Genome Editing for Mucopolysaccharidoses




Abstract
1. Introduction: Therapeutic Principles in Mucopolysaccharidoses
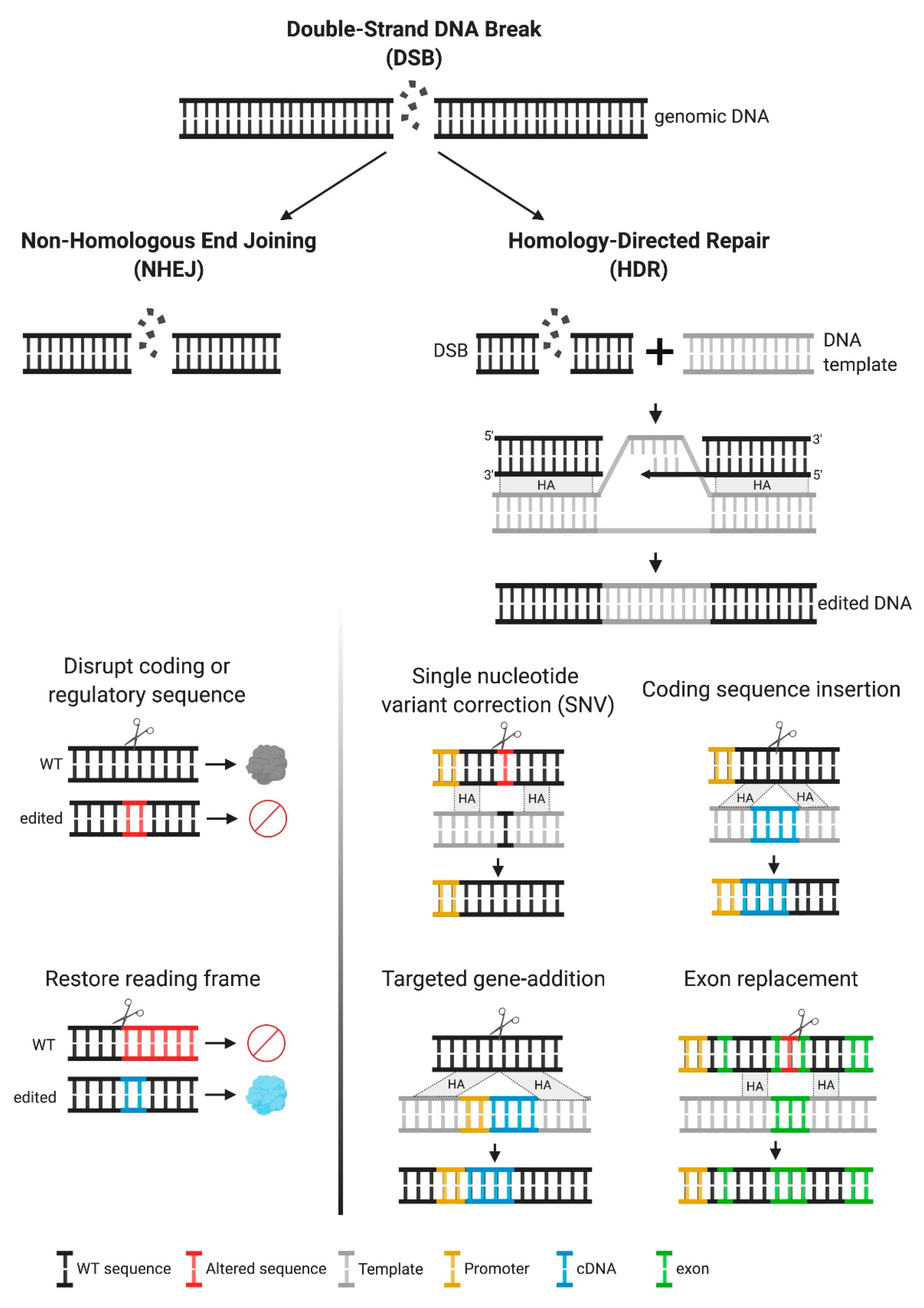
2. Gene Editing: The basics
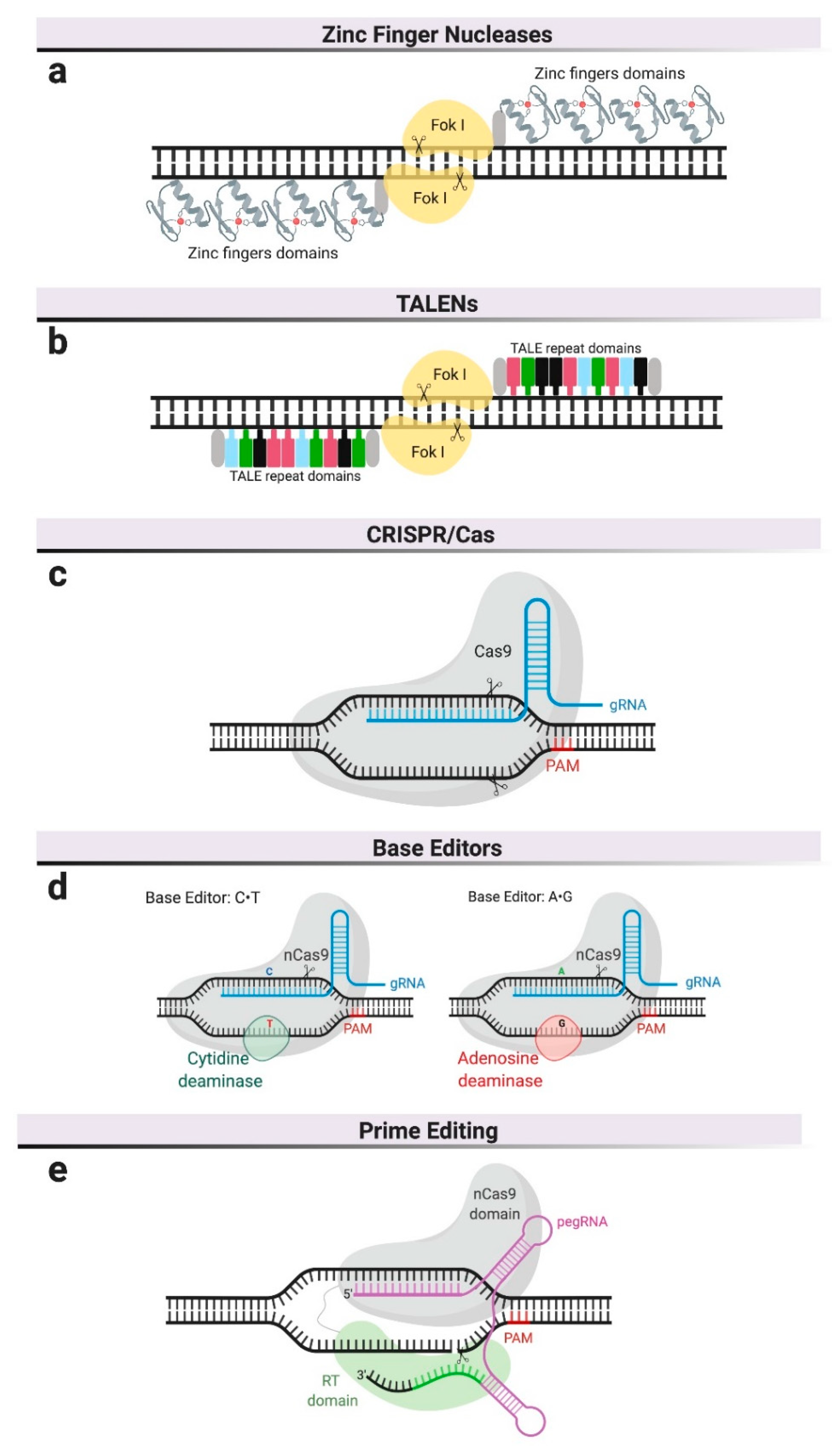
2.1. Genome Editing Platforms
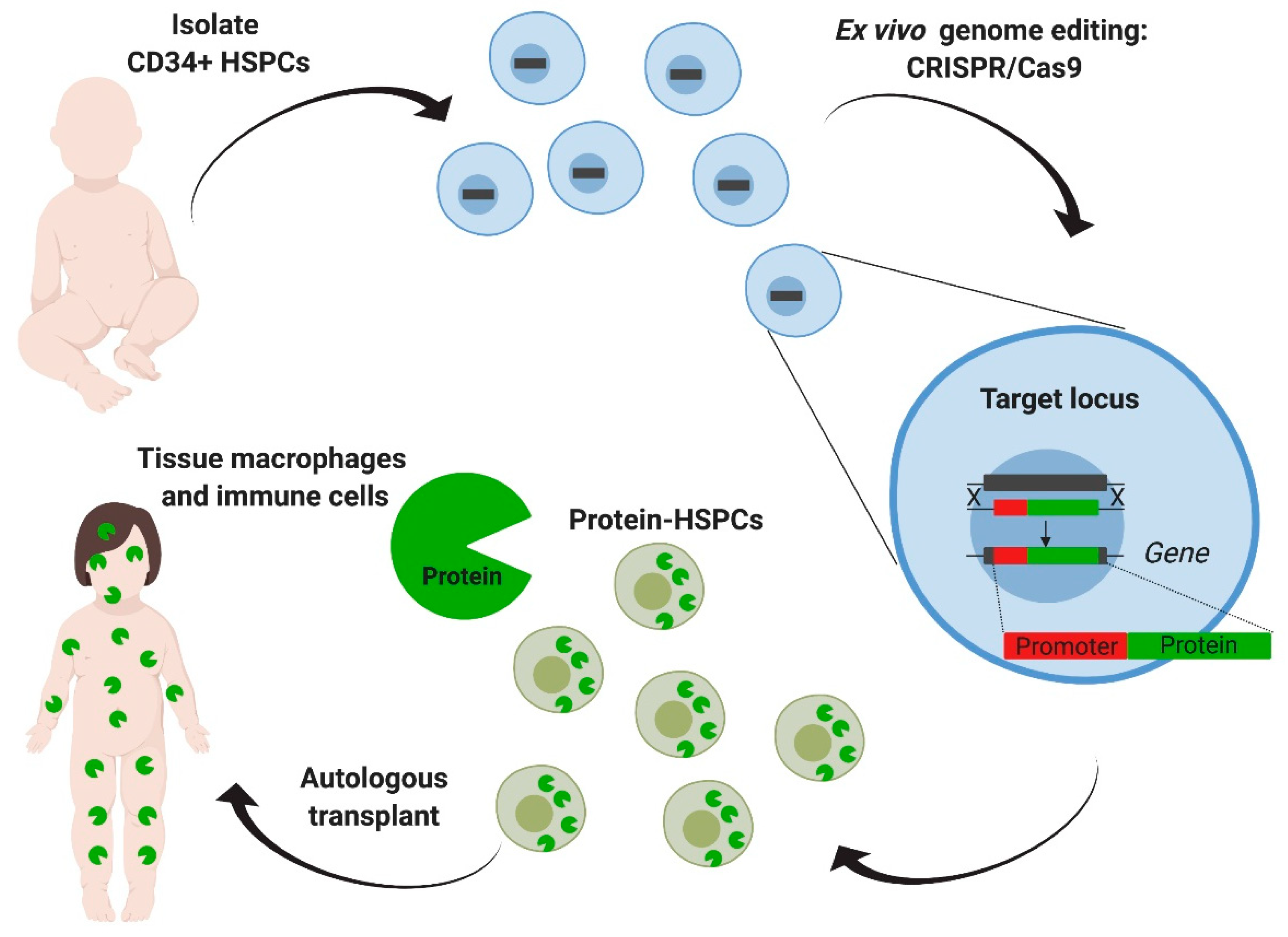
2.2. Multiple Genetic Modifications and Their Therapeutic Applications
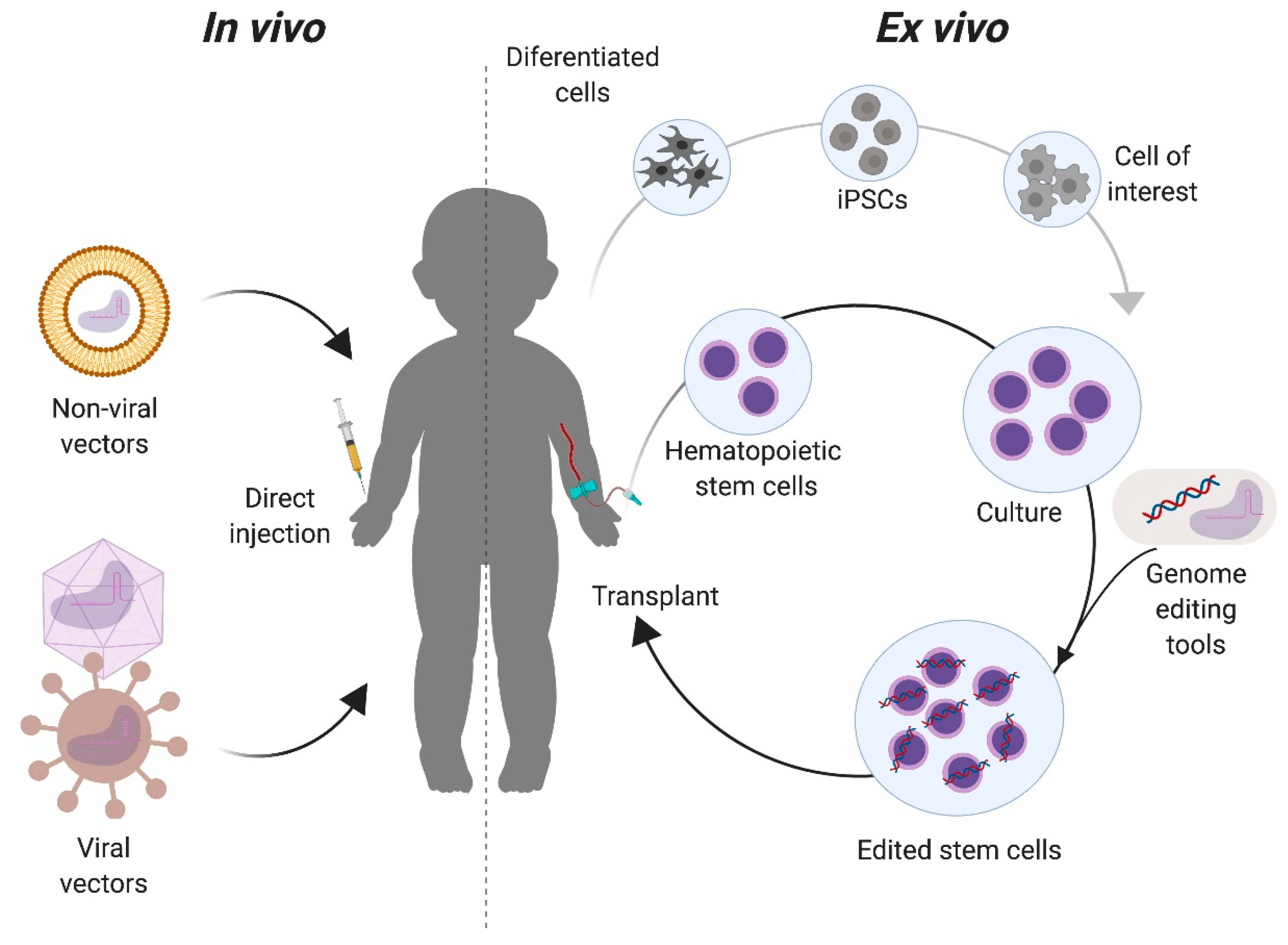
2.3. Delivery Platforms: Ex Vivo vs. In Vivo Genome Editing
3. Genome Editing vs. Other Therapeutic Approaches in MPS Disorders
3.1. Enzyme Replacement Therapy (ERT)
3.2. Substrate Reduction Therapy
3.3. In Vivo Gene Therapy with Adeno-Associated Viruses (AAV)
3.4. Allogeneic Hematopoietic Stem Cell Transplantation
3.5. Ex Vivo Lentiviral Modification of Hematopoietic Stem and Progenitor Cells
4. From Proof of Concept Studies in Animal Models to Clinical Trials
4.1. In Vivo Approaches
4.2. Ex Vivo Approaches
5. Challenges to the Clinical Adaptation of Genome Editing in MPSs
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Giugliani, R.; Iii, I.I. Mucopolysacccharidoses: From understanding to treatment, a century of discoveries. Genet. Mol. Boil. 2012, 35, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Hřebíček, M.; Mrázová, L.S.; Seyrantepe, V.; Durand, S.; Roslin, N.M.; Nosková, L.; Hartmannová, H.; Ivanek, R.; Čížková, A.; Poupetova, H.; et al. Mutations in TMEM76* Cause Mucopolysaccharidosis IIIC (Sanfilippo C Syndrome). Am. J. Hum. Genet. 2006, 79, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Fratantoni, J.C.; Hall, C.W.; Neufeld, E.F. Hurler and Hunter syndromes: Mutual correction of the defect in cultured fibroblasts. Science 1968, 162, 570–572. [Google Scholar] [CrossRef]
- Baldo, G.; Giugliani, R.; Matte, U. Gene delivery strategies for the treatment of mucopolysaccharidoses. Expert Opin. Drug Deliv. 2014, 11, 449–459. [Google Scholar] [CrossRef]
- Beck, M. Treatment strategies for lysosomal storage disorders. Dev. Med. Child Neurol. 2018, 60, 13–18. [Google Scholar] [CrossRef]
- Christensen, C.L.; Ashmead, R.E.; Choy, F.Y.M. Cell and Gene Therapies for Mucopolysaccharidoses: Base Editing and Therapeutic Delivery to the CNS. Diseases 2019, 7, 47. [Google Scholar] [CrossRef]
- Bak, R.O.; Gomez-Ospina, N.; Porteus, M.H. Gene Editing on Center Stage. Trends Genet. 2018, 34, 600–611. [Google Scholar] [CrossRef]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; East, A.; Cheng, A.; Lin, S.; Ma, E.; Doudna, J. RNA-programmed genome editing in human cells. eLife 2013, 2, e00471. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Güell, M.; Dicarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef]
- Kim, D.; Lim, K.; Kim, S.T.; Yoon, S.H.; Kim, K.; Ryu, S.M.; Kim, J.S. Genome-wide target specificities of CRISPR RNA-guided programmable deaminases. Nat. Biotechnol. 2017, 35, 475–480. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Psatha, N.; Reik, A.; Phelps, S.; Zhou, Y.; Dalas, D.; Yannaki, E.; Levasseur, D.N.; Urnov, F.D.; Holmes, M.C.; Papayannopoulou, T. Disruption of the BCL11A Erythroid Enhancer Reactivates Fetal Hemoglobin in Erythroid Cells of Patients with beta-Thalassemia Major. Mol. Ther. Methods Clin. Dev. 2018, 10, 313–326. [Google Scholar] [CrossRef]
- Amoasii, L.; Long, C.; Li, H.; Mireault, A.A.; Shelton, J.M.; Sanchez-Ortiz, E.; McAnally, J.R.; Bhattacharyya, S.; Schmidt, F.; Grimm, D.; et al. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci. Transl. Med. 2017, 9, 418. [Google Scholar] [CrossRef]
- Ousterout, D.G.; Perez-Pinera, P.; Thakore, P.I.; Kabadi, A.M.; Brown, M.T.; Qin, X.; Fedrigo, O.; Mouly, V.; Tremblay, J.P.; Gersbach, C.A. Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne muscular dystrophy patients. Mol Ther. 2013, 21, 1718–1726. [Google Scholar] [CrossRef]
- Dever, D.P.; Bak, R.O.; Reinisch, A.; Camarena, J.; Washington, G.; Nicolas, C.E.; Pavel-Dinu, M.; Saxena, N.; Wilkens, A.B.; Mantri, S.; et al. CRISPR/Cas9 beta-globin gene targeting in human haematopoietic stem cells. Nature 2016, 539, 384–389. [Google Scholar] [CrossRef] [PubMed]
- DeWitt, M.A.; Magis, W.; Bray, N.L.; Wang, T.; Berman, J.R.; Urbinati, F.; Heo, S.J.; Mitros, T.; Munoz, D.P.; Boffelli, D.; et al. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci. Transl. Med. 2016, 8, 360ra134. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Anguela, X.M.; Doyon, Y.; Wechsler, T.; DeKelver, R.C.; Sproul, S.; Paschon, D.E.; Miller, J.C.; Davidson, R.J.; Shivak, D.; et al. In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood 2015, 126, 1777–1784. [Google Scholar] [CrossRef] [PubMed]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef]
- Robert, F.; Barbeau, M.; Ethier, S.; Dostie, J.; Pelletier, J. Pharmacological inhibition of DNA-PK stimulates Cas9-mediated genome editing. Genome Med. 2015, 7, 93. [Google Scholar] [CrossRef]
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015, 33, 538–542. [Google Scholar] [CrossRef]
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kuhn, R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef]
- Yu, S.; Song, Z.; Luo, J.; Dai, Y.; Li, N. Over-expression of RAD51 or RAD54 but not RAD51/4 enhances extra-chromosomal homologous recombination in the human sarcoma (HT-1080) cell line. J. Biotechnol. 2011, 154, 21–24. [Google Scholar] [CrossRef]
- Charpentier, M.; Khedher, A.H.Y.; Menoret, S.; Brion, A.; Lamribet, K.; Dardillac, E.; Boix, C.; Perrouault, L.; Tesson, L.; Geny, S.; et al. CtIP fusion to Cas9 enhances transgene integration by homology-dependent repair. Nat. Commun. 2018, 9, 1133. [Google Scholar] [CrossRef]
- Schiroli, G.; Conti, A.; Ferrari, S.; Della Volpe, L.; Jacob, A.; Albano, L.; Beretta, S.; Calabria, A.; Vavassori, V.; Gasparini, P.; et al. Precise Gene Editing Preserves Hematopoietic Stem Cell Function following Transient p53-Mediated DNA Damage Response. Cell Stem Cell 2019, 24, 551–565.e8. [Google Scholar] [CrossRef]
- Yang, D.; Scavuzzo, M.A.; Chmielowiec, J.; Sharp, R.; Bajic, A.; Borowiak, M. Enrichment of G2/M cell cycle phase in human pluripotent stem cells enhances HDR-mediated gene repair with customizable endonucleases. Sci. Rep. 2016, 6, 21264. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 2014, 3, e04766. [Google Scholar] [CrossRef] [PubMed]
- Savic, N.; Ringnalda, F.C.; Lindsay, H.; Berk, C.; Bargsten, K.; Li, Y.; Neri, D.; Robinson, M.D.; Ciaudo, C.; Hall, J.; et al. Covalent linkage of the DNA repair template to the CRISPR-Cas9 nuclease enhances homology-directed repair. Elife 2018, 7, e33761. [Google Scholar] [CrossRef]
- Dever, D.P.; Porteus, M.H. The changing landscape of gene editing in hematopoietic stem cells: A step towards Cas9 clinical translation. Curr. Opin. Hematol. 2017, 24, 481–488. [Google Scholar] [CrossRef]
- Mussolino, C.; Alzubi, J.; Pennucci, V.; Turchiano, G.; Cathomen, T. Genome and Epigenome Editing to Treat Disorders of the Hematopoietic System. Hum. Gene Ther. 2017, 28, 1105–1115. [Google Scholar] [CrossRef]
- Hasilik, A.; Neufeld, E.F. Biosynthesis of lysosomal enzymes in fibroblasts. Phosphorylation of mannose residues. J. Biol. Chem. 1980, 255, 4946–4950. [Google Scholar]
- Kakkis, E.D.; Muenzer, J.; Tiller, G.E.; Waber, L.; Belmont, J.; Passage, M.; Izykowski, B.; Phillips, J.; Doroshow, R.; Walot, I.; et al. Enzyme-replacement therapy in mucopolysaccharidosis I. N. Engl. J. Med. 2001, 344, 182–188. [Google Scholar] [CrossRef]
- Muenzer, J.; Wraith, J.E.; Beck, M.; Giugliani, R.; Harmatz, P.; Eng, C.M.; Vellodi, A.; Martin, R.; Ramaswami, U.; Gucsavas-Calikoglu, M.; et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet. Med. 2006, 8, 465–473. [Google Scholar] [CrossRef]
- Hendriksz, C.J.; Burton, B.; Fleming, T.R.; Harmatz, P.; Hughes, D.; Jones, S.A.; Lin, S.P.; Mengel, E.; Scarpa, M.; Valayannopoulos, V.; et al. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): A phase 3 randomised placebo-controlled study. J. Inherit. Metab. Dis. 2014, 37, 979–990. [Google Scholar] [CrossRef]
- Harmatz, P.; Ketteridge, D.; Giugliani, R.; Guffon, N.; Teles, E.L.; Miranda, M.C.; Yu, Z.F.; Swiedler, S.J.; Hopwood, J.J.; Group, M.V.S. Direct comparison of measures of endurance, mobility, and joint function during enzyme-replacement therapy of mucopolysaccharidosis VI (Maroteaux-Lamy syndrome): Results after 48 weeks in a phase 2 open-label clinical study of recombinant human N-acetylgalactosamine 4-sulfatase. Pediatrics 2005, 115, e681–e689. [Google Scholar] [PubMed]
- Harmatz, P.; Whitley, C.B.; Waber, L.; Pais, R.; Steiner, R.; Plecko, B.; Kaplan, P.; Simon, J.; Butensky, E.; Hopwood, J.J. Enzyme replacement therapy in mucopolysaccharidosis VI (Maroteaux-Lamy syndrome). J. Pediatr. 2004, 144, 574–580. [Google Scholar] [CrossRef]
- Harmatz, P.; Whitley, C.B.; Wang, R.Y.; Bauer, M.; Song, W.; Haller, C.; Kakkis, E. A novel Blind Start study design to investigate vestronidase alfa for mucopolysaccharidosis VII, an ultra-rare genetic disease. Mol. Genet. Metab. 2018, 123, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, R.H. Enzyme replacement therapy for lysosomal storage diseases. Curr. Opin. Pediatr. 2011, 23, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lozier, J.; Johnson, G.; Kirshner, S.; Verthelyi, D.; Pariser, A.; Shores, E.; Rosenberg, A. Neutralizing antibodies to therapeutic enzymes: Considerations for testing, prevention and treatment. Nat. Biotechnol. 2008, 26, 901–908. [Google Scholar] [CrossRef]
- Boado, R.J.; Pardridge, W.M. Brain and Organ Uptake in the Rhesus Monkey in Vivo of Recombinant Iduronidase Compared to an Insulin Receptor Antibody–Iduronidase Fusion Protein. Mol. Pharm. 2017, 14, 1271–1277. [Google Scholar] [CrossRef]
- Sonoda, H.; Morimoto, H.; Yoden, E.; Koshimura, Y.; Kinoshita, M.; Golovina, G.; Takagi, H.; Yamamoto, R.; Minami, K.; Mizoguchi, A.; et al. A Blood-Brain-Barrier-Penetrating Anti-human Transferrin Receptor Antibody Fusion Protein for Neuronopathic Mucopolysaccharidosis II. Mol. Ther. 2018, 26, 1366–1374. [Google Scholar] [CrossRef]
- Dickson, P.I. Novel treatments and future perspectives: Outcomes of intrathecal drug delivery. Int. J. Clin. Pharmacol. Ther. 2009, 47 (Suppl. 1), S124–S127. [Google Scholar]
- Wyatt, K.; Henley, W.; Anderson, L.; Anderson, R.; Nikolaou, V.; Stein, K.; Klinger, L.; Hughes, D.; Waldek, S.; Lachmann, R.; et al. The effectiveness and cost-effectiveness of enzyme and substrate replacement therapies: A longitudinal cohort study of people with lysosomal storage disorders. Health Technol. Assess. 2012, 16, 1–543. [Google Scholar] [CrossRef]
- Arruda, V.R.; Favaro, P.; Finn, J.D. Strategies to Modulate Immune Responses: A New Frontier for Gene Therapy. Mol. Ther. 2009, 17, 1492–1503. [Google Scholar] [CrossRef]
- Coutinho, M.F.; Santos, J.I.; Alves, S. Less Is More: Substrate Reduction Therapy for Lysosomal Storage Disorders. Int. J. Mol. Sci. 2016, 17, 1065. [Google Scholar] [CrossRef] [PubMed]
- Lukina, E.; Watman, N.; Arreguin, E.A.; Dragosky, M.; Iastrebner, M.; Rosenbaum, H.; Phillips, M.; Pastores, G.M.; Kamath, R.S.; Rosenthal, D.I.; et al. Improvement in hematological, visceral, and skeletal manifestations of Gaucher disease type 1 with oral eliglustat tartrate (Genz-112638) treatment: 2-year results of a phase 2 study. Blood 2010, 116, 4095–4098. [Google Scholar] [CrossRef]
- Pastores, G.M.; Barnett, N.L.; Kolodny, E.H. An open-label, noncomparative study of miglustat in type I Gaucher disease: Efficacy and tolerability over 24 months of treatment. Clin. Ther. 2005, 27, 1215–1227. [Google Scholar] [CrossRef]
- Guffon, N.; Bin-Dorel, S.; Decullier, E.; Paillet, C.; Guitton, J.; Fouilhoux, A. Evaluation of Miglustat Treatment in Patients with Type III Mucopolysaccharidosis: A Randomized, Double-Blind, Placebo-Controlled Study. J. Pediatr. 2011, 159, 838–844.e1. [Google Scholar] [CrossRef]
- Derrick-Roberts, A.L.K.; Jackson, M.R.; Pyragius, C.E.; Byers, S. Substrate Deprivation Therapy to Reduce Glycosaminoglycan Synthesis Improves Aspects of Neurological and Skeletal Pathology in MPS I Mice. Diseases 2017, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Malinowska, M.; Wilkinson, F.L.; Bennett, W.; Langford-Smith, K.J.; O’Leary, H.A.; Jakobkiewicz-Banecka, J.; Wynn, R.; Wraith, J.E.; Wegrzyn, G.; Bigger, B.W. Genistein reduces lysosomal storage in peripheral tissues of mucopolysaccharide IIIB mice. Mol. Genet. Metab. 2009, 98, 235–242. [Google Scholar] [CrossRef]
- de Ruijter, J.; Valstar, M.J.; Narajczyk, M.; Wegrzyn, G.; Kulik, W.; Ijlst, L.; Wagemans, T.; van der Wal, W.M.; Wijburg, F.A. Genistein in Sanfilippo disease: A randomized controlled crossover trial. Ann. Neurol. 2012, 71, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.M.; Amato, D.; Hollak, C.E.; Luzy, C.; Silkey, M.; Giorgino, R.; Steiner, R.D. Evaluation of miglustat as maintenance therapy after enzyme therapy in adults with stable type 1 Gaucher disease: A prospective, open-label non-inferiority study. Orphanet J. Rare Dis. 2012, 7, 102. [Google Scholar] [CrossRef]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther. Methods Clin. Dev. 2018, 8, 87–104. [Google Scholar] [CrossRef]
- Deyle, D.R.; Russell, D.W. Adeno-associated virus vector integration. Curr. Opin. Mol. Ther 2009, 11, 442–447. [Google Scholar]
- Mitchell, R.; Nivison-Smith, I.; Anazodo, A.; Tiedemann, K.; Shaw, P.J.; Teague, L.; Fraser, C.J.; Carter, T.L.; Tapp, H.; Alvaro, F.; et al. Outcomes of haematopoietic stem cell transplantation for inherited metabolic disorders: A report from the Australian and New Zealand Children’s Haematology Oncology Group and the Australasian Bone Marrow Transplant Recipient Registry. Pediatr. Transplant. 2013, 17, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Okuyama, T.; Suzuki, Y.; Sakai, N.; Takakura, H.; Sawada, T.; Tanaka, T.; Otomo, T.; Ohashi, T.; Ishige-Wada, M.; et al. Long-term efficacy of hematopoietic stem cell transplantation on brain involvement in patients with mucopolysaccharidosis type II: A nationwide survey in Japan. Mol. Genet. Metab. 2012, 107, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Wynn, R.F.; Wraith, J.E.; Mercer, J.; O’Meara, A.; Tylee, K.; Thornley, M.; Church, H.J.; Bigger, B.W. Improved metabolic correction in patients with lysosomal storage disease treated with hematopoietic stem cell transplant compared with enzyme replacement therapy. J. Pediatr. 2009, 154, 609–611. [Google Scholar] [CrossRef] [PubMed]
- Muenzer, J.; Fisher, A. Advances in the treatment of mucopolysaccharidosis type I. N. Engl. J. Med. 2004, 350, 1932–1934. [Google Scholar] [CrossRef]
- Shapiro, E.; Guler, O.E.; Rudser, K.; Delaney, K.; Bjoraker, K.; Whitley, C.; Tolar, J.; Orchard, P.; Provenzale, J.; Thomas, K.M. An exploratory study of brain function and structure in mucopolysaccharidosis type I: Long term observations following hematopoietic cell transplantation (HCT). Mol. Genet. Metab. 2012, 107, 116–121. [Google Scholar] [CrossRef]
- Weisstein, J.S.; Delgado, E.; Steinbach, L.S.; Hart, K.; Packman, S. Musculoskeletal manifestations of Hurler syndrome: Long-term follow-up after bone marrow transplantation. J. Pediatr. Orthop. 2004, 24, 97–101. [Google Scholar] [CrossRef]
- Martins, A.M.; Dualibi, A.P.; Norato, D.; Takata, E.T.; Santos, E.S.; Valadares, E.R.; Porta, G.; de Luca, G.; Moreira, G.; Pimentel, H.; et al. Guidelines for the Management of Mucopolysaccharidosis Type I. J. Pediatr. 2009, 155, S32–S46. [Google Scholar] [CrossRef]
- Montano, A.M.; Lock-Hock, N.; Steiner, R.D.; Graham, B.H.; Szlago, M.; Greenstein, R.; Pineda, M.; Gonzalez-Meneses, A.; Coker, M.; Bartholomew, D.; et al. Clinical course of sly syndrome (mucopolysaccharidosis type VII). J. Med. Genet. 2016, 53, 403–418. [Google Scholar] [CrossRef]
- Yabe, H.; Tanaka, A.; Chinen, Y.; Kato, S.; Sawamoto, K.; Yasuda, E.; Shintaku, H.; Suzuki, Y.; Orii, T.; Tomatsu, S. Hematopoietic stem cell transplantation for Morquio A syndrome. Mol. Genet. Metab. 2016, 117, 84–94. [Google Scholar] [CrossRef]
- Shapiro, E.G.; Lockman, L.A.; Balthazor, M.; Krivit, W. Neuropsychological outcomes of several storage diseases with and without bone marrow transplantation. J. Inherit. Metab. Dis. 1995, 18, 413–429. [Google Scholar] [CrossRef]
- Vellodi, A.; Young, E.; Cooper, A.; Lidchi, V.; Winchester, B.; Wraith, J.E. Long-term follow-up following bone marrow transplantation for Hunter disease. J. Inherit. Metab. Dis. 1999, 22, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A. Hematopoietic Stem Cell Gene Therapy for Storage Disease: Current and New Indications. Mol. Ther. 2017, 25, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Visigalli, I.; Delai, S.; Ferro, F.; Cecere, F.; Vezzoli, M.; Sanvito, F.; Chanut, F.; Benedicenti, F.; Spinozzi, G.; Wynn, R.; et al. Preclinical testing of the safety and tolerability of LV-mediated above normal alpha-L-iduronidase expression in murine and human hematopoietic cells using toxicology and biodistribution GLP studies. Hum. Gene Ther. 2016, 27, 813–829. [Google Scholar] [CrossRef]
- Visigalli, I.; Delai, S.; Politi, L.S.; Di Domenico, C.; Cerri, F.; Mrak, E.; D’Isa, R.; Ungaro, D.; Stok, M.; Sanvito, F.; et al. Gene therapy augments the efficacy of hematopoietic cell transplantation and fully corrects mucopolysaccharidosis type I phenotype in the mouse model. Blood 2010, 116, 5130–5139. [Google Scholar] [CrossRef]
- Gentner, B.; Bernardo, M.; Zonari, E.; Tucci, F.; Fumagalli, F.; Redaelli, D.; Acquati, S.; Silvani, P.; Parini, R.; La Marca, G.; et al. Ex-Vivo Gene Therapy for Hurler Disease: Initial Results from a Phase I/II Clinical Study. Mol. Ther 2019, 27, 1. [Google Scholar]
- Biasco, L.; Rothe, M.; Schott, J.W.; Schambach, A. Integrating Vectors for Gene Therapy and Clonal Tracking of Engineered Hematopoiesis. Hematol. Oncol. Clin. N. Am. 2017, 31, 737–752. [Google Scholar] [CrossRef]
- Laoharawee, K.; DeKelver, R.C.; Podetz-Pedersen, K.M.; Rohde, M.; Sproul, S.; Nguyen, H.O.; Nguyen, T.; St Martin, S.J.; Ou, L.; Tom, S.; et al. Dose-Dependent Prevention of Metabolic and Neurologic Disease in Murine MPS II by ZFN-Mediated In Vivo Genome Editing. Mol. Ther. 2018, 26, 1127–1136. [Google Scholar] [CrossRef]
- Ou, L.; DeKelver, R.C.; Rohde, M.; Tom, S.; Radeke, R.; St Martin, S.J.; Santiago, Y.; Sproul, S.; Przybilla, M.J.; Koniar, B.L.; et al. ZFN-Mediated In Vivo Genome Editing Corrects Murine Hurler Syndrome. Mol. Ther. 2019, 27, 178–187. [Google Scholar] [CrossRef]
- Muenzer, J.; Prada, C.E.; Burton, B.; Lau, H.A.; Ficicioglu, C.; Wong Po Foo, C.; Vaidya, S.A.; Whitley, C.B.; Harmatz, P. CHAMPIONS: A phase 1/2 clinical trial with dose escalation of SB-913 ZFN-mediated in vivo human genome editing for treatment of MPS II (Hunter syndrome). Mol. Genet. Metab. 2019, 126, S104. [Google Scholar] [CrossRef]
- Sheridan, C. Sangamo’s landmark genome editing trial gets mixed reception. Nat. Biotechnol. 2018, 36, 907–908. [Google Scholar] [CrossRef]
- Harmatz, P.; Lau, H.E.; Heldermon, C.; Leslie, N.; Wong Po Foo, C.; Vaidya, S.A.; Whitley, C.B. EMPOWERS: A phase 1/2 clinical trial of SB-318 ZFN-mediated in vivo human genome editing for treatment of MPS I (Hurler syndrome). Mol. Genet. Metab. 2019, 126, S68. [Google Scholar] [CrossRef]
- de Carvalho, T.G.; Schuh, R.; Pasqualim, G.; Pellenz, F.M.; Filippi-Chiela, E.C.; Giugliani, R.; Baldo, G.; Matte, U. CRISPR-Cas9-mediated gene editing in human MPS I fibroblasts. Gene 2018, 678, 33–37. [Google Scholar] [CrossRef]
- Poletto, E.; Pasqualim, G.; Giugliani, R.; Matte, U.; Baldo, G. Worldwide distribution of common IDUA pathogenic variants. Clin. Genet. 2018, 94, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Schuh, R.S.; Poletto, E.; Pasqualim, G.; Tavares, A.M.V.; Meyer, F.S.; Gonzalez, E.A.; Giugliani, R.; Matte, U.; Teixeira, H.F.; Baldo, G. In vivo genome editing of mucopolysaccharidosis I mice using the CRISPR/Cas9 system. J Control Release 2018, 288, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Schuh, R.S.; Gonzalez, E.A.; Tavares, A.M.V.; Seolin, B.G.; Elias, L.S.; Vera, L.N.P.; Kubaski, F.; Poletto, E.; Giugliani, R.; Teixeira, H.F.; et al. Neonatal nonviral gene editing with the CRISPR/Cas9 system improves some cardiovascular, respiratory, and bone disease features of the mucopolysaccharidosis I phenotype in mice. Gene Ther. 2019. [CrossRef]
- Wang, D.; Li, J.; Song, C.Q.; Tran, K.; Mou, H.; Wu, P.H.; Tai, P.W.L.; Mendonca, C.A.; Ren, L.; Wang, B.Y.; et al. Cas9-mediated allelic exchange repairs compound heterozygous recessive mutations in mice. Nat. Biotechnol. 2018, 36, 839–842. [Google Scholar] [CrossRef]
- Aldenhoven, M.; Jones, S.A.; Bonney, D.; Borrill, R.E.; Coussons, M.; Mercer, J.; Bierings, M.B.; Versluys, B.; van Hasselt, P.M.; Wijburg, F.A.; et al. Hematopoietic cell transplantation for mucopolysaccharidosis patients is safe and effective: Results after implementation of international guidelines. Boil. Blood Marrow Transplant. 2015, 21, 1106–1109. [Google Scholar] [CrossRef]
- Aldenhoven, M.; Kurtzberg, J. Cord blood is the optimal graft source for the treatment of pediatric patients with lysosomal storage diseases: Clinical outcomes and future directions. Cytotherapy 2015, 17, 765–774. [Google Scholar] [CrossRef]
- Taylor, M.; Khan, S.; Stapleton, M.; Wang, J.; Chen, J.; Wynn, R.; Yabe, H.; Chinen, Y.; Boelens, J.J.; Mason, R.W.; et al. Hematopoietic Stem Cell Transplantation for Mucopolysaccharidoses: Past, Present, and Future. Boil. Blood Marrow Transplant. 2019, 25, e226–e246. [Google Scholar] [CrossRef]
- Gomez-Ospina, N.; Scharenberg, S.G.; Mostrel, N.; Bak, R.O.; Mantri, S.; Quadros, R.M.; Gurumurthy, C.B.; Lee, C.; Bao, G.; Suarez, C.J.; et al. Human genome-edited hematopoietic stem cells phenotypically correct Mucopolysaccharidosis type I. Nat. Commun. 2019, 10, 4045. [Google Scholar] [CrossRef]
- Hendel, A.; Bak, R.O.; Clark, J.T.; Kennedy, A.B.; Ryan, D.E.; Roy, S.; Steinfeld, I.; Lunstad, B.D.; Kaiser, R.J.; Wilkens, A.B.; et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015, 33, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Miki, T.; Vazquez, L.; Yanuaria, L.; Lopez, O.; Garcia, I.M.; Ohashi, K.; Rodriguez, N.S. Induced Pluripotent Stem Cell Derivation and Ex Vivo Gene Correction Using a Mucopolysaccharidosis Type 1 Disease Mouse Model. Stem Cells Int. 2019, 2019, 6978303. [Google Scholar] [CrossRef] [PubMed]
- Schuh, R.S.; de Carvalho, T.G.; Giugliani, R.; Matte, U.; Baldo, G.; Teixeira, H.F. Gene editing of MPS I human fibroblasts by co-delivery of a CRISPR/Cas9 plasmid and a donor oligonucleotide using nanoemulsions as nonviral carriers. Eur. J. Pharm. Biopharm. 2018, 122, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Cradick, T.J.; Qiu, P.; Lee, C.M.; Fine, E.J.; Bao, G. COSMID: A Web-based Tool for Identifying and Validating CRISPR/Cas Off-target Sites. Mol. Ther. Nucleic Acids 2014, 3, e214. [Google Scholar] [CrossRef] [PubMed]
- Frock, R.L.; Hu, J.; Meyers, R.M.; Ho, Y.J.; Kii, E.; Alt, F.W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol. 2015, 33, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, R.; Lombardo, A.; Arens, A.; Miller, J.C.; Genovese, P.; Kaeppel, C.; Nowrouzi, A.; Bartholomae, C.C.; Wang, J.; Friedman, G.; et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat. Biotechnol. 2011, 29, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Park, S.W.; Kim, J.H.; Lee, S.H.; Kim, D.; Koo, T.; Kim, K.E.; Kim, J.H.; Kim, J.S. Genome surgery using Cas9 ribonucleoproteins for the treatment of age-related macular degeneration. Genome Res. 2017, 27, 419–426. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef]
- Kim, D.; Bae, S.; Park, J.; Kim, E.; Kim, S.; Yu, H.R.; Hwang, J.; Kim, J.I.; Kim, J.S. Digenome-seq: Genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat. Methods 2015, 12, 237–243. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Nguyen, N.T.; Malagon-Lopez, J.; Topkar, V.V.; Aryee, M.J.; Joung, J.K. CIRCLE-seq: A highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat. Methods 2017, 14, 607–614. [Google Scholar] [CrossRef]
- Mirzazadeh, R.; Kallas, T.; Bienko, M.; Crosetto, N. Genome-Wide Profiling of DNA Double-Strand Breaks by the BLESS and BLISS Methods. Methods Mol. Biol. 2018, 1672, 167–194. [Google Scholar] [PubMed]
- Giannoukos, G.; Ciulla, D.M.; Marco, E.; Abdulkerim, H.S.; Barrera, L.A.; Bothmer, A.; Dhanapal, V.; Gloskowski, S.W.; Jayaram, H.; Maeder, M.L.; et al. UDiTaS, a genome editing detection method for indels and genome rearrangements. BMC Genom. 2018, 19, 212. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef]
- Vakulskas, C.A.; Dever, D.P.; Rettig, G.R.; Turk, R.; Jacobi, A.M.; Collingwood, M.A.; Bode, N.M.; McNeill, M.S.; Yan, S.; Camarena, J.; et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018, 24, 1216–1224. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, C.T.; Deshpande, P.S.; Dever, D.P.; Camarena, J.; Lemgart, V.T.; Cromer, M.K.; Vakulskas, C.A.; Collingwood, M.A.; Zhang, L.; Bode, N.M.; et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019, 25, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Simhadri, V.L.; McGill, J.; McMahon, S.; Wang, J.; Jiang, H.; Sauna, Z.E. Prevalence of Pre-existing Antibodies to CRISPR-Associated Nuclease Cas9 in the USA Population. Mol. Ther. Methods Clin. Dev. 2018, 10, 105–112. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pre-Clinical Studies in Cell Models | ||||||||||
| Disease | Affected Gene | Targeted Gene | Platform | Cell Type | Delivery Method | Genetic Modification | Reference | |||
| MPS I | IDUA | IDUA | CRISPR/Cas9 | Human fibroblasts | Plasmid-Liposome complex | SNV correction | [82] | |||
| MPS I | IDUA | IDUA | CRISPR/Cas9 | Human fibroblasts | Plasmid-Liposome complex | SNV correction | [93] | |||
| MPS I | IDUA | IDUA | CRISPR/Cas9 | mouse iPSCs | Plasmid-Liposome complex | Precise deletion | [92] | |||
| Pre-Clinical Studies in Murine Models | ||||||||||
| Disease | Affected Gene | Targeted Gene | Platform | In Vivo Vs. Ex Vivo | Cargo and Vehicle | Genetic Modification | Reference | |||
| MPS I | IDUA | CCR5 | CRISPR/Cas9 | ex vivo | RNP/AAV6 | Gene addition | [90] | |||
| MPS I | IDUA | ROSA26 | CRISPR/Cas9 | in vivo | Liposome and plasmid vectors, IV | Gene addition | [84] | |||
| MPS I | IDUA | IDUA | CRISPR/Cas9 | in vivo | 2 AAV9 vectors, IV | SNV correction | [86] | |||
| MPS I | IDUA | ALB | ZFNs | in vivo | 3 AAV2/8 vectors, IV | Gene addition | [78] | |||
| MPS II | IDS | ALB | ZFNs | in vivo | 3 AAV2/8 vectors, IV | Gene addition | [77] | |||
| Clinical Trials | ||||||||||
| Disease | Affected Gene | Targeted Gene | Platform | In Vivo vs. Ex Vivo | Cargo and Vehicle | Genetic Modification | Company | Trial Name | Clinicaltrials.gov Identifier | |
| MPS I | IDUA | CCR5 | CRISPR/Cas9 | ex vivo | RNP/AAV6 | Gene addition | Stanford University | in the pipeline | ||
| MPS I | IDUA | ALB | ZFNs | in vivo | 3 AAV2/6 vectors, IV | Gene addition | Sangamo therapeutics | SB-318 | NCT02702115 | |
| MPS II | IDS | ALB | ZFNs | in vivo | 3 AAV2/6 vectors, IV | Gene addition | Sangamo therapeutics | SB-913 | NCT3041324 | |
| IV: intravenous | ||||||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poletto, E.; Baldo, G.; Gomez-Ospina, N. Genome Editing for Mucopolysaccharidoses. Int. J. Mol. Sci. 2020, 21, 500. https://doi.org/10.3390/ijms21020500
Poletto E, Baldo G, Gomez-Ospina N. Genome Editing for Mucopolysaccharidoses. International Journal of Molecular Sciences. 2020; 21(2):500. https://doi.org/10.3390/ijms21020500
Chicago/Turabian StylePoletto, Edina, Guilherme Baldo, and Natalia Gomez-Ospina. 2020. "Genome Editing for Mucopolysaccharidoses" International Journal of Molecular Sciences 21, no. 2: 500. https://doi.org/10.3390/ijms21020500
APA StylePoletto, E., Baldo, G., & Gomez-Ospina, N. (2020). Genome Editing for Mucopolysaccharidoses. International Journal of Molecular Sciences, 21(2), 500. https://doi.org/10.3390/ijms21020500