Solid-Phase Synthesis and In-Silico Analysis of Iron-Binding Catecholato Chelators

 , , ,
, , ,
Abstract
1. Introduction
2. Results and Discussion
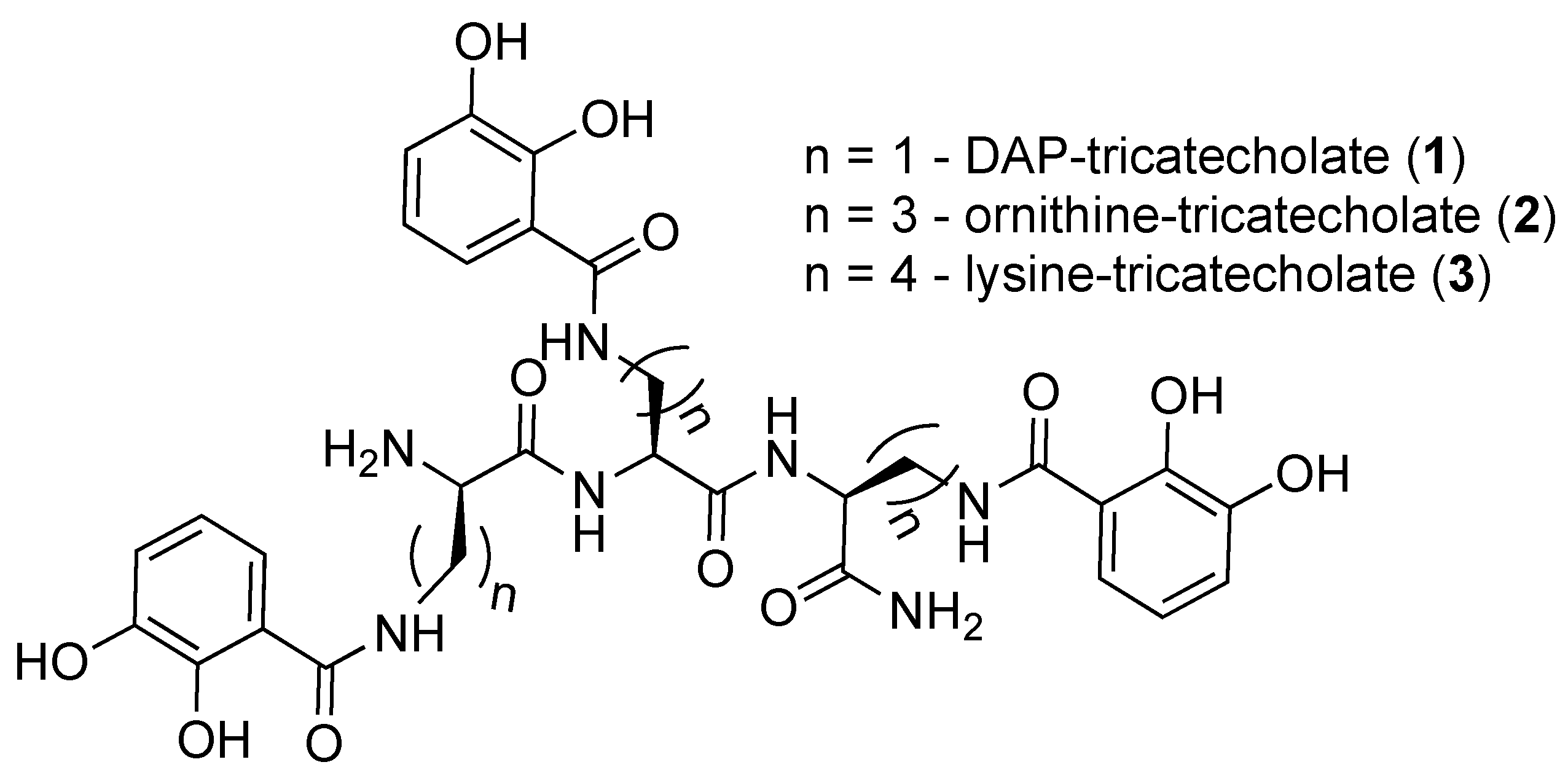
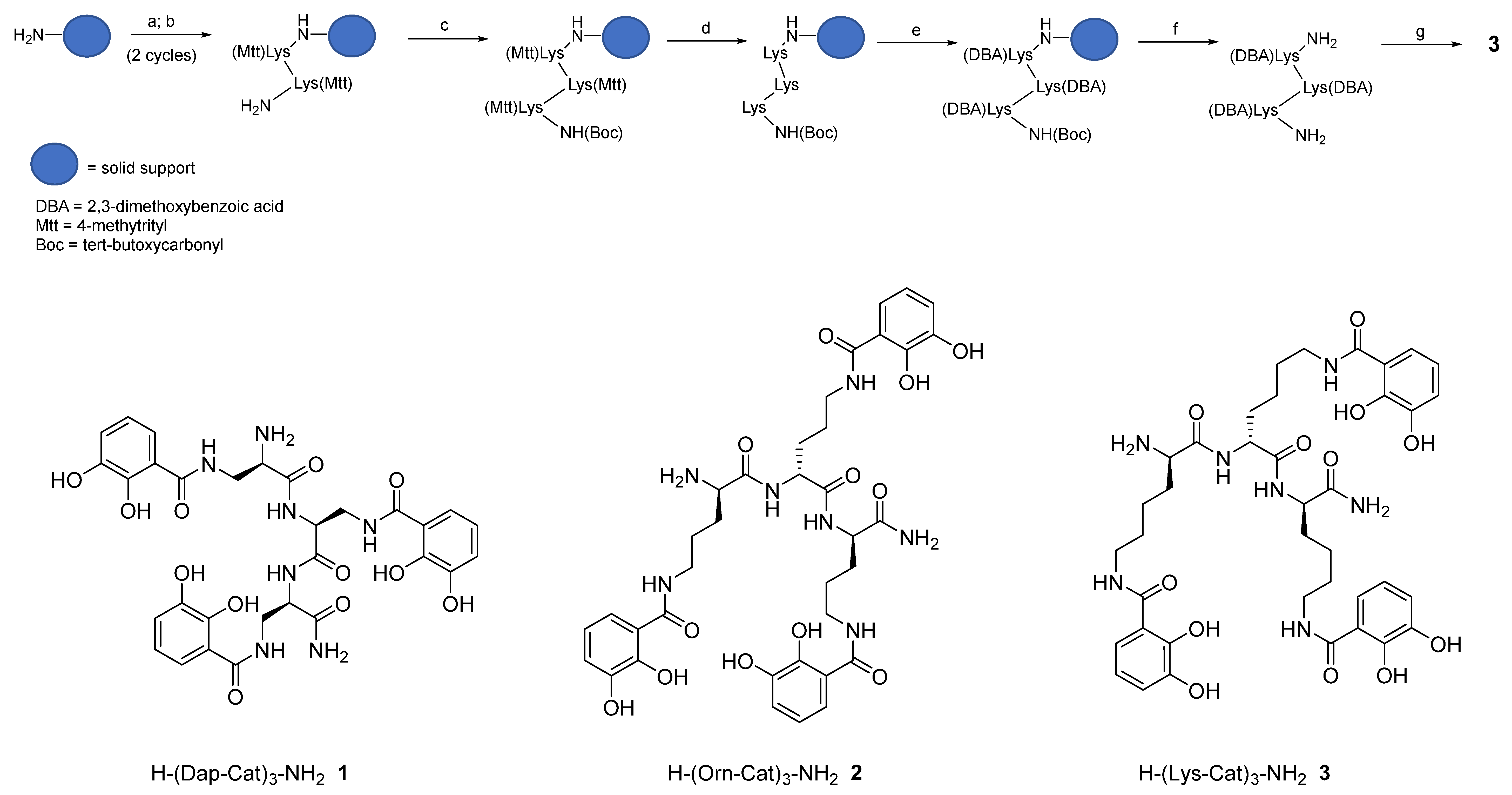
2.1. Synthesis of Tricatecholato Ligands Attached to a Peptide Backbone
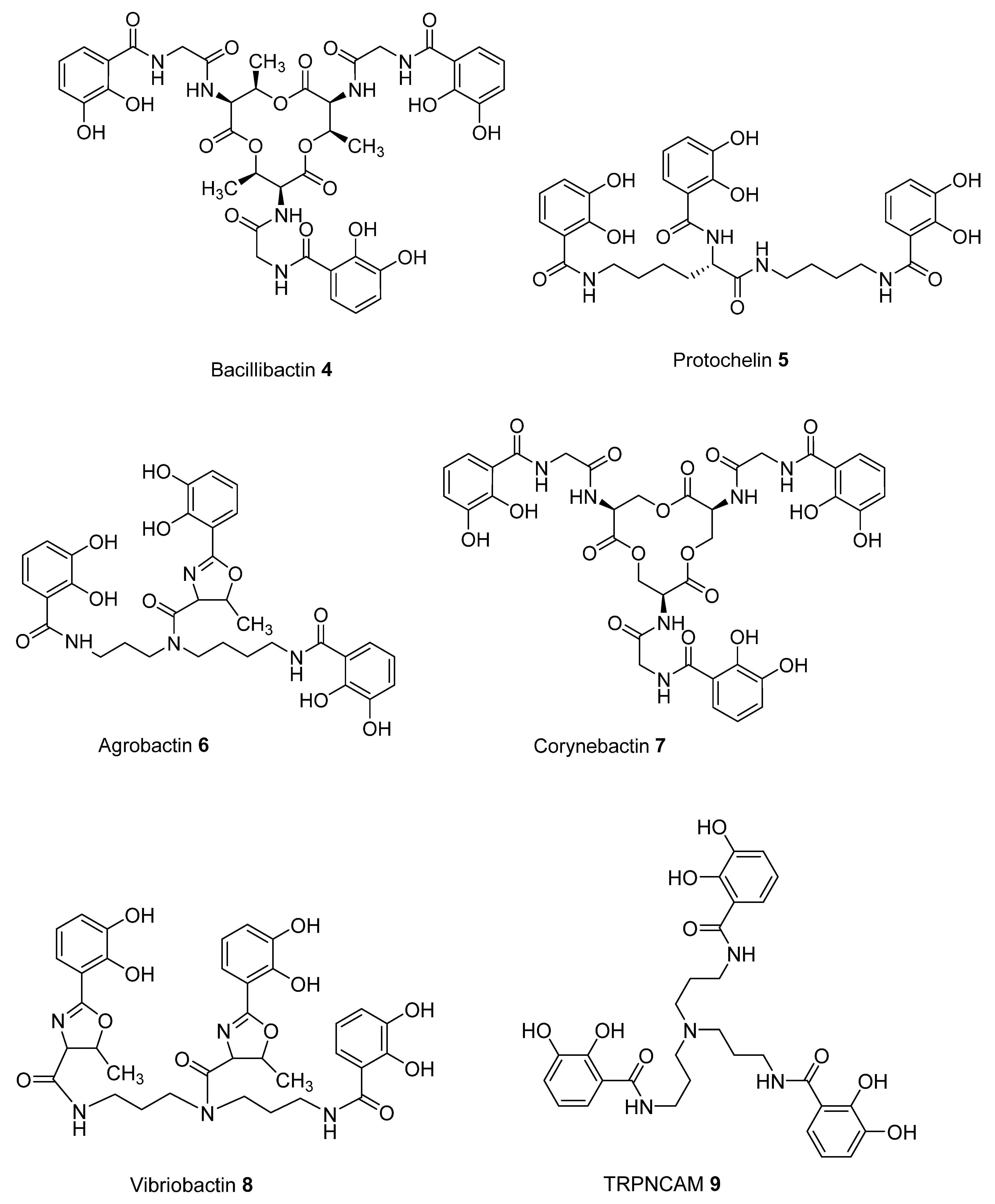
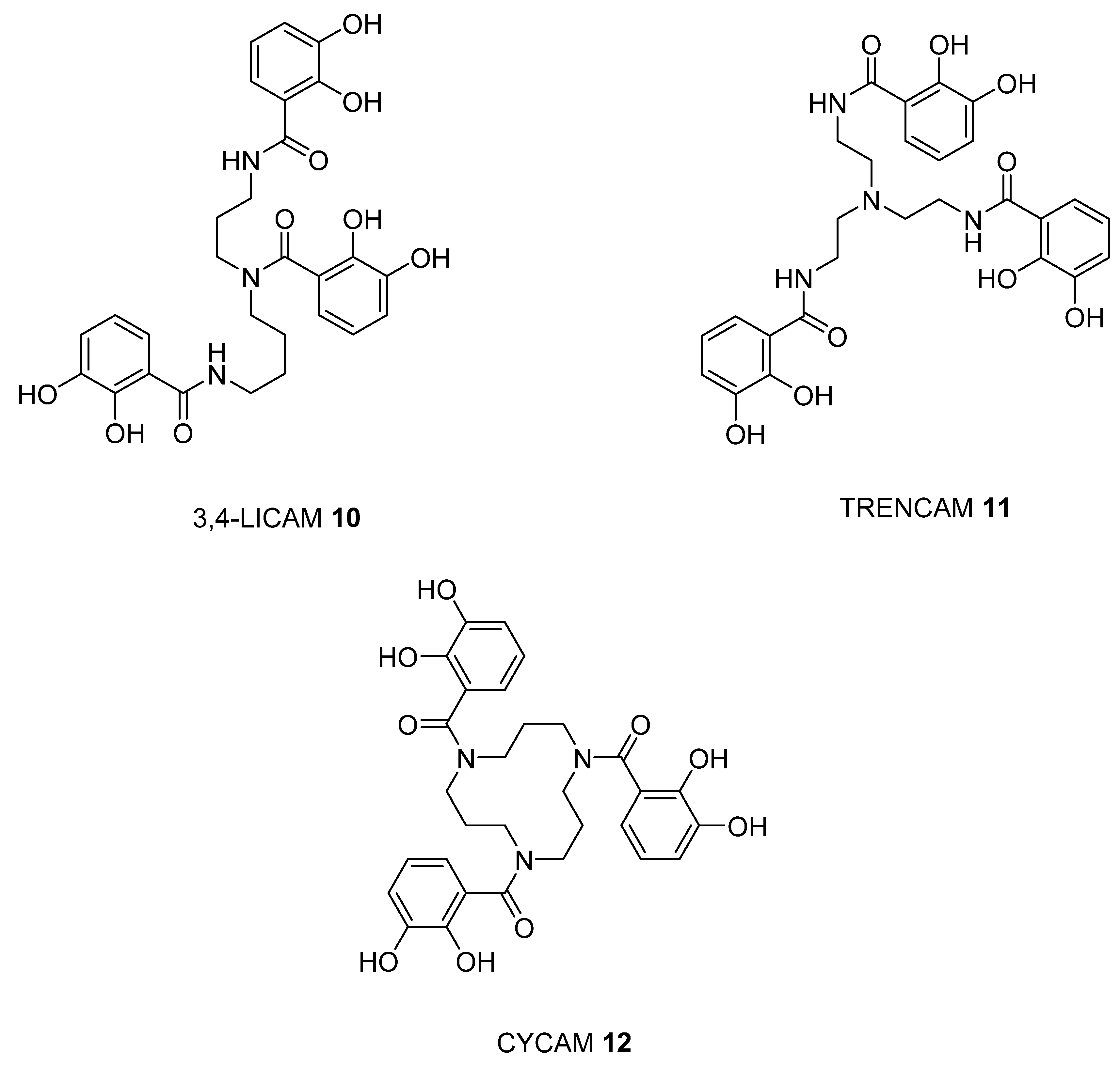
2.2. Analysis of iron(III) Complexes of Tricatecholato Structures
2.3. Analysis of iron(III) Complexes of Hypothetical Tricatecholato Structures
3. Materials and Methods
3.1. Structural Analysis of iron(III) Complexes
3.2. Materials
3.3. Solid Phase Synthesis
3.4. Deprotection Reactions
3.5. Peptide-Resin Cleavage Procedure
3.6. Method for Determination of pFe3+
3.7. Instrumentation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DFT | Density Function Theory |
| RMSD | roots mean square difference |
| OXYMA | ethyl hydroxyimino cyanoacetate |
| DIC | N,N’-diisopropylcarbodiimide |
| DBA | 2,3-dimethoxybenzoic acid |
| PyOxP | O-[(1-cyano-2-ethoxy-2-oxoethylidene)amino]-oxytri(pyrrolidin-1-yl)phosphonium hexafluorophosphate |
| DIPEA | N,N’-diisopropylethylamine |
| UFF | Universal Force Field |
References
- Crumbliss, A.L.; Harrington, J.M. Iron sequestration by small molecules: Thermodynamic and kinetic studies of natural siderophores and synthetic model compounds. Med. Chem. 2009, 61, 179–250. [Google Scholar] [CrossRef]
- Hider, R.C.; Kong, X. Chemistry and biology of siderophores. Nat. Prod. Rep. 2010, 27, 637–657. [Google Scholar] [CrossRef]
- Zhou, Y.J.; Zhang, M.X.; Hider, R.C.; Zhou, T. In vitro antimicrobial activity of hydroxypyridinone hexadentate-based dendrimeric chelators alone and in combination with norfloxacin. FEMS Microbiol. Lett. 2014, 355, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Gorden, A.E.V.; Xu, J.; Raymond, K.N.; Durbin, P. Rational Design of Sequestering Agents for Plutonium and Other Actinides. Chem. Rev. 2003, 103, 4207–4282. [Google Scholar] [CrossRef] [PubMed]
- Pierre, V.C.; Melchior, M.; Doble, D.M.J.; Raymond, K.N. Toward Optimized High-Relaxivity MRI Agents: Thermodynamic Selectivity of Hydroxypyridonate/Catecholate Ligands1. Inorg. Chem. 2004, 43, 8520–8525. [Google Scholar] [CrossRef] [PubMed]
- Grazina, R.; Gano, L.; Šebestík, J.; Santos, M.A. New tripodal hydroxypyridinone based chelating agents for Fe(III), Al(III) and Ga(III): Synthesis, physico-chemical properties and bioevaluation. J. Inorg. Biochem. 2009, 103, 262–273. [Google Scholar] [CrossRef]
- Berry, D.J.; Ma, Y.; Ballinger, J.R.; Tavaré, R.; Koers, A.; Sunassee, K.; Zhou, T.; Nawaz, S.; Mullen, G.E.D.; Hider, R.C.; et al. Efficient bifunctional gallium-68 chelators for positron emission tomography: Tris(hydroxypyridinone) ligands. Chem. Commun. 2011, 47, 7068–7070. [Google Scholar] [CrossRef]
- Imbert, D.; Thomas, F.; Baret, P.; Serratrice, G.; Gaude, D.; Pierre, J.-L.; Laulhère, J.-P. Synthesis and iron(III) complexing ability of CacCAM, a new analog of enterobactin possessing a free carboxylic anchor arm. Comparative studies with TRENCAM. New J. Chem. 2000, 24, 281–288. [Google Scholar] [CrossRef]
- Piyamongkol, S.; Zhou, T.; Liu, Z.D.; Khodr, H.H.; Hider, R.C. Design and characterisation of novel hexadentate 3-hydroxypyridin-4-one ligands. Tetrahedron Lett. 2005, 46, 1333–1336. [Google Scholar] [CrossRef]
- Harris, W.R.; Carrano, C.J.; Raymond, K.N. Spectrophotometric determination of the proton dependent stability constant of ferric enterobaction. J. Amer. Chem. Soc. 1979, 101, 2213–2214. [Google Scholar] [CrossRef]
- Hay, B.P.; Dixon, D.A.; Vargas, R.; Garza, J.; Raymond, K.N. Structural criteria for the rational design of selective ligands. 3. Quantitative structure-stability relationship for iron(III) complexation by tris-catecholamide siderophores. Inorg. Chem. 2001, 40, 3922–3935. [Google Scholar] [CrossRef] [PubMed]
- Bühl, M.; Kabrede, H. Geometries of Transition-Metal Complexes from Density-Functional Theory. J. Chem. Theory Comput. 2006, 2, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Cilibrizzi, A.; Abbate, V.; Chen, Y.-L.; Ma, Y.; Zhou, T.; Hider, R.C. Hydroxypyridinone Journey into Metal Chelation. Chem. Rev. 2018, 118, 7657–7701. [Google Scholar] [CrossRef] [PubMed]
- Heistand, R.H.; Roe, A.L.; Que, L. Dioxygenase Models. Crystal Structures of [N,N’,-(1,2-Phenylene)bis(salicylideniminato)](catecholato-O)iron(III) and μ-(1,4-Benzenediolato-O,O’)-bis[N,N’-ethylenebis(salicylideniminato)iron(III)]. Inorg. Chem. 1982, 21, 676–681. [Google Scholar] [CrossRef]
- KCL Report to Zede Pharm. 2017; Copies of the report available on application to R.C.Hider.
- Subirós-Funosas, R.; Prohens, R.; Barbas, R.; El-Faham, A.; Albericio, F. Oxyma: An Efficient Additive for Peptide Synthesis to Replace the Benzotriazole-Based HOBt and HOAt with a Lower Risk of Explosion. Chem. Eur. J. 2009, 15, 9394–9403. [Google Scholar] [CrossRef] [PubMed]
- Subirós-Funosas, R.; El-Faham, A.; Albericio, F. PyOxP and PyOxB: The Oxyma-based novel family of phosphonium salts. Org. Biomol. Chem. 2010, 8, 3665. [Google Scholar] [CrossRef]
- Ma, Y.; Hider, R.C.; Zhou, T. pFe3+ determination of multidentate ligands by a fluorescence assay. Analyst 2015, 140, 3603–3606. [Google Scholar] [CrossRef]
- Hanwell, M.D.; E Curtis, D.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Chemin. 2012, 4, 17. [Google Scholar] [CrossRef]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Parr, R.G. Density Functional Theory. Annu. Rev. Phys. Chem. 1983, 34, 631–656. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 2, 73–78. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Kaiser, E.; Colescott, R.L.; Bossinger, C.D.; Cook, P.I. Aqueous Microwave-Assisted Solid-Phase Synthesis Using Boc-Amino Acid Nanoparticles. Anal Biochem. 1970, 34, 595–598. [Google Scholar] [CrossRef]
- Li, D.; Elbert, D.L. The kinetics of the removal of the N-methyltrityl (Mtt) group during the synthesis of branched peptides. J. Pept. Res. 2002, 60, 300–303. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
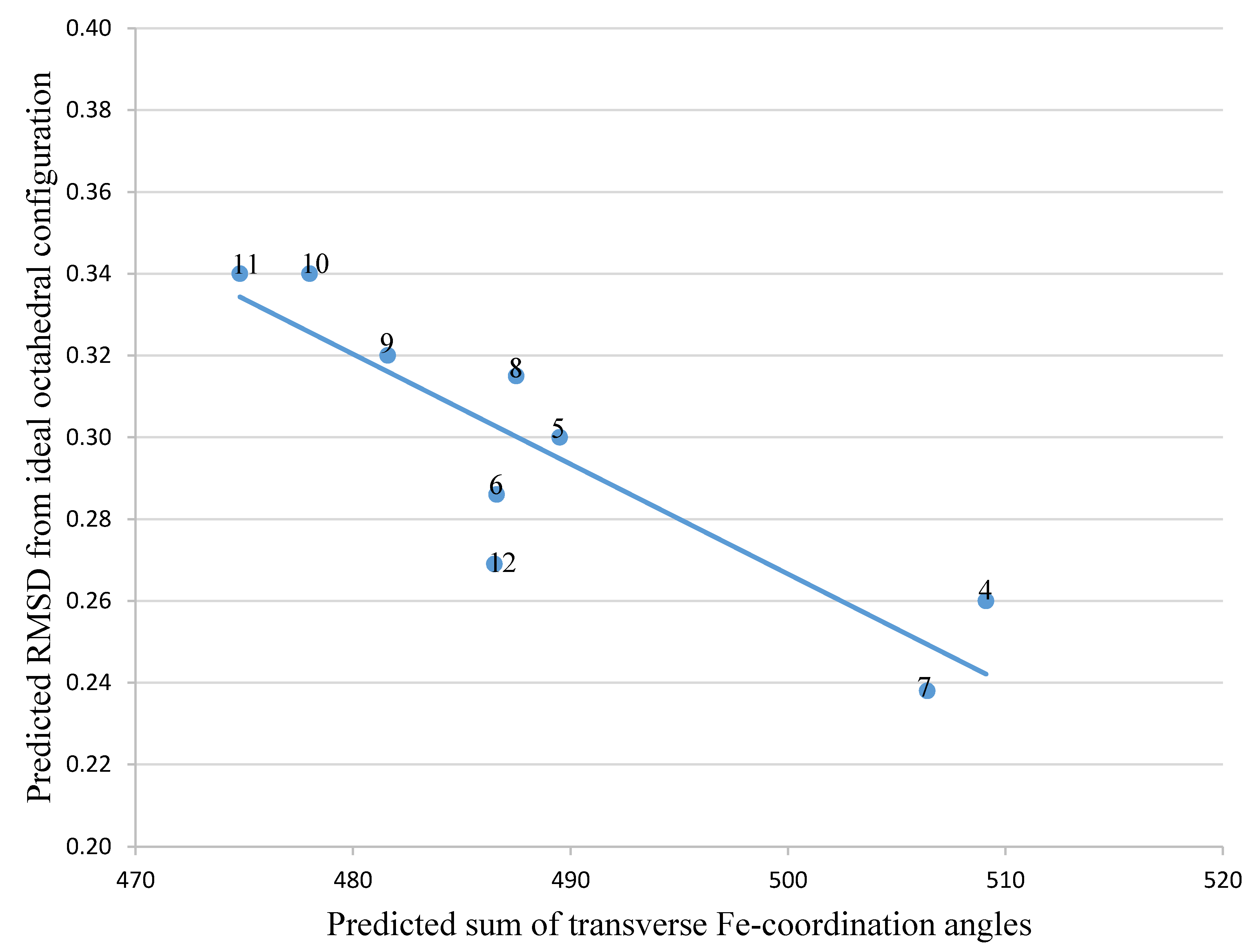
| Ligand in FeL Complex | Sum of Angles 2 | RMSD 1 |
|---|---|---|
| 4 | 509.1 | 0.260 |
| 5 | 489.5 | 0.300 |
| 6 | 486.6 | 0.286 |
| 7 | 506.4 | 0.238 |
| 8 | 487.5 | 0.315 |
| 9 | 481.6 | 0.320 |
| 10 | 478.0 | 0.340 |
| 11 | 474.8 | 0.340 |
| 12 | 486.5 | 0.269 |
| Ligand in FeL Complex | Sum of Angles 2 | RMSD 1 |
|---|---|---|
| 1 | 489.4 | 0.270 |
| 2 | 502.9 | 0.230 |
| 3 | 466.2 | 0.450 |
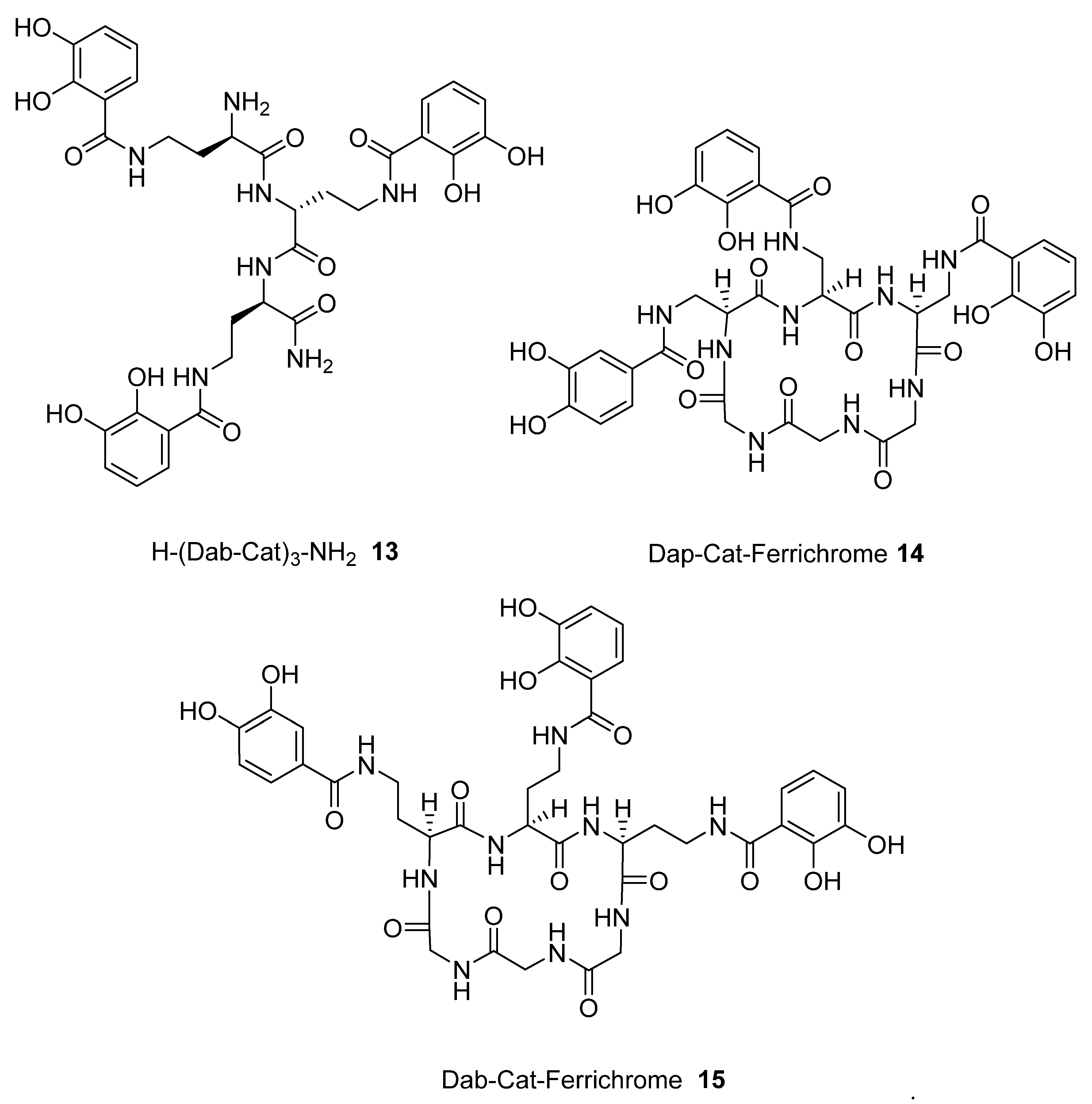
| 13 | 501.0 | 0.230 |
| 14 | 480.2 | 0.307 |
| 15 | 488.5 | 0.309 |
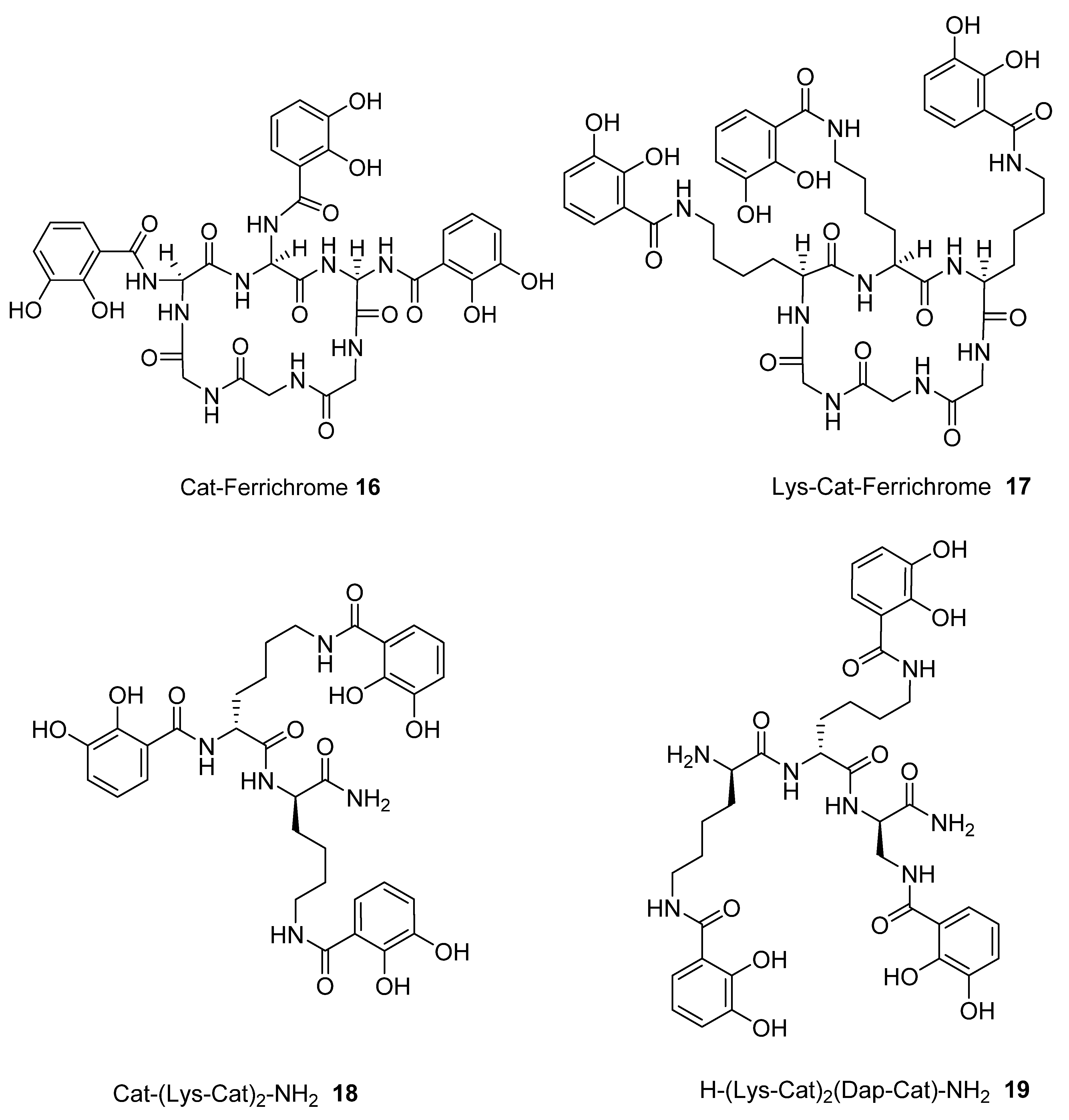
| 16 | 482.9 | 0.291 |
| 17 | 494.3 | 0.280 |
| 18 | 477.4 | 0.350 |
| 19 | 498.9 | 0.230 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gacesa, R.; Tripodi, A.A.P.; Cilibrizzi, A.; Leggio, A.; Hider, R.; Abbate, V. Solid-Phase Synthesis and In-Silico Analysis of Iron-Binding Catecholato Chelators. Int. J. Mol. Sci. 2020, 21, 7498. https://doi.org/10.3390/ijms21207498
Gacesa R, Tripodi AAP, Cilibrizzi A, Leggio A, Hider R, Abbate V. Solid-Phase Synthesis and In-Silico Analysis of Iron-Binding Catecholato Chelators. International Journal of Molecular Sciences. 2020; 21(20):7498. https://doi.org/10.3390/ijms21207498
Chicago/Turabian StyleGacesa, Ranko, Andrea A. P. Tripodi, Agostino Cilibrizzi, Antonella Leggio, Robert Hider, and Vincenzo Abbate. 2020. "Solid-Phase Synthesis and In-Silico Analysis of Iron-Binding Catecholato Chelators" International Journal of Molecular Sciences 21, no. 20: 7498. https://doi.org/10.3390/ijms21207498
APA StyleGacesa, R., Tripodi, A. A. P., Cilibrizzi, A., Leggio, A., Hider, R., & Abbate, V. (2020). Solid-Phase Synthesis and In-Silico Analysis of Iron-Binding Catecholato Chelators. International Journal of Molecular Sciences, 21(20), 7498. https://doi.org/10.3390/ijms21207498