The Role of CYP450 Drug Metabolism in Precision Cardio-Oncology


Abstract

1. Introduction
2. CYP450 Class of Enzymes
2.1. Phase I Enzymes
2.2. Phase II Enzymes
2.3. Substrates, Inducers and Inhibitors
3. Drug-Drug Interactions
4. Precision Cardio-Oncology
4.1. Variability in Concentration and Activity
4.2. Interindividual and Genetic Variability
4.3. Genomic Profiling
4.3.1. Genomic Variation in CYP450
4.3.2. Genomic Variation in CYP2C19

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotype | Example Genotypes | Enzyme Activity |
|---|---|---|
| Ultra-rapid metabolizer (UM) | *1/*17 *17/*17 | Normal or increased |
| Extensive metabolizer (EM) | *1/*1 (wild type) | Normal |
| Intermediate metabolizer (IM) | *1/*2 *1/*3 *2/*17 *3/17 | Intermediate Likely intermediate Likely intermediate |
| Poor metabolizer (PM) | *2/*2 *3/*3 *2/*3 | Low or absent |
4.3.3. Genomic Variation in CYP2D6
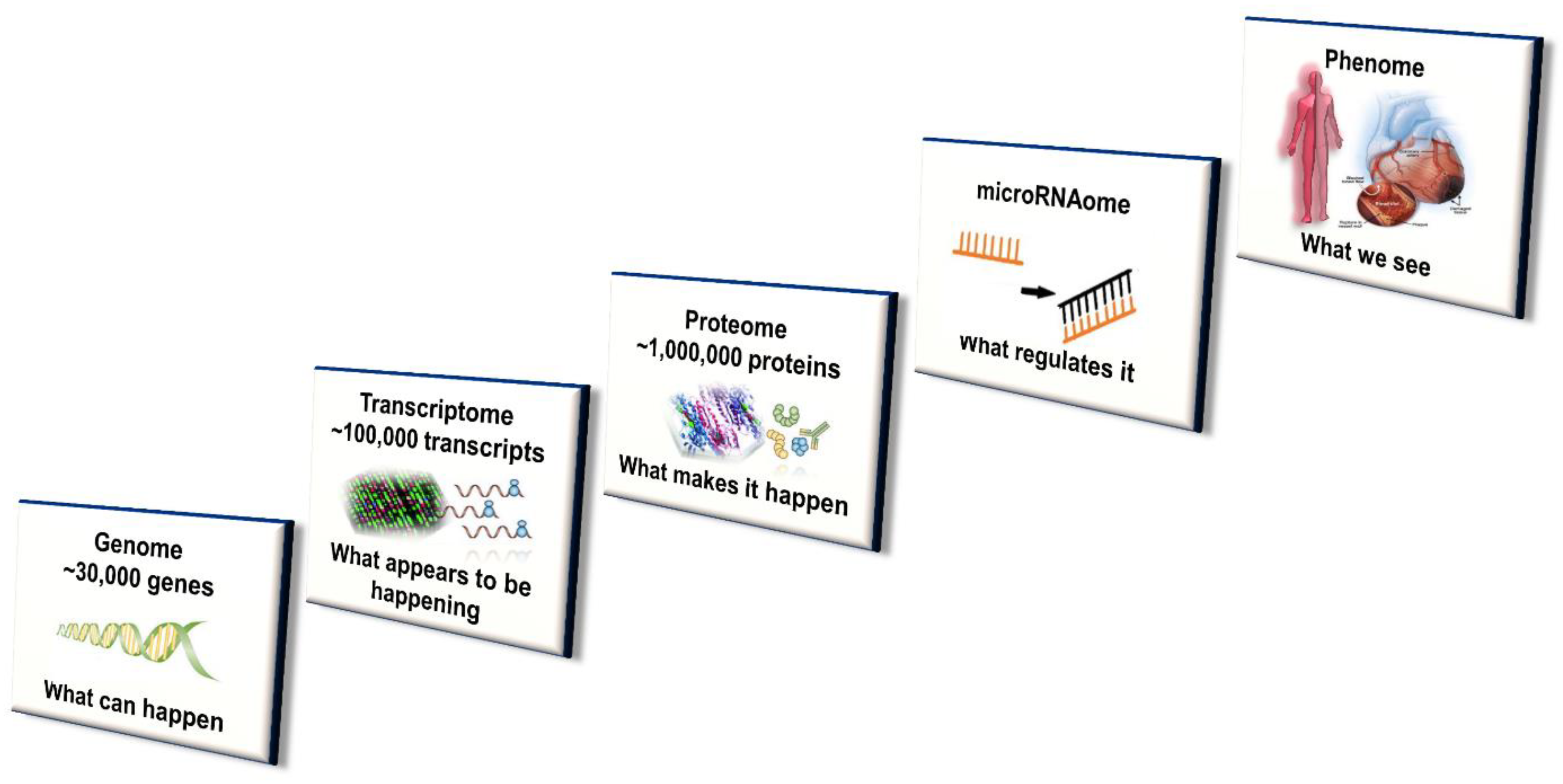
4.4. Systems Approach
4.4.1. Transcriptomics
4.4.2. Epigenomics
4.4.3. Proteomics
4.4.4. Metabolomics
4.4.5. Microbiomics
4.4.6. MicroRNAomics
4.4.7. Small RNA Therapeutics
4.4.8. Integration of ‘Omics’
5. Clinical Implementation
5.1. Pharmacogenomics
5.1.1. Master Regulators
5.1.2. Warfarin
5.1.3. Clopidogrel
5.2. Modified P*3 Pathway
6. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Lynch, T.; Price, A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am. Fam. Physician 2007, 76, 391–396. [Google Scholar]
- Undevia, S.; Gomez-Abuin, G.; Ratain, M. Pharmacokinetic Variability of Anticancer Agents. Available online: www.medscape.com/viewarticle/506712_1 (accessed on 12 September 2019).
- Chaudhary, K.R.; Batchu, S.N.; Seubert, J.M. Cytochrome P450 enzymes and the heart. IUBMB Life 2009, 61, 954–960. [Google Scholar] [CrossRef]
- Carreca, I.; Balducci, L. Oral chemotherapy of cancer in the elderly. Am. J. Cancer 2002, 1, 101–108. [Google Scholar] [CrossRef]
- DeMario, M.D.; Ratain, M.J. Oral chemotherapy: Rationale and future directions. J. Clin. Oncol. 1998, 16, 2557–2567. [Google Scholar] [CrossRef]
- Brown, S.-A.; Pereira, N. Pharmacogenomic Impact of CYP2C19 Variation on Clopidogrel Therapy in Precision Cardiovascular Medicine. J. Pers. Med. 2018, 8, 8. [Google Scholar] [CrossRef]
- Kutsuno, Y.; Itoh, T.; Tukey, R.H.; Fujiwara, R. Glucuronidation of drugs and drug-induced toxicity in humanized UDP-glucuronosyltransferase 1 mice. Drug Metab. Dispos. 2014, 42, 1146–1152. [Google Scholar] [CrossRef]
- Li, A.P.; Kaminski, D.L.; Rasmussen, A. Substrates of human hepatic cytochrome P450 3A4. Toxicology 1995, 104, 1–8. [Google Scholar] [CrossRef]
- Evans, W.E.; Relling, M.V. Pharmacogenomics: Translating functional genomics into rational therapeutics. Science 1999, 286, 487–491. [Google Scholar] [CrossRef]
- Wrighton, S.A.; Brian, W.R.; Sari, M.A.; Iwasaki, M.; Guengerich, F.P.; Raucy, J.L.; Molowa, D.T.; Vandenbranden, M. Studies on the expression and metabolic capabilities of human liver cytochrome P450IIIA5 (HLp3). Mol. Pharmacol. 1990, 38, 207–213. [Google Scholar]
- Shimada, T.; Yamazaki, H.; Mimura, M.; Inui, Y.; Guengerich, F.P. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: Studies with liver microsomes of 30 Japanese and 30 Caucasians. J. Pharmacol. Exp. Ther. 1994, 270, 414–423. [Google Scholar]
- Watkins, P.B.; Wrighton, S.A.; Schuetz, E.G.; Molowa, D.T.; Guzelian, P.S. Identification of glucocorticoid-inducible cytochromes P-450 in the intestinal mucosa of rats and man. J. Clin. Investig. 1987, 80, 1029–1036. [Google Scholar] [CrossRef]
- Kolars, J.C.; Schmiedlin-Ren, P.; Schuetz, J.D.; Fang, C.; Watkins, P.B. Identification of rifampin-inducible P450IIIA4 (CYP3A4) in human small bowel enterocytes. J. Clin. Investig. 1992, 90, 1871–1878. [Google Scholar] [CrossRef]
- Wei, Z.; Jiang, S.; Zhang, Y.; Wang, X.; Peng, X.; Meng, C.; Liu, Y.; Wang, H.; Guo, L.; Qin, S.; et al. The effect of microRNAs in the regulation of human CYP3A4: A systematic study using a mathematical model. Sci. Rep. 2014, 4, 4283. [Google Scholar] [CrossRef]
- Tracy, T.S.; Chaudhry, A.S.; Prasad, B.; Thummel, K.E.; Schuetz, E.G.; Zhong, X.B.; Tien, Y.C.; Jeong, H.; Pan, X.; Shireman, L.M.; et al. Interindividual Variability in Cytochrome P450-Mediated Drug Metabolism. Drug Metab. Dispos. 2016, 44, 343–351. [Google Scholar] [CrossRef]
- Westlind, A.; Löfberg, L.; Tindberg, N.; Andersson, T.B.; Ingelman-Sundberg, M. Interindividual differences in hepatic expression of CYP3A4: Relationship to genetic polymorphism in the 5’-upstream regulatory region. Biochem. Biophys. Res. Commun. 1999, 259, 201–205. [Google Scholar] [CrossRef]
- Lamba, J.K.; Lin, Y.S.; Schuetz, E.G.; Thummel, K.E. Genetic contribution to variable human CYP3A-mediated metabolism. Adv. Drug Deliv. Rev. 2002, 54, 1271–1294. [Google Scholar] [CrossRef]
- Hart, S.N.; Wang, S.; Nakamoto, K.; Wesselman, C.; Li, Y.; Zhong, X.B. Genetic polymorphisms in cytochrome P450 oxidoreductase influence microsomal P450-catalyzed drug metabolism. Pharm. Genom. 2008, 18, 11–24. [Google Scholar] [CrossRef]
- Court, M.H. Interindividual variability in hepatic drug glucuronidation: Studies into the role of age, sex, enzyme inducers, and genetic polymorphism using the human liver bank as a model system. Drug Metab. Rev. 2010, 42, 209–224. [Google Scholar] [CrossRef]
- Foti, R.S.; Dalvie, D.K. Cytochrome P450 and Non-Cytochrome P450 Oxidative Metabolism: Contributions to the Pharmacokinetics, Safety, and Efficacy of Xenobiotics. Drug Metab. Dispos. 2016, 44, 1229–1245. [Google Scholar] [CrossRef]
- Tornio, A.; Filppula, A.M.; Kailari, O.; Neuvonen, M.; Nyrönen, T.H.; Tapaninen, T.; Neuvonen, P.J.; Niemi, M.; Backman, J.T. Glucuronidation converts clopidogrel to a strong time-dependent inhibitor of CYP2C8: A phase II metabolite as a perpetrator of drug-drug interactions. Clin. Pharmacol. Ther. 2014, 96, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, R.; Yoda, E.; Tukey, R.H. Species differences in drug glucuronidation: Humanized UDP-glucuronosyltransferase 1 mice and their application for predicting drug glucuronidation and drug-induced toxicity in humans. Drug Metab. Pharm. 2018, 33, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Diao, X.; Pang, X.; Xie, C.; Guo, Z.; Zhong, D.; Chen, X. Bioactivation of 3-n-butylphthalide via sulfation of its major metabolite 3-hydroxy-NBP: Mediated mainly by sulfotransferase 1A1. Drug Metab. Dispos. 2014, 42, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-Q.; James, M. Inhibition of Sulfotransferases by Xenobiotics. Curr. Drug Metab. 2006, 7, 83–104. [Google Scholar] [CrossRef]
- Gamage, N.; Barnett, A.; Hempel, N.; Duggleby, R.G.; Windmill, K.F.; Martin, J.L.; McManus, M.E. Human sulfotransferases and their role in chemical metabolism. Toxicol. Sci. 2006, 90, 5–22. [Google Scholar] [CrossRef]
- Ipe, J.; Lu, J.; Nguyen, A. P450 Drug Interactions—Flockhart Table™. Available online: https://drug-interactions.medicine.iu.edu/MainTable.aspx (accessed on 5 August 2019).
- Chang, H.M.; Okwuosa, T.M.; Scarabelli, T.; Moudgil, R.; Yeh, E.T.H. Cardiovascular Complications of Cancer Therapy: Best Practices in Diagnosis, Prevention, and Management: Part 2. J. Am. Coll. Cardiol. 2017, 70, 2552–2565. [Google Scholar] [CrossRef]
- Mosarla, R.C.; Vaduganathan, M.; Qamar, A.; Moslehi, J.; Piazza, G.; Giugliano, R.P. Anticoagulation Strategies in Patients With Cancer: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 1336–1349. [Google Scholar] [CrossRef]
- van Eijk, M.; Boosman, R.J.; Schinkel, A.H.; Huitema, A.D.R.; Beijnen, J.H. Cytochrome P450 3A4, 3A5, and 2C8 expression in breast, prostate, lung, endometrial, and ovarian tumors: Relevance for resistance to taxanes. Cancer Chemother. Pharmacol. 2019, 84, 487–499. [Google Scholar] [CrossRef]
- Gunes, A.; Coskun, U.; Boruban, C.; Gunel, N.; Babaoglu, M.O.; Sencan, O.; Bozkurt, A.; Rane, A.; Hassan, M.; Zengil, H.; et al. Inhibitory effect of 5-fluorouracil on cytochrome P450 2C9 activity in cancer patients. Basic Clin. Pharmacol. Toxicol. 2006, 98, 197–200. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Lathia, C.; Frye, R.F.; Schuchter, L.; Redlinger, M.; Rosen, M.; O’Dwyer, P.J. Interaction of sorafenib and cytochrome P450 isoenzymes in patients with advanced melanoma: A phase I/II pharmacokinetic interaction study. Cancer Chemother. Pharmacol. 2011, 68, 1111–1118. [Google Scholar] [CrossRef]
- Chang, H.M.; Moudgil, R.; Scarabelli, T.; Okwuosa, T.M.; Yeh, E.T.H. Cardiovascular Complications of Cancer Therapy: Best Practices in Diagnosis, Prevention, and Management: Part 1. J. Am. Coll. Cardiol. 2017, 70, 2536–2551. [Google Scholar] [CrossRef] [PubMed]
- Bullock, K.E.; Petros, W.P.; Younis, I.; Uronis, H.E.; Morse, M.A.; Blobe, G.C.; Zafar, S.Y.; Gockerman, J.P.; Lager, J.J.; Truax, R.; et al. A Phase I Study of Bevacizumab (B) in Combination with Everolimus (E) and Erlotinib (E) in Advanced Cancer (BEE). Cancer Chemother. Pharmacol. 2014, 67, 465–474. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, L.; Christopher, L.J.; Cui, D.; Li, W.; Iyer, R.; Humphreys, W.G.; Zhang, D. Identification of the human enzymes involved in the oxidative metabolism of dasatinib: An effective approach for determining metabolite formation kinetics. Drug Metab. Dispos. Biol. Fate Chem. 2008. [Google Scholar] [CrossRef] [PubMed]
- Colburn, D.E.; Giles, F.J.; Oladovich, D.; Smith, J.A. In vitro evaluation of cytochrome P450-mediated drug interactions between cytarabine, idarubicin, itraconazole and caspofungin. Hematology 2004, 9, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Quintanilha, J.C.F.; de Sousa, V.M.; Visacri, M.B.; Amaral, L.S.; Santos, R.M.M.; Zambrano, T.; Salazar, L.A.; Moriel, P. Involvement of cytochrome P450 in cisplatin treatment: Implications for toxicity. Cancer Chemother. Pharmacol. 2017, 80, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Murayama, N.; van Beuningen, R.; Suemizu, H.; Guillouzo, C.G.; Shibata, N.; Yajima, K.; Utoh, M.; Shimizu, M.; Chesné, C.; Nakamura, M.; et al. Thalidomide increases human hepatic cytochrome P450 3A enzymes by direct activation of the pregnane X receptor. Chem. Res. Toxicol. 2014, 27, 304–308. [Google Scholar] [CrossRef]
- Lawrence, S.; Nguyen, D.; Bowen, C.; Richards-Peterson, L.; Sordos, K. The metabolic drug-drug interaction profile of Dabrafenib: In vitro investigations and quantitative extrapolation of the P450-mediated DDI risk. Drug Metab. Dispos. 2014, 42, 1180–1190. [Google Scholar] [CrossRef]
- Zhang, W.; Heinzmann, D.; Grippo, J.F. Clinical Pharmacokinetics of Vemurafenib. Clin. Pharm. 2017, 56, 1033–1043. [Google Scholar] [CrossRef]
- Whirl-Carrillo, M.; McDonagh, E.M.; Hebert, J.M.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Altman, R.B.; Klein, T.E. Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 2012, 92, 414–417. [Google Scholar] [CrossRef]
- van Leeuwen, R.W.; Brundel, D.H.; Neef, C.; van Gelder, T.; Mathijssen, R.H.; Burger, D.M.; Jansman, F.G. Prevalence of potential drug-drug interactions in cancer patients treated with oral anticancer drugs. Br. J. Cancer 2013, 108, 1071–1078. [Google Scholar] [CrossRef]
- Zukkoor, S.; Thohan, V. Drug-Drug Interactions of Common Cardiac Medications and Chemotherapeutic Agents. Available online: www.acc.org/latest-in-cardiology/articles/2018/12/21/09/52/drug-drug-interactions-of-common-cardiac-medications-and-chemotherapeutic-agents (accessed on 25 August 2019).
- Sasu-Tenkoramaa, J.; Fudin, J. Drug Interactions in Cancer Patients Requiring Concomitant Chemotherapy and Analgesics. Prac. Pain Manag. 2013, 13, 50–64. [Google Scholar]
- Kennedy, C.; Brewer, L.; Williams, D. Drug interactions. Medicine 2016, 44, 422–426. [Google Scholar] [CrossRef]
- Lexicomp Medication Database. Lexicomp® Online™, 2013.
- Yamazaki, S.; Johnson, T.R.; Smith, B.J. Prediction of Drug-Drug Interactions with Crizotinib as the CYP3A Substrate Using a Physiologically Based Pharmacokinetic Model. Drug Metab. Dispos. 2015, 43, 1417–1429. [Google Scholar] [CrossRef] [PubMed]
- Khozin, S.; Blumenthal, G.M.; Zhang, L.; Tang, S.; Brower, M.; Fox, E.; Helms, W.; Leong, R.; Song, P.; Pan, Y.; et al. FDA approval: Ceritinib for the treatment of metastatic anaplastic lymphoma kinase-positive non-small cell lung cancer. Clin. Cancer Res. 2015, 21, 2436–2439. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Gao, Y.; Zheng, M.; Xu, T.; Schoenhagen, P.; Zhaohui, J. Recent Progress and Market Analysis of Anticoagulant Drugs. J. Thorac. Dis. 2018, 10, 2011–2025. [Google Scholar] [CrossRef] [PubMed]
- Al-Samkari, H.; Connors, J.M. The Role of Direct Oral Anticoagulants in Treatment of Cancer-Associated Thrombosis. Cancers 2018, 10, 271. [Google Scholar] [CrossRef] [PubMed]
- Engman, C.A.; Zacharski, L.R. Low molecular weight heparins as extended prophylaxis against recurrent thrombosis in cancer patients. J. Natl. Compr. Cancer Netw. 2008, 6, 637–645. [Google Scholar] [CrossRef]
- Lyman, G.H.; Khorana, A.A.; Kuderer, N.M.; Lee, A.Y.; Arcelus, J.I.; Balaban, E.P.; Clarke, J.M.; Flowers, C.R.; Francis, C.W.; Gates, L.E.; et al. Venous thromboembolism prophylaxis and treatment in patients with cancer: American Society of Clinical Oncology clinical practice guideline update. J. Clin. Oncol. 2013, 31, 2189–2204. [Google Scholar] [CrossRef]
- Farge, D.; Debourdeau, P.; Beckers, M.; Baglin, C.; Bauersachs, R.M.; Brenner, B.; Brilhante, D.; Falanga, A.; Gerotzafias, G.T.; Haim, N.; et al. International clinical practice guidelines for the treatment and prophylaxis of venous thromboembolism in patients with cancer. J. Thromb. Haemost. 2013, 11, 56–70. [Google Scholar] [CrossRef]
- Kearon, C.; Akl, E.; Ornelas, J.; Blaivas, A.; Jimenez, D.; Bounameaux, H.; Huisman, M.; King, C.; Morris, T.; Sood, N.; et al. Correction to Grade in: Antithrombotic Therapy for VTE Disease: CHEST Guideline and Expert Panel Report. Chest 2016, 150, 315–352. [Google Scholar] [CrossRef]
- Samuelson Bannow, B.T.; Lee, A.; Khorana, A.A.; Zwicker, J.I.; Noble, S.; Ay, C.; Carrier, M. Management of Cancer-Associated Thrombosis in Patients with Thrombocytopenia: Guidance from the SSC of the ISTH. J. Thromb. Haemost. 2018, 16, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Short, N.J.; Connors, J.M. New oral anticoagulants and the cancer patient. Oncologist 2014, 19, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Lakkireddy, D.; Karst, E.; Mahapatra, S.; Winterfield, J.; Mansour, M. Lower Adherence Direct Oral Anticoagulants Use Is Associated With Increased Risk Of Thromboembolic Events Than Warfarin—Understanding The Real-World Performance Of Systemic Anticoagulation In Atrial Fibrillation. In Proceedings of the Heart Rhythm Society’s 39th Annual Scientific Sessions, Boston, MA, USA, 9–12 May 2018. [Google Scholar]
- Burn, J.; Pirmohamed, M. Correction: Direct Oral Anticoagulants versus Warfarin: Is New Always Better than the Old? Open Heart 2018, 5, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, V.; Kalow, W.; Tang, B.K.; Paterson, A.D.; Walker, S.E.; Endrenyi, L.; Kashuba, A.D. Evaluation of the genetic component of variability in CYP3A4 activity: A repeated drug administration method. Pharmacogenetics 2000, 10, 373–388. [Google Scholar] [CrossRef] [PubMed]
- Helsby, N.A.; Lo, W.Y.; Sharples, K.; Riley, G.; Murray, M.; Spells, K.; Dzhelai, M.; Simpson, A.; Findlay, M. CYP2C19 pharmacogenetics in advanced cancer: Compromised function independent of genotype. Br. J. Cancer 2008, 99, 1251–1255. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Nakajima, M.; Takagi, S.; Taniya, T.; Yokoi, T. MicroRNA regulates the expression of human cytochrome P450 1B1. Cancer Res. 2006, 66, 9090–9098. [Google Scholar] [CrossRef]
- Shimada, T.; Hayes, C.L.; Yamazaki, H.; Amin, S.; Hecht, S.S.; Guengerich, F.P.; Sutter, T.R. Activation of chemically diverse procarcinogens by human cytochrome P-450 1B1. Cancer Res. 1996, 56, 2979–2984. [Google Scholar]
- Murray, G.I.; Taylor, M.C.; McFadyen, M.C.; McKay, J.A.; Greenlee, W.F.; Burke, M.D.; Melvin, W.T. Tumor-specific expression of cytochrome P450 CYP1B1. Cancer Res. 1997, 57, 3026–3031. [Google Scholar]
- Newbold, R.R.; Liehr, J.G. Induction of uterine adenocarcinoma in CD-1 mice by catechol estrogens. Cancer Res. 2000, 60, 235–237. [Google Scholar]
- Han, X.; Liehr, J.G. DNA single-strand breaks in kidneys of Syrian hamsters treated with steroidal estrogens: Hormone-induced free radical damage preceding renal malignancy. Carcinogenesis 1994, 15, 997–1000. [Google Scholar] [CrossRef]
- Genomic profiling. In NCI Dictionary of Cancer Terms; National Cancer Institute at the National Institutes of Health. Available online: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/genomic-profiling (accessed on 20 September 2019).
- Puckelwartz, M.J.; McNally, E.M. Genetic profiling for risk reduction in human cardiovascular disease. Genes 2014, 5, 214–234. [Google Scholar] [CrossRef] [PubMed]
- Pharmacogene Variation Consortium (PharmVar). Available online: https://www.pharmvar.org/ (accessed on 20 September 2019).
- Weinshilboum, R. Inheritance and drug response. N. Engl. J. Med. 2003, 348, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Scott, S.A.; Sangkuhl, K.; Stein, C.M.; Hulot, J.S.; Mega, J.L.; Roden, D.M.; Klein, T.E.; Sabatine, M.S.; Johnson, J.A.; Shuldiner, A.R.; et al. Clinical Pharmacogenetics Implementation Consortium guidelines for CYP2C19 genotype and clopidogrel therapy: 2013 update. Clin. Pharmacol. Ther. 2013, 94, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.Y.; Pickle, L.W.; Meyer, P.S.; Woosley, R.L. Salivary analysis for determination of dextromethorphan metabolic phenotype. Clin. Pharmacol. Ther. 1991, 49, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Bertilsson, L.; Dahl, M.; Dalén, P.; Al-Shurbaji, A. Molecular genetics of CYP2D6: Clinical relevance with focus on psychotropic drugs. Br. J. Clin. Pharmacol. 2002, 53, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Dahl, M.-L.; Johansson, I.; Palmertz, M.P.; Ingelman-Sundberg, M.; Sjöqvist, F. Analysis of the CYP2D6 gene in relation to debrisoquin and desipramine hydroxylation in a Swedish population. Clin. Pharmacol. Ther. 1992, 51, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Sachse, C.; Brockmöller, J.; Bauer, S.; Roots, I. Cytochrome P450 2D6 variants in a Caucasian population: Allele frequencies and phenotypic consequences. Am. J. Hum. Genet. 1997, 60, 284–295. [Google Scholar]
- Zanger, U.M.; Fischer, J.; Raimundo, S.; Stüven, T.; Evert, B.O.; Schwab, M.; Eichelbaum, M. Comprehensive analysis of the genetic factors determining expression and function of hepatic CYP2D6. Pharmacogenetics 2001, 11, 573–585. [Google Scholar] [CrossRef]
- Gaedigk, A.; Simon, S.; Pearce, R.; Bradford, L.; Kennedy, M.; Leeder, J. The CYP2D6 Activity Score: Translating Genotype Information into a Qualitative Measure of Phenotype. Clin. Pharmacol. Ther. 2007, 83, 234–242. [Google Scholar] [CrossRef]
- Dreyfuss, A.D.; Bravo, P.E.; Koumenis, C.; Ky, B. Precision Cardio-Oncology. J. Nucl. Med. 2019, 60, 443–450. [Google Scholar] [CrossRef]
- Brown, S.A.; Sandhu, N.; Herrmann, J. Systems biology approaches to adverse drug effects: The example of cardio-oncology. Nat. Rev. Clin. Oncol. 2015, 12, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Cappola, T.; Margulies, K. Functional Genomics Applied to Cardiovascular Medicine. Circulation 2011, 124, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Lamba, V.; Ghodke, Y.; Guan, W.; Tracy, T.S. microRNA-34a is associated with expression of key hepatic transcription factors and cytochromes P450. Biochem. Biophys. Res. Commun. 2014, 445, 404–411. [Google Scholar] [CrossRef]
- Lin, J.H.; Lu, A.Y. Interindividual variability in inhibition and induction of cytochrome P450 enzymes. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 535–567. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.L.; Bhargava, P.; Cherrouk, I.; Marshall, J.L.; A Flockhart, D.; Wainer, I.W. A discordance of the cytochrome P450 2C19 genotype and phenotype in patients with advanced cancer. Br. J. Clin. Pharmacol. 2000, 49, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.F.; Schneider, V.M.; Frye, C.S.; Feldman, A.M. Plasma levels of TNF-alpha and IL-6 are inversely related to cytochrome P450-dependent drug metabolism in patients with congestive heart failure. J. Card. Fail. 2002, 8, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Ishizawa, Y.; Yasui-Furukori, N.; Takahata, T.; Sasaki, M.; Tateishi, T. The effect of aging on the relationship between the cytochrome P450 2C19 genotype and omeprazole pharmacokinetics. Clin. Pharm. 2005, 44, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Li, X.; Yu, Q.; Liu, Y.; Wang, Y.; Song, H.; Cui, H.; Du, W.; Fei, X.; Liu, J.; et al. Association of P2Y12 Gene Promoter DNA Methylation with the Risk of Clopidogrel Resistance in Coronary Artery Disease Patients. BioMed Res. Int. 2014, 2014, 1–8. [Google Scholar]
- Li, X.G.; Ma, N.; Wang, B.; Li, X.Q.; Mei, S.H.; Zhao, K.; Wang, Y.J.; Li, W.; Zhao, Z.G.; Sun, S.S.; et al. The impact of P2Y12 promoter DNA methylation on the recurrence of ischemic events in Chinese patients with ischemic cerebrovascular disease. Sci. Rep. 2016, 6, 34570. [Google Scholar] [CrossRef]
- Caruso, R.; Rocchiccioli, S.; Gori, A.M.; Cecchettini, A.; Giusti, B.; Parodi, G.; Cozzi, L.; Marcucci, R.; Parolini, M.; Romagnuolo, I.; et al. Inflammatory and antioxidant pattern unbalance in “clopidogrel-resistant” patients during acute coronary syndrome. Mediat. Inflamm. 2015, 2015, 710123. [Google Scholar] [CrossRef]
- Goodacre, R. Metabolomics of a superorganism. J. Nutr. 2007, 137, 259S–266S. [Google Scholar] [CrossRef] [PubMed]
- Senthong, V.; Wang, Z.; Li, X.S.; Fan, Y.; Wu, Y.; Tang, W.H.W.; Hazen, S.L. Intestinal Microbiota-Generated Metabolite Trimethylamine- N- Oxide and 5-Year Mortality Risk in Stable Coronary Artery Disease: The Contributory Role of Intestinal Microbiota in a COURAGE-Like Patient Cohort. J. Am. Hear. Assoc. 2016, 5, e002816. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Wang, Z.; Tang, W.H.W.; Hazen, S.L. Gut Microbe-Generated Trimethylamine N-Oxide From Dietary Choline is Prothrombotic in Subects. Circulation 2017, 135, 1671–1673. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.A.M.; Aronoff, D.M. The influence of non-steroidal anti-inflammatory drugs on the gut microbiome. Clin. Microbiol. Infect. 2016, 22, e171–e178. [Google Scholar] [CrossRef] [PubMed]
- Bentwich, I.; Avniel, A.; Karov, Y.; Aharonov, R.; Gilad, S.; Barad, O.; Barzilai, A.; Einat, P.; Einav, U.; Meiri, E.; et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat. Genet. 2005, 37, 766–770. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef]
- Wu, S.; Huang, S.; Ding, J.; Zhao, Y.; Liang, L.; Liu, T.; Zhan, R.; He, X. Multiple microRNAs modulate p21Cip1/Waf1 expression by directly targeting its 3′ untranslated region. Oncogene 2010, 29, 2302–2308. [Google Scholar] [CrossRef]
- Kim, C.H.; Kim, H.K.; Rettig, R.L.; Kim, J.; Lee, E.T.; Aprelikova, O.; Choi, I.J.; Munroe, D.J.; Green, J.E. miRNA signature associated with outcome of gastric cancer patients following chemotherapy. BMC Med. Genom. 2011, 4, 79. [Google Scholar] [CrossRef]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [PubMed]
- Carnell, D.M.; Smith, R.E.; Daley, F.M.; Barber, P.R.; Hoskin, P.J.; Wilson, G.D.; Murray, G.I.; Everett, S.A. Target validation of cytochrome P450 CYP1B1 in prostate carcinoma with protein expression in associated hyperplastic and premalignant tissue. Int. J. Radiat Oncol. Biol. Phys. 2004, 58, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Dang, Y.; Luo, D.; Rong, M.; Chen, G. Underexpression of miR-34a in hepatocellular carcinoma and its contribution towards enhancement of proliferating inhibitory effects of agents targeting c-MET. PLoS ONE 2013, 8, e61054. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.; Hossain, G.S.; Kocerha, J. The Potential for microRNA Therapeutics and Clinical Research. Front. Genet. 2019, 10, 478. [Google Scholar] [CrossRef]
- Delivering the promise of RNA therapeutics. Nat. Med. 2019, 25, 1321. [CrossRef]
- Di Fusco, D.; Dinallo, V.; Marafini, I.; Figliuzzi, M.M.; Romano, B.; Monteleone, G. Antisense Oligonucleotide: Basic Concepts and Therapeutic Application in Inflammatory Bowel Disease. Front Pharmacol. 2019, 10, 305. [Google Scholar] [CrossRef]
- Kalra, P.; Dhiman, A.; Cho, W.C.; Bruno, J.G.; Sharma, T.K. Simple Methods and Rational Design for Enhancing Aptamer Sensitivity and Specificity. Front Mol Biosci. 2018, 5, 41. [Google Scholar] [CrossRef]
- Soldevilla, M.M.; Meraviglia-Crivelli de Caso, D.; Menon, A.P.; Pastor, F. Aptamer-iRNAs as Therapeutics for Cancer Treatment. Pharmaceuticals (Basel) 2018, 11, 108. [Google Scholar] [CrossRef]
- Li, J.; Wu, C.; Wang, W.; He, Y.; Elkayam, E.; Joshua-Tor, L. Structurally modulated codelivery of siRNA and Argonaute 2 for enhanced RNA interference. Proc. Natl. Acad. Sci. USA 2018, 115, E2696–E2705. [Google Scholar] [CrossRef]
- Lieberman, J. Tapping the RNA world for therapeutics. Nat. Struct. Mol. Biol. 2018, 25, 357–364. [Google Scholar] [CrossRef]
- Owens, J. Determining druggability. Nat. Rev. Drug Discov. 2007, 6, 187. [Google Scholar] [CrossRef]
- Dance, A. Drug Discovery Techniques Open the Door to RNA-targeted Drugs. The Scientist: Exploring Life, Inspiring Innovation. 1 June 2019. Available online: https://www.the-scientist.com/lab-tools/drug-discovery-techniques-open-the-door-to-rna-targeted-drugs-65903 (accessed on 20 September 2019).
- Peer, D.; Lieberman, J. Special delivery: Targeted therapy with small RNAs. Gene Ther. 2011, 18, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, G.R. Drug metabolism and variability among patients in drug response. N. Engl. J. Med. 2005, 352, 2211–2221. [Google Scholar] [CrossRef] [PubMed]
- Dainis, A.M.; Ashley, E.A. Cardiovascular Precision Medicine in the Genomics Era. JACC Basic Transl. Sci 2018, 3, 313–326. [Google Scholar] [CrossRef]
- Phillips, K.A.; Veenstra, D.L.; Oren, E.; Lee, J.K.; Sadee, W. Potential role of pharmacogenomics in reducing adverse drug reactions: A systematic review. JAMA 2001, 286, 2270–2279. [Google Scholar] [CrossRef]
- Bradford, L.D. CYP2D6 allele frequency in European Caucasians, Asians, Africans and their descendants. Pharmacogenomics 2002, 3, 229–243. [Google Scholar] [CrossRef]
- Bernard, S.; Neville, K.; Nguyen, A.; Flockhart, D. Inter-ethnic differences in genetic polymorphisms of CYP2D6 in the U.S. population: Clinical implications. Oncologist 2006, 11, 126–135. [Google Scholar] [CrossRef]
- Abraham, B.K.; Adithan, C.; Mohanasundaram, J.; Shashindran, C.H.; Koumaravelou, K.; Asad, M. Genetic polymorphism of CYP2D6 in Tamil population. Eur. J. Clin. Pharmacol. 2001, 56, 849–850. [Google Scholar] [CrossRef]
- Cavallari, L.H.; Mason, D.L. Cardiovascular Pharmacogenomics—Implications for Patients With CKD. Adv. Chronic Kidney Dis. 2016, 23, 82–90. [Google Scholar] [CrossRef]
- Johnson, J.A.; Gong, L.; Whirl-Carrillo, M.; Gage, B.F.; Scott, S.A.; Stein, C.M.; Anderson, J.L.; Kimmel, S.E.; Lee, M.T.; Pirmohamed, M.; et al. Clinical Pharmacogenetics Implementation Consortium Guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin. Pharmacol. Ther. 2011, 90, 625–629. [Google Scholar] [CrossRef]
- Cavallari, L.H.; Perera, M.A. The future of warfarin pharmacogenetics in under-represented minority groups. Future Cardiol. 2012, 8, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, S.E.; French, B.; Kasner, S.E.; Johnson, J.A.; Anderson, J.L.; Gage, B.F.; Rosenberg, Y.D.; Eby, C.S.; Madigan, R.A.; McBane, R.B.; et al. A pharmacogenetic versus a clinical algorithm for warfarin dosing. N. Engl. J. Med. 2013, 369, 2283–2293. [Google Scholar] [CrossRef] [PubMed]
- Pirmohamed, M.; Burnside, G.; Eriksson, N.; Jorgensen, A.L.; Toh, C.H.; Nicholson, T.; Kesteven, P.; Christersson, C.; Wahlström, B.; Stafberg, C.; et al. A randomized trial of genotype-guided dosing of warfarin. N. Engl. J. Med. 2013, 369, 2294–2303. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, A.; Schulman, S.; Witt, D.M.; Vandvik, P.O.; Fish, J.; Kovacs, M.J.; Svensson, P.J.; Veenstra, D.L.; Crowther, M.; Guyatt, G.H. Evidence-based management of anticoagulant therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012, 141, e152S–e184S. [Google Scholar] [CrossRef] [PubMed]
- Gage, B.F.; Bass, A.R.; Lin, H.; Woller, S.C.; Stevens, S.M.; Al-Hammadi, N.; Li, J.; Rodríguez, T.; Miller, J.P.; McMillin, G.A.; et al. Effect of Genotype-Guided Warfarin Dosing on Clinical Events and Anticoagulation Control Among Patients Undergoing Hip or Knee Arthroplasty: The GIFT Randomized Clinical Trial. JAMA 2017, 318, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Meschia, J.F.; Bushnell, C.; Boden-Albala, B.; Braun, L.T.; Bravata, D.M.; Chaturvedi, S.; Creager, M.A.; Eckel, R.H.; Elkind, M.S.; Fornage, M.; et al. Guidelines for the primary prevention of stroke: A statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2014, 45, 3754–3832. [Google Scholar] [CrossRef] [PubMed]
- Nutescu, E.; Duarte, J.; Cheng, W.; Sarangpur, S.; Gor, D.; Drozda, K.; Galanter, W.; Stamos, T.; Peace, D.; Garofalo, J.; et al. Abstract 16119: Novel Genotype Guided Personalized Warfarin Service Improves Outcomes in an Ethnically Diverse Population. CORE 2. EPIDEMIOLOGY AND PREVENTION OF CV DISEASE: PHYSIOLOGY, PHARMACOLOGY AND LIFESTYLE. Circulation 2014, 130, A16119. [Google Scholar]
- Kim, K.; Lee, T.A.; Touchette, D.R.; DiDomenico, R.J.; Ardati, A.K.; Walton, S.M. Contemporary Trends in Oral Antiplatelet Agent Use in Patients Treated with Percutaneous Coronary Intervention for Acute Coronary Syndrome. J. Manag. Care Spec. Pharm. 2017, 23, 57–63. [Google Scholar] [CrossRef]
- Mauskopf, J.A.; Graham, J.B.; Bae, J.P.; Ramaswamy, K.; Zagar, A.J.; Magnuson, E.A.; Cohen, D.J.; Meadows, E.S. Cost-effectiveness of prasugrel in a US managed care population. J. Med. Econ. 2012, 15, 166–174. [Google Scholar] [CrossRef]
- Coleman, C.I.; Limone, B.L. Cost-effectiveness of universal and platelet reactivity assay-driven antiplatelet therapy in acute coronary syndrome. Am. J. Cardiol. 2013, 112, 355–362. [Google Scholar] [CrossRef]
- Patel, V.; Lin, F.J.; Ojo, O.; Rao, S.; Yu, S.; Zhan, L.; Touchette, D.R. Cost-utility analysis of genotype-guided antiplatelet therapy in patients with moderate-to-high risk acute coronary syndrome and planned percutaneous coronary intervention. Pharm. Pract. 2014, 12, 438. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Claassens, D.M.F.; Vos, G.J.A.; Bergmeijer, T.O.; Hermanides, R.S.; van ‘t Hof, A.W.J.; van der Harst, P.; Barbato, E.; Morisco, C.; Tjon Joe Gin, R.M.; Asselbergs, F.W.; et al. A Genotype-Guided Strategy for Oral P2Y. N. Engl. J. Med. 2019, 381, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Koski, R.; Kennedy, B. Comparative Review of Oral P2Y. Pharm Ther. 2018, 43, 352–357. [Google Scholar]
- Roden, D.M. Clopidogrel Pharmacogenetics—Why the Wait? N. Engl. J. Med. 2019, 381, 1677–1678. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Yin, T.; Li, Y.; Song, L.-Q.; Yu, J.; Si, R.; Zhang, Y.-M.; He, Y.; Guo, W.-Y.; Wang, H.-C. CYP2C19 polymorphisms and coronary heart disease risk factors synergistically impact clopidogrel response variety after percutaneous coronary intervention. Coron. Artery Dis. 2014, 25, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.D.; van Schaik, R.H.; Rivory, L.P.; Ten Tije, A.J.; Dinh, K.; Graveland, W.J.; Schenk, P.W.; Charles, K.A.; Clarke, S.J.; Carducci, M.A.; et al. Factors affecting cytochrome P-450 3A activity in cancer patients. Clin. Cancer Res. 2004, 10, 8341–8350. [Google Scholar] [CrossRef] [PubMed]
- Slaviero, K.A.; Clarke, S.J.; Rivory, L.P. Inflammatory response: An unrecognised source of variability in the pharmacokinetics and pharmacodynamics of cancer chemotherapy. Lancet. Oncol. 2003, 4, 224–232. [Google Scholar] [CrossRef]
- Shedlofsky, S.I.; Israel, B.C.; McClain, C.J.; Hill, D.B.; Blouin, R.A. Endotoxin administration to humans inhibits hepatic cytochrome P450-mediated drug metabolism. J. Clin. Investig. 1994, 94, 2209–2214. [Google Scholar] [CrossRef]
- Brown, S.-A.; Nhola, L.; Herrmann, J. Cardiovascular Toxicities of Small Molecule Tyrosine Kinase Inhibitors: An Opportunity for Systems-Based Approaches. Clin. Pharmacol. Ther. 2017, 101, 65–80. [Google Scholar] [CrossRef]
- Jones, L.W.; Haykowsky, M.J.; Swartz, J.J.; Douglas, P.S.; Mackey, J.R. Early breast cancer therapy and cardiovascular injury. J. Am. Coll. Cardiol. 2007, 50, 1435–1441. [Google Scholar] [CrossRef]
- Garcia-Pavia, P.; Kim, Y.; Restrepo-Cordoba, M.A.; Lunde, I.G.; Wakimoto, H.; Smith, A.M.; Toepfer, C.N.; Getz, K.; Gorham, J.; Patel, P.; et al. Genetic Variants Associated With Cancer Therapy-Induced Cardiomyopathy. Circulation 2019, 140, 31–41. [Google Scholar] [CrossRef] [PubMed]
- McGowan, J.V.; Chung, R.; Maulik, A.; Piotrowska, I.; Walker, J.M.; Yellon, D.M. Anthracycline Chemotherapy and Cardiotoxicity. Cardiovasc. Drugs Ther. 2017, 31, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Saidi, A.; Alharethi, R. Management of chemotherapy induced cardiomyopathy. Curr. Cardiol. Rev. 2011, 7, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Pasipoularides, A. Implementing genome-driven personalized cardiology in clinical practice. J. Mol. Cell Cardiol. 2018, 115, 142–157. [Google Scholar] [CrossRef]



| Enzyme | Upper Limit of Normal Percentage of Total Hepatic CYP450 (%) |
|---|---|
| CYP3A4 | 37 |
| CYP3A5 | 1 |
| CYP2C9 | 29 |
| CYP1A2 | 16.3 |
| CYP2A6 | 14 |
| CYP2B6 | 5.3 |
| CYP2D6 | 4.3 |
| CYP2C19 | 3.8 |
| Enzyme | Substrate Characteristics | Examples of Drugs Relevant to Cardiovascular Care | Inhibitors | Inducers |
|---|---|---|---|---|
| CYP3A4 and CYP3A5 | Large & lipophilic molecules, with very diverse structures; includes over 50% of all clinically used drugs CCBs Statins Taxanes (paclitaxel and docetaxel) Sorafenib Dasatinib Cyclophosphamide Proteosome inhibitors (e.g., Bortezomib) Everolimus Cytarabine Dabrafenib Vemurafenib Irinotecan Imatinib/Ibrutinib | Antiarrhythmics: quinidine-3-OH (not 3A5) Calcium Channel Blockers: amlodipine, diltiazem, felodipine, lercanidipine, nifedipine2, nisoldipine, nitrendipine, verapamil HMG CoA Reductase Inhibitors: atorvastatin, cerivastatin, lovastatin, NOT pravastatin, NOT rosuvastatin, simvastatin Propranolol Cilostazol Eplerenone Fentanyl Lidocaine Others: Ondansetron Caffeine-trimethyluric acid Sorafenib | Strong: (Protease Inhibitors) indinavir, nelfinavir, ritonavir, saquinavir, (Antibacterials): clarithromycin, erythromycin, telithromycin; chloramphenicol (Antifungal): itraconazole, ketoconazole, fluconazole, voriconazole (Antidepressant): nefazodone (Vasopressin antagonist): conivaptan Moderate: (Antiemetic): aprepitant (Antibacterials): erythromycin, (Antifungal): fluconazole, (nDP-CCB): verapamil, diltiazem. Mibefradil (Immune modulating agents): Cyclosporine, Tacrolimus (Tyrosine kinase inhibitors -TKIs): Nilotinib, Iapatinib (Hormonal agents): Enzalutamide, Bicalutamide (Chemotherapy): Sorafenib (Misc.): grapefruit juice, starfruit Weak: (H2 blockers): cimetidine (Topoisomerase inhibitors): Etoposide (Anthracyclines): Idarubicine (Alkylating agents): Cyclophosphamide (Antibacterials): ciprofloxacin, norfloxacin (Antifungal): voriconazole, ketoconazole, itraconazole, posaconazole, fluconazole (NNRTI): delavirdine (Antiarrhythmics): amiodarone (SSRIs): fluvoxamine, norfluoxetine (Protease Inhibitors): boceprevir, telaprevir (OCP): * gestodene, mifepristone (Chemotherapy): imatinib (Misc.): starfruit | (NNRTI): efavirenz, nevirapine, efavirenz/emtricitabine/tenofovir (GABA-Agonists): barbiturates, phenobarbital, (Anti-epileptics): carbamazepine, oxcarbazepine, phenytoin (Non-steroidal Anti-androgen): enzalutamide (Antibiotics): rifabutin, rifampin (Misc.): glucocorticoids, modafinil, St. John’s Wort (Thiazolidinedione): pioglitazone troglitazone (Antimitotic agents): Paclitaxel (TKIs): Vemurafenib, Dabrafenib (Hormonal agents): enzalutamide (Angiogenesis inhibitor): Thalidomide (BRAF inhibitor): Vemurafenib |
| CYP2C9 | Relatively large and weakly acidic molecules; includes antimalarials and oral antidiabetics Fluvastatin Nateglinide phenytoin-4-OH2 rosiglitazone | Angiotensin II Blockers: losartan irbesartan, valsartan Torsemide S-Warfarin Fluvastatin Rosiglitazone Others: NSAIDs, Sulfonylureas | Strong: fluconazole2 Moderate: amiodarone (NNRTI): efavirenz (Fibrate): fenofibrate (Antifungal): fluconazole, voriconazole (Statin): fluvastatin, lovastatin (SSRI): fluvoxamine2, paroxetine, sertraline (Antibiotic): isoniazid, metronidazole * phenylbutazone, sulfamethoxazole * sulfaphenazole, (Chemotherapeutic): teniposide, 5-flourouracil (Leukotrieine receptor antag LTRA): zafirlukast | (Non-steroidal Anti-androgen): enzalutamide (~3A4/5/7, 2C19) (NNRTI) Nevirapine (Antibiotics): Rifampin (Antiepileptics): phenobarbital, * secobarbital, carbamazepine (Misc.): St. John’s Wort (~3A4,5,7) |
| CYP2C8 | Relatively large and weakly acidic molecules; includes antimalarials and oral antidiabetics Docetaxel Imatinib/Ibrutinib | Torsemide Cerivastatin Amiodarone (n) Thiazoladinedione (Pioglitazone, Rosiglitazone) Others: Repaglinide | Strong: gemfibrozil Moderate: trimethoprim (Thiazolidinediones): glitazones, (LTRA): montelukast (Plant flavonoid): quercetin (found in fruits, vegetables, leaves and grains; red onions and kale) | Rifampin |
| CYP2E1 | Small, generally neutral and hydrophilic, planar molecules; includes aliphatic alcohols and halogenated alkanes Cisplatin | Ethanol | Disulfiram | Ethanol Isoniazid |
| CYP1A2 | Planar, aromatic, polyaromatic and heterocyclic amides and amines | Caffeine Naproxen Ondansetron | Strong: fluvoxamine, ciprofloxacin Moderate: Vemurafenib Weak: cimetidine amiodarone, efavirenz, fluoroquinolones, fluvoxamine, furafylline1, interferon, * methoxsalen, * mibefradil, ticlopidine | (Food): broccoli, brussels sprouts, char-grilled meat (AEDs): carbamazepine (Diabetic meds) insulin (Misc.): Modafinil (Antibiotic): Nafcillin, Rifampin (PPI): Omeprazole (Toxins): tobacco |
| CYP2A6 | Nonplanar low molecular weight molecules usually with 2 hydrogen bond acceptors; includes ketones and nitrosamines | |||
| CYP2D6 | Basic molecules with protonatable nitrogen atom(4–7) Å from the metabolism site; includes many plant alkaloids and antidepressants Proteosome inhibitors | Ondansetron | Strong: bupropion, cinacalcet, fluoxetine, paroxetine, quinidine Moderate: duloxetine, sertraline, terbinafine, sorafenib Weak: amiodarone, cimetidine (NSAID): celecoxib (Antihistamine): chlorpheniramine, * clemastine, diphenhydramine, doxepin, histamine H1 receptor antagonists, hydroxyzine, promethazine, tripelennamine (Antipsychotic): chlorpromazine (SSRI): citalopram, escitalopram (TCA): clomipramine (ChemoRx): doxorubicin, imatinib (Antimalarial): halofantrine (Antipsychotic): haloperidol, levomepromazine, perphenazine (Opioids): methadone (DA agonist, Prokinetic): metoclopramide * mibefradil (Vasopressor): midodrine * moclobemide (H2 blocker): ranitidine (protease inhibitor): ritonavir (Antiplatelet): ticlopidine (Misc.): cocaine | Dexamethasone Rifampin |
| CYP2B6 | Neutral or weakly basic, mostly lipophilic non-planar molecules with 1 to 2 hydrogen bond acceptors; includes anesthetics, insecticides and herbicides cyclophosphamide | N/A | Antiplatelets: clopidogrel, ticlopidine2, Antifungal: voriconazole, Chemotherapeutic: thiotepa | Artemisinin (AED): Carbamazepine, Phenobarbital, Phenytoin (NNRTI): Efavirenz, Nevirapine Rifampin (induces every listed CYP enzyme except 2E1) |
| CYP2C19 | Neutral or weakly basic molecules or amides with 2 or 3 hydrogen bond acceptors; includes most proton pump inhibitors Proteosome inhibitors cyclophosphamide | Clopidogrel Labetalol Propranolol R-warfarin→8-OH Others: PPIs: Esomeprazole Lansoprazole Omeprazole2 Pantoprazole | (PPIs): esomeprazole, lansoprazole, omeprazole2, pantoprazole (Antibiotic): chloramphenicol, isoniazid (Antifungal): ketoconazole, voriconazole (H2 blocker): cimetidine (SSRI): fluoxetine, fluvoxamine (NSAID): indomethacin (Dopaminergic): modafinil oral contraceptives (Antiepileptics): oxcarbazepine topiramate (Antiplatelet): ticlopidine (Chemotherapy): sorafenib (Misc.): probenecid | (AED): carbamazepine (NNRTI): efavirenz (Protease Inhibitor): ritonavir (Non-steroidal Anti-androgen): enzalutamide (~3A4/5/7, 2C9) (NNRTI) (OCP): norethindrone (Misc.): prednisone, St. John’s Wort (~3A4/5/7, 2C9) (Antibiotics): Rifampicin |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fatunde, O.A.; Brown, S.-A. The Role of CYP450 Drug Metabolism in Precision Cardio-Oncology. Int. J. Mol. Sci. 2020, 21, 604. https://doi.org/10.3390/ijms21020604
Fatunde OA, Brown S-A. The Role of CYP450 Drug Metabolism in Precision Cardio-Oncology. International Journal of Molecular Sciences. 2020; 21(2):604. https://doi.org/10.3390/ijms21020604
Chicago/Turabian StyleFatunde, Olubadewa A., and Sherry-Ann Brown. 2020. "The Role of CYP450 Drug Metabolism in Precision Cardio-Oncology" International Journal of Molecular Sciences 21, no. 2: 604. https://doi.org/10.3390/ijms21020604
APA StyleFatunde, O. A., & Brown, S.-A. (2020). The Role of CYP450 Drug Metabolism in Precision Cardio-Oncology. International Journal of Molecular Sciences, 21(2), 604. https://doi.org/10.3390/ijms21020604