Association of α/β-Hydrolase D16B with Bovine Conception Rate and Sperm Plasma Membrane Lipid Composition

 ,
,
Abstract
1. Introduction
2. Results
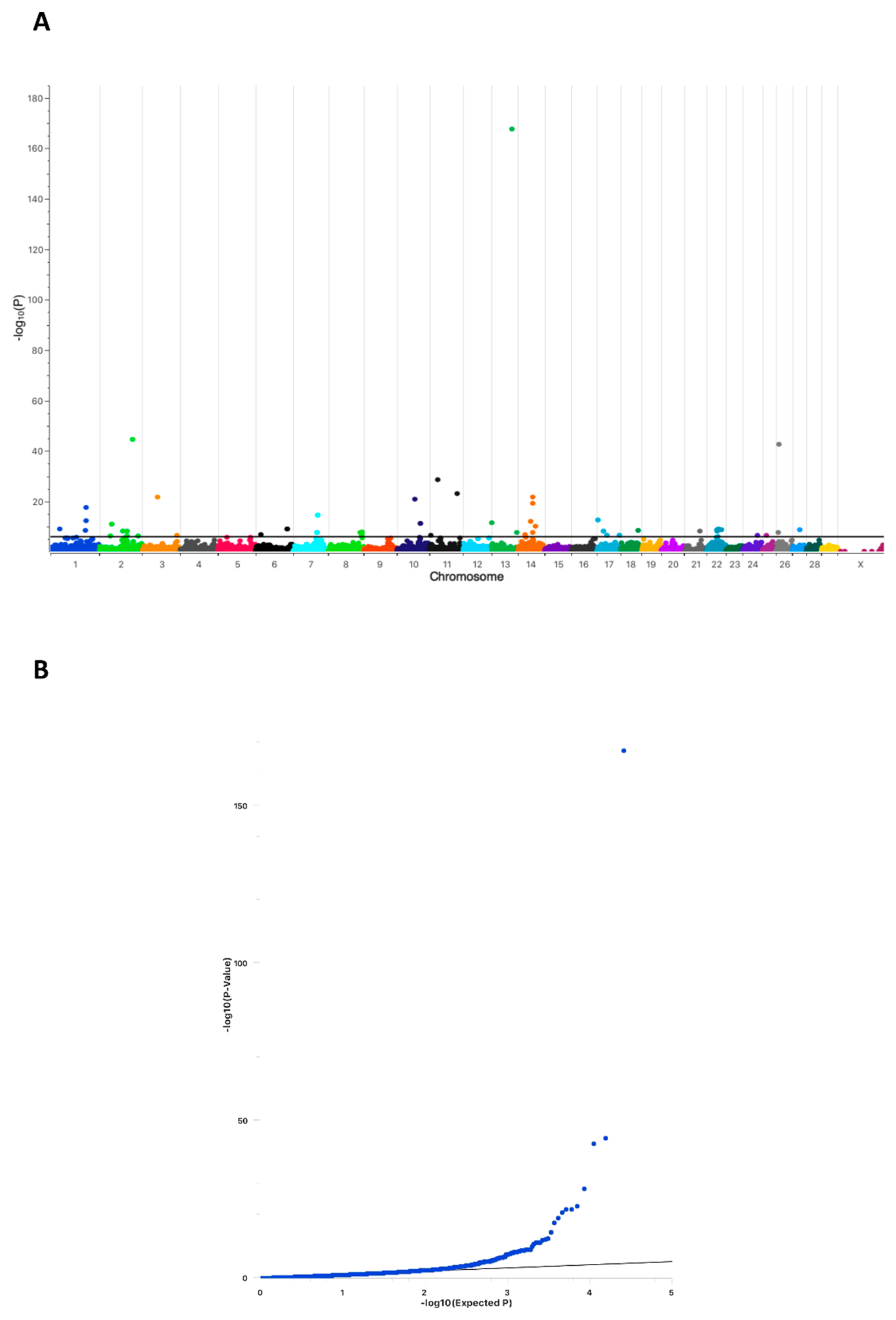
2.1. Conception Ability of Sires Is Highly Associated with a Chromosomal Region on Bovine Chromosome 13
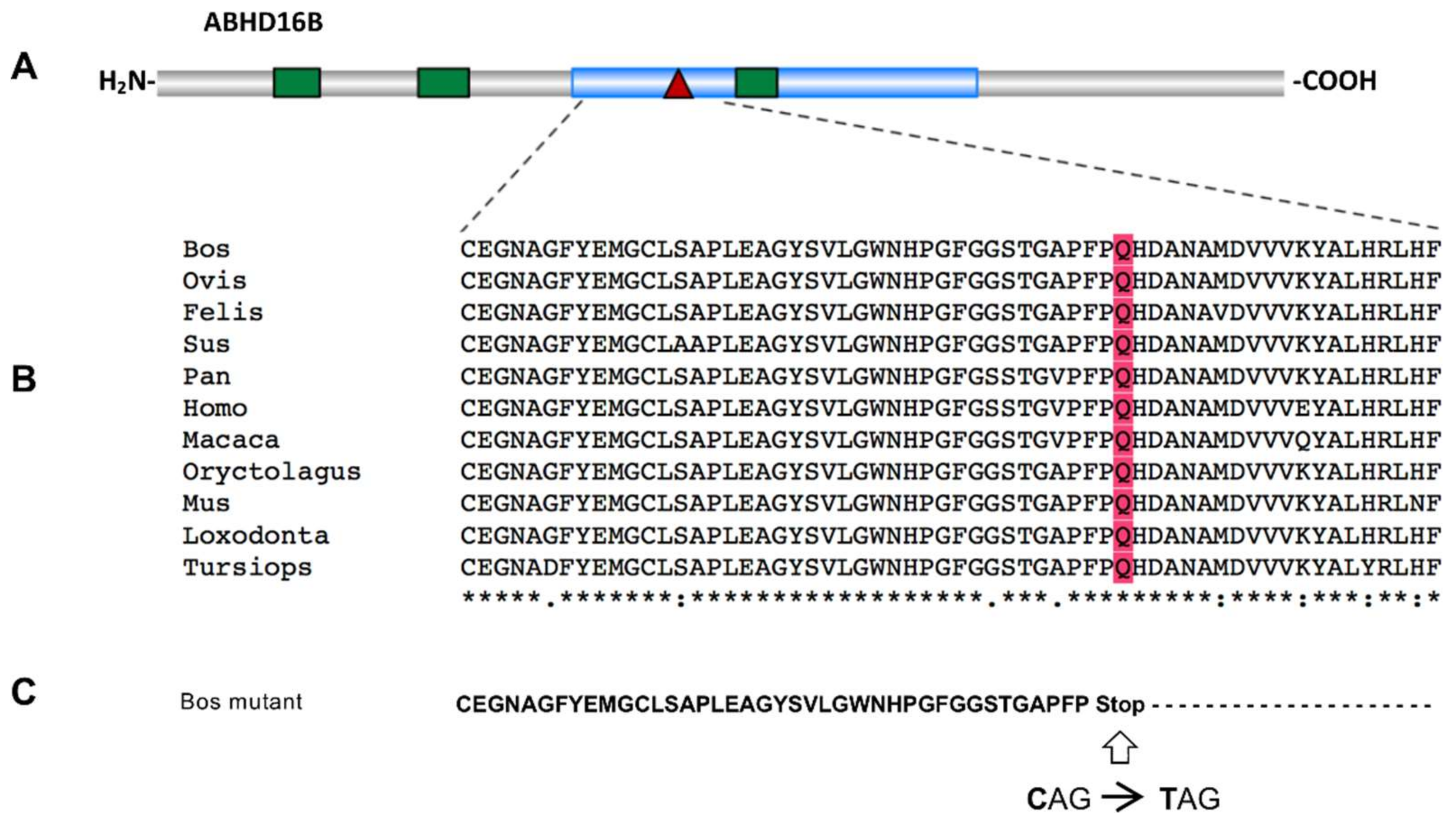
2.2. Whole-Genome Sequencing Reveals Two Potential Protein-Altering Variants Upstream the Associated Position on BTA13
2.3. Verification and Validation of the Nonsense Variant g.54429815G>A (ABHD16B) in the Holstein Population
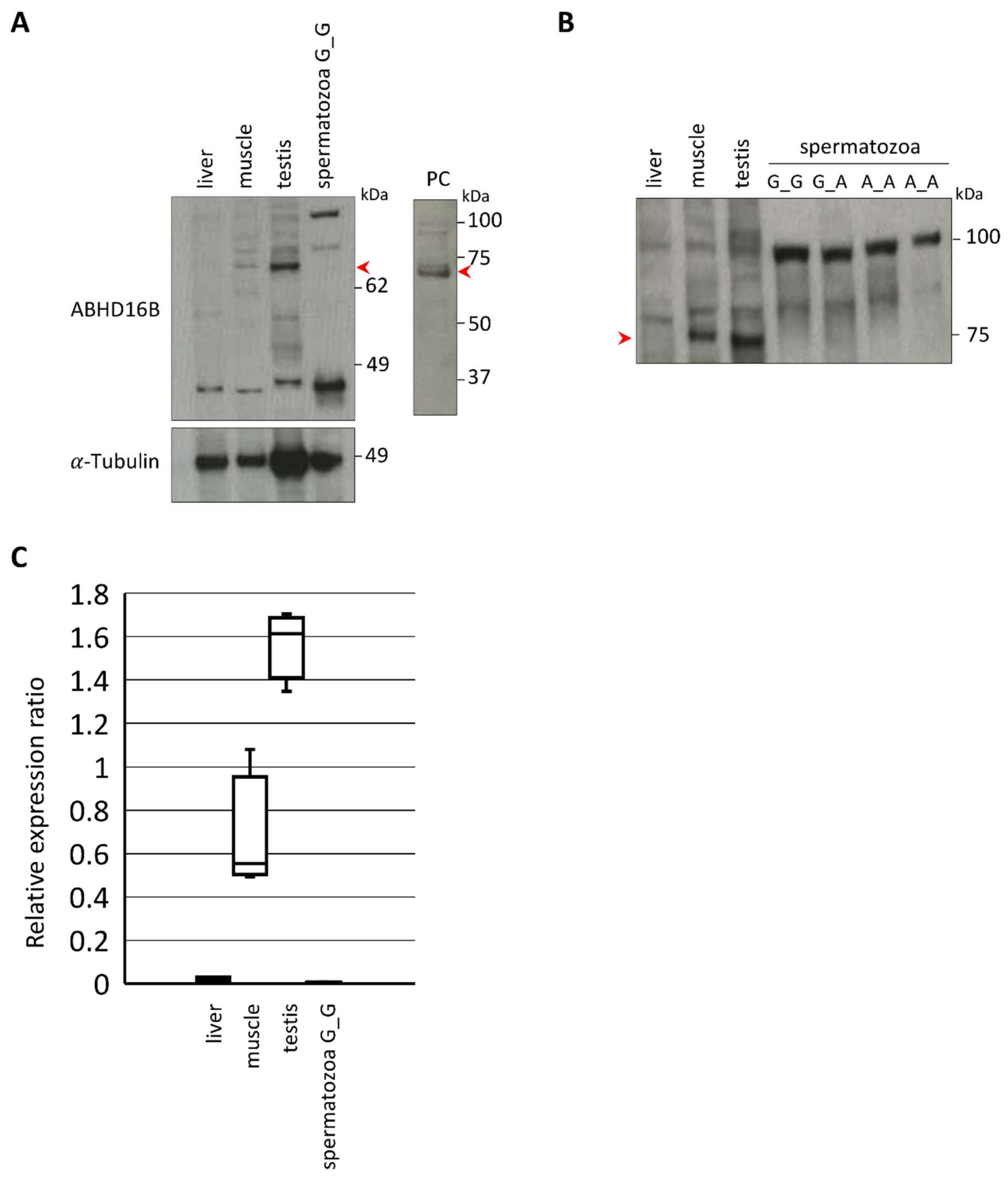
2.4. Expression and Tissue Distribution of ABHD16B
2.5. ABHD16B Is Expressed in Testis but not in Spermatozoa
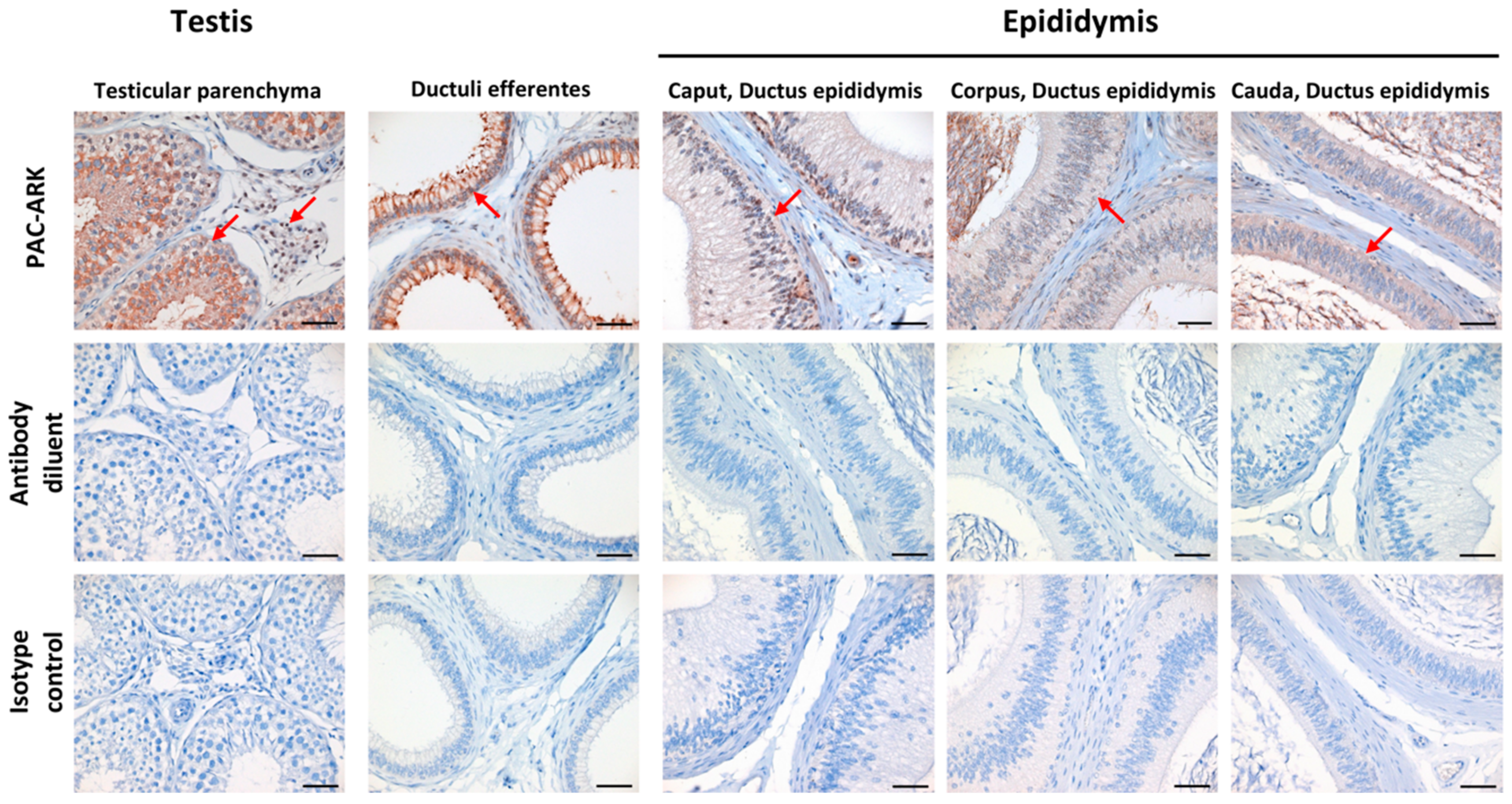
2.6. Immunohistochemical Analysis Revealed ABHD16B Expression in Testis and Epididymis
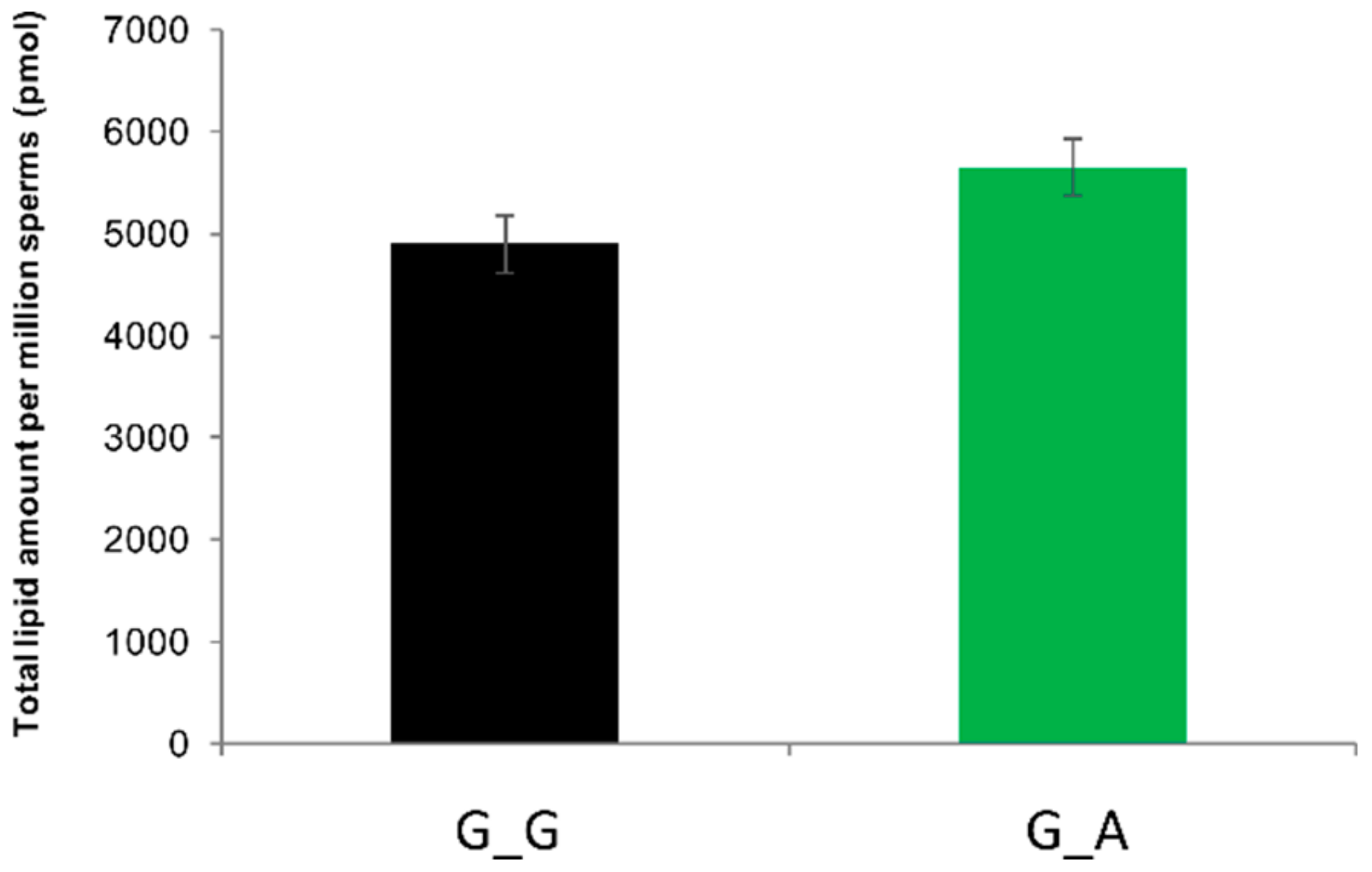
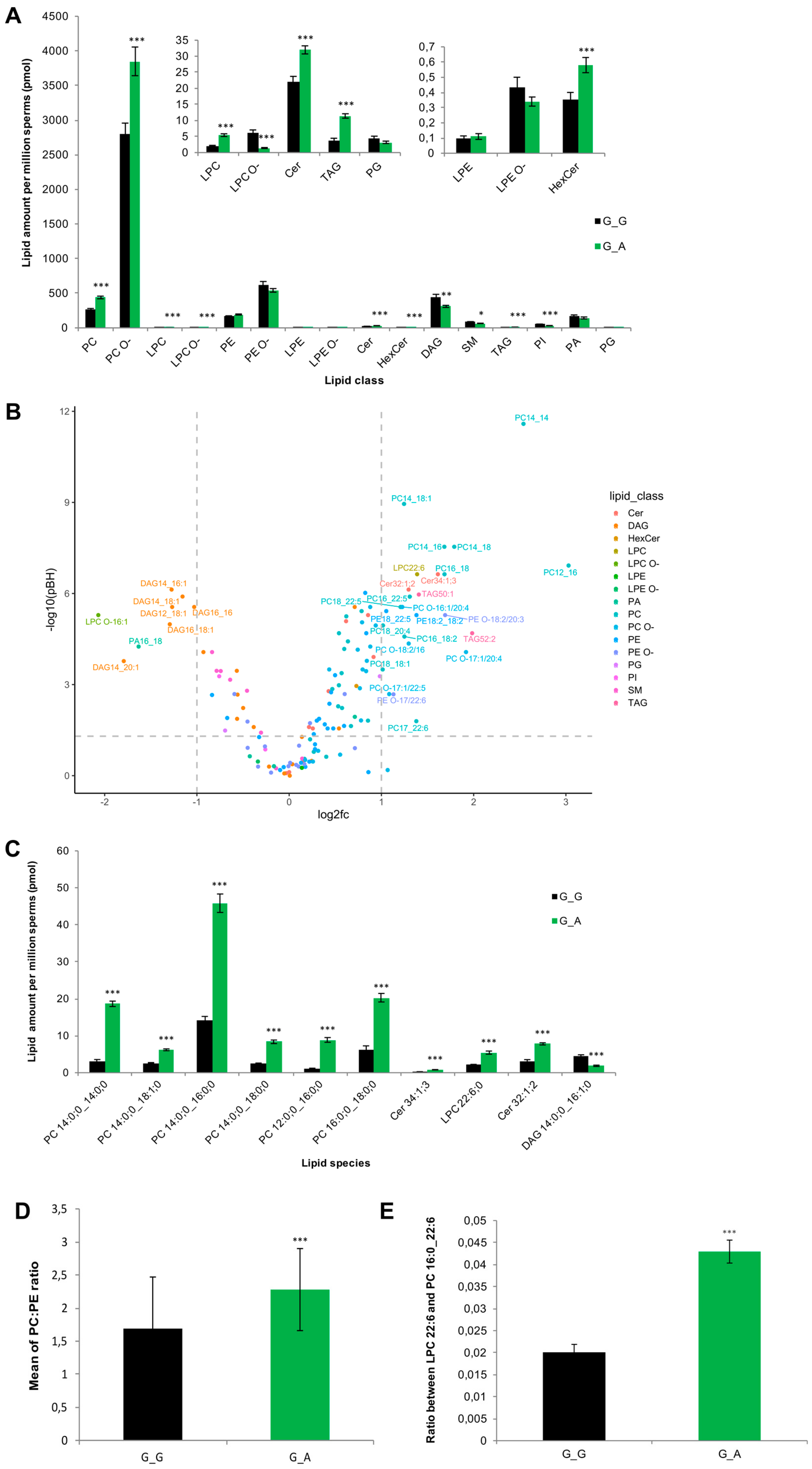
2.7. ABHD16B Is Involved in Lipid Metabolism and Influences Sperm Plasma Membrane Lipid Composition
3. Discussion
4. Materials and Methods
4.1. Ethical Statement
4.2. Genome Wide Association Analysis (GWAS)
4.3. Next Generation Sequencing of Tarantino and Its Parents
4.4. Genotyping of SNP rs468948776 (ABHD16B)
4.5. Western Blotting
4.6. Immunohistochemistry of Testes
4.7. Lipidomics of Wild Type and Heterozygous Spermatozoa
4.7.1. Semen Collection for Lipidome Analysis
4.7.2. Lipid Extraction for Mass Spectrometry Lipidomics
4.7.3. MS Data Acquisition
4.7.4. Data Analysis and Post-Processing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sakaguchi, M. Practical aspects of the fertility of dairy cattle. J. Reprod. Dev. 2011, 57, 17–33. [Google Scholar] [CrossRef]
- Veerkamp, R.F.; Beerda, B. Genetics and genomics to improve fertility in high producing dairy cows. Theriogenology 2007, 68 (Suppl. 1), S266–S273. [Google Scholar] [CrossRef]
- Lucy, M.C. Fertility in high-producing dairy cows: Reasons for decline and corrective strategies for sustainable improvement. Soc. Reprod. Fertil. Suppl. 2007, 64, 237–254. [Google Scholar] [CrossRef] [PubMed]
- Olynk, N.J.; Wolf, C.A. Economic analysis of reproductive management strategies on US commercial dairy farms. J. Dairy Sci. 2008, 91, 4082–4091. [Google Scholar] [CrossRef]
- Harstine, B.R.; Utt, M.D.; DeJarnette, J.M. Review: Integrating a semen quality control program and sire fertility at a large artificial insemination organization. Animal 2018, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yaniz, J.L.; Soler, C.; Alquezar-Baeta, C.; Santolaria, P. Toward an integrative and predictive sperm quality analysis in Bos taurus. Anim. Reprod. Sci. 2017, 181, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Morrell, J.M.; Nongbua, T.; Valeanu, S.; Lima Verde, I.; Lundstedt-Enkel, K.; Edman, A.; Johannisson, A. Sperm quality variables as indicators of bull fertility may be breed dependent. Anim. Reprod. Sci. 2017, 185, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Puglisi, R.; Gaspa, G.; Balduzzi, D.; Severgnini, A.; Vanni, R.; Macciotta, N.; Galli, A. Genomewide analysis of bull sperm quality and fertility traits. Reprod. Domest. Anim. 2016, 51, 840–843. [Google Scholar] [CrossRef]
- Kastelic, J.P. Understanding and evaluating bovine testes. Theriogenology 2014, 81, 18–23. [Google Scholar] [CrossRef]
- Amann, R.P.; DeJarnette, J.M. Impact of genomic selection of AI dairy sires on their likely utilization and methods to estimate fertility: A paradigm shift. Theriogenology 2012, 77, 795–817. [Google Scholar] [CrossRef]
- Kealey, C.G.; MacNeil, M.D.; Tess, M.W.; Geary, T.W.; Bellows, R.A. Genetic parameter estimates for scrotal circumference and semen characteristics of Line 1 Hereford bulls. J. Anim. Sci. 2006, 84, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Feugang, J.M.; Rodriguez-Osorio, N.; Kaya, A.; Wang, H.; Page, G.; Ostermeier, G.C.; Topper, E.K.; Memili, E. Transcriptome analysis of bull spermatozoa: Implications for male fertility. Reprod. Biomed. Online 2010, 21, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Selvaraju, S.; Parthipan, S.; Somashekar, L.; Kolte, A.P.; Krishnan Binsila, B.; Arangasamy, A.; Ravindra, J.P. Occurrence and functional significance of the transcriptome in bovine (Bos taurus) spermatozoa. Sci. Rep. 2017, 7, 42392. [Google Scholar] [CrossRef] [PubMed]
- Lalancette, C.; Thibault, C.; Bachand, I.; Caron, N.; Bissonnette, N. Transcriptome analysis of bull semen with extreme nonreturn rate: Use of suppression-subtractive hybridization to identify functional markers for fertility. Biol. Reprod. 2008, 78, 618–635. [Google Scholar] [CrossRef] [PubMed]
- Samanta, L.; Swain, N.; Ayaz, A.; Venugopal, V.; Agarwal, A. Post-Translational Modifications in sperm Proteome: The Chemistry of Proteome diversifications in the Pathophysiology of male factor infertility. Biochim. Biophys. Acta 2016, 1860, 1450–1465. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, G.; Swain, N.; Samanta, L. Sperm Proteome: What Is on the Horizon? Reprod. Sci. 2015, 22, 638–653. [Google Scholar] [CrossRef]
- Legare, C.; Droit, A.; Fournier, F.; Bourassa, S.; Force, A.; Cloutier, F.; Tremblay, R.; Sullivan, R. Investigation of male infertility using quantitative comparative proteomics. J. Proteome Res. 2014, 13, 5403–5414. [Google Scholar] [CrossRef]
- Gan, H.; Cai, T.; Lin, X.; Wu, Y.; Wang, X.; Yang, F.; Han, C. Integrative proteomic and transcriptomic analyses reveal multiple post-transcriptional regulatory mechanisms of mouse spermatogenesis. Mol. Cell. Proteom. 2013, 12, 1144–1157. [Google Scholar] [CrossRef]
- Panner Selvam, M.K.; Agarwal, A. Update on the proteomics of male infertility: A systematic review. Arab J. Urol. 2018, 16, 103–112. [Google Scholar] [CrossRef]
- Abdollahi-Arpanahi, R.; Morota, G.; Penagaricano, F. Predicting bull fertility using genomic data and biological information. J. Dairy Sci. 2017, 100, 9656–9666. [Google Scholar] [CrossRef]
- Suchocki, T.; Szyda, J. Genome-wide association study for semen production traits in Holstein-Friesian bulls. J. Dairy Sci. 2015, 98, 5774–5780. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.F.; Schnabel, R.D.; Sutovsky, P. Review: Genomics of bull fertility. Animal 2018, 12, s172–s183. [Google Scholar] [CrossRef] [PubMed]
- Penagaricano, F.; Weigel, K.A.; Khatib, H. Genome-wide association study identifies candidate markers for bull fertility in Holstein dairy cattle. Anim. Genet. 2012, 43 (Suppl. 1), 65–71. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Penagaricano, F. Unravelling the genomic architecture of bull fertility in Holstein cattle. BMC Genet. 2016, 17, 143. [Google Scholar] [CrossRef] [PubMed]
- Ogorevc, J.; Dovc, P.; Kunej, T. Comparative genomics approach to identify candidate genetic loci for male fertility. Reprod. Domest. Anim. 2011, 46, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Whiston, R.; Finlay, E.K.; McCabe, M.S.; Cormican, P.; Flynn, P.; Cromie, A.; Hansen, P.J.; Lyons, A.; Fair, S.; Lonergan, P.; et al. A dual targeted beta-defensin and exome sequencing approach to identify, validate and functionally characterise genes associated with bull fertility. Sci. Rep. 2017, 7, 12287. [Google Scholar] [CrossRef]
- Pausch, H.; Kolle, S.; Wurmser, C.; Schwarzenbacher, H.; Emmerling, R.; Jansen, S.; Trottmann, M.; Fuerst, C.; Gotz, K.U.; Fries, R. A nonsense mutation in TMEM95 encoding a nondescript transmembrane protein causes idiopathic male subfertility in cattle. PLoS Genet. 2014, 10, e1004044. [Google Scholar] [CrossRef]
- Pausch, H.; Venhoranta, H.; Wurmser, C.; Hakala, K.; Iso-Touru, T.; Sironen, A.; Vingborg, R.K.; Lohi, H.; Soderquist, L.; Fries, R.; et al. A frameshift mutation in ARMC3 is associated with a tail stump sperm defect in Swedish Red (Bos taurus) cattle. BMC Genet. 2016, 17, 49. [Google Scholar] [CrossRef][Green Version]
- Fernandez-Fuertes, B.; Laguna-Barraza, R.; Fernandez-Gonzalez, R.; Gutierrez-Adan, A.; Blanco-Fernandez, A.; O’Doherty, A.M.; Di Fenza, M.; Kelly, A.K.; Kolle, S.; Lonergan, P. Subfertility in bulls carrying a nonsense mutation in transmembrane protein 95 is due to failure to interact with the oocyte vestments. Biol. Reprod. 2017, 97, 50–60. [Google Scholar] [CrossRef]
- Dai, L.; Xu, Y.; Yu, W.; Liu, S.; Gao, Y.; Zhang, L.; Yuan, B.; Chen, J.; Ma, T.; Zhang, J. Naturally occurring genetic mutations in the 5’-upstream regulatory region of bovine FSHB generate a novel cis-regulatory element that affects its expression. Anim. Genet. 2015, 46, 693–696. [Google Scholar] [CrossRef]
- Lord, C.C.; Thomas, G.; Brown, J.M. Mammalian alpha beta hydrolase domain (ABHD) proteins: Lipid metabolizing enzymes at the interface of cell signaling and energy metabolism. Biochim. Biophys. Acta 2013, 1831, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Wan, E.S.; Qiu, W.; Baccarelli, A.; Carey, V.J.; Bacherman, H.; Rennard, S.I.; Agusti, A.; Anderson, W.H.; Lomas, D.A.; DeMeo, D.L. Systemic steroid exposure is associated with differential methylation in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2012, 186, 1248–1255. [Google Scholar] [CrossRef] [PubMed]
- Kurima, K.; Ebrahim, S.; Pan, B.; Sedlacek, M.; Sengupta, P.; Millis, B.A.; Cui, R.; Nakanishi, H.; Fujikawa, T.; Kawashima, Y.; et al. TMC1 and TMC2 Localize at the Site of Mechanotransduction in Mammalian Inner Ear Hair Cell Stereocilia. Cell Rep. 2015, 12, 1606–1617. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Geleoc, G.S.; Asai, Y.; Horwitz, G.C.; Kurima, K.; Ishikawa, K.; Kawashima, Y.; Griffith, A.J.; Holt, J.R. TMC1 and TMC2 are components of the mechanotransduction channel in hair cells of the mammalian inner ear. Neuron 2013, 79, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Carrell, D.T.; Cairns, B.; Aston, K.I.; Jenkins, T.; Smith, A.D.; Uren, P.J.; Horsager, A. Methods of Identifying Male Fertility Status and Embryo Quality. 2019. Available online: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017024311&_cid=P22-K5CGNG-22960-1. (accessed on 16 January 2020).
- Moghbelinejad, S.; Mozdarania, H.; Ghoraeian, P.; Asadi, R. Basic and clinical genetic studies on male infertility in Iran during 2000–2016: A review. Int. J. Reprod. Biomed. (Yazd) 2018, 16, 131–148. [Google Scholar] [CrossRef]
- Krausz, C.; Riera-Escamilla, A. Genetics of male infertility. Nat. Rev. Urol. 2018, 15, 369–384. [Google Scholar] [CrossRef]
- Bracke, A.; Peeters, K.; Punjabi, U.; Hoogewijs, D.; Dewilde, S. A search for molecular mechanisms underlying male idiopathic infertility. Reprod. Biomed. Online 2018, 36, 327–339. [Google Scholar] [CrossRef]
- Tuttelmann, F.; Ruckert, C.; Ropke, A. Disorders of spermatogenesis: Perspectives for novel genetic diagnostics after 20 years of unchanged routine. Med. Genet. 2018, 30, 12–20. [Google Scholar] [CrossRef]
- Li, G.; Penagaricano, F.; Weigel, K.A.; Zhang, Y.; Rosa, G.; Khatib, H. Comparative genomics between fly, mouse, and cattle identifies genes associated with sire conception rate. J. Dairy Sci. 2012, 95, 6122–6129. [Google Scholar] [CrossRef]
- Blaschek, M.; Kaya, A.; Zwald, N.; Memili, E.; Kirkpatrick, B.W. A whole-genome association analysis of noncompensatory fertility in Holstein bulls. J. Dairy Sci. 2011, 94, 4695–4699. [Google Scholar] [CrossRef] [PubMed]
- Fortes, M.R.; Reverter, A.; Kelly, M.; McCulloch, R.; Lehnert, S.A. Genome-wide association study for inhibin, luteinizing hormone, insulin-like growth factor 1, testicular size and semen traits in bovine species. Andrology 2013, 1, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Hoglund, J.K.; Sahana, G.; Guldbrandtsen, B.; Lund, M.S. Validation of associations for female fertility traits in Nordic Holstein, Nordic Red and Jersey dairy cattle. BMC Genet. 2014, 15, 8. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, P.; Amorin, R.; Han, Y.; Penagaricano, F. Whole-genome scan reveals significant non-additive effects for sire conception rate in Holstein cattle. BMC Genet. 2018, 19, 14. [Google Scholar] [CrossRef]
- Nani, J.P.; Rezende, F.M.; Penagaricano, F. Predicting male fertility in dairy cattle using markers with large effect and functional annotation data. BMC Genom. 2019, 20, 258. [Google Scholar] [CrossRef]
- Wei, W.; Luo, W.; Wu, F.; Peng, X.; Zhang, Y.; Zhang, M.; Zhao, Y.; Su, N.; Qi, Y.; Chen, L.; et al. Deep Coverage Proteomics Identifies More Low-Abundance Missing Proteins in Human Testis Tissue with Q-Exactive HF Mass Spectrometer. J. Proteome Res. 2016, 15, 3988–3997. [Google Scholar] [CrossRef]
- Chalmel, F.; Lardenois, A.; Evrard, B.; Mathieu, R.; Feig, C.; Demougin, P.; Gattiker, A.; Schulze, W.; Jegou, B.; Kirchhoff, C.; et al. Global human tissue profiling and protein network analysis reveals distinct levels of transcriptional germline-specificity and identifies target genes for male infertility. Hum. Reprod. 2012, 27, 3233–3248. [Google Scholar] [CrossRef]
- Miller, M.R.; Mannowetz, N.; Iavarone, A.T.; Safavi, R.; Gracheva, E.O.; Smith, J.F.; Hill, R.Z.; Bautista, D.M.; Kirichok, Y.; Lishko, P.V. Unconventional endocannabinoid signaling governs sperm activation via the sex hormone progesterone. Science 2016, 352, 555–559. [Google Scholar] [CrossRef]
- Rejraji, H.; Sion, B.; Prensier, G.; Carreras, M.; Motta, C.; Frenoux, J.-M.; Vericel, E.; Grizard, G.; Vernet, P.; Drevet, J.R. Lipid remodeling of murine epididymosomes and spermatozoa during epididymal maturation. Biol. Reprod. 2006, 74, 1104–1113. [Google Scholar] [CrossRef]
- Martínez, P.; Morros, A. Membrane lipid dynamics during human sperm capacitation. Front. Biosci. 1996, 1, d103–d117. [Google Scholar]
- Merrill, A.; Sweeley, C.C. Sphingolipids: Metabolism and cell signalling. New Compr. Biochem. 1996, 31, 309–339. [Google Scholar]
- Fonseca, B.; Costa, M.; Almada, M.; Correia-da-Silva, G.; Teixeira, N. Endogenous cannabinoids revisited: A biochemistry perspective. Prostaglandins Other Lipid Mediat. 2013, 102, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Cerolini, S.; Kelso, K.; Noble, R.; Speake, B.; Pizzi, F.; Cavalchini, L. Relationship between spermatozoan lipid composition and fertility during aging of chickens. Biol. Reprod. 1997, 57, 976–980. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bjorkgren, I.; Gylling, H.; Turunen, H.; Huhtaniemi, I.; Strauss, L.; Poutanen, M.; Sipila, P. Imbalanced lipid homeostasis in the conditional Dicer1 knockout mouse epididymis causes instability of the sperm membrane. FASEB J. 2015, 29, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Min-Seok, R.; Kawamata, Y.; Nakamura, H.; Ohta, A.; Takagi, M. Isolation and characterization of ECT1 gene encoding CTP: Phosphoethanolamine cytidylyltransferase of Saccharomyces cerevisiae. J. Biochem. 1996, 120, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Agellon, L.B.; Allen, T.M.; Umeda, M.; Jewell, L.; Mason, A.; Vance, D.E. The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell Metab. 2006, 3, 321–331. [Google Scholar] [CrossRef]
- Glander, H.J.; Schiller, J.; Suss, R.; Paasch, U.; Grunewald, S.; Arnhold, J. Deterioration of spermatozoal plasma membrane is associated with an increase of sperm lyso-phosphatidylcholines. Andrologia 2002, 34, 360–366. [Google Scholar] [CrossRef]
- Fuchs, B.; Muller, K.; Goritz, F.; Blottner, S.; Schiller, J. Characteristic oxidation products of choline plasmalogens are detectable in cattle and roe deer spermatozoa by MALDI-TOF mass spectrometry. Lipids 2007, 42, 991–998. [Google Scholar] [CrossRef]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.Y.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef]
- Vilhjalmsson, B.J.; Nordborg, M. The nature of confounding in genome-wide association studies. Nat. Rev. Genet. 2013, 14, 1–2. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43. [Google Scholar] [CrossRef]
- Schutz, E.; Von Ahsen, N. Spreadsheet software for thermodynamic melting point prediction of oligonucleotide hybridization with and without mismatches. Biotechniques 1999, 27, 1218–1222, 1224. [Google Scholar] [CrossRef] [PubMed]
- Von Ahsen, N.; Oellerich, M.; Armstrong, V.W.; Schutz, E. Application of a thermodynamic nearest-neighbor model to estimate nucleic acid stability and optimize probe design: Prediction of melting points of multiple mutations of apolipoprotein B-3500 and factor V with a hybridization probe genotyping assay on the LightCycler. Clin. Chem. 1999, 45, 2094–2101. [Google Scholar] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Pursel, V.G.; Gerrits, R.J. Total phospholipid and phospholipid fatty acids of ejaculated and epididymal semen and seminal vesicle fluids of boars. J. Anim. Sci. 1972, 35, 398–403. [Google Scholar] [CrossRef][Green Version]
- Angrimani, D.S.R.; Nichi, M.; Losano, J.D.A.; Lucio, C.F.; Veiga, G.A.L.; Franco, M.V.M.J.; Vannucchi, C.I. Fatty acid content in epididymal fluid and spermatozoa during sperm maturation in dogs. J. Anim. Sci. Biotechnol. 2017, 8, 18. [Google Scholar] [CrossRef]
- Sampaio, J.L.; Gerl, M.J.; Klose, C.; Ejsing, C.S.; Beug, H.; Simons, K.; Shevchenko, A. Membrane lipidome of an epithelial cell line. Proc. Natl. Acad. Sci. USA 2011, 108, 1903–1907. [Google Scholar] [CrossRef]
- Ejsing, C.S.; Sampaio, J.L.; Surendranath, V.; Duchoslav, E.; Ekroos, K.; Klemm, R.W.; Simons, K.; Shevchenko, A. Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc. Natl. Acad. Sci. USA 2009, 106, 2136–2141. [Google Scholar] [CrossRef]
- Surma, M.A.; Herzog, R.; Vasilj, A.; Klose, C.; Christinat, N.; Morin-Rivron, D.; Simons, K.; Masoodi, M.; Sampaio, J.L. An automated shotgun lipidomics platform for high throughput, comprehensive, and quantitative analysis of blood plasma intact lipids. Eur. J. Lipid Sci. Technol. 2015, 117, 1540–1549. [Google Scholar] [CrossRef]
- Herzog, R.; Schuhmann, K.; Schwudke, D.; Sampaio, J.L.; Bornstein, S.R.; Schroeder, M.; Shevchenko, A. LipidXplorer: A software for consensual cross-platform lipidomics. PLoS ONE 2012, 7, e29851. [Google Scholar] [CrossRef] [PubMed]
- Herzog, R.; Schwudke, D.; Schuhmann, K.; Sampaio, J.L.; Bornstein, S.R.; Schroeder, M.; Shevchenko, A. A novel informatics concept for high-throughput shotgun lipidomics based on the molecular fragmentation query language. Genome Biol. 2011, 12, R8. [Google Scholar] [CrossRef] [PubMed]
- Zorn, C. Shapiro-Wilk test. In The SAGE Encyclopedia of Social Science Research Methods; Sage Publications, Inc.: Thousand Oaks, CA, USA, 2004; pp. 1030–1031. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sire/ID | NRdeva) | No. of First Inseminations |
|---|---|---|
| Tarantino | −29 | 412 |
| 19_39644 | −27 | 402 |
| 19_39643 | −25 | 364 |
| 05_34345 | −9 | 412 |
| 04_44565 | −4 | 421 |
| 04_39067 | −3 | 315 |
| 04_43327 | −3 | 424 |
| 04_40476 | −2 | 407 |
| 04_41962 | −2 | 640 |
| 04_37666 | −2 | 571 |
| Gene | Primer Name | Sequence (5’->3’) | Probe Name | Sequence (5’->3’) |
|---|---|---|---|---|
| ABHD16B | ABHD16B_FRET_f | ACCCGGGCTTCGGGGGCAGC | ABHD16B_FRET_Pro | [Cy5]CGTTCCCTCAGCATGATG[Phos] |
| ABHD16B_FRET_r | GCGTACTTGACCACCACGTC | ABHD16B_FRET_Anc | GGGGCAGCACGGGCG[Flc] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shan, S.; Xu, F.; Bleyer, M.; Becker, S.; Melbaum, T.; Wemheuer, W.; Hirschfeld, M.; Wacker, C.; Zhao, S.; Schütz, E.; et al. Association of α/β-Hydrolase D16B with Bovine Conception Rate and Sperm Plasma Membrane Lipid Composition. Int. J. Mol. Sci. 2020, 21, 627. https://doi.org/10.3390/ijms21020627
Shan S, Xu F, Bleyer M, Becker S, Melbaum T, Wemheuer W, Hirschfeld M, Wacker C, Zhao S, Schütz E, et al. Association of α/β-Hydrolase D16B with Bovine Conception Rate and Sperm Plasma Membrane Lipid Composition. International Journal of Molecular Sciences. 2020; 21(2):627. https://doi.org/10.3390/ijms21020627
Chicago/Turabian StyleShan, Shuwen, Fangzheng Xu, Martina Bleyer, Svenja Becker, Torben Melbaum, Wilhelm Wemheuer, Marc Hirschfeld, Christin Wacker, Shuhong Zhao, Ekkehard Schütz, and et al. 2020. "Association of α/β-Hydrolase D16B with Bovine Conception Rate and Sperm Plasma Membrane Lipid Composition" International Journal of Molecular Sciences 21, no. 2: 627. https://doi.org/10.3390/ijms21020627
APA StyleShan, S., Xu, F., Bleyer, M., Becker, S., Melbaum, T., Wemheuer, W., Hirschfeld, M., Wacker, C., Zhao, S., Schütz, E., & Brenig, B. (2020). Association of α/β-Hydrolase D16B with Bovine Conception Rate and Sperm Plasma Membrane Lipid Composition. International Journal of Molecular Sciences, 21(2), 627. https://doi.org/10.3390/ijms21020627