Crystal Structure of Non-Structural Protein 10 from Severe Acute Respiratory Syndrome Coronavirus-2

 , ,
, ,
Abstract

1. Introduction
2. Results and Discussion
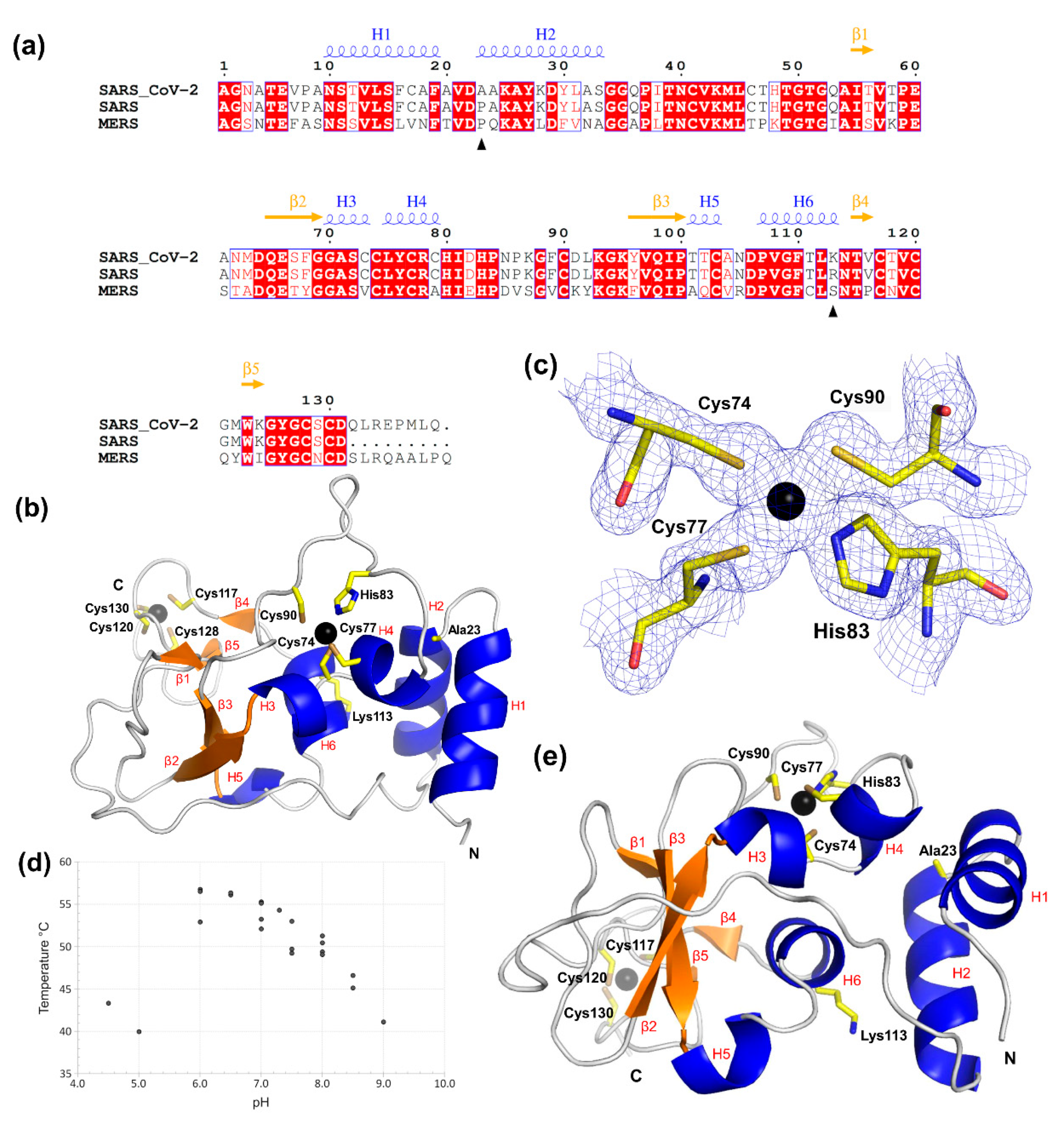
2.1. SARS-CoV-2 nsp10 Is a Zinc-Finger Protein of the LIM Type Containing Two Zinc-Finger Motifs
2.2. SARS-CoV-2 nsp10 Exhibits Close Similarity to Its SARS Homologue
2.3. Nsp10′s Absence in Prokaryotes and Eukaryotes Makes It a Unique Viral Protein
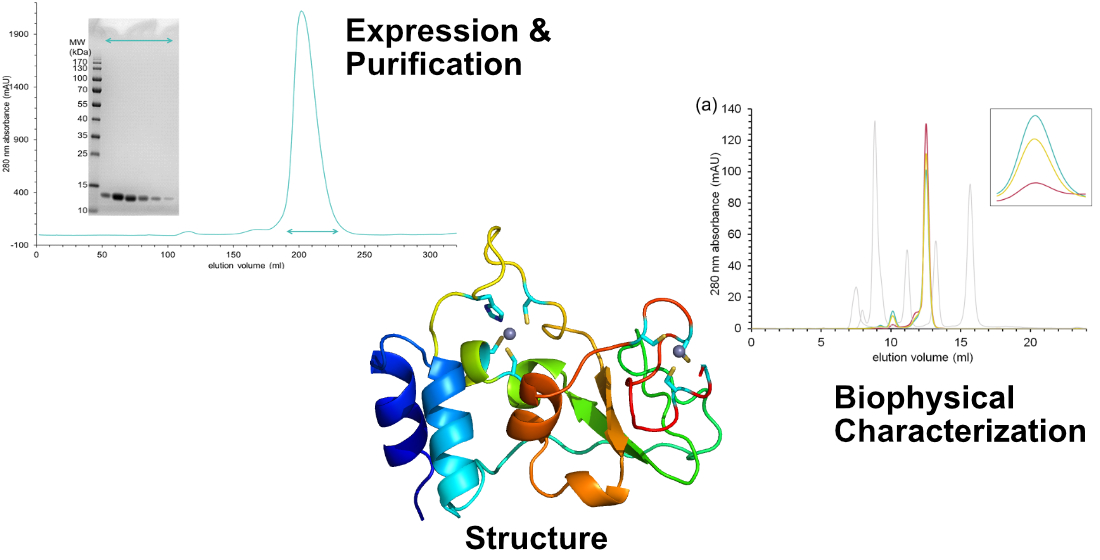
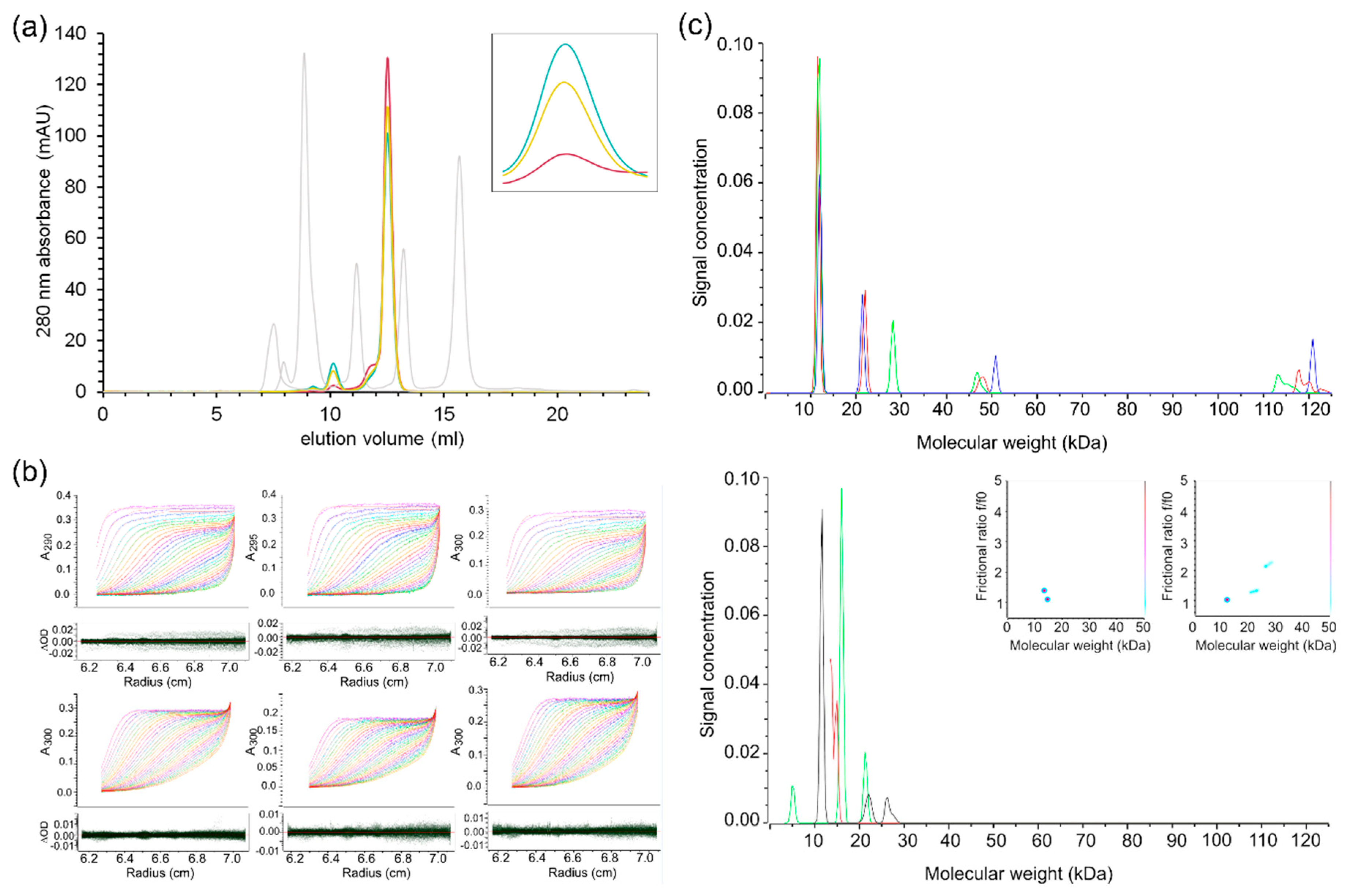
2.4. Characterisation of SARS-CoV-2 nsp10′s Behaviour in Solution Reveals Differences to Its SARS Homologue
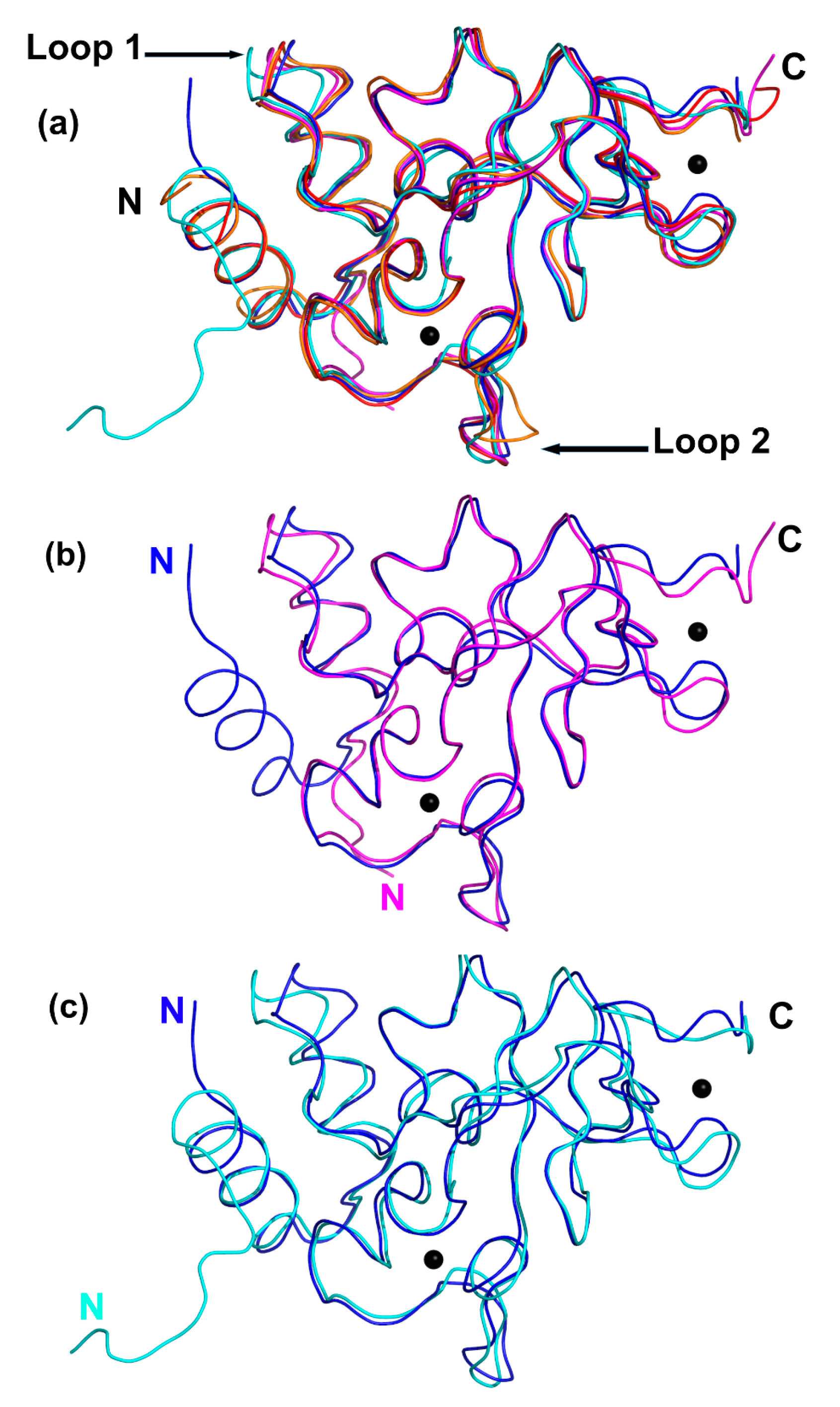
2.5. Comparison between the Unbound and Complex-Bound Forms of nsp10 Reveals Subtle Differences
2.6. SARS-CoV-2 nsp10, as a Central Player, Represents a Potential Antiviral Drug Target
3. Materials and Methods
3.1. Design of the SARS-CoV-2 nsp10 Expression Plasmid
3.2. Nsp10 Expression and Purification
3.3. Crystallisation of nsp10
3.4. Data Collection, Structure Determination and Refinement
3.5. Analytical Size Exclusion Chromatography
3.6. Thermal Shift Assays
3.7. Analytical Ultracentrifugation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Å | Angstrom |
| CoV | Coronavirus |
| DPA | Dipicolinic acid |
| DTT | Dithiothreitol |
| EDTA | Ethylenediaminetetraacetic acid |
| ExoN | 3′-to-5′ exoribonuclease |
| IPTG | Isopropyl-d-thiogalactopyranoside |
| MERS | Middle East Respiratory Syndrome |
| MHV | Murine Hepatitis virus |
| MTase | Methyltransferase |
| Nsp | Non-structural protein |
| ORF | Open reading frame |
| PDB | Protein Data Bank |
| RdRp | RNA-dependent RNA polymerase |
| RMSD | Root Mean Square Deviation |
| RTC | Replication transcription complex |
| SAM | S-adenosyl-L-methionine |
| SARS | Severe Acute Respiratory Syndrome |
| SEC | Size-Exclusion Chromatography |
| TCEP | Tris (2-carboxyethyl) phosphine |
References
- Can Chan-Yeung, M.; Xu, R.H. SARS: Epidemiology. Respirology 2020, 8, S9–S14. [Google Scholar] [CrossRef] [PubMed]
- WHO 2020: MERS Situation Update, January 2020. Available online: http://www.emro.who.int/pandemic-epidemic-diseases/mers-cov/mers-situation-update-january-2020.html (accessed on 14 June 2020).
- Guy, R.K.; DiPaola, R.S.; Romanelli, F.; Dutch, R.E. Rapid repurposing of drugs for COVID-19. Science 2020, 368, 829–830. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Subissi, J.; Imbert, I.; Ferron, F.; Collet, A.; Coutard, B.; Decroly, E.; Canard, B. SARS-CoV ORF1b-encoded Non-structural Proteins 12-16: Replicative Enzymes as Antiviral Targets. Antivir. Res. 2014, 101, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Chan, J.F.W.; Azhar, E.I.; Hui, D.S.C.; Yuen, K.-Y. Coronaviruses—Drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef]
- Graci, J.D.; Cameron, C.E. Mechanisms of action of ribavirin against distinct viruses. Rev. Med. Virol. 2006, 16, 37–48. [Google Scholar] [CrossRef]
- Ferron, F.; Subissi, L.; De Morais, A.T.S.; Le, N.T.T.; Sevajol, M.; Gluais, L.; Decroly, E.; Vonrhein, C.; Bricogne, G.; Canard, B.; et al. Structural and molecular basis of mismatch correction and ribavirin excision from coronavirus RNA. Proc. Natl. Acad. Sci. USA 2017, 162–171. [Google Scholar] [CrossRef]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural basis for the inhibition of the RNA-Dependent RNA Polymerase from SARS-CoV-2 by Remdesivir. Science 2020, 368, 1499–1504. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19—Preliminary Report. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Villar, J.; Ferrando, C.; Martínez, D.; Ambrós, A.; Muñoz, T.; Soler, J.A.; Aguilar, G.; Alba, F.; González-Higueras, E.; Conesa, L.A.; et al. Dexamethasone treatment for the acute respiratory distress syndrome: A multicentre, randomised controlled trial. Lancet Respir. Med. 2020, 8, 267–276. [Google Scholar] [CrossRef]
- Chen, Y.; Cai, H.; Pan, J.; Xiang, N.; Tien, P.; Ahola, T.; Guo, D. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc. Natl. Acad. Sci. USA 2009, 106, 3484–3489. [Google Scholar] [CrossRef] [PubMed]
- Bouvet, M.; Debarnot, C.; Imbert, I.; Selisko, B.; Snijder, E.J.; Canard, B.; Decroly, E. In Vitro Reconstitution of SARS-Coronavirus mRNA Cap Methylation. PLoS Pathog. 2010, 6, e1000863. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, D. Molecular mechanisms of coronavirus RNA capping and Methylation. Virol. Sin. 2016, 31, 3–11. [Google Scholar] [CrossRef]
- Donaldson, E.F.; Sims, A.C.; Graham, R.L.; Denison, M.R.; Baric, R.S. Murine Hepatitis virus replicase protein nsp10 is a critical regulator of viral RNA synthesis. J. Virol. 2007, 81, 6456–6468. [Google Scholar] [CrossRef]
- Ke, M.; Chen, Y.; Wu, A.; Sun, Y.; Su, C.; Wu, H.; Jin, X.; Tao, J.; Wang, Y.; Ma, X.; et al. Short peptides derived from the interaction domain of SARS coronavirus non-structural protein nsp10 can suppress the 2′-O-methyltransferase activity of nsp10/nsp16 complex. Virus Res. 2012, 167, 322–328. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, Y.; Wu, A.; Xu, S.; Pan, R.; Zeng, C.; Jin, X.; Ge, X.; Shi, Z.; Ahola, T.; et al. Coronavirus nsp10/nsp16 Methyltransferase Can Be Targeted by nsp10-Derived Peptide In Vitro and In Vivo To Reduce Replication and Pathogenesis. J. Virol. 2015, 89, 8416–8427. [Google Scholar] [CrossRef]
- Cassandri, M.; Smirnov, A.; Novelli, F.; Pitolli, C.; Agostini, M.; Malewicz, M.; Melino, G.; Raschellà, G. Zinc-finger proteins in health and disease. Cell Death Discov. 2017, 3, 17071. [Google Scholar] [CrossRef]
- Bombarda, E.; Grell, E.; Roques, B.P.; Mély, Y. Molecular Mechanism of the Zn2+-Induced Folding of the Distal CCHC Finger Motif of the HIV-1 Nucleocapsid Protein. Biophys. J. 2007, 93, 208–217. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Holm, L.; Laakso, L.M. Dali server update. Nucl. Acids Res. 2016, 44, W351–W355. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucl. Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed]
- Heinig, M.; Frishman, D. STRIDE: A Web server for secondary structure assignment from known atomic coordinates of proteins. Nucl. Acids Res. 2004, 32, W500–W502. [Google Scholar] [CrossRef]
- Joseph, J.S.; Saikatendu, K.S.; Subramanian, V.; Neuman, B.W.; Brooun, A.; Griffith, M. Crystal Structure of Non-structural Protein 10 from the Severe Acute Respiratory Syndrome Coronavirus Reveals a Novel Fold with Two Zinc-Binding Motifs. J. Virol. 2006, 80, 7894–7901. [Google Scholar] [CrossRef]
- Viswanathan, T.; Arya, S.; Chan, S.-H.; Qi, S.; Dai, N.; Misra, A.; Park, J.-G.; Oladunni, F.; Kovalskyy, D.; Hromas, R.A.; et al. Structural basis of RNA cap modification by SARS CoV-2. Nat. Commun. 2020, 11, 3718. [Google Scholar] [CrossRef]
- Krafcikova, P.; Silhan, J.; Nencka, R.; Boura, E. Structural analysis of the SARS-CoV-2 methyltransferase complex involved in RNA cap creation bound to sinefungin. Nat. Commun. 2020, 11, 3717. [Google Scholar] [CrossRef]
- Züst, R.; Cervantes-Barragan, L.; Habjan, M.; Maier, R.; Neuman, B.W.; Ziebuhr, J.; Szretter, K.J.; Baker, S.C.; Barchet, W.; Diamond, M.S.; et al. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 2011, 12, 137–143. [Google Scholar] [CrossRef]
- Su, D.; Lou, Z.; Joachimiak, A.; Zhang, X.C.; Bartlam, M.; Zhang, R.; Joachimiak, A.; Zhang, X.C.; Bartlam, M.; Rao, Z.; et al. Dodecamer Structure of Severe Acute Respiratory Syndrome Coronavirus Nonstructural Protein nsp10. J. Virol. 2006, 80, 7902–7908. [Google Scholar] [CrossRef]
- Ma, Y.; Wu, L.; Shaw, N.; Gao, Y.; Wang, J.; Sun, Y.; Lou, Z.; Yan, L.; Zhang, R.; Rao, Z.; et al. Structural basis and functional analysis of the SARS coronavirus nsp14–nsp10 complex. Proc. Natl. Acad. Sci. USA 2015, 112, 9436–9441. [Google Scholar] [CrossRef]
- Bouvet, M.; Imbert, I.; Subissi, J.; Gluais, L.; Canard, B.; Decroly, E. RNA 3′-end mismatch excision by the severe acute respiratory syndrome coronavirus nonstructural protein nsp10/nsp14 exoribonuclease complex. Proc. Natl. Acad. Sci. USA 2012, 109, 9372–9377. [Google Scholar] [CrossRef] [PubMed]
- Eckerle, L.D.; Lu, X.; Sperry, S.M.; Choi, L.; Denison, M.R. High fidelity of murine hepatitis virus replication is decreased in nsp14 exoribonuclease mutants. J. Virol. 2007, 81, 12135–12144. [Google Scholar] [CrossRef] [PubMed]
- Pruijssers, A.J.; Denison, M.R. Nucleoside analogues for the treatment of coronavirus infections. Curr. Opin. Virol. 2019, 35, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.P.H.; Armstrong, J.; Davenport, A.P.; Davies, J.; Faccenda, E.; Harding, S.D.; Levi-Schaffer, F.; Maguire, J.J.; Pawson, A.J.; Southan, C.; et al. A rational roadmap for SARS-CoV-2/COVID-19 pharmacotherapeutic research and development. Br. J. Pharmacol. 2020, 1–25. [Google Scholar] [CrossRef]
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.; Cowtan, K. Features and Development of Coot. Acta Cryst D 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D 2012, 68, 352–367. [Google Scholar] [CrossRef]
- Demeler, B. UltraScan A Comprehensive Modern Analytical Ultracentrifugation: Techniques and Methods. Data Analysis Software Package for Analytical Ultracentrifugation Experiments; Scott, D.J., Harding, S.E., Rowe, A.J., Eds.; Royal Society of Chemistry: London, UK, 2005; pp. 210–229. [Google Scholar] [CrossRef]
- Brookes, E.; Cao, W.; Demeler, B. A two-dimensional spectrum analysis for sedimentation velocity experiments of mixtures with heterogeneity in molecular weight and shape. Eur. Biophys. J. 2010, 39, 405–414. [Google Scholar] [CrossRef]
- Brookes, E.; Demeler, B. Genetic Algorithm Optimization for obtaining accurate Molecular Weight Distributions from Sedimentation Velocity Experiments. Analytical Ultracentrifugation VIII, C. Wandrey and H.; Cölfen, Eds. Springer. Prog. Colloid Polym. Sci. 2006, 131, 78–82. [Google Scholar] [CrossRef]
- Demeler, B.; Brookes, E. Monte Carlo analysis of sedimentation experiments. Colloid Polym. Sci. 2008, 286, 129–137. [Google Scholar] [CrossRef]
- Brookes, E.H.; Rocco, M. Recent advances in the UltraScan SOlution MOdeller (US-SOMO) hydrodynamic and small-angle scattering data analysis and simulation suite. Eur. Biophys. J. 2018, 47, 855–864. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection and Refinement Statistics | SARS-CoV-2 nsp10 (PDB ID 6ZPE) |
|---|---|
| Wavelength (Å) | 0.976 |
| Resolution range (Å) | 75.39–1.58 (1.61–0.58) |
| Space group | I213 |
| Unit cell parameters (Å; °) | a = b = c = 106.62 Å; α = β = γ = 90 |
| Matthew coefficient (Å3/Da) | 3.81 |
| Molecules per asymmetric unit | 1 |
| Total reflections | 277,522 (14,080) |
| Unique reflections | 27,706 (1391) |
| Multiplicity | 10.0 (10.1) |
| Completeness (%) | 99.9 (100.0) |
| Mean I/sigma(I) | 21.1 (2.0) |
| Wilson B-factor | 29.95 |
| R-meas (%) | 4.7 (128.3) |
| CC1/2 (%) | 99.9 (70.9) |
| Reflections used in refinement | 27,702 (2782) |
| Rcryst/Rfree (%) | 15.2/16.0 |
| Total no. of non-hydrogen atoms (protein) | 1122 |
| No. of protein/ligand/solvent residues | 127/11/169 |
| RMSD bond lengths, bond angles (Å; °) | 0.012/1.09 |
| Ramachandran favored/allowed/outliers/rotamer outliers (%) | 98.4/1.6/0.0/0.0 |
| Clashscore | 2.7 |
| Average B-factor/protein/ligands/solvent | 38.0/35.9/55.8/48.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rogstam, A.; Nyblom, M.; Christensen, S.; Sele, C.; Talibov, V.O.; Lindvall, T.; Rasmussen, A.A.; André, I.; Fisher, Z.; Knecht, W.; et al. Crystal Structure of Non-Structural Protein 10 from Severe Acute Respiratory Syndrome Coronavirus-2. Int. J. Mol. Sci. 2020, 21, 7375. https://doi.org/10.3390/ijms21197375
Rogstam A, Nyblom M, Christensen S, Sele C, Talibov VO, Lindvall T, Rasmussen AA, André I, Fisher Z, Knecht W, et al. Crystal Structure of Non-Structural Protein 10 from Severe Acute Respiratory Syndrome Coronavirus-2. International Journal of Molecular Sciences. 2020; 21(19):7375. https://doi.org/10.3390/ijms21197375
Chicago/Turabian StyleRogstam, Annika, Maria Nyblom, Signe Christensen, Celeste Sele, Vladimir O. Talibov, Therese Lindvall, Anna Andersson Rasmussen, Ingemar André, Zoë Fisher, Wolfgang Knecht, and et al. 2020. "Crystal Structure of Non-Structural Protein 10 from Severe Acute Respiratory Syndrome Coronavirus-2" International Journal of Molecular Sciences 21, no. 19: 7375. https://doi.org/10.3390/ijms21197375
APA StyleRogstam, A., Nyblom, M., Christensen, S., Sele, C., Talibov, V. O., Lindvall, T., Rasmussen, A. A., André, I., Fisher, Z., Knecht, W., & Kozielski, F. (2020). Crystal Structure of Non-Structural Protein 10 from Severe Acute Respiratory Syndrome Coronavirus-2. International Journal of Molecular Sciences, 21(19), 7375. https://doi.org/10.3390/ijms21197375