The Mitochondrial Ca2+ Overload via Voltage-Gated Ca2+ Entry Contributes to an Anti-Melanoma Effect of Diallyl Trisulfide


Abstract
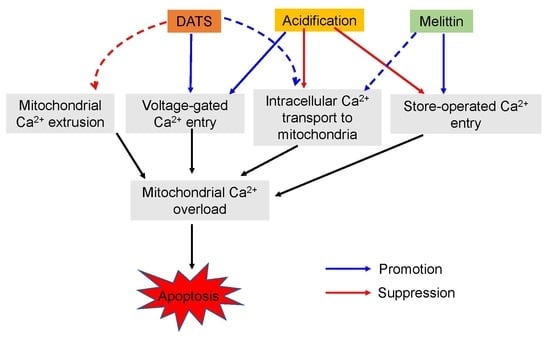
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
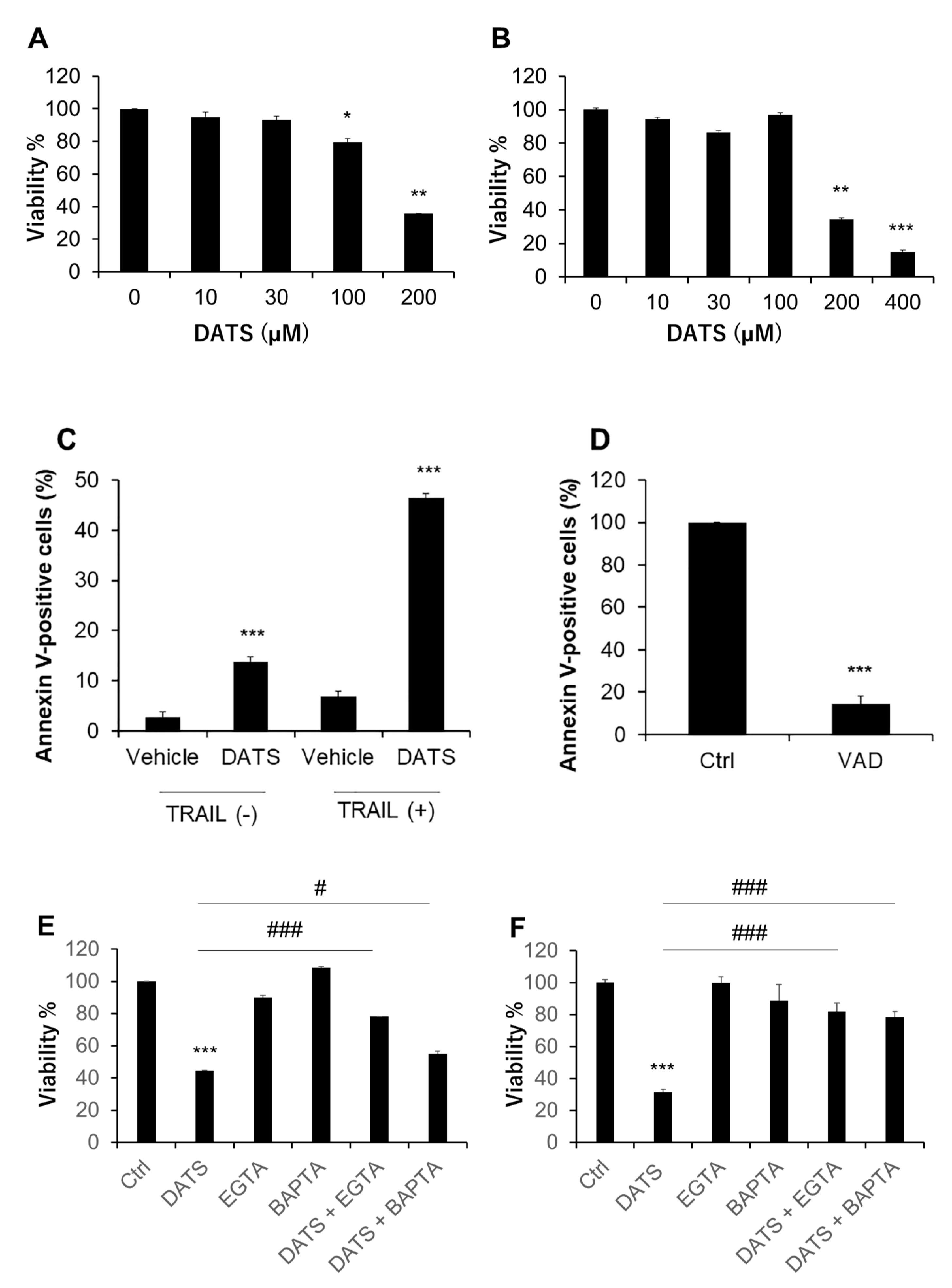
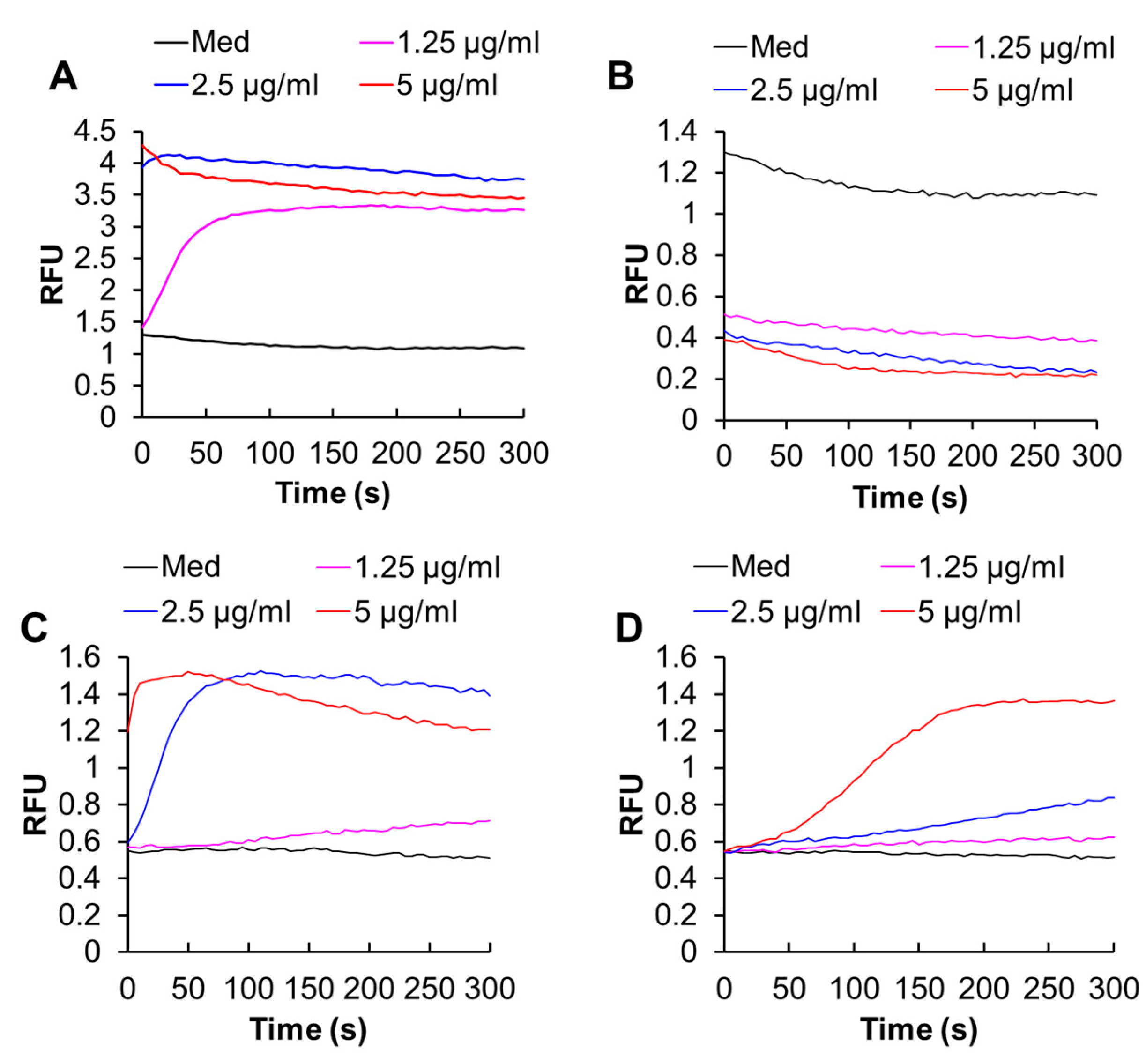
2.1. DATS Reduces Cell Viability and Increases Apoptosis in a Ca2+- and Caspase-Dependent Manner
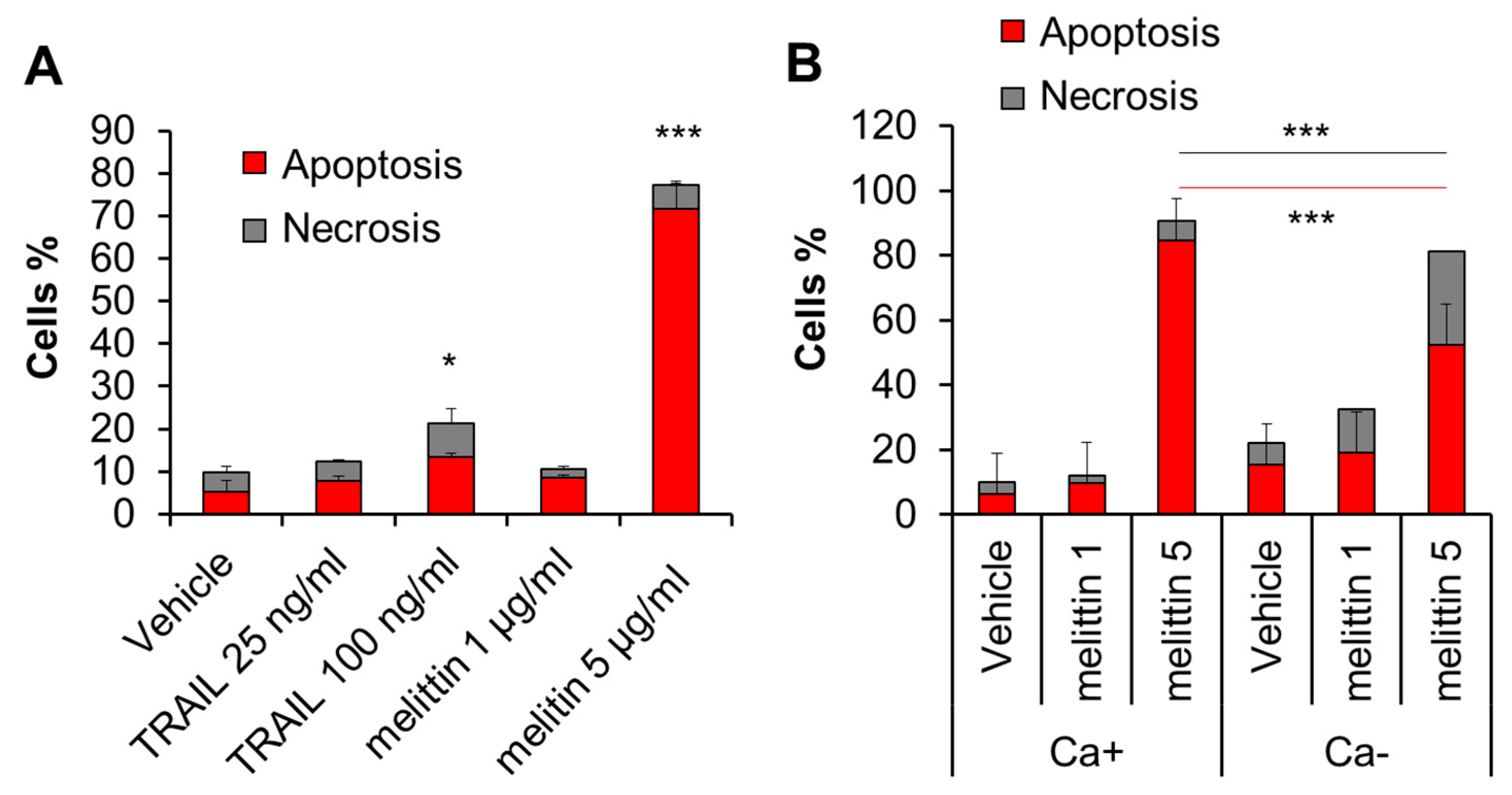
2.2. Melittin Exhibits Anti-Melanoma Effect in a Ca2+-Dependent Manner
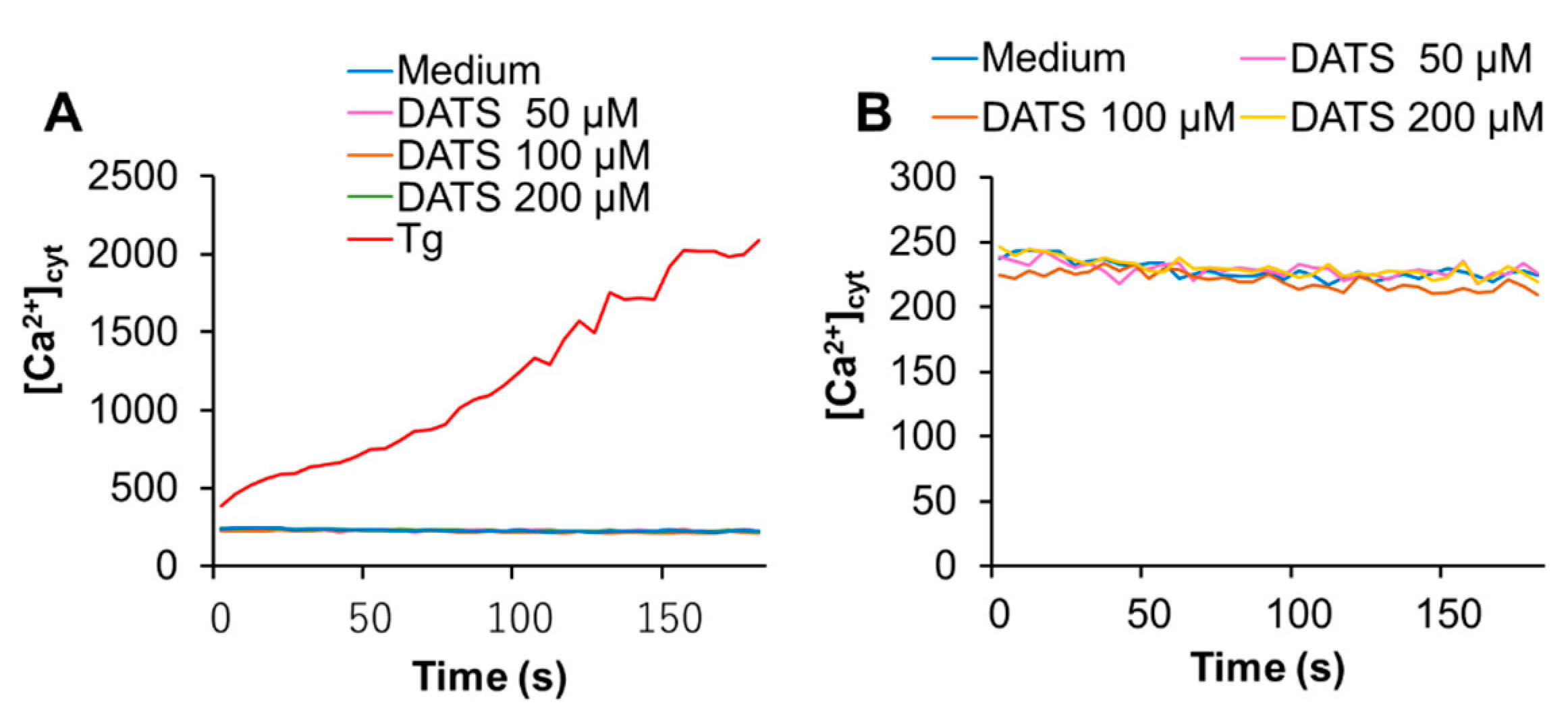
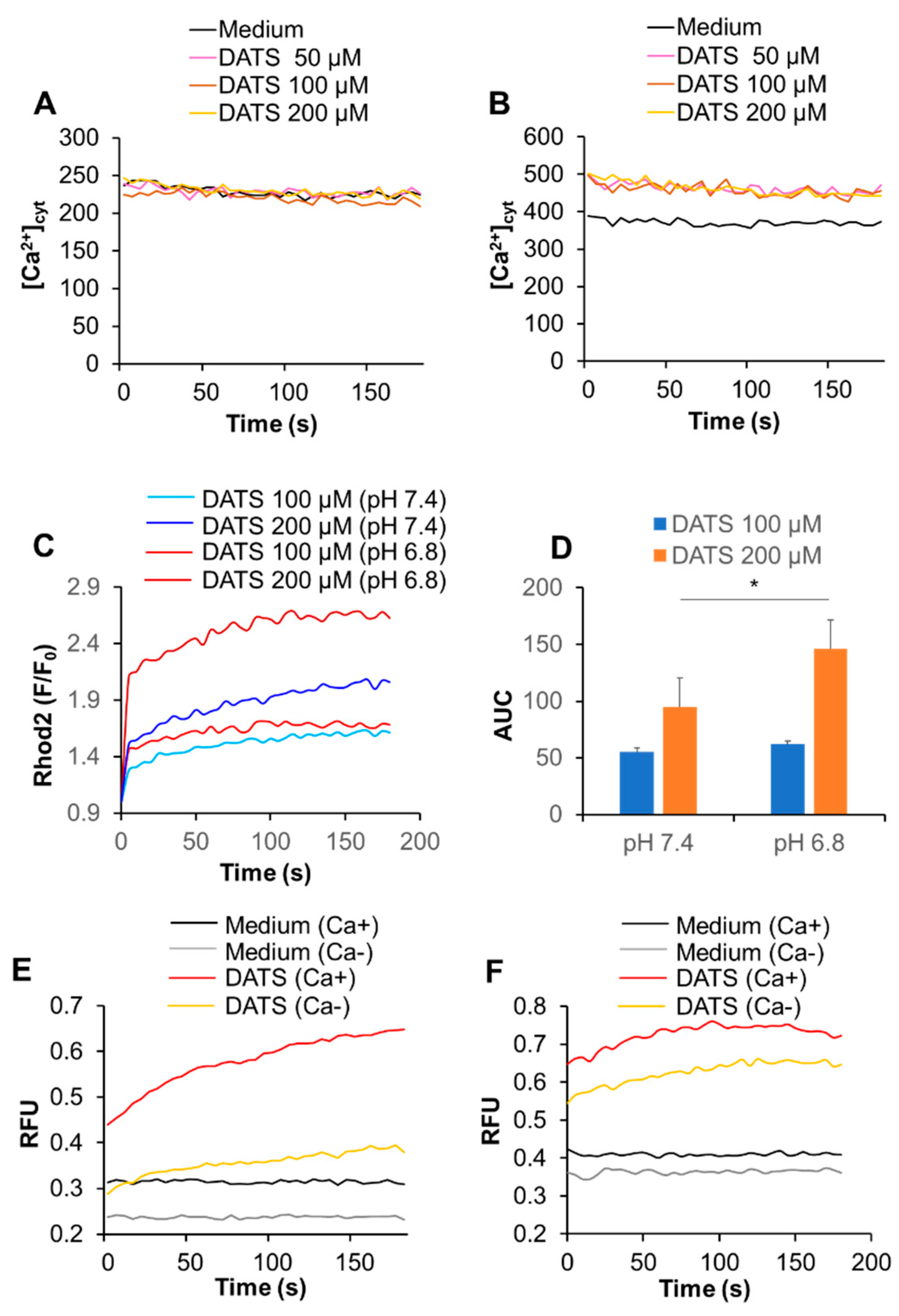
2.3. DATS Increases [Ca2+]mit without Increasing [Ca2+]cyt
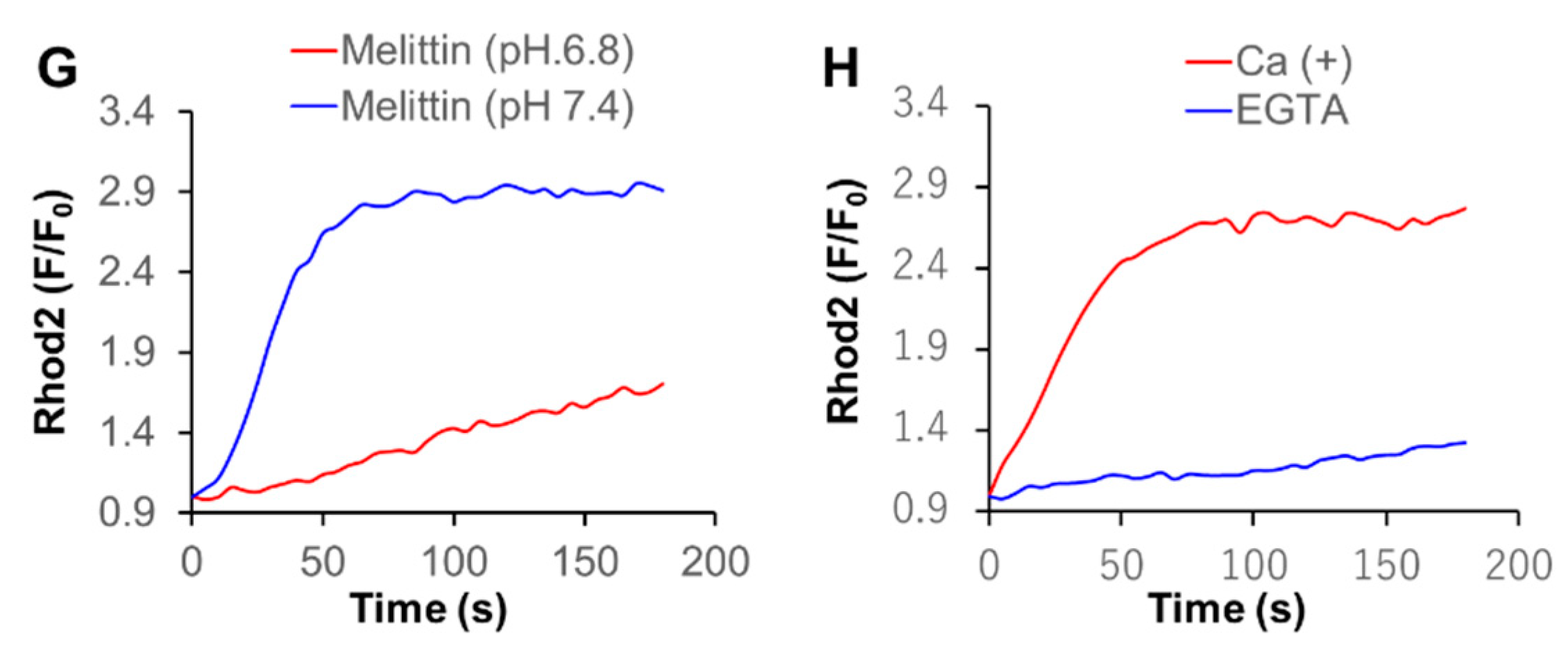
2.4. Melittin Increases Both [Ca2+]cyt and [Ca2+]mit in an Extracellular Ca2+-Dependent Manner
2.5. Acidification Has a Reciprocal Effect on Ca2+mit Overload Caused by DATS and Melittin
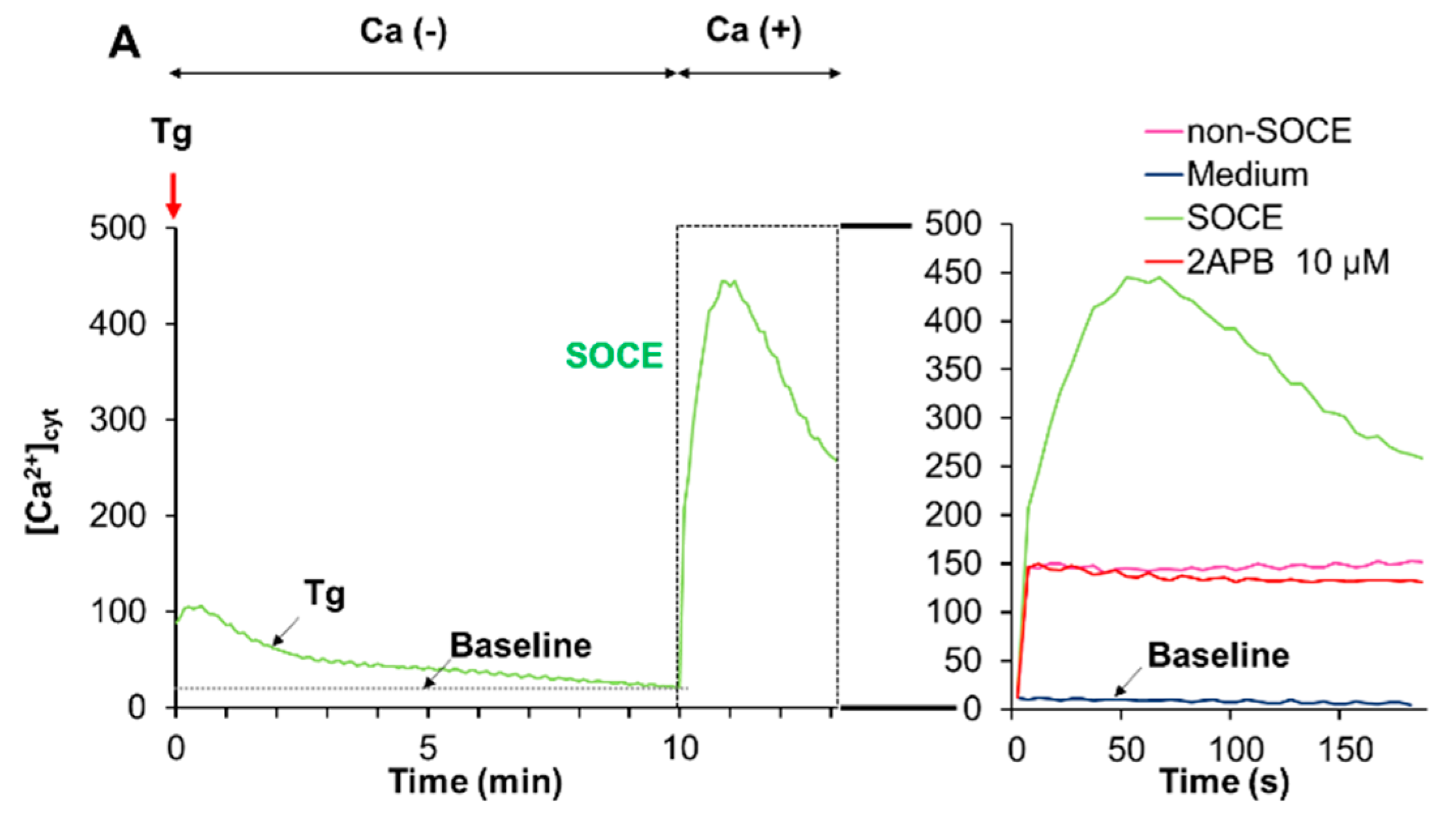
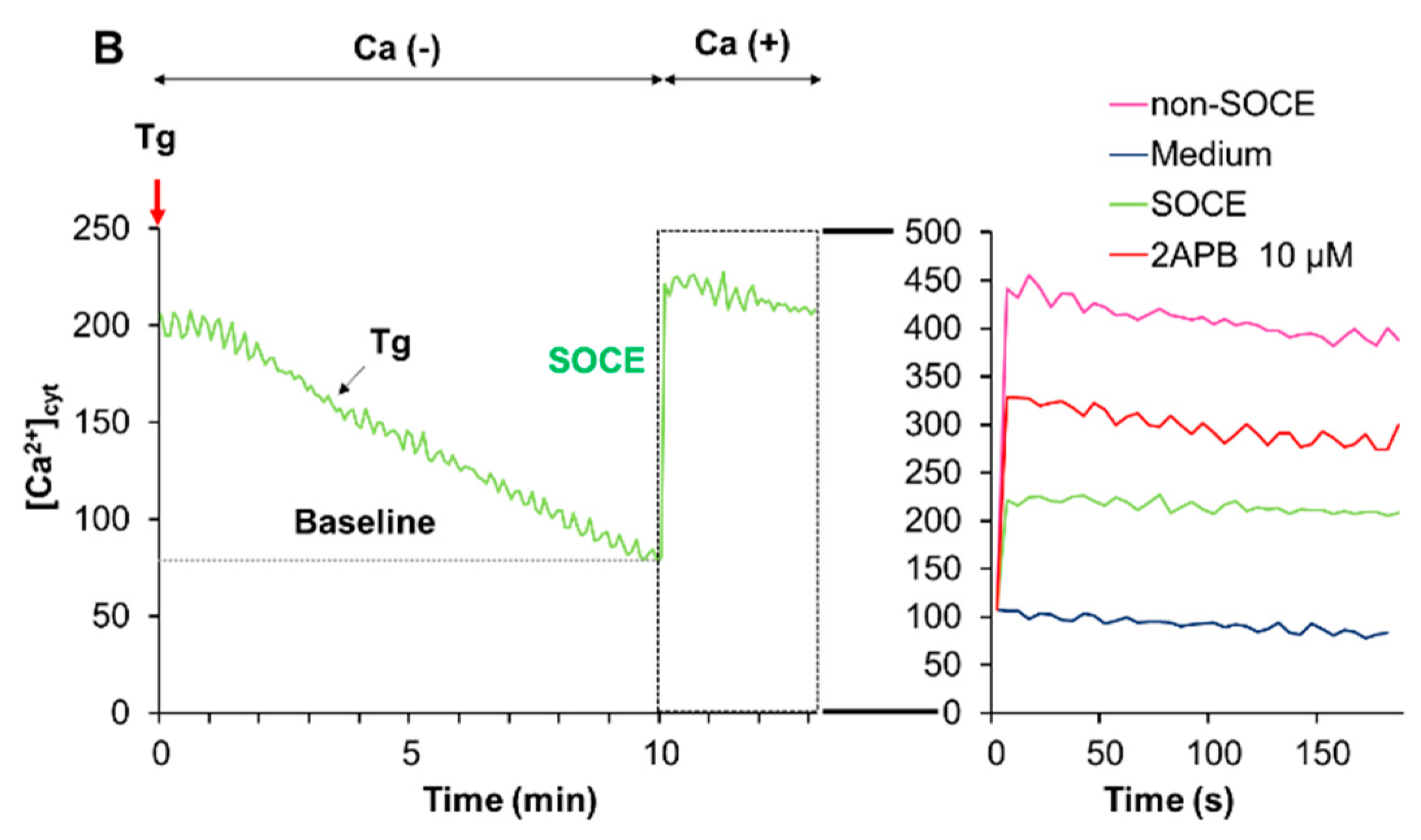
2.6. Acidification Mitigates SOCE
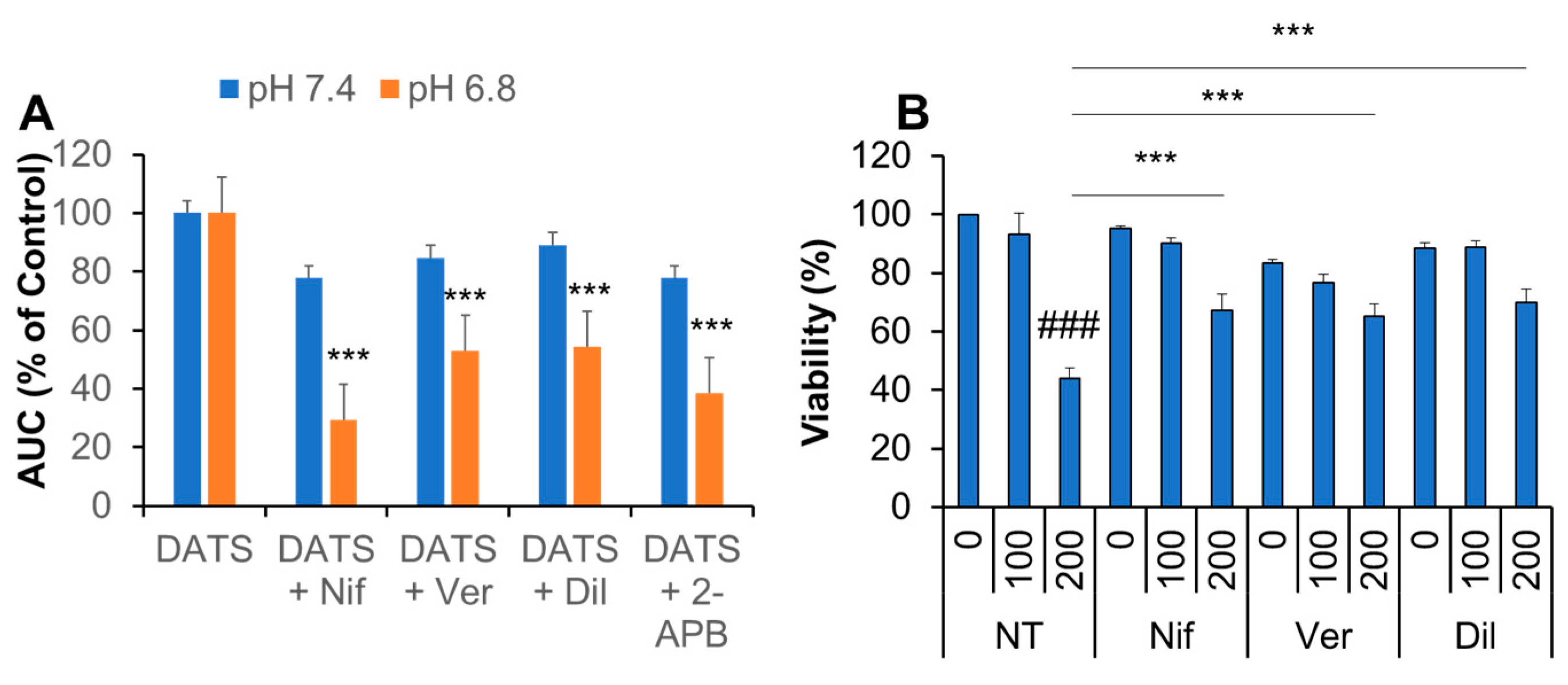
2.7. Ca2+ Channel Blockers Inhibit Ca2+mit Overload and Cell Death Caused by DATS
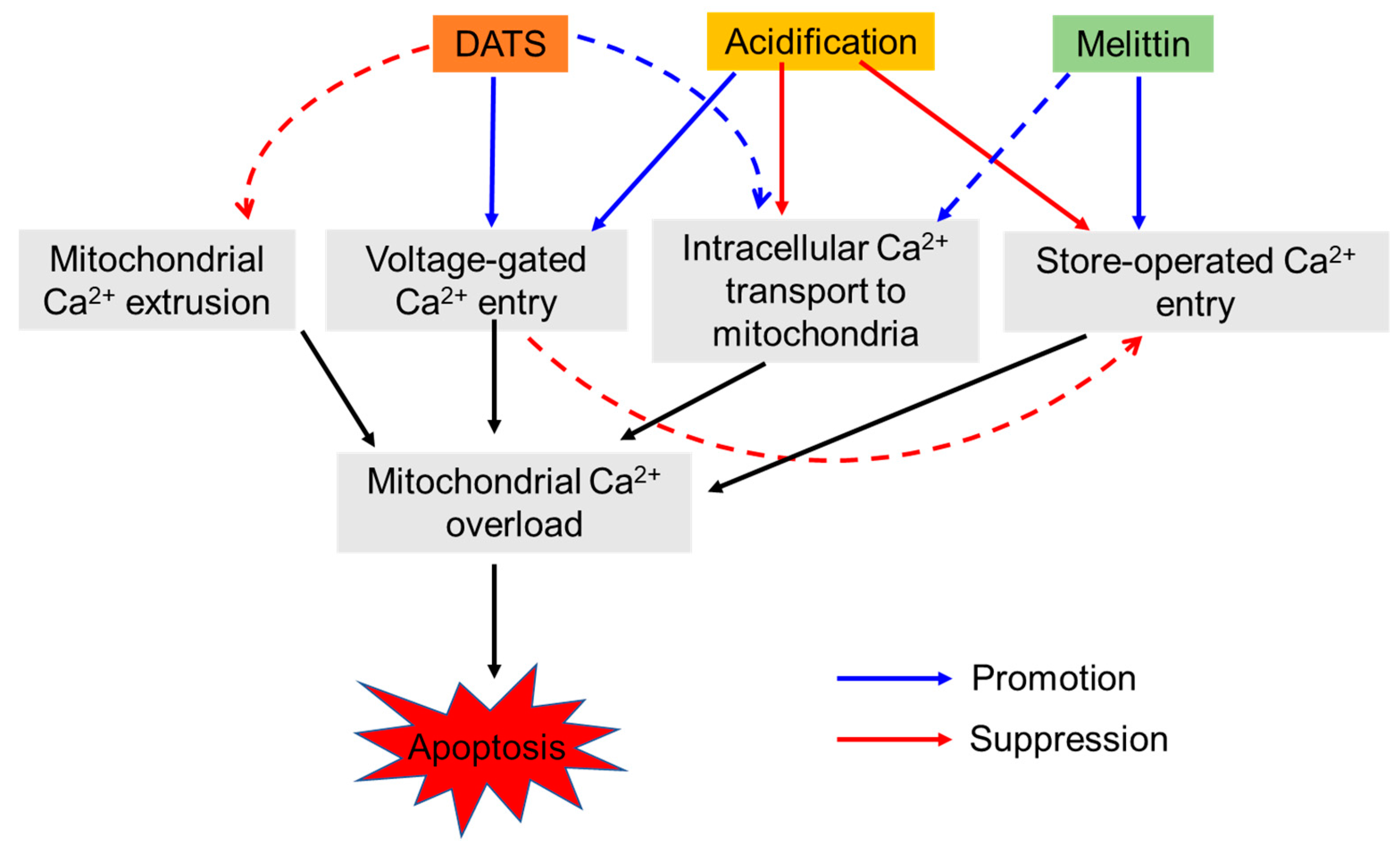
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Cell Death Assay
4.5. Intracellular Ca2+ Measurements
4.6. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tapiero, H.; Townsend, D.M.; Tew, K.D. Organosulfur compounds from alliaceae in the prevention of human pathologies. Biomed. Pharmacother. 2004, 58, 183–193. [Google Scholar] [CrossRef]
- Kodera, Y.; Suzuki, A.; Imada, O.; Kasuga, S.; Sumioka, I.; Kanezawa, A.; Taru, N.; Fujikawa, M.; Nagae, S.; Masamoto, K.; et al. Physical, chemical, and biological properties of s-allylcysteine, an amino acid derived from garlic. J. Agric. Food Chem. 2002, 50, 622–632. [Google Scholar] [CrossRef]
- Schäfer, G.; Kaschula, C.H. The immunomodulation and anti-inflammatory effects of garlic organosulfur compounds in cancer chemoprevention. Anticancer Agents Med. Chem. 2014, 14, 233–240. [Google Scholar] [CrossRef]
- Sun, S.Y.; Hail, N., Jr.; Lotan, R. Apoptosis as a novel target for cancer chemoprevention. J. Natl. Cancer Inst. 2002, 96, 662–672. [Google Scholar] [CrossRef]
- Hail, N., Jr.; Lotan, R. Cancer chemoprevention and mitochondria: Targeting apoptosis in transformed cells via the disruption of mitochondrial bioenergetics/redox state. Mol. Nutr. Food Res. 2009, 53, 49–67. [Google Scholar] [CrossRef]
- Belman, S. Onion and garlic oils inhibit tumor promotion. Carcinogenesis 1983, 4, 1063–1065. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Pao, J.; Lin, S.; Sheen, L.Y. Molecular mechanisms of garlic-derived allyl sulfides in the inhibition of skin cancer progression. Ann. N. Y. Acad. Sci. 2012, 1271, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Banik, N.L.; Ray, S.K. Garlic compounds generate reactive oxygen species leading to activation of stress kinases and cysteine proteases for apoptosis in human glioblastoma T98G and U87MG cells. Cancer 2007, 110, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, S.; Banik, N.L.; Patel, S.J.; Ray, S.K. Garlic compounds induced calpain and intrinsic caspase cascade for apoptosis in human malignant neuroblastoma SH-SY5Y cells. Apoptosis 2007, 12, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Powolny, A.A.; Singh, S.V. Multitargeted prevention and therapy of cancer by diallyl trisulfide and related Allium vegetable-derived organosulfur compounds. Cancer Lett. 2008, 269, 305–314. [Google Scholar] [CrossRef]
- Busch, C.; Jacob, C.; Anwar, A.; Burkholz, T.; Aicha Ba, L.; Cerella, C.; Diederich, M.; Brandt, W.; Wessjohann, L.; Montenarh, M. Diallylpolysulfides induce growth arrest and apoptosis. Int. J. Oncol. 2010, 36, 743–749. [Google Scholar] [PubMed]
- Shankar, S.; Chen, Q.; Ganapathy, S.; Singh, K.P.; Srivastava, R.K. Diallyl trisulfide increases the effectiveness of TRAIL and inhibits prostate cancer growth in an orthotopic model: Molecular mechanisms. Mol. Cancer Ther. 2008, 7, 2328–2338. [Google Scholar] [CrossRef] [PubMed]
- Suzuki-Karasaki, Y.; Suzuki-Karasaki, M.; Uchida, M.; Ochiai, T. Depolarization Controls TRAIL-Sensitization and Tumor-Selective Killing of Cancer Cells: Crosstalk with ROS. Front. Oncol. 2014, 4, 128. [Google Scholar] [CrossRef] [PubMed]
- Murai, M.; Inoue, T.; Suzuki-Karasaki, M.; Ochiai, T.; Ra, C.; Nishida, S.; Suzuki-Karasaki, Y. Diallyl trisulfide sensitizes human melanoma cells to TRAIL-induced cell death by promoting endoplasmic reticulum-mediated apoptosis. Int. J. Oncol. 2012, 41, 2029–2037. [Google Scholar] [CrossRef]
- Dyer, M.J.; MacFarlane, M.; Cohen, G.M. Barriers to effective TRAIL-targeted therapy of malignancy. J. Clin. Oncol. 2007, 25, 4505–4506. [Google Scholar] [CrossRef]
- Dimberg, L.Y.; Anderson, C.K.; Camidge, R.; Behbakht, K.; Thorburn, A.; Ford, H.L. On the TRAIL to successful cancer therapy? Predicting and counteracting resistance against TRAIL-based therapeutics. Oncogene 2013, 32, 1341–1350. [Google Scholar] [CrossRef]
- Mellier, G.; Pervaiz, S. The three Rs along the TRAIL: Resistance, re-sensitization and reactive oxygen species (ROS). Free Radic. Res. 2012, 46, 996–1003. [Google Scholar] [CrossRef]
- Inoue, T.; Suzuki-Karasaki, Y. Mitochondrial superoxide mediates mitochondrial and endoplasmic reticulum dysfunctions in TRAIL-induced apoptosis in Jurkat cells. Free Radic. Biol. Med. 2013, 61, 273–284. [Google Scholar] [CrossRef]
- Suzuki-Karasaki, M.; Ochiai, T.; Suzuki-Karasaki, Y. Crosstalk between mitochondrial ROS and depolarization in the potentiation of TRAIL-induced apoptosis in human tumor cells. Int. J. Oncol. 2014, 44, 616–628. [Google Scholar] [CrossRef]
- Gualdani, R.; de Clippele, M.; Ratbi, I.; Gailly, P.; Tajeddine, N. Store-Operated Calcium Entry Contributes to Cisplatin-Induced Cell Death in Non-Small Cell Lung Carcinoma. Cancers 2019, 11, 430. [Google Scholar] [CrossRef]
- Zhao, M.; Brunk, U.T.; Eaton, J.W. Delayed oxidant-induced cell death involves activation of phospholipase A2. FEBS Lett. 2001, 509, 399–404. [Google Scholar] [CrossRef]
- Oršolić, N. Bee venom in cancer therapy. Cancer Metastasis Rev. 2012, 31, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.T.; Cheng, H.H.; Huang, C.J.; Chang, H.C.; Chi, C.C.; Su, H.H.; Hsu, S.S.; Wang, J.L.; Chen, I.S.; Liu, S.I.; et al. Phospholipase A2-independent Ca2+ entry and subsequent apoptosis induced by melittin in human MG63 osteosarcoma cells. Life Sci. 2007, 80, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V. Melittin-induced hyperactivation of phospholipase A2 activity and calcium influx in ras-transformed cells. Oncogene 1993, 8, 939–947. [Google Scholar] [PubMed]
- Smani, T.; Zakharov, S.I.; Leno, E.; Csutora, P.; Trepakova, E.S.; Bolotina, V.M. Ca2+-independent phospholipase A2 is a novel determinant of store-operated Ca2+ entry. J. Biol. Chem. 2003, 278, 11909–11915. [Google Scholar] [CrossRef] [PubMed]
- Smani, T.; Zakharov, S.I.; Csutora, P.; Leno, E.; Trepakova, E.S.; Bolotina, V.M. A novel mechanism for the store-operated calcium influx pathway. Nat. Cell Biol. 2004, 6, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Whitton, B.; Okamoto, H.; Packham, G.; Crabb, S.J. Vacuolar ATPase as a potential therapeutic target and mediator of treatment resistance in cancer. Cancer Med. 2018, 7, 3800–3811. [Google Scholar] [CrossRef]
- Pamarthy, S.; Kulshrestha, A.; Katara, G.K.; Beaman, K.D. The curious case of vacuolar ATPase: Regulation of signaling pathways. Mol. Cancer 2018, 17, 41. [Google Scholar] [CrossRef]
- Nicotera, P.; Zhivotovsky, B.; Orrenius, S. Nuclear calcium transport and the role of calcium in apoptosis. Cell Calcium 1994, 16, 279–288. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Zhu, L.P.; Yu, X.D.; Ling, S.; Brown, R.A.; Kuo, T.H. Mitochondrial Ca2+ homeostasis in the regulation of apoptotic and necrotic cell deaths. Cell Calcium 2000, 28, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Kania, E.; Roest, G.; Vervliet, T.; Parys, J.B.; Bultynck, G. IP3 Receptor-Mediated Calcium Signaling and Its Role in Autophagy in Cancer. Front. Oncol. 2017, 7, 140. [Google Scholar] [CrossRef] [PubMed]
- Pedriali, G.; Rimessi, A.; Sbano, L.; Giorgi, C.; Wieckowski, M.R.; Previati, M.; Pinton, P. Regulation of Endoplasmic Reticulum-Mitochondria Ca2+ Transfer and Its Importance for Anti-Cancer Therapies. Front. Oncol. 2017, 7, 180. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pozzan, T. Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, M.; Pellegrini, L. The coming of age of the mitochondria-ER contact: A matter of thickness. Cell Death Differ. 2016, 23, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Pinton, P. The mitochondrial calcium uniporter complex: Molecular components, structure and physiopathological implications. J. Physiol. 2014, 592, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Takata, N.; Ohshima, Y.; Suzuki-Karasaki, M.; Yoshida, Y.; Tokuhashi, Y.; Suzuki-Karasaki, Y. Mitochondrial Ca2+ removal amplifies TRAIL cytotoxicity toward apoptosis-resistant tumor cells via promotion of multiple cell death modalities. Int. J. Oncol. 2017, 51, 193–203. [Google Scholar] [CrossRef]
- Brand, M.D. Electroneutral efflux of Ca2+ from liver mitochondria. Biochem. J. 1985, 225, 413–419. [Google Scholar] [CrossRef]
- Altschuld, R.A.; Hohl, C.M.; Castillo, L.C.; Garleb, A.A.; Starling, R.C.; Brierley, G.P. Cyclosporin inhibits mitochondrial calcium efflux in isolated adult rat ventricular cardiomyocytes. Am. J. Physiol. 1992, 262, 1699–1704. [Google Scholar]
- Bernardi, P.; von Stockum, S. The permeability transition pore as a Ca2+ release channel: New answers to an old question. Cell Calcium 2012, 52, 22–27. [Google Scholar] [CrossRef]
- Di Lisa, F.; Carpi, A.; Giorgio, V.; Bernardi, P. The mitochondrial permeability transition pore and cyclophilin D in cardioprotection. Biochim. Biophys. Acta 2011, 1813, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Aguilar, M.; Baines, C.P. Structural mechanisms of cyclophilin D-dependent control of the mitochondrial permeability transition pore. Biochim. Biophys. Acta 2014, 1850, 2041–2047. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, Y.; Takata, N.; Suzuki-Karasaki, M.; Yoshida, Y.; Tokuhashi, Y.; Suzuki-Karasaki, Y. Disrupting mitochondrial Ca2+ homeostasis causes tumor-selective TRAIL sensitization through mitochondrial network abnormalities. Int. J. Oncol. 2017, 51, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Bergmeier, W.; Weidinger, C.; Zee, I.; Feske, S. Emerging roles of store-operated Ca2⁺ entry through STIM and ORAI proteins in immunity, hemostasis, and cancer. Channels 2013, 7, 379–391. [Google Scholar] [CrossRef]
- Jardin, I.; Rosado, J.A. STIM and calcium channel complexes in cancer. Biochim. Biophys. Acta 2016, 1863, 1418–1426. [Google Scholar] [CrossRef]
- Chalmers, S.B.; Monteith, G.R. ORAI channels and cancer. Cell Calcium 2018, 74, 160–167. [Google Scholar] [CrossRef]
- Deak, A.T.; Blass, S.; Khan, M.J.; Groschner, L.N.; Waldeck-Weiermair, M.; Hallström, S.; Graier, W.F.; Malli, R. RIP3-mediated STIM1 oligomerization requires intact mitochondrial Ca2+ uptake. J. Cell Sci. 2014, 127, 2944–2955. [Google Scholar] [CrossRef]
- Elustondo, P.A.; Nichols, M.; Robertson, G.S.; Pavlov, E.V. Mitochondrial Ca2+ uptake pathways. J. Bioenerg. Biomembr. 2017, 49, 113–119. [Google Scholar] [CrossRef]
- Kato, Y.; Ozawa, S.; Tsukuda, M.; Kubota, E.; Miyazaki, K.; St-Pierre, Y.; Hata, R. Acidic extracellular pH increases calcium influx-triggered phospholipase D activity along with acidic sphingomyelinase activation to induce matrix metalloproteinase-9 expression in mouse metastatic melanoma. FEBS. J. 2007, 274, 3171–3183. [Google Scholar] [CrossRef]
- Wang, Y.; Deng, X.; Mancarella, S.; Hendron, E.; Eguchi, S.; Soboloff, J.; Tang, X.D.; Gill, D.L. The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science 2010, 330, 105–109. [Google Scholar] [CrossRef]
- Dionisio, N.; Smani, T.; Woodard, G.E.; Castellano, A.; Salido, G.M.; Rosado, J.A. Homer proteins mediate the interaction between STIM1 and Cav1.2 channels. Biochim. Biophys. Acta 2015, 1853, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Hajnóczky, G.; Robb-Gaspers, L.D.; Seitz, M.B.; Thomas, A.P. Decoding of cytosolic calcium oscillations in the mitochondria. Cell 1995, 82, 415–424. [Google Scholar] [CrossRef]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakagawa, C.; Suzuki-Karasaki, M.; Suzuki-Karasaki, M.; Ochiai, T.; Suzuki-Karasaki, Y. The Mitochondrial Ca2+ Overload via Voltage-Gated Ca2+ Entry Contributes to an Anti-Melanoma Effect of Diallyl Trisulfide. Int. J. Mol. Sci. 2020, 21, 491. https://doi.org/10.3390/ijms21020491
Nakagawa C, Suzuki-Karasaki M, Suzuki-Karasaki M, Ochiai T, Suzuki-Karasaki Y. The Mitochondrial Ca2+ Overload via Voltage-Gated Ca2+ Entry Contributes to an Anti-Melanoma Effect of Diallyl Trisulfide. International Journal of Molecular Sciences. 2020; 21(2):491. https://doi.org/10.3390/ijms21020491
Chicago/Turabian StyleNakagawa, Chinatsu, Manami Suzuki-Karasaki, Miki Suzuki-Karasaki, Toyoko Ochiai, and Yoshihiro Suzuki-Karasaki. 2020. "The Mitochondrial Ca2+ Overload via Voltage-Gated Ca2+ Entry Contributes to an Anti-Melanoma Effect of Diallyl Trisulfide" International Journal of Molecular Sciences 21, no. 2: 491. https://doi.org/10.3390/ijms21020491
APA StyleNakagawa, C., Suzuki-Karasaki, M., Suzuki-Karasaki, M., Ochiai, T., & Suzuki-Karasaki, Y. (2020). The Mitochondrial Ca2+ Overload via Voltage-Gated Ca2+ Entry Contributes to an Anti-Melanoma Effect of Diallyl Trisulfide. International Journal of Molecular Sciences, 21(2), 491. https://doi.org/10.3390/ijms21020491