular Domains of SR Proteins in Subnuclear Localization and Alternative Splicing Specificity. J. Cell Biol. 1997, 138, 225–238. [Google Scholar] [CrossRef]
- Lai, M.-C.; Tarn, W.-Y. Hypophosphorylated ASF/SF2 Binds TAP and Is Present in Messenger Ribonucleoproteins. J. Biol. Chem. 2004, 279, 31745–31749. [Google Scholar] [CrossRef]
- Zhou, Z.; Fu, X.-D. Regulation of Splicing by SR Proteins and SR Protein-Specific Kinases. Chromosoma 2013, 122, 191–207. [Google Scholar] [CrossRef]
- Ding, S.; Shi, J.; Qian, W.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.-X.; Liu, F. Regulation of Alternative Splicing of Tau Exon 10 by 9G8 and Dyrk1A. Neurobiol. Aging 2012, 33, 1389–1399. [Google Scholar] [CrossRef]
- Smith, B.E.; Wang, S.L.; Jaime-Figueroa, S.; Harbin, A.; Wang, J.; Hamman, B.D.; Crews, C.M. Differential PROTAC Substrate Specificity Dictated by Orientation of Recruited E3 Ligase. Nat. Commun. 2019, 10, 131. [Google Scholar] [CrossRef]
- Shi, J.; Qian, W.; Yin, X.; Iqbal, K.; Grundke-Iqbal, I.; Gu, X.; Ding, F.; Gong, C.-X.; Liu, F. Cyclic AMP-Dependent Protein Kinase Regulates the Alternative Splicing of Tau Exon 10: A Mechanism Involved in Tau Pathology of Alzheimer Disease. J. Biol. Chem. 2011, 286, 14639–14648. [Google Scholar] [CrossRef]
- Kvissel, A.-K.; Ørstavik, S.; Eikvar, S.; Brede, G.; Jahnsen, T.; Collas, P.; Akusjärvi, G.; Skålhegg, B.S. Involvement of the Catalytic Subunit of Protein Kinase A and of HA95 in Pre-MRNA Splicing. Exp. Cell Res. 2007, 313, 2795–2809. [Google Scholar] [CrossRef] [PubMed]
- Simarro, M.; Mauger, D.; Rhee, K.; Pujana, M.A.; Kedersha, N.L.; Yamasaki, S.; Cusick, M.E.; Vidal, M.; Garcia-Blanco, M.A.; Anderson, P. Fas-Activated Serine/Threonine Phosphoprotein (FAST) Is a Regulator of Alternative Splicing. Proc. Natl. Acad. Sci. USA 2007, 104, 11370–11375. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Wang, Q.; Kennedy, C.J.; Silver, P.A. An Alternative Splicing Network Links Cell-Cycle Control to Apoptosis. Cell 2010, 142, 625–636. [Google Scholar] [CrossRef]
- Weg-Remers, S. Regulation of Alternative Pre-MRNA Splicing by the ERK MAP-Kinase Pathway. EMBO J. 2001, 20, 4194–4203. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, Y.; Letwin, K.; Tannock, L.; Bernstein, A.; Pawson, T. A Mammalian Protein Kinase with Potential for Serine/Threonine and Tyrosine Phosphorylation Is Related to Cell Cycle Regulators. EMBO J. 1991, 10, 317–325. [Google Scholar] [CrossRef]
- Howell, B.W.; Afar, D.E.; Lew, J.; Douville, E.M.; Icely, P.L.; Gray, D.A.; Bell, J.C. STY, a Tyrosine-Phosphorylating Enzyme with Sequence Homology to Serine/Threonine Kinases. Mol. Cell. Biol. 1991, 11, 568–572. [Google Scholar] [CrossRef][Green Version]
- UniProt Consortium. UniProt. Available online: https://www.uniprot.org/ (accessed on 19 March 2020).
- Bullock, A.N.; Das, S.; Debreczeni, J.É.; Rellos, P.; Fedorov, O.; Niesen, F.H.; Guo, K.; Papagrigoriou, E.; Amos, A.L.; Cho, S.; et al. Kinase Domain Insertions Define Distinct Roles of CLK Kinases in SR Protein Phosphorylation. Structure 2009, 17, 352–362. [Google Scholar] [CrossRef]
- Keri, G.; Orfi, L.; Eros, D.; Hegymegi-Barakonyi, B.; Szantai-Kis, C.; Horvath, Z.; Waczek, F.; Marosfalvi, J.; Szabadkai, I.; Pato, J.; et al. Signal Transduction Therapy with Rationally Designed Kinase Inhibitors. Curr. Signal Transduct. Ther. 2006, 1, 67–95. [Google Scholar] [CrossRef]
- Knighton, D.; Zheng, J.; Ten Eyck, L.; Ashford, V.; Xuong, N.; Taylor, S.; Sowadski, J. Crystal Structure of the Catalytic Subunit of Cyclic Adenosine Monophosphate-Dependent Protein Kinase. Science 1991, 253, 407–414. [Google Scholar] [CrossRef]
- Grant, S.K. Therapeutic Protein Kinase Inhibitors. Cell. Mol. Life Sci. 2009, 66, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Chaikuad, A. RCSB PDB—3NR9: Structure of Human CDC2-Like Kinase 2 (CLK2). Available online: https://www.rcsb.org/structure/3NR9 (accessed on 28 August 2020).
- Zheng, G. RCSB PDB—1ATP: 2.2 Angstrom Refined Crystal Structure of the Catalytic Subunit of cAMP-Dependent Protein Kinase Complexed with MNATP and a Peptide Inhibitor. Available online: https://www.rcsb.org/structure/1ATP (accessed on 28 August 2020).
- Lee, J.Y.; Yun, J.-S.; Kim, W.-K.; Chun, H.-S.; Jin, H.; Cho, S.; Chang, J.H. Structural Basis for the Selective Inhibition of Cdc2-Like Kinases by CX-4945. BioMed Res. Int. 2019, 2019, 6125068. [Google Scholar] [CrossRef] [PubMed]
- Schröder, M.; Bullock, A.N.; Fedorov, O.; Bracher, F.; Chaikuad, A.; Knapp, S. DFG-1 Residue Controls Inhibitor Binding Mode and Affinity Providing a Basis for Rational Design of Kinase Inhibitor Selectivity. J. Med. Chem. 2020, 0c00898. [Google Scholar] [CrossRef] [PubMed]
- Němec, V.; Hylsová, M.; Maier, L.; Flegel, J.; Sievers, S.; Ziegler, S.; Schröder, M.; Berger, B.; Chaikuad, A.; Valčíková, B.; et al. Furo[3,2-b]Pyridine: A Privileged Scaffold for Highly Selective Kinase Inhibitors and Effective Modulators of the Hedgehog Pathway. Angew. Chem. Int. Ed. 2019, 58, 1062–1066. [Google Scholar] [CrossRef]
- Bank, R.P.D. RCSB PDB—6YTW: CLK3 Bound with Benzothiazole Tg003 (Cpd 2). Available online: https://www.rcsb.org/structure/6YTW (accessed on 2 October 2020).
- Aubol, B.E.; Plocinik, R.M.; Hagopian, J.C.; Ma, C.-T.; McGlone, M.L.; Bandyopadhyay, R.; Fu, X.-D.; Adams, J.A. Partitioning RS Domain Phosphorylation in an SR Protein through the CLK and SRPK Protein Kinases. J. Mol. Biol. 2013, 425, 2894–2909. [Google Scholar] [CrossRef]
- Ghosh, G.; Adams, J.A. Phosphorylation Mechanism and Structure of Serine-Arginine Protein Kinases: SRPK Structure and Mechanism. FEBS J. 2011, 278, 587–597. [Google Scholar] [CrossRef]
- Papagrigoriou, E. RCSB PDB—2EU9: Crystal Structure of CLK3. Available online: https://www.rcsb.org/structure/2EU9 (accessed on 28 August 2020).
- Prasad, J.; Manley, J.L. Regulation and Substrate Specificity of the SR Protein Kinase Clk/Sty. Mol. Cell. Biol. 2003, 23, 4139–4149. [Google Scholar] [CrossRef][Green Version]
- Nayler, O.; Stamm, S.; Ullrich, A. Characterization and Comparison of Four Serine- and Arginine-Rich (SR) Protein Kinases. Biochem. J. 1997, 326, 693–700. [Google Scholar] [CrossRef]
- Colwill, K.; Pawson, T.; Andrews, B.; Prasad, J.; Manley, J.L.; Bell, J.C.; Duncan, P.I. The Clk/Sty Protein Kinase Phosphorylates SR Splicing Factors and Regulates Their Intranuclear Distribution. EMBO J. 1996, 15, 265–275. [Google Scholar] [CrossRef]
- Aubol, B.E.; Wu, G.; Keshwani, M.M.; Movassat, M.; Fattet, L.; Hertel, K.J.; Fu, X.-D.; Adams, J.A. Release of SR Proteins from CLK1 by SRPK1: A Symbiotic Kinase System for Phosphorylation Control of Pre-MRNA Splicing. Mol. Cell 2016, 63, 218–228. [Google Scholar] [CrossRef]
- Hochberg-Laufer, H.; Neufeld, N.; Brody, Y.; Nadav-Eliyahu, S.; Ben-Yishay, R.; Shav-Tal, Y. Availability of Splicing Factors in the Nucleoplasm Can Regulate the Release of MRNA from the Gene after Transcription. PLOS Genet. 2019, 15, e1008459. [Google Scholar] [CrossRef] [PubMed]
- Shin, C.; Manley, J.L. The SR Protein SRp38 Represses Splicing in M Phase Cells. Cell 2002, 111, 407–417. [Google Scholar] [CrossRef]
- Shin, C.; Feng, Y.; Manley, J.L. Dephosphorylated SRp38 Acts as a Splicing Repressor in Response to Heat Shock. Nature 2004, 427, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Mermoud, J.E.; Cohen, P.; Lamond, A.I. Ser/Thr-Specific Protein Phosphatases Are Required for Both Catalytic Steps of Pre-MRNA Splicing. Nucleic Acids Res. 1992, 20, 5263–5269. [Google Scholar] [CrossRef]
- Mermoud, J.E.; Cohen, P.T.; Lamond, A.I. Regulation of Mammalian Spliceosome Assembly by a Protein Phosphorylation Mechanism. EMBO J. 1994, 13, 5679–5688. [Google Scholar] [CrossRef]
- Stojdl, D.F.; Bell, J.C. SR Protein Kinases: The Splice of Life. Biochem. Cell Biol. 1999, 77, 6. [Google Scholar] [CrossRef]
- Huang, Y.; Gattoni, R.; Stévenin, J.; Steitz, J.A. SR Splicing Factors Serve as Adapter Proteins for TAP-Dependent MRNA Export. Mol. Cell 2003, 11, 837–843. [Google Scholar] [CrossRef]
- Graveley, B.R. Sorting out the Complexity of SR Protein Functions. RNA 2000, 6, 1197–1211. [Google Scholar] [CrossRef]
- Muraki, M.; Ohkawara, B.; Hosoya, T.; Onogi, H.; Koizumi, J.; Koizumi, T.; Sumi, K.; Yomoda, J.; Murray, M.V.; Kimura, H.; et al. Manipulation of Alternative Splicing by a Newly Developed Inhibitor of Clks. J. Biol. Chem. 2004, 279, 24246–24254. [Google Scholar] [CrossRef]
- Johnson, K.; Smith, K. Molecular Cloning of a NovelHuman CdcZ/CDC28-like Protein Kinase. J. Biol. Chem. 1991, 25, 3402–3407. [Google Scholar]
- Lew, J.; Qi, Z.; Huang, Q.-Q.; Paudel, H.; Matsuura, I.; Matsushita, M.; Zhu, X.; Wang, J.H. Structure, Function, and Regulation of Neuronal Cdc2-like Protein Kinase. Neurobiol. Aging 1995, 16, 263–268. [Google Scholar] [CrossRef]
- Hanes, J.; von der Kammer, H.; Klaudiny, J.; Scheit, K.H. Characterization by CDNA Cloning of Two New Human Protein Kinases. J. Mol. Biol. 1994, 244, 665–672. [Google Scholar] [CrossRef] [PubMed]
- George, A.; Aubol, B.E.; Fattet, L.; Adams, J.A. Disordered Protein Interactions for an Ordered Cellular Transition: Cdc2-like Kinase 1 Is Transported to the Nucleus via Its Ser–Arg Protein Substrate. J. Biol. Chem. 2019, 294, 9631–9641. [Google Scholar] [CrossRef] [PubMed]
- Duncan, P.I.; Howell, B.W.; Marius, R.M.; Drmanic, S.; Douville, E.M.J.; Bell, J.C. Alternative Splicing of STY, a Nuclear Dual Specificity Kinase. J. Biol. Chem. 1995, 270, 21524–21531. [Google Scholar] [CrossRef]
- Duncan, P.I.; Stojdl, D.F.; Marius, R.M.; Scheit, K.H.; Bell, J.C. The Clk2 and Clk3 Dual-Specificity Protein Kinases Regulate the Intranuclear Distribution of SR Proteins and Influence Pre-MRNA Splicing. Exp. Cell Res. 1998, 241, 300–308. [Google Scholar] [CrossRef]
- Haltenhof, T.; Kotte, A.; De Bortoli, F.; Schiefer, S.; Meinke, S.; Emmerichs, A.-K.; Petermann, K.K.; Timmermann, B.; Imhof, P.; Franz, A.; et al. A Conserved Kinase-Based Body-Temperature Sensor Globally Controls Alternative Splicing and Gene Expression. Mol. Cell 2020, 78, 57–69.e4. [Google Scholar] [CrossRef]
- Petsalaki, E.; Zachos, G. Clks 1, 2 and 4 Prevent Chromatin Breakage by Regulating the Aurora B-Dependent Abscission Checkpoint. Nat. Commun. 2016, 7, 11451. [Google Scholar] [CrossRef]
- Lopez Soto, E.J.; Gandal, M.J.; Gonatopoulos-Pournatzis, T.; Heller, E.A.; Luo, D.; Zheng, S. Mechanisms of Neuronal Alternative Splicing and Strategies for Therapeutic Interventions. J. Neurosci. 2019, 39, 8193–8199. [Google Scholar] [CrossRef]
- Wang, E.; Aifantis, I. RNA Splicing and Cancer. Trends Cancer 2020, 6, 631–644. [Google Scholar] [CrossRef]
- Ule, J.; Blencowe, B.J. Alternative Splicing Regulatory Networks: Functions, Mechanisms, and Evolution. Mol. Cell 2019, 76, 329–345. [Google Scholar] [CrossRef]
- Tam, B.Y.; Chiu, K.; Chung, H.; Bossard, C.; Nguyen, J.D.; Creger, E.; Eastman, B.W.; Mak, C.C.; Ibanez, M.; Ghias, A.; et al. The CLK Inhibitor SM08502 Induces Anti-Tumor Activity and Reduces Wnt Pathway Gene Expression in Gastrointestinal Cancer Models. Cancer Lett. 2019, S0304383519304732. [Google Scholar] [CrossRef] [PubMed]
- Glatz, D.C.; Rujescu, D.; Tang, Y.; Berendt, F.J.; Hartmann, A.M.; Faltraco, F.; Rosenberg, C.; Hulette, C.; Jellinger, K.; Hampel, H.; et al. The Alternative Splicing of Tau Exon 10 and Its Regulatory Proteins CLK2 and TRA2-BETA1 Changes in Sporadic Alzheimer’s Disease. J. Neurochem. 2006, 96, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A.M.; Rujescu, D.; Giannakouros, T.; Nikolakaki, E.; Goedert, M.; Mandelkow, E.-M.; Gao, Q.S.; Andreadis, A.; Stamm, S. Regulation of Alternative Splicing of Human Tau Exon 10 by Phosphorylation of Splicing Factors. Mol. Cell. Neurosci. 2001, 18, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front. Aging Neurosci. 2019, 11, 204. [Google Scholar] [CrossRef] [PubMed]
- Uzor, S.; Zorzou, P.; Bowler, E.; Porazinski, S.; Wilson, I.; Ladomery, M. Autoregulation of the Human Splice Factor Kinase CLK1 through Exon Skipping and Intron Retention. Gene 2018, 670, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Keshwani, M.M.; Hailey, K.L.; Aubol, B.E.; Fattet, L.; McGlone, M.L.; Jennings, P.A.; Adams, J.A. Nuclear Protein Kinase CLK1 Uses a Non-Traditional Docking Mechanism to Select Physiological Substrates. Biochem. J. 2015, 472, 329–338. [Google Scholar] [CrossRef]
- Tang, Z.; Luca, M.; Portillio, J.; Ngo, B.; Chang, C.; Wen, T.; Murray, J.; Carr, A. LAMMER Kinase Kic1 Is Involved in Pre-MRNA Processing. Exp. Cell Res. 2011, 317, 2308–2320. [Google Scholar] [CrossRef]
- Giannakouros, T.; Nikolakaki, E.; Mylonis, I.; Georgatsou, E. Serine-Arginine Protein Kinases: A Small Protein Kinase Family with a Large Cellular Presence: Serine-Arginine Protein Kinases. FEBS J. 2011, 278, 570–586. [Google Scholar] [CrossRef]
- Aubol, B.E.; Keshwani, M.M.; Fattet, L.; Adams, J.A. Mobilization of a Splicing Factor through a Nuclear Kinase–Kinase Complex. Biochem. J. 2018, 475, 677–690. [Google Scholar] [CrossRef]
- Aubol, B.E.; Fattet, L.; Adams, J.A. A Conserved Sequence Motif Bridges Two Protein Kinases for Enhanced Phosphorylation and Nuclear Function of a Splicing Factor. FEBS J. 2020, 15351. [Google Scholar] [CrossRef]
- Aubol, B.E.; Plocinik, R.M.; Keshwani, M.M.; McGlone, M.L.; Hagopian, J.C.; Ghosh, G.; Fu, X.-D.; Adams, J.A. N-Terminus of the Protein Kinase CLK1 Induces SR Protein Hyperphosphorylation. Biochem. J. 2014, 462, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Conaway, L.; Rutherford Bethard, J.; Al-Ayoubi, A.M.; Thompson Bradley, A.; Zheng, H.; Weed, S.A.; Eblen, S.T. Phosphorylation of the Alternative MRNA Splicing Factor 45 (SPF45) by Clk1 Regulates Its Splice Site Utilization, Cell Migration and Invasion. Nucleic Acids Res. 2013, 41, 4949–4962. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Patel, N.A.; Watson, J.E.; Apostolatos, H.; Kleiman, E.; Hanson, O.; Hagiwara, M.; Cooper, D.R. Akt2 Regulation of Cdc2-Like Kinases (Clk/Sty), Serine/Arginine-Rich (SR) Protein Phosphorylation, and Insulin-Induced Alternative Splicing of PKCβII Messenger Ribonucleic Acid. Endocrinology 2009, 150, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Saldivia, M.; Rao, S.P.S.; Fang, E.; Myburgh, E.; Brown, E.; Wollman, A.J.M.; Ritchie, R.; Lakhsminarayana, S.B.; Chen, Y.L.; Patra, D.; et al. Targeting the Trypanosome Kinetochore with CLK1 Protein Kinase Inhibitors. Nat. Microbiol. 2019. preprint. [Google Scholar] [CrossRef]
- Sun, Q.-Z.; Lin, G.-F.; Li, L.-L.; Jin, X.-T.; Huang, L.-Y.; Zhang, G.; Yang, W.; Chen, K.; Xiang, R.; Chen, C.; et al. Discovery of Potent and Selective Inhibitors of Cdc2-Like Kinase 1 (CLK1) as a New Class of Autophagy Inducers. J. Med. Chem. 2017, 60, 6337–6352. [Google Scholar] [CrossRef]
- Fant, X.; Durieu, E.; Chicanne, G.; Payrastre, B.; Sbrissa, D.; Shisheva, A.; Limanton, E.; Carreaux, F.; Bazureau, J.-P.; Meijer, L. Cdc-Like/Dual-Specificity Tyrosine Phosphorylation–Regulated Kinases Inhibitor Leucettine L41 Induces MTOR-Dependent Autophagy: Implication for Alzheimer’s Disease. Mol. Pharmacol. 2014, 85, 441–450. [Google Scholar] [CrossRef]
- Tabata, M.; Rodgers, J.T.; Hall, J.A.; Lee, Y.; Jedrychowski, M.P.; Gygi, S.P.; Puigserver, P. Cdc2-Like Kinase 2 Suppresses Hepatic Fatty Acid Oxidation and Ketogenesis Through Disruption of the PGC-1α and MED1 Complex. Diabetes 2014, 63, 1519–1532. [Google Scholar] [CrossRef]
- Hatting, M.; Rines, A.K.; Luo, C.; Tabata, M.; Sharabi, K.; Hall, J.A.; Verdeguer, F.; Trautwein, C.; Puigserver, P. Adipose Tissue CLK2 Promotes Energy Expenditure during High-Fat Diet Intermittent Fasting. Cell Metab. 2017, 25, 428–437. [Google Scholar] [CrossRef]
- Nam, S.Y.; Seo, H.H.; Park, H.S.; An, S.; Kim, J.-Y.; Yang, K.H.; Kim, C.S.; Jeong, M.; Jin, Y.-W. Phosphorylation of CLK2 at Serine 34 and Threonine 127 by AKT Controls Cell Survival after Ionizing Radiation. J. Biol. Chem. 2010, 285, 31157–31163. [Google Scholar] [CrossRef]
- Moeslein, F.M.; Myers, M.P.; Landreth, G.E. The CLK Family Kinases, CLK1 and CLK2, Phosphorylate and Activate the Tyrosine Phosphatase, PTP-1B. J. Biol. Chem. 1999, 274, 26697–26704. [Google Scholar] [CrossRef]
- Kulkarni, P.; Jolly, M.K.; Jia, D.; Mooney, S.M.; Bhargava, A.; Kagohara, L.T.; Chen, Y.; Hao, P.; He, Y.; Veltri, R.W.; et al. Phosphorylation-Induced Conformational Dynamics in an Intrinsically Disordered Protein and Potential Role in Phenotypic Heterogeneity. Proc. Natl. Acad. Sci. USA 2017, 114, E2644–E2653. [Google Scholar] [CrossRef] [PubMed]
- Menegay, H.; Moeslein, F.; Landreth, G. The Dual Specificity Protein Kinase CLK3 Is Abundantly Expressed in Mature Mouse Spermatozoa. Exp. Cell Res. 1999, 253, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, K.; Adachi, S.; Natsume, T.; Iwakiri, J.; Terai, G.; Asai, K.; Hirose, T. Lnc RNA -dependent Nuclear Stress Bodies Promote Intron Retention through SR Protein Phosphorylation. EMBO J. 2020, 39. [Google Scholar] [CrossRef] [PubMed]
- Gui, J.-F.; Lane, W.S.; Fu, X.-D. A Serine Kinase Regulates Intracellular Localization of Splicing Factors in the Cell Cycle. Nature 1994, 369, 678–682. [Google Scholar] [CrossRef]
- Dostie, J.; Lejbkowicz, F.; Sonenberg, N. Nuclear Eukaryotic Initiation Factor 4e (Eif4e) Colocalizes with Splicing Factors in Speckles. J. Cell Biol. 2000, 148, 239–246. [Google Scholar] [CrossRef]
- Sacco-Bubulya, P.; Spector, D.L. Disassembly of Interchromatin Granule Clusters Alters the Coordination of Transcription and Pre-MRNA Splicing. J. Cell Biol. 2002, 156, 425–436. [Google Scholar] [CrossRef]
- Lai, M.-C.; Lin, R.-I.; Tarn, W.-Y. Differential Effects of Hyperphosphorylation on Splicing Factor SRp55. Biochem. J. 2003, 371, 937–945. [Google Scholar] [CrossRef]
- Duncan, P.I.; Stojdl, D.F.; Marius, R.M.; Bell, J.C. In Vivo Regulation of Alternative Pre-MRNA Splicing by the Clk1 Protein Kinase. Mol. Cell. Biol. 1997, 17, 5996–6001. [Google Scholar] [CrossRef]
- Rieder, D.; Ploner, C.; Krogsdam, A.M.; Stocker, G.; Fischer, M.; Scheideler, M.; Dani, C.; Amri, E.-Z.; Müller, W.G.; McNally, J.G.; et al. Co-Expressed Genes Prepositioned in Spatial Neighborhoods Stochastically Associate with SC35 Speckles and RNA Polymerase II Factories. Cell. Mol. Life Sci. 2014, 71, 1741–1759. [Google Scholar] [CrossRef]
- García-Sacristán, A.; Fernández-Nestosa, M.J.; Hernández, P.; Schvartzman, J.B.; Krimer, D.B. Protein Kinase Clk/STY Is Differentially Regulated during Erythroleukemia Cell Differentiation: A Bias toward the Skipped Splice Variant Characterizes Postcommitment Stages. Cell Res. 2005, 15, 495–503. [Google Scholar] [CrossRef][Green Version]
- Alfonso, J.; Frick, L.R.; Silberman, D.M.; Palumbo, M.L.; Genaro, A.M.; Frasch, A.C. Regulation of Hippocampal Gene Expression Is Conserved in Two Species Subjected to Different Stressors and Antidepressant Treatments. Biol. Psychiatry 2006, 59, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Virgirinia, R.P.; Jahan, N.; Okada, M.; Takebayashi-Suzuki, K.; Yoshida, H.; Nakamura, M.; Akao, H.; Yoshimoto, Y.; Fatchiyah, F.; Ueno, N.; et al. Cdc2-like Kinase 2 (Clk2) Promotes Early Neural Development in Xenopus Embryos. Dev. Growth Differ. 2019, 61, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Kim, J.H.; Carver, K.; Su, Y.; Weremowicz, S.; Mulvey, L.; Yamamoto, S.; Brennan, C.; Mei, S.; Long, H.; et al. CLK2 Is an Oncogenic Kinase and Splicing Regulator in Breast Cancer. Cancer Res. 2015, 75, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Cui, X.; Hu, Q.; Chen, X.; Zhou, P. CLK3 Is A Direct Target of MiR-144 and Contributes To Aggressive Progression in Hepatocellular Carcinoma. OncoTargets Ther. 2019, 12, 9201–9213. [Google Scholar] [CrossRef] [PubMed]
- Quaresma, P.G.F.; Weissmann, L.; Zanotto, T.M.; Santos, A.C.; de Matos, A.H.B.; Furigo, I.C.; Simabuco, F.M.; Donato, J., Jr.; Bittencourt, J.C.; Lopes-Cendes, I.; et al. Cdc2-like Kinase 2 in the Hypothalamus Is Necessary to Maintain Energy Homeostasis. Int. J. Obes. 2017, 41, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.T.; Haas, W.; Gygi, S.P.; Puigserver, P. Cdc2-like Kinase 2 Is an Insulin-Regulated Suppressor of Hepatic Gluconeogenesis. Cell Metab. 2010, 11, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Srebrow, A. The Connection between Splicing and Cancer. J. Cell Sci. 2006, 119, 2635–2641. [Google Scholar] [CrossRef]
- Escobar-Hoyos, L.; Knorr, K.; Abdel-Wahab, O. Aberrant RNA Splicing in Cancer. Annu. Rev. Cancer Biol. 2019, 3, 167–185. [Google Scholar] [CrossRef]
- El Marabti, E.; Younis, I. The Cancer Spliceome: Reprograming of Alternative Splicing in Cancer. Front. Mol. Biosci. 2018, 5, 80. [Google Scholar] [CrossRef]
- Graubert, T.A.; Shen, D.; Ding, L.; Okeyo-Owuor, T.; Lunn, C.L.; Shao, J.; Krysiak, K.; Harris, C.C.; Koboldt, D.C.; Larson, D.E.; et al. Recurrent Mutations in the U2AF1 Splicing Factor in Myelodysplastic Syndromes. Nat. Genet. 2012, 44, 53–57. [Google Scholar] [CrossRef]
- Bonnal, S.C.; López-Oreja, I.; Valcárcel, J. Roles and Mechanisms of Alternative Splicing in Cancer—Implications for Care. Nat. Rev. Clin. Oncol. 2020, 17, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Dvinge, H.; Kim, E.; Abdel-Wahab, O.; Bradley, R.K. RNA Splicing Factors as Oncoproteins and Tumour Suppressors. Nat. Rev. Cancer 2016, 16, 413–430. [Google Scholar] [CrossRef] [PubMed]
- Iwai, K.; Yaguchi, M.; Nishimura, K.; Yamamoto, Y.; Tamura, T.; Nakata, D.; Dairiki, R.; Kawakita, Y.; Mizojiri, R.; Ito, Y.; et al. Anti-tumor Efficacy of a Novel CLK Inhibitor via Targeting RNA Splicing and MYC-dependent Vulnerability. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Di Maira, G.; Gentilini, A.; Pastore, M.; Caligiuri, A.; Piombanti, B.; Raggi, C.; Rovida, E.; Lewinska, M.; Andersen, J.B.; Borgo, C.; et al. The Protein Kinase CK2 Contributes to the Malignant Phenotype of Cholangiocarcinoma Cells. Oncogenesis 2019, 8, 61. [Google Scholar] [CrossRef]
- Intervention Dynamic Trial Listing Page. Available online: https://www.cancer.gov/about-cancer/treatment/clinical-trials/intervention/silmitasertib-sodium (accessed on 28 August 2020).
- Park, S.Y.; Piao, Y.; Thomas, C.; Fuller, G.N.; de Groot, J.F. Cdc2-like Kinase 2 Is a Key Regulator of the Cell Cycle via FOXO3a/P27 in Glioblastoma. Oncotarget 2016, 7, 26793–26805. [Google Scholar] [CrossRef]
- Wong, R.; Balachandran, A.; Mao, A.Y.; Dobson, W.; Gray-Owen, S.; Cochrane, A. Differential Effect of CLK SR Kinases on HIV-1 Gene Expression: Potential Novel Targets for Therapy. Retrovirology 2011, 8, 47. [Google Scholar] [CrossRef]
- Lee, K.-Y.; Clark, A.W.; Rosales, J.L.; Chapman, K.; Fung, T.; Johnston, R.N. Elevated Neuronal Cdc2-like Kinase Activity in the Alzheimer Disease Brain. Neurosci. Res. 1999, 34, 21–29. [Google Scholar] [CrossRef]
- Irwin, D.J. Tauopathies as Clinicopathological Entities. Parkinsonism Relat. Disord. 2016, 22, S29–S33. [Google Scholar] [CrossRef]
- Ledesma, M.D.; Correas, I.; Avila, J.; Diaz-Nido, J. Implication of Brain Cdc2 and MAP2 Kinases in the Phosphorylation of Tau Protein in Alzheimer’s Disease. FEBS Lett. 1992, 308, 218–224. [Google Scholar] [CrossRef]
- Jain, P.; Karthikeyan, C.; Moorthy, N.S.; Waiker, D.; Jain, A.; Trivedi, P. Human CDC2-Like Kinase 1 (CLK1): A Novel Target for Alzheimer’s Disease. Curr. Drug Targets 2014, 15, 539–550. [Google Scholar] [CrossRef]
- Sako, Y.; Ninomiya, K.; Okuno, Y.; Toyomoto, M.; Nishida, A.; Koike, Y.; Ohe, K.; Kii, I.; Yoshida, S.; Hashimoto, N.; et al. Development of an Orally Available Inhibitor of CLK1 for Skipping a Mutated Dystrophin Exon in Duchenne Muscular Dystrophy. Sci. Rep. 2017, 7, 46126. [Google Scholar] [CrossRef] [PubMed]
- Artarini, A.; Meyer, M.; Shin, Y.J.; Huber, K.; Hilz, N.; Bracher, F.; Eros, D.; Orfi, L.; Keri, G.; Goedert, S.; et al. Regulation of Influenza A Virus MRNA Splicing by CLK1. Antivir. Res. 2019, 168, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Bidinosti, M.; Botta, P.; Krüttner, S.; Proenca, C.C.; Stoehr, N.; Bernhard, M.; Fruh, I.; Mueller, M.; Bonenfant, D.; Voshol, H.; et al. CLK2 Inhibition Ameliorates Autistic Features Associated with SHANK3 Deficiency. Science 2016, 351, 1199–1203. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.M.; Sanchez-Azqueta, A.; Janha, O.; Flannery, E.L.; Mahindra, A.; Mapesa, K.; Char, A.B.; Sriranganadane, D.; Brancucci, N.M.B.; Antonova-Koch, Y.; et al. Validation of the Protein Kinase Pf CLK3 as a Multistage Cross-Species Malarial Drug Target. Science 2019, 365, 1682. [Google Scholar] [CrossRef] [PubMed]
- Mahindra, A.; Janha, O.; Mapesa, K.; Sanchez-Azqueta, A.; Alam, M.M.; Amambua-Ngwa, A.; Nwakanma, D.C.; Tobin, A.B.; Jamieson, A.G. Development of Potent Pf CLK3 Inhibitors Based on TCMDC-135051 as a New Class of Antimalarials. J. Med. Chem. 2020, 63, 9300–9315. [Google Scholar] [CrossRef]
- Kubori, T.; Hyakutake, A.; Nagai, H. Legionella Translocates an E3 Ubiquitin Ligase That Has Multiple U-Boxes with Distinct Functions. Mol. Microbiol. 2008, 67, 1307–1319. [Google Scholar] [CrossRef] [PubMed]
- Kallen, J.; Bergsdorf, C.; Arnaud, B.; Bernhard, M.; Brichet, M.; Cobos-Correa, A.; Elhajouji, A.; Freuler, F.; Galimberti, I.; Guibourdenche, C.; et al. X-ray Structures and Feasibility Assessment of CLK2 Inhibitors for Phelan-McDermid Syndrome. ChemMedChem 2018, 13, 1997–2007. [Google Scholar] [CrossRef]
- Mott, B.T.; Tanega, C.; Shen, M.; Maloney, D.J.; Shinn, P.; Leister, W.; Marugan, J.J.; Inglese, J.; Austin, C.P.; Misteli, T.; et al. Evaluation of Substituted 6-Arylquinazolin-4-Amines as Potent and Selective Inhibitors of Cdc2-like Kinases (Clk). Bioorg. Med. Chem. Lett. 2009, 19, 6700–6705. [Google Scholar] [CrossRef]
- Funnell, T.; Tasaki, S.; Oloumi, A.; Araki, S.; Kong, E.; Yap, D.; Nakayama, Y.; Hughes, C.S.; Cheng, S.-W.G.; Tozaki, H.; et al. CLK-Dependent Exon Recognition and Conjoined Gene Formation Revealed with a Novel Small Molecule Inhibitor. Nat. Commun. 2017, 8, 7. [Google Scholar] [CrossRef]
- Mallinger, A.; Schiemann, K.; Rink, C.; Stieber, F.; Calderini, M.; Crumpler, S.; Stubbs, M.; Adeniji-Popoola, O.; Poeschke, O.; Busch, M.; et al. Discovery of Potent, Selective, and Orally Bioavailable Small-Molecule Modulators of the Mediator Complex-Associated Kinases CDK8 and CDK19. J. Med. Chem. 2016, 59, 1078–1101. [Google Scholar] [CrossRef]
- Zhu, D.; Xu, S.; Deyanat-Yazdi, G.; Peng, S.X.; Barnes, L.A.; Narla, R.K.; Tran, T.; Mikolon, D.; Ning, Y.; Shi, T.; et al. Synthetic Lethal Strategy Identifies a Potent and Selective TTK and CLK1/2 Inhibitor for Treatment of Triple-Negative Breast Cancer with a Compromised G1–S Checkpoint. Mol. Cancer Ther. 2018, 17, 1727–1738. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, A.; Tanega, C.; Shen, M.; Mott, B.; Bougie, J.; Nguyen, D.-T.; Misteli, T.; Auld, D.; Maloney, D.; Thomas, C. An Inhibitor of the Cdc2-like Kinase 4 (Clk4). In Probe Reports from the NIH Molecular Libraries Program [Internet]; National Center for Biotechnology Information: Bethesda, MD, USA, 2010. [Google Scholar]
- Kim, H.; Choi, K.; Kang, H.; Lee, S.-Y.; Chi, S.-W.; Lee, M.-S.; Song, J.; Im, D.; Choi, Y.; Cho, S. Identification of a Novel Function of CX-4945 as a Splicing Regulator. PLoS ONE 2014, 9, e94978. [Google Scholar] [CrossRef] [PubMed]
- Battistutta, R.; Cozza, G.; Pierre, F.; Papinutto, E.; Lolli, G.; Sarno, S.; O’Brien, S.E.; Siddiqui-Jain, A.; Haddach, M.; Anderes, K.; et al. Unprecedented Selectivity and Structural Determinants of a New Class of Protein Kinase CK2 Inhibitors in Clinical Trials for the Treatment of Cancer. Biochemistry 2011, 50, 8478–8488. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, O.; Huber, K.; Eisenreich, A.; Filippakopoulos, P.; King, O.; Bullock, A.N.; Szklarczyk, D.; Jensen, L.J.; Fabbro, D.; Trappe, J.; et al. Specific CLK Inhibitors from a Novel Chemotype for Regulation of Alternative Splicing. Chem. Biol. 2011, 18, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Tahtouh, T.; Elkins, J.M.; Filippakopoulos, P.; Soundararajan, M.; Burgy, G.; Durieu, E.; Cochet, C.; Schmid, R.S.; Lo, D.C.; Delhommel, F.; et al. Selectivity, Cocrystal Structures, and Neuroprotective Properties of Leucettines, a Family of Protein Kinase Inhibitors Derived from the Marine Sponge Alkaloid Leucettamine B. J. Med. Chem. 2012, 55, 9312–9330. [Google Scholar] [CrossRef]
- Coombs, T.C.; Tanega, C.; Shen, M.; Wang, J.L.; Auld, D.S.; Gerritz, S.W.; Schoenen, F.J.; Thomas, C.J.; Aubé, J. Small-Molecule Pyrimidine Inhibitors of the Cdc2-like (Clk) and Dual Specificity Tyrosine Phosphorylation-Regulated (Dyrk) Kinases: Development of Chemical Probe ML315. Bioorg. Med. Chem. Lett. 2013, 23, 3654–3661. [Google Scholar] [CrossRef]
- Schmitt, C.; Kail, D.; Mariano, M.; Empting, M.; Weber, N.; Paul, T.; Hartmann, R.W.; Engel, M. Design and Synthesis of a Library of Lead-Like 2,4-Bisheterocyclic Substituted Thiophenes as Selective Dyrk/Clk Inhibitors. PLoS ONE 2014, 9, e87851. [Google Scholar] [CrossRef]
- Prak, K.; Kriston-Vizi, J.; Chan, A.W.E.; Luft, C.; Costa, J.R.; Pengo, N.; Ketteler, R. Benzobisthiazoles Represent a Novel Scaffold for Kinase Inhibitors of CLK Family Members. Biochemistry 2016, 55, 608–617. [Google Scholar] [CrossRef]
- Araki, S.; Dairiki, R.; Nakayama, Y.; Murai, A.; Miyashita, R.; Iwatani, M.; Nomura, T.; Nakanishi, O. Inhibitors of CLK Protein Kinases Suppress Cell Growth and Induce Apoptosis by Modulating Pre-MRNA Splicing. PLoS ONE 2015, 10, e0116929. [Google Scholar] [CrossRef]
- Shi, Y.; Park, J.; Lagisetti, C.; Zhou, W.; Sambucetti, L.C.; Webb, T.R. A Triple Exon-Skipping Luciferase Reporter Assay Identifies a New CLK Inhibitor Pharmacophore. Bioorg. Med. Chem. Lett. 2017, 27, 406–412. [Google Scholar] [CrossRef]
- T3-CLK. A Chemical Probe for CLK Kinases. SGC Chem. Probes. Available online: https://www.thesgc.org/chemical-probes/T3-CLK (accessed on 12 October 2020).
- Walter, A.; Chaikuad, A.; Helmer, R.; Loaëc, N.; Preu, L.; Ott, I.; Knapp, S.; Meijer, L.; Kunick, C. Molecular Structures of Cdc2-like Kinases in Complex with a New Inhibitor Chemotype. PLoS ONE 2018, 13, e0196761. [Google Scholar] [CrossRef] [PubMed]
- Tazarki, H.; Zeinyeh, W.; Esvan, Y.J.; Knapp, S.; Chatterjee, D.; Schröder, M.; Joerger, A.C.; Khiari, J.; Josselin, B.; Baratte, B.; et al. New Pyrido[3,4-g]Quinazoline Derivatives as CLK1 and DYRK1A Inhibitors: Synthesis, Biological Evaluation and Binding Mode Analysis. Eur. J. Med. Chem. 2019, 166, 304–317. [Google Scholar] [CrossRef] [PubMed]
- SGC-CLK-1. A Chemical Probe for CLK1, CLK2, and CLK4. SGC Chem. Probes. Available online: https://www.thesgc.org/chemical-probes/SGC-CLK-1 (accessed on 12 October 2020).
- MU1210. A Chemical Probe for CLK Kinases. SGC Chem. Probes. Available online: https://www.thesgc.org/chemical-probes/MU1210 (accessed on 12 October 2020).
- Rosenthal, A.S.; Tanega, C.; Shen, M.; Mott, B.T.; Bougie, J.M.; Nguyen, D.-T.; Misteli, T.; Auld, D.S.; Maloney, D.J.; Thomas, C.J. Potent and Selective Small Molecule Inhibitors of Specific Isoforms of Cdc2-like Kinases (Clk) and Dual Specificity Tyrosine-Phosphorylation-Regulated Kinases (Dyrk). Bioorg. Med. Chem. Lett. 2011, 21, 3152–3158. [Google Scholar] [CrossRef] [PubMed]
- Frye, S.V. The Art of the Chemical Probe. Nat. Chem. Biol. 2010, 6, 159–161. [Google Scholar] [CrossRef]
- Workman, P.; Collins, I. Probing the Probes: Fitness Factors for Small Molecule Tools. Chem. Biol. 2010, 17, 561–577. [Google Scholar] [CrossRef]
- Knapp, S.; Arruda, P.; Blagg, J.; Burley, S.; Drewry, D.H.; Edwards, A.; Fabbro, D.; Gillespie, P.; Gray, N.S.; Kuster, B.; et al. A Public-Private Partnership to Unlock the Untargeted Kinome. Nat. Chem. Biol. 2013, 9, 3–6. [Google Scholar] [CrossRef]































| CLK Form (Uniprot Ref.) | Amino Acid Residues | kDa | Amino Acids in the ATP Phosphates’ Binding Region |
|---|---|---|---|
| CLK1 (P49759) | 484 | 57 | 167–175 (LGEGAFGKV) |
| CLK2 (P49760) | 499 | 60 | 169–177 (LGEGTFGRV) |
| CLK3 (P49761) | 638 | 73 | 310–318 (LGEGTFGKV) |
| CLK4 (Q9HAZ1) | 481 | 57 | 165–173 (LGEGAFGKV) |
| Kinases | Kd (nM) |
|---|---|
| CLK4 | 70.0 |
| DYRK1A | 7.8 |
| CLK1 | 75.0 |
| DYRK2 | 450.0 |
| HIPK1 | 320.0 |
| CLK3 | 1100.0 |
| IRAK1 | 930.0 |
| HIPK3 | 230.0 |
| DYRK1B | 140.0 |
| CLK2 | 360.0 |
| TAOK1 | 1700.0 |
| TYK2 | n.t |
| GSK3A | 550.0 |
| Kinase |  MU1210 Activity [nM] |  MU140 Activity [nM] (Negative Control) | ||
|---|---|---|---|---|
| Biochemical IC50 | Cellular Ki | Biochemical IC50 | Cellular Ki | |
| CLK1 | 8 | 84 | >3000 | >10,000 |
| CLK2 | 20 | 91 | >10,000 | >10,000 |
| CLK3 | >3000 | - | >3000 | - |
| CLK4 | 12 | 23 | >10,000 | >10,000 |
| DYRK1A | 213 | 6580 | >3000 | >10,000 |
| DYRK1B | 956 | >10,000 | >3000 | >10,000 |
| DYRK2 | 1309 | 1700 | >10,000 | >10,000 |
| HIPK1 | 187 | - | >10,000 | - |
| HIPK2 | 29 | >10,000 | >10,000 | - |
| HIPK3 | 159 | - | >3000 | - |
| HIPK4 | - | 5410 | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martín Moyano, P.; Němec, V.; Paruch, K. Cdc-Like Kinases (CLKs): Biology, Chemical Probes, and Therapeutic Potential. Int. J. Mol. Sci. 2020, 21, 7549. https://doi.org/10.3390/ijms21207549
Martín Moyano P, Němec V, Paruch K. Cdc-Like Kinases (CLKs): Biology, Chemical Probes, and Therapeutic Potential. International Journal of Molecular Sciences. 2020; 21(20):7549. https://doi.org/10.3390/ijms21207549
Chicago/Turabian StyleMartín Moyano, Paula, Václav Němec, and Kamil Paruch. 2020. "Cdc-Like Kinases (CLKs): Biology, Chemical Probes, and Therapeutic Potential" International Journal of Molecular Sciences 21, no. 20: 7549. https://doi.org/10.3390/ijms21207549
APA StyleMartín Moyano, P., Němec, V., & Paruch, K. (2020). Cdc-Like Kinases (CLKs): Biology, Chemical Probes, and Therapeutic Potential. International Journal of Molecular Sciences, 21(20), 7549. https://doi.org/10.3390/ijms21207549