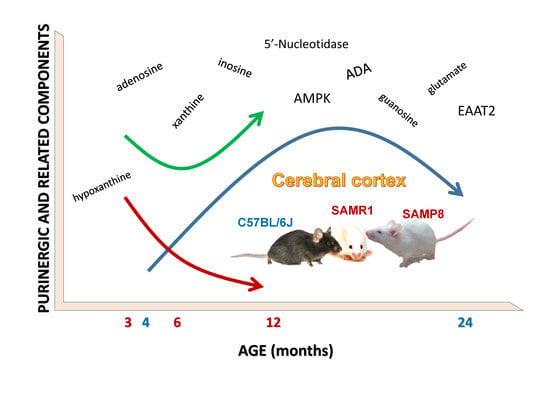
Adenosine Metabolism in the Cerebral Cortex from Several Mice Models during Aging



Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
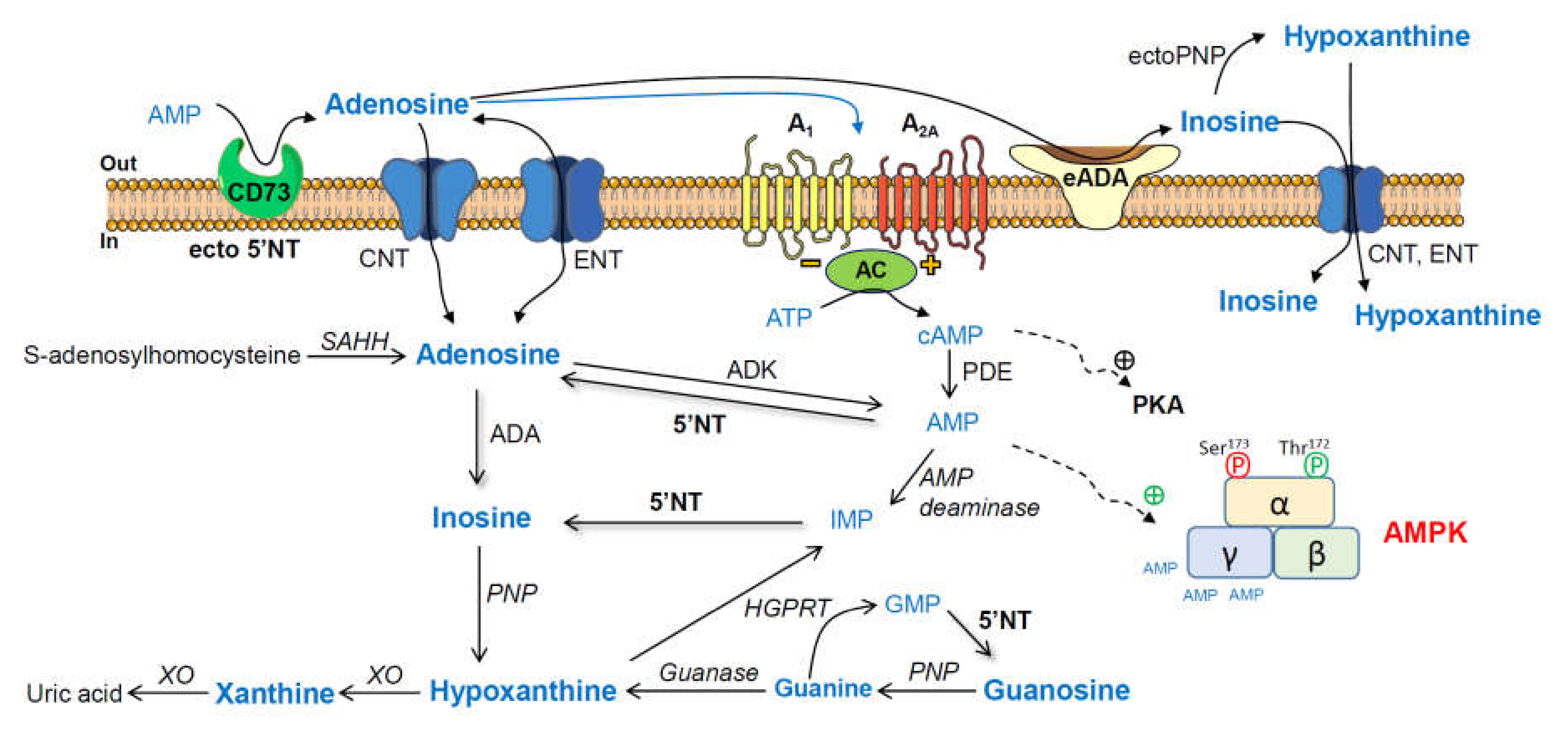
1. Introduction
2. Results
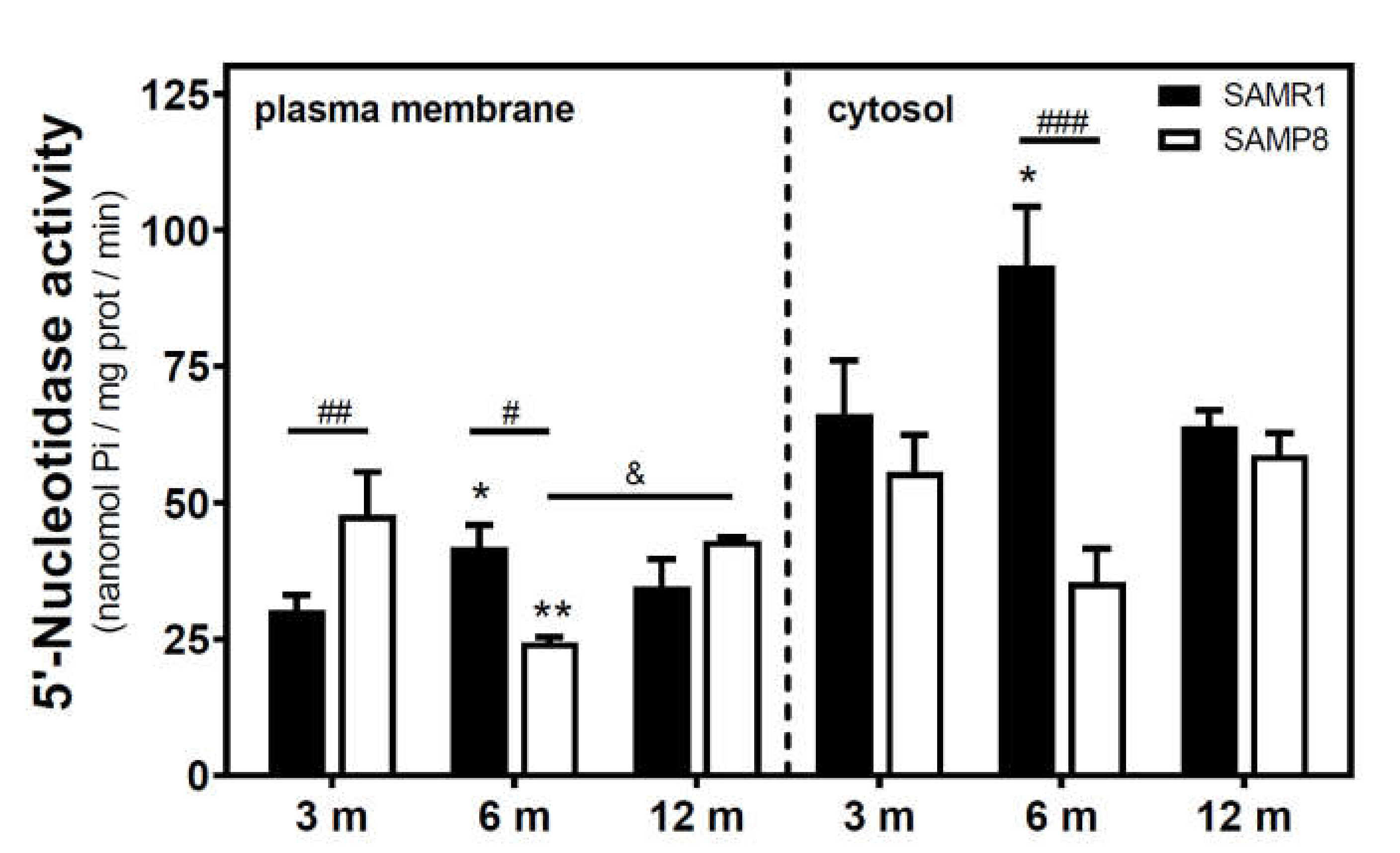
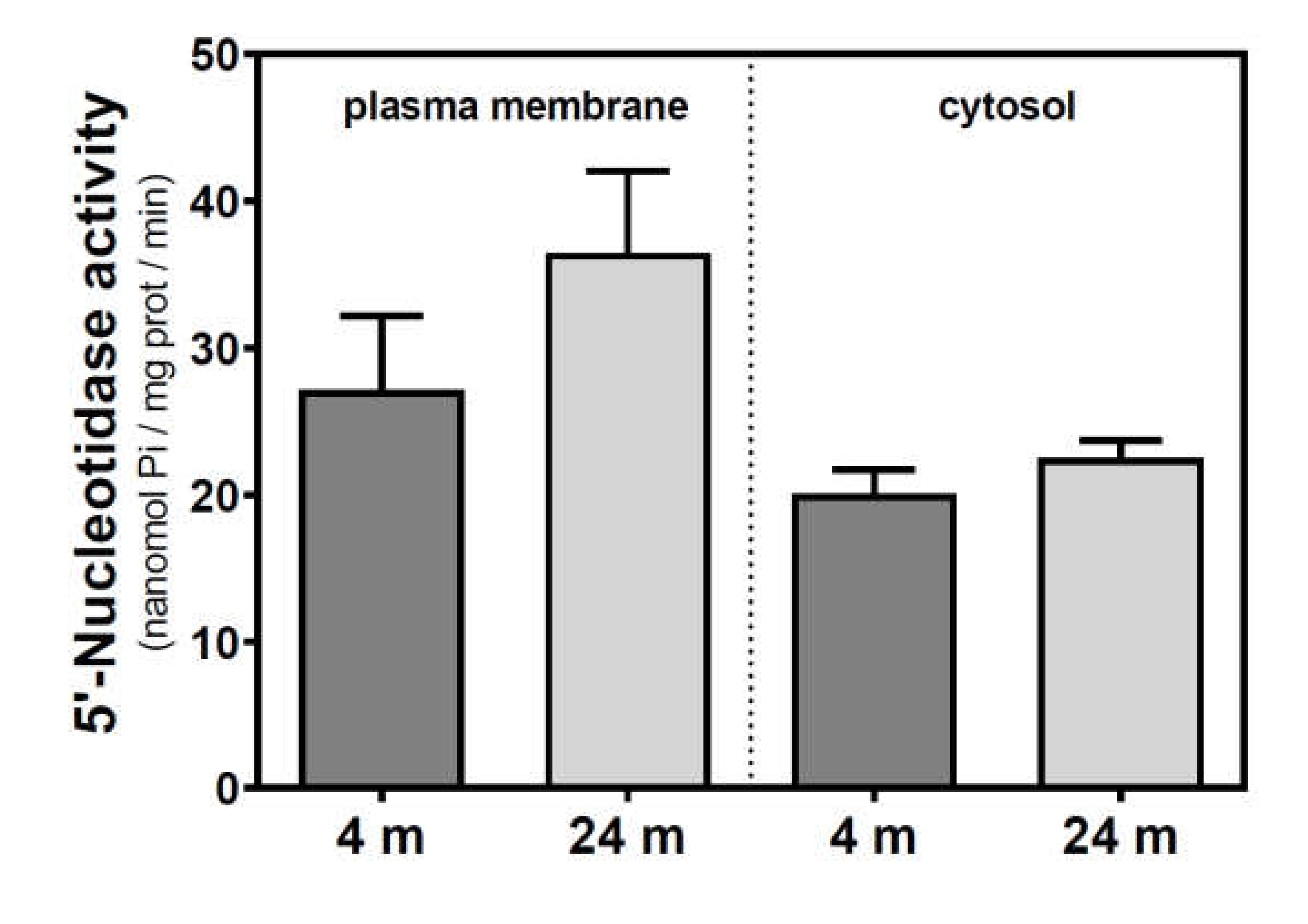
2.1. Modulation of 5′-Nucleotidase Activity during Aging in the Cerebral Cortex from SAM and C57BL/6J Mice
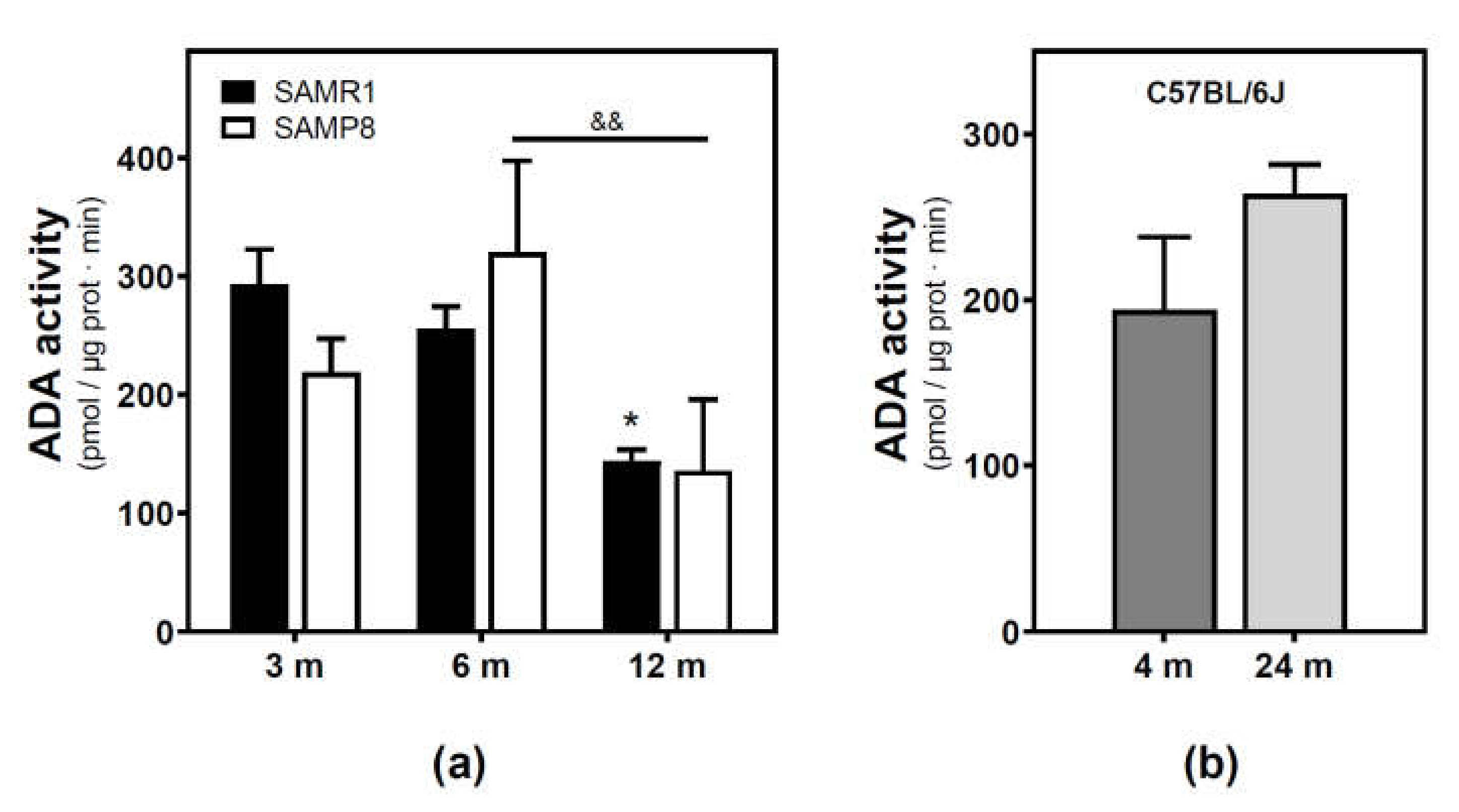
2.2. Modulation of Adenosine Deaminase Activity during Aging in the Cerebral Cortex from SAM and C57BL/6J Mice
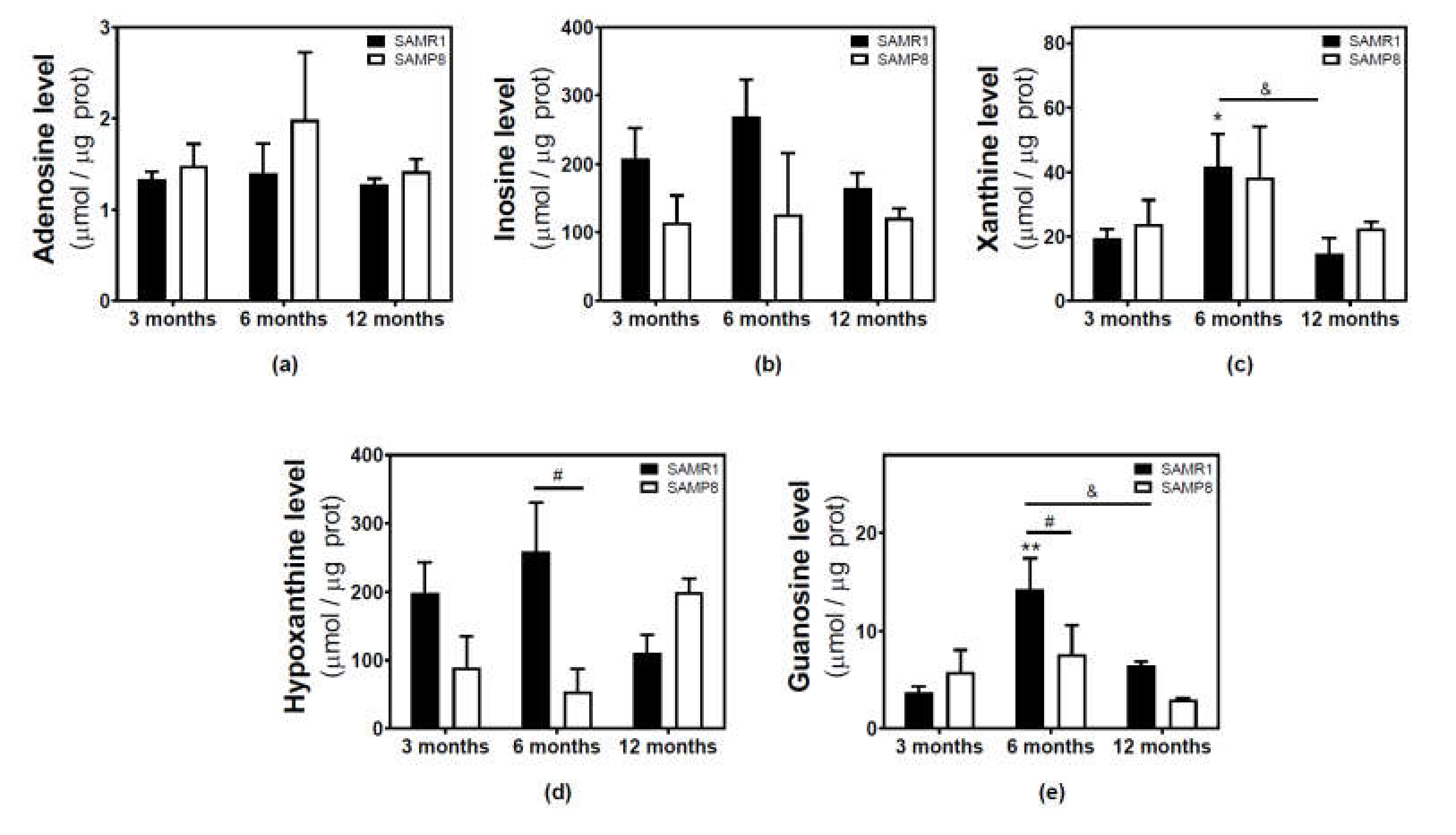
2.3. Modulation of Adenosine and Related Metabolite Levels during Aging in the Cerebral Cortex from SAM and C57BL/6J Mice
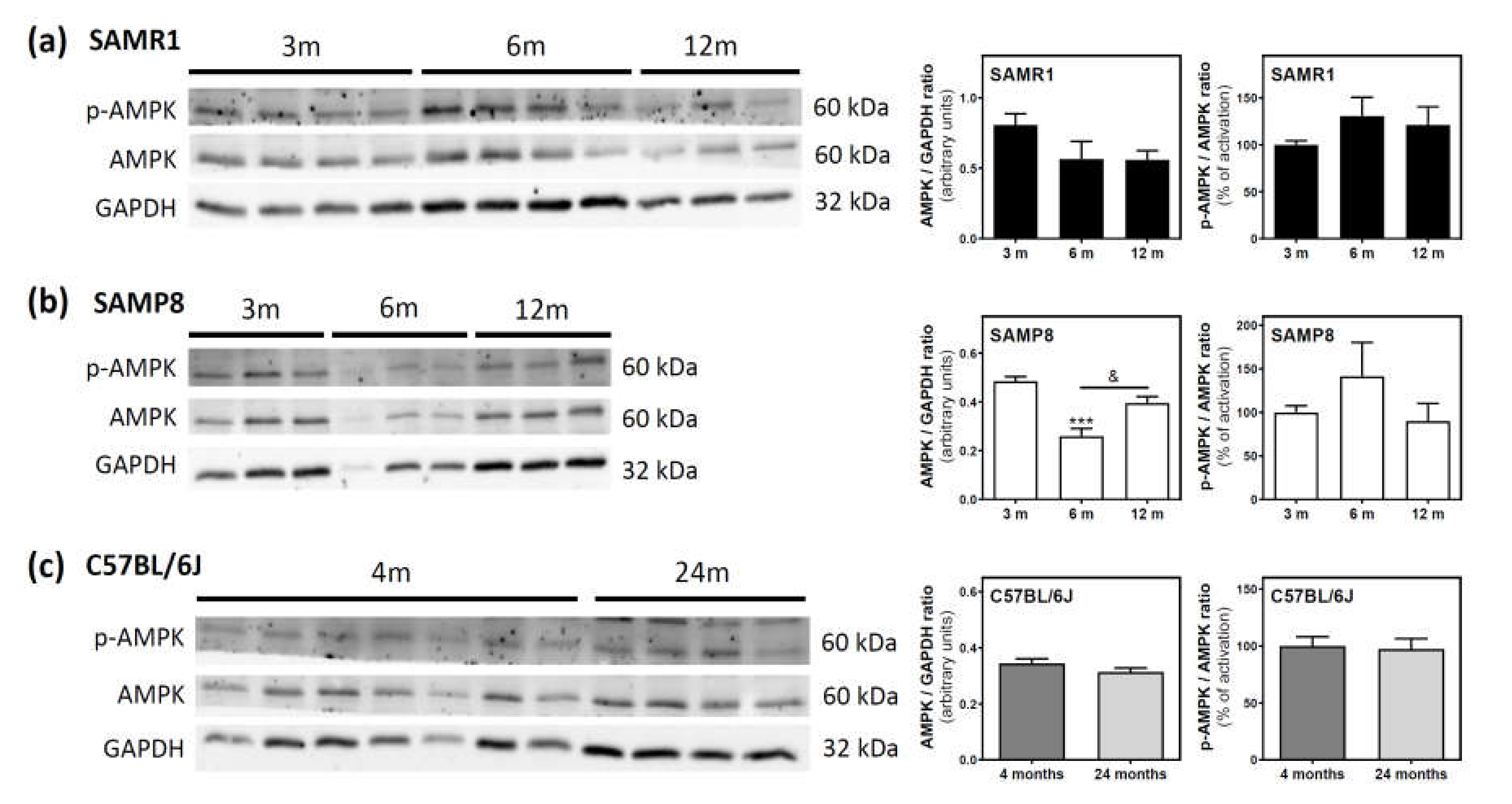
2.4. AMPK Activation State during Aging in the Cerebral Cortex from SAM and C57BL/6J Mice
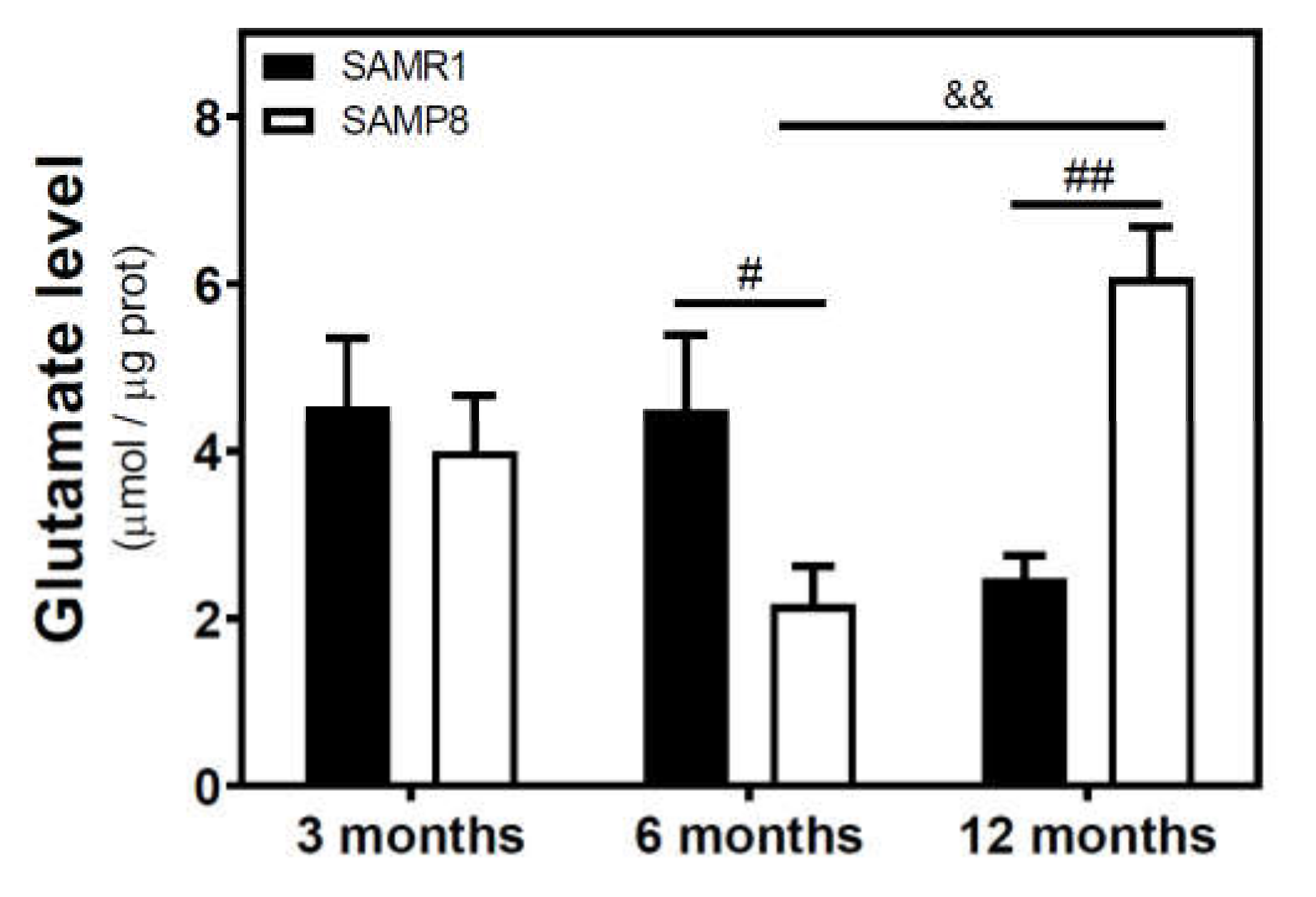
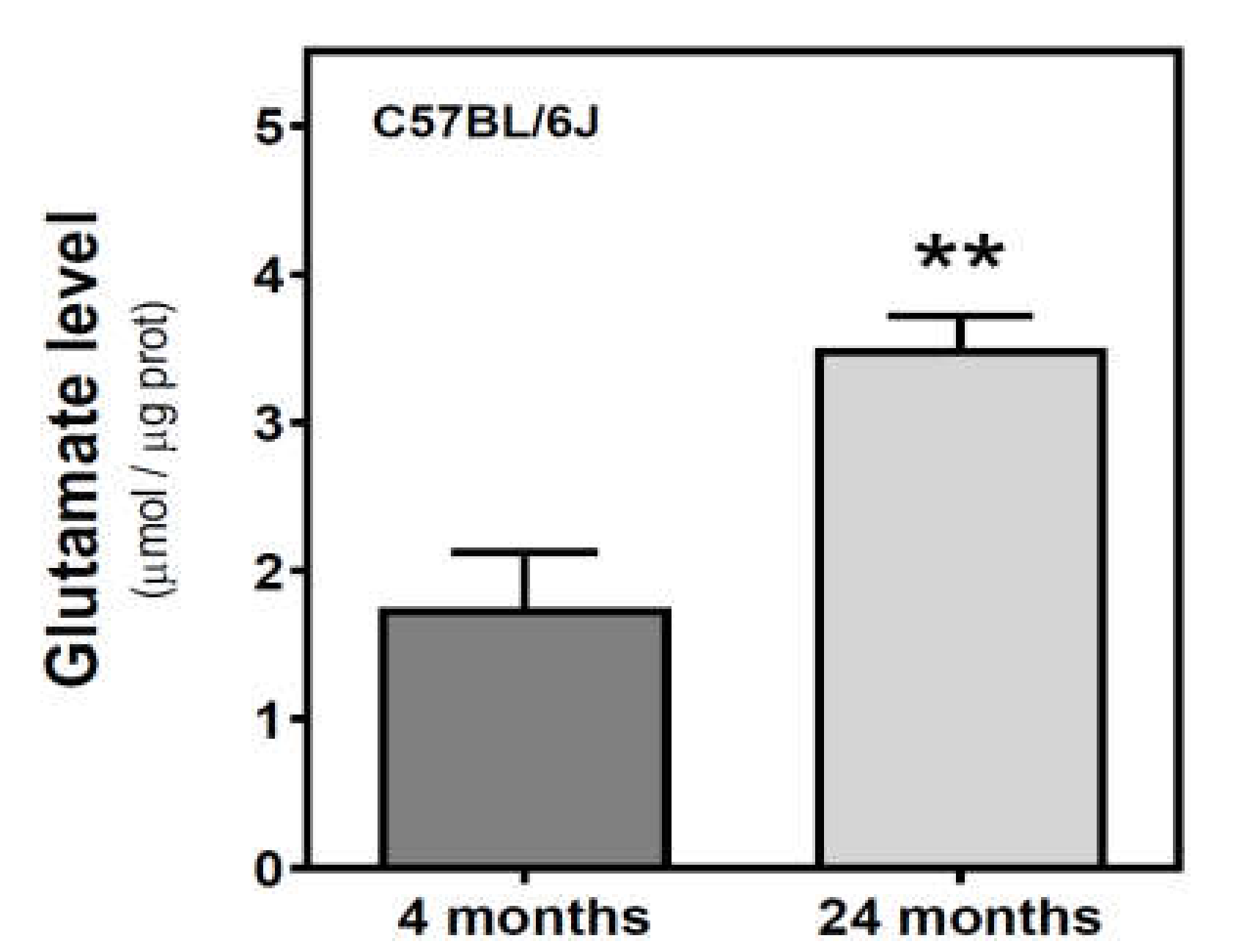
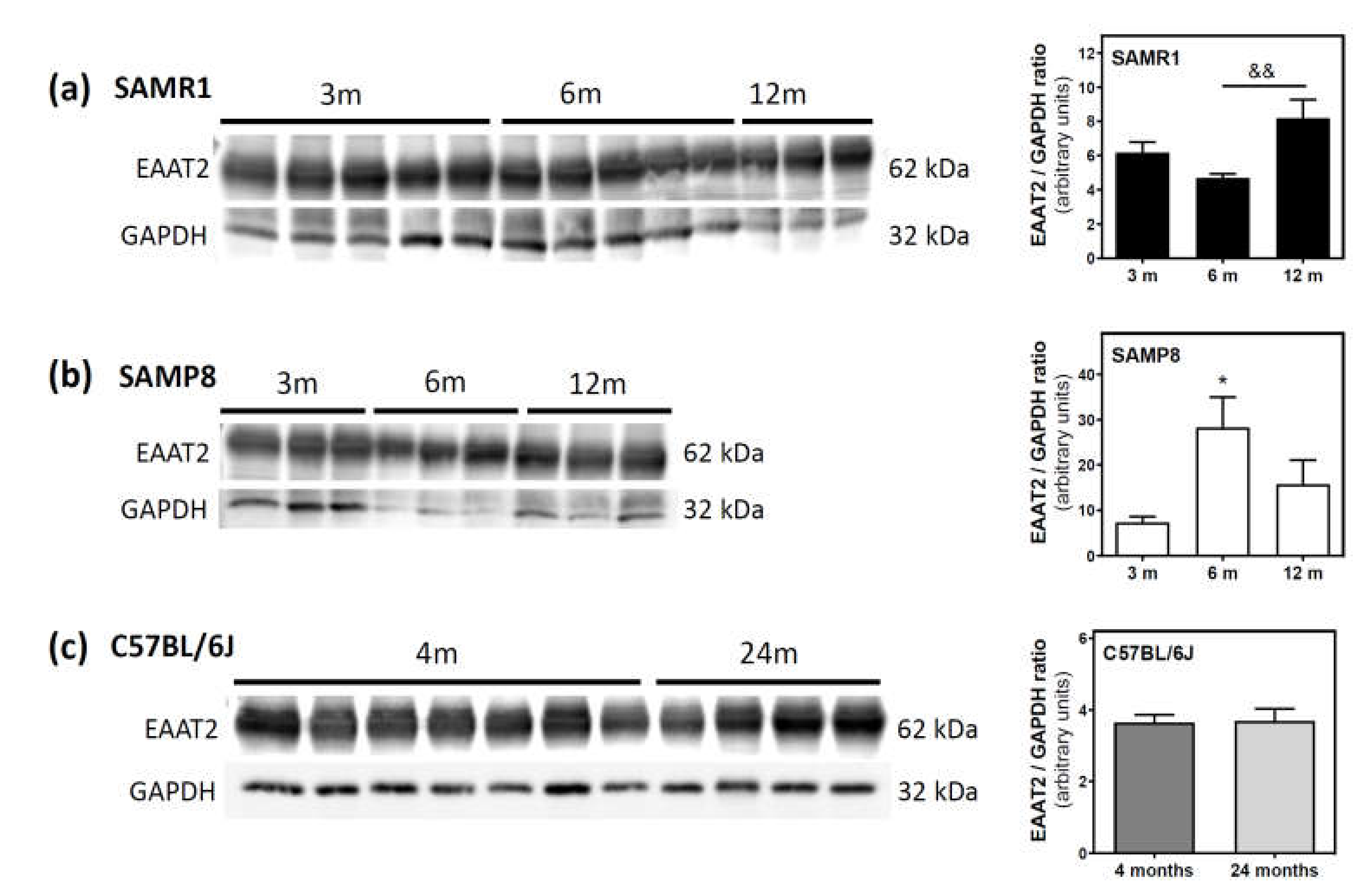
2.5. Glutamate and Excitatory Amino Acid Transporter 2 Levels during Aging in the Cerebral Cortex from SAM and C57BL/6J Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Plasma Membrane Isolation
4.3. 5’-Nucleotidase Activity Assay
4.4. Adenosine and Related Metabolite Levels Quantification by HPLC
4.5. Immunodetection of p-AMPK and AMPK by Western Blotting
4.6. Glutamate Level Quantification
4.7. Adenosine Deaminase Activity Assay
4.8. Statistical and Data Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fredholm, B.B.; Ijzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors-An update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-F.; Lee, C.-F.; Chern, Y. Adenosine receptor neurobiology: Overview. Int. Rev. Neurobiol. 2014, 119, 1–49. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Fredholm, B.B.; Verkhratsky, A. Adenosine and ATP receptors in the brain. Curr. Top. Med. Chem. 2011, 11, 973–1011. [Google Scholar] [CrossRef] [PubMed]
- Albasanz, J.L.; Perez, S.; Barrachina, M.; Ferrer, I.; Martín, M.; Martín, M. Research article: Up-regulation of adenosine receptors in the frontal cortex in Alzheimer’s disease. Brain Pathol. 2008, 18, 211–219. [Google Scholar] [CrossRef]
- Blum, D.; Chern, Y.; Domenici, M.R.; Buée, L.; Lin, C.-Y.; Rea, W.; Ferré, S.; Popoli, P. The role of adenosine tone and adenosine receptors in Huntington’s disease. J. Caffeine Adenosine Res. 2018, 8, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Angulo-Pueyo, E.; Ecasado, V.; Mallol, J.; Canela, E.I.; Viñals, F.; Ferrer, I.; Lluis, C.; Franco, R. A1 adenosine receptors accumulate in neurodegenerative structures in Alzheimer disease and mediate both amyloid precursor protein processing and tau phosphorylation and translocation. Brain Pathol. 2003, 13, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Rial, D.; Canas, P.M.; Yoo, J.-H.; Li, W.; Zhou, X.; Wang, Y.; Van Westen, G.J.; Payen, M.-P.; Augusto, E.; et al. Optogenetic activation of intracellular adenosine A2A receptor signaling in the hippocampus is sufficient to trigger CREB phosphorylation and impair memory. Mol. Psychiatry 2015, 20, 1339–1349. [Google Scholar] [CrossRef]
- Franco, R.; Rivas-Santisteban, R.; Casanovas, M.; Lillo, A.; Saura, C.A.; Navarro, G. Adenosine A2A receptor antagonists affects NMDA glutamate receptor function. potential to address neurodegeneration in Alzheimer’s disease. Cells 2020, 9, 1075. [Google Scholar] [CrossRef]
- Rial, D.; Lara, D.R.; Cunha, R.A. The adenosine neuromodulation system in schizophrenia. Int. Rev. Neurobiol. 2014, 119, 395–449. [Google Scholar] [CrossRef]
- Matos, M.; Shen, H.-Y.; Augusto, E.; Wang, Y.; Wei, C.J.; Wang, Y.T.; Agostinho, P.; Boison, D.; Cunha, R.A.; Chen, J.-F. Deletion of adenosine A2A receptors from astrocytes disrupts glutamate homeostasis leading to psychomotor and cognitive impairment: Relevance to schizophrenia. Biol. Psychiatry 2015, 78, 763–764. [Google Scholar] [CrossRef]
- Villar-Menéndez, I.; Díaz-Sánchez, S.; Blanch, M.; Albasanz, J.L.; Pereira-Veiga, T.; Monje, A.; Planchat, L.M.; Ferrer, I.; Martín, M.; Barrachina, M.; et al. Reduced striatal adenosine A2A receptor levels define a molecular subgroup in schizophrenia. J. Psychiatry Res. 2014, 51, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Andrés, P.; Albasanz, J.L.; Ferrer, I.; Martín, M. Purine-related metabolites and their converting enzymes are altered in frontal, parietal and temporal cortex at early stages of Alzheimer’s disease pathology. Brain Pathol. 2018, 28, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Dale, N. Purinergic signalling during development and ageing. Purinergic Signal. 2015, 11, 277–305. [Google Scholar] [CrossRef] [PubMed]
- Cunha, R.A. How does adenosine control neuronal dysfunction and neurodegeneration. J. Neurochem. 2016, 139, 1019–1055. [Google Scholar] [CrossRef] [PubMed]
- Mackiewicz, M.; Nikonova, E.V.; Zimmermann, J.E.; Romer, M.A.; Cater, J.; Galante, R.J.; Pack, A.I. Age-related changes in adenosine metabolic enzymes in sleep/wake regulatory areas of the brain. Neurobiol. Aging 2006, 27, 351–360. [Google Scholar] [CrossRef]
- Zimmermann, H.; Zebisch, M.; Sträter, N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. 2012, 8, 437–502. [Google Scholar] [CrossRef] [PubMed]
- Cortes, A.; Gracia, E.; Moreno, E.; Mallol, J.; Lluís, C.; Canela, E.I.; Ecasado, V. Moonlighting adenosine deaminase: A target protein for drug development. Med. Res. Rev. 2014, 35, 85–125. [Google Scholar] [CrossRef]
- Jeong, S. Molecular and cellular basis of neurodegeneration in Alzheimer’s disease. Mol. Cells 2017, 40, 613–620. [Google Scholar] [CrossRef]
- Hof, P.R.; Morrison, J.H. The aging brain: Morphomolecular senescence of cortical circuits. Trends Neurosci. 2004, 27, 607–613. [Google Scholar] [CrossRef]
- Fjell, A.M.; McEvoy, L.; Holland, M.; Dale, A.M.; Walhovd, K.B.; Initiative, A.D.N. What is normal in normal aging? Effects of aging, amyloid and Alzheimer’s disease on the cerebral cortex and the hippocampus. Prog. Neurobiol. 2014, 117, 20–40. [Google Scholar] [CrossRef]
- Giannakopoulos, P.; Hof, P.R.; Michel, J.-P.; Guimon, J.; Bouras, C. Cerebral cortex pathology in aging and Alzheimer’s disease: A quantitative survey of large hospital-based geriatric and psychiatric cohorts. Brain Res. Rev. 1997, 25, 217–245. [Google Scholar] [CrossRef]
- Niccoli, T.; Partridge, L. Ageing as a risk factor for disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef] [PubMed]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. ROS, cell senescence, and novel molecular mechanisms in aging and age-related diseases. Oxidative Med. Cell. Longev. 2016, 2016, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Flurkey, K.; Currer, J.; Harrison, D. The Mouse in Aging Research. In The Mouse in Biomedical Research, 2nd Edition; Fox, J.G., Ed.; Elsevier: Amsterdam, The Netherlands, 2007; pp. 637–672. [Google Scholar]
- Busquets, O.; Ettcheto, M.; Pallás, M.; Beas-Zarate, C.; Verdaguer, E.; Auladell, C.; Folch, J.; Camins, A. Long-term exposition to a high fat diet favors the appearance of β-amyloid depositions in the brain of C57BL/6J mice. A potential model of sporadic Alzheimer’s disease. Mech. Ageing Dev. 2017, 162, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Takanashi, Y.; Sugita, K.; Miyazawa, M.; Yanagihara, R.; Yasuda, K.; Onouchi, H.; Kawabe, N.; Nakata, M.; Yamamoto, Y.; et al. Endogenous reactive oxygen species cause astrocyte defects and neuronal dysfunctions in the hippocampus: A new model for aging brain. Aging Cell 2016, 16, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Ingram, D.K. Murine models of brain aging and age-related neurodegenerative diseases. Behav. Brain Res. 1997, 85, 1–25. [Google Scholar] [CrossRef]
- Liu, B.; Liu, J.; Shi, J.-S. SAMP8 Mice as a model of age-related cognition decline with underlying mechanisms in Alzheimer’s disease. J. Alzheimer’s Dis. 2020, 75, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Hosokawa, M.; Higuchi, K. Senescence-accelerated mouse (SAM): A novel murine model of senescence. Exp. Gerontol. 1997, 32, 105–109. [Google Scholar] [CrossRef]
- Akiguchi, I.; Pallàs, M.; Takahashi, R.; Unno, K.; Higuchi, K.; Hosokawa, M.; Budka, H.; Akiyama, H.; Ueno, M.; Han, J.; et al. SAMP8 mice as a neuropathological model of accelerated brain aging and dementia: Toshio Takeda’s legacy and future directions. Neuropathology 2017, 37, 293–305. [Google Scholar] [CrossRef]
- Pallas, M.; Camins, A.; Smith, M.A.; Perry, G.; Lee, H.G.; Casadesus, G. From aging to Alzheimer’s disease: Unveiling “the switch” with the senescence-accelerated mouse model (SAMP8). J. Alzheimers Dis. 2008, 15, 615–624. [Google Scholar] [CrossRef]
- Morley, J.E.; Farr, S.A.; Kumar, V.B.; Armbrecht, H.J. The SAMP8 mouse: A model to develop therapeutic interventions for Alzheimer’s disease. Curr. Pharm. Des. 2012, 18, 1123–1130. [Google Scholar] [CrossRef]
- Carter, T.A.; Greenhall, J.A.; Yoshida, S.; Fuchs, S.; Helton, R.; Swaroop, A.; Lockhart, D.J.; Barlow, C. Mechanisms of aging in senescence-accelerated mice. Genome Biol. 2005, 6, R48. [Google Scholar] [CrossRef] [PubMed]
- Yuan, R.; Tsaih, S.-W.; Petkova, S.B.; De Evsikova, C.M.; Xing, S.; Marion, M.A.; Bogue, M.A.; Mills, K.D.; Peters, L.L.; Bult, C.J.; et al. Aging in inbred strains of mice: Study design and interim report on median lifespans and circulating IGF1 levels. Aging Cell 2009, 8, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Kunstyr, I.; Leuenberger, H.-G.W. Gerontological data of C57BL/6J mice. I. Sex differences in survival curves. J. Gerontol. 1975, 30, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Sengupta, P. Men and mice: Relating their ages. Life Sci. 2016, 152, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Changarathil, G.; Ramirez, K.; Isoda, H.; Sada, A.; Yanagisawa, H. Wild-type and SAMP8 mice show age-dependent changes in distinct stem cell compartments of the interfollicular epidermis. PLoS ONE 2019, 14, e0215908. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Melgar, A.; Albasanz, J.L.; Palomera-Ávalos, V.; Pallàs, M.; Martín, M. Resveratrol modulates and reverses the age-related effect on adenosine-mediated signalling in SAMP8 mice. Mol. Neurobiol. 2018, 56, 2881–2895. [Google Scholar] [CrossRef]
- Castillo-Sarmiento, C.A.; Albasanz, J.L.; León, D.; Jordan, J.; Pallás, M.; Camins, A.; Martín, M. Age-related expression of adenosine receptors in brain from the senescence-accelerated mouse. Exp. Gerontol. 2009, 44, 453–461. [Google Scholar] [CrossRef]
- De Oliveira, P.G.; Ramos, M.L.S.; Amaro, A.J.; Dias, R.; Vieira, S.I. Gi/o-protein coupled receptors in the aging brain. Front. Aging Neurosci. 2019, 11, 89. [Google Scholar] [CrossRef]
- Cunha, R.A.; Constantino, M.D.; Sebastião, A.M.; Ribeiro, J.A. Modification of A-i and A2a adenosine receptor binding in aged striatum, hippocampus and cortex of the rat. NeuroReport 1995, 6, 1583. [Google Scholar] [CrossRef]
- Cunha, R.A. Overactivity of neuronal adenosine A2A receptors accelerates neurodegeneration. Brain 2019, 142, 3323–3324. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W.; Ceruti, S.; Maria, A. Adenosine receptors and neurological disease: Neuroprotection and neurodegeneration. cGMP: Gener. Eff. Ther. Implic. 2009, 193, 535–587. [Google Scholar] [CrossRef]
- Kelly, M.P. Cyclic nucleotide signaling changes associated with normal aging and age-related diseases of the brain. Cell. Signal. 2018, 42, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Hadem, I.K.H.; Majaw, T.; Kharbuli, B.; Sharma, R. Beneficial effects of dietary restriction in aging brain. J. Chem. Neuroanat. 2019, 95, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Morgunova, G.V.; Klebanov, A.A. Age-related AMP-activated protein kinase alterations: From cellular energetics to longevity. Cell Biochem. Funct. 2019, 37, 169–176. [Google Scholar] [CrossRef]
- López-Otín, C.; Galluzzi, L.; Freije, J.M.P.; Madeo, F.; Kroemer, G. Metabolic control of longevity. Cell 2016, 166, 802–821. [Google Scholar] [CrossRef]
- Peixoto, C.A.; De Oliveira, W.H.; Araújo, S.M.D.R.; Nunes, A.K.S. AMPK activation: Role in the signaling pathways of neuroinflammation and neurodegeneration. Exp. Neurol. 2017, 298, 31–41. [Google Scholar] [CrossRef]
- Liu, Y.-J.; Chern, Y. AMPK-mediated regulation of neuronal metabolism and function in brain diseases. J. Neurogenet. 2015, 29, 50–58. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Zimmermann, H.R.; Yang, W.; Kasica, N.P.; Zhou, X.; Wang, X.; Beckelman, B.C.; Lee, J.; Furdui, C.M.; Keene, C.D.; Ma, T. Brain-specific repression of AMPKα1 alleviates pathophysiology in Alzheimer’s model mice. J. Clin. Investig. 2020. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2017, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Aymerich, I.; Foufelle, F.; Ferré, P.; Casado, F.J.; Pastor-Anglada, M. Extracellular adenosine activates AMP-dependent protein kinase (AMPK). J. Cell Sci. 2006, 119, 1612–1621. [Google Scholar] [CrossRef]
- Aw, D.K.L.; Sinha, R.A.; Xie, S.Y.; Yen, P.M. Differential AMPK phosphorylation by glucagon and metformin regulates insulin signaling in human hepatic cells. Biochem. Biophys. Res. Commun. 2014, 447, 569–573. [Google Scholar] [CrossRef]
- Fredholm, B.B.; Chen, J.-F.; Masino, S.A.; Vaugeois, J.-M. Actions of adenosine at its receptors in the CNS: Insights from knockouts and drugs. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 385–412. [Google Scholar] [CrossRef] [PubMed]
- Leon-Navarro, J.L.A.A.M.M.D.A.; Albasanz, J.L.; Martín, M. Functional cross-talk between adenosine and metabotropic glutamate receptors. Curr. Neuropharmacol. 2019, 17, 422–437. [Google Scholar] [CrossRef] [PubMed]
- Segovia, G.; Porras, A.; Del Arco, A.; Mora, F. Glutamatergic neurotransmission in aging: A critical perspective. Mech. Ageing Dev. 2001, 122, 1–29. [Google Scholar] [CrossRef]
- Kumar, A.; Foster, T. Alteration in NMDA Receptor mediated glutamatergic neurotransmission in the hippocampus during senescence. Neurochem. Res. 2018, 44, 38–48. [Google Scholar] [CrossRef]
- Tzschentke, T.M. Glutamatergic mechanisms in different disease states: Overview and therapeutical implications-An introduction. Amino Acids 2002, 23, 147–152. [Google Scholar] [CrossRef]
- Gleichmann, M.; Chow, V.W.; Mattson, M.P. Homeostatic disinhibition in the aging brain and Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 24, 15–24. [Google Scholar] [CrossRef]
- Treyer, V.; Gietl, A.F.; Suliman, H.; Gruber, E.; Meyer, R.; Buchmann, A.; Johayem, A.; Unschuld, P.G.; Nitsch, R.M.; Buck, A.; et al. Reduced uptake of [11C]- ABP688, a PET tracer for metabolic glutamate receptor 5 in hippocampus and amygdala in Alzheimer’s dementia. Brain Behav. 2020, 10, e01632. [Google Scholar] [CrossRef] [PubMed]
- Albasanz, J.L.; Dalfó, E.; Ferrer, I.; Martín, M.; Martín, M. Impaired metabotropic glutamate receptor/phospholipase C signaling pathway in the cerebral cortex in Alzheimer’s disease and dementia with Lewy bodies correlates with stage of Alzheimer’s-disease-related changes. Neurobiol. Dis. 2005, 20, 685–693. [Google Scholar] [CrossRef]
- Sánchez-Melgar, A.; Albasanz, J.L.; Pallás, M.; Martin, M. Resveratrol Differently Modulates Group I metabotropic glutamate receptors depending on age in SAMP8 mice. ACS Chem. Neurosci. 2020, 11, 1770–1780. [Google Scholar] [CrossRef] [PubMed]
- Boison, D.; Yegutkin, G.G. Adenosine metabolism: Emerging concepts for cancer therapy. Cancer Cell 2019, 36, 582–596. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, Z.; Juhasz, G.; Dobolyi, A.; Bobest, M.; Papp, V.; Takáts, L.; Kekesi, K.A. Gender-and age-dependent changes in nucleoside levels in the cerebral cortex and white matter of the human brain. Brain Res. Bull. 2010, 81, 579–584. [Google Scholar] [CrossRef]
- Bellé, L.P.; De Bona, K.S.; Abdalla, F.H.; Pimentel, V.C.; Pigatto, A.S.; Moretto, M.B. Comparative evaluation of adenosine deaminase activity in cerebral cortex and hippocampus of young and adult rats: Effect of garlic extract (Allium sativum L.) on their susceptibility to heavy metal exposure. Basic Clin. Pharmacol. Toxicol. 2009, 104, 408–413. [Google Scholar] [CrossRef]
- Shoji, H.; Miyakawa, T. Age-related behavioral changes from young to old age in male mice of a C57BL/6J strain maintained under a genetic stability program. Neuropsychopharmacol. Rep. 2019, 39, 100–118. [Google Scholar] [CrossRef]
- Dong, Y.; Guo, C.; Chen, D.; Chen, S.; Peng, Y.; Song, H.; Shi, J. Association between age-related hearing loss and cognitive decline in C57BL/6J mice. Mol. Med. Rep. 2018, 18, 1726–1732. [Google Scholar] [CrossRef]
- Ma, L.; Wang, R.; Dong, W.; Zhao, Z. Caloric restriction can improve learning and memory in C57/BL mice probably via regulation of the AMPK signaling pathway. Exp. Gerontol. 2018, 102, 28–35. [Google Scholar] [CrossRef]
- Liu, F.; Benashski, S.E.; Persky, R.; Xu, Y.; Li, J.; McCullough, L.D. Age-related changes in AMP-activated protein kinase after stroke. AGE 2011, 34, 157–168. [Google Scholar] [CrossRef]
- Baranowski, B.J.; Bott, K.N.; MacPherson, R.E.K. Evaluation of neuropathological effects of a high-fat high-sucrose diet in middle-aged male C57BL6/J mice. Physiol. Rep. 2018, 6, e13729. [Google Scholar] [CrossRef]
- Kim, H.-S.; Moon, S.; Paik, J.-H.; Shin, D.W.; Kim, L.S.; Park, C.-S.; Ha, J.; Kang, J.-H. Activation of the 5′-AMP-Activated Protein Kinase in the Cerebral Cortex of Young Senescence-Accelerated P8 mice and association with GSK3β-and PP2A-dependent inhibition of p-tau396 expression. J. Alzheimer’s Dis. 2015, 46, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Davies, P.; Dickson, D.W.; Marambaud, P. AMPK is abnormally activated in tangle-and pre-tangle-bearing neurons in Alzheimer’s disease and other tauopathies. Acta Neuropathol. 2010, 121, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A. Brain glucose metabolism: Integration of energetics with function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef] [PubMed]
- Fujibayashi, Y.; Waki, A.; Wada, K.; Ueno, M.; Magata, Y.; Yonekura, Y.; Konishi, J.; Takeda, T.; Yokoyama, A. differential aging pattern of cerebral accumulation of radiolabeled glucose and amino acid in the senescence accelerated mouse (SAM), a new model for the study of memory impairment. Biol. Pharm. Bull. 1994, 17, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Inoue, A.; Kurokawa, T.; Ishibashi, S. Early changes in glucose metabolism in the cerebrum of senescence accelerated mouse: Involvement of glucose transporter. Brain Res. 1994, 637, 133–138. [Google Scholar] [CrossRef]
- Ohta, H.; Nishikawa, H.; Hirai, K.; Kato, K.; Miyamoto, M. Relationship of impaired brain glucose metabolism to learning deficit in the senescence-accelerated mouse. Neurosci. Lett. 1996, 217, 37–40. [Google Scholar] [CrossRef]
- Kondziella, D.; Hammer, J.; Sletvold, O.; Sonnewald, U. The pentylenetetrazole-kindling model of epilepsy in SAMP8 mice: Glial-neuronal metabolic interactions. Neurochem. Int. 2003, 43, 629–637. [Google Scholar] [CrossRef]
- Kondziella, D.; Bidar, A.; Urfjell, B.; Sletvold, O.; Sonnewald, U. The pentylenetetrazole-kindling model of epilepsy in SAMP8 mice: Behavior and metabolism. Neurochem. Int. 2002, 40, 413–418. [Google Scholar] [CrossRef]
- Liu, H.-W.; Chan, Y.-C.; Wei, C.-C.; Chen, Y.-A.; Wang, M.-F.; Chang, S.-J. An alternative model for studying age-associated metabolic complications: Senescence-accelerated mouse prone 8. Exp. Gerontol. 2017, 99, 61–68. [Google Scholar] [CrossRef]
- Zhang, X.; Li, G.; Guo, L.; Nie, K.; Jia, Y.; Zhao, L.; Yu, J. Age-related alteration in cerebral blood flow and energy failure is correlated with cognitive impairment in the senescence-accelerated prone mouse strain 8 (SAMP8). Neurol. Sci. 2013, 34, 1917–1924. [Google Scholar] [CrossRef] [PubMed]
- García-Matas, S.; Gutierrez-Cuesta, J.; Coto-Montes, A.; Rubio-Acero, R.; Díez-Vives, C.; Camins, A.; Pallàs, M.; Sanfeliu, C.; Cristòfol, R. Dysfunction of astrocytes in senescence-accelerated mice SAMP8 reduces their neuroprotective capacity. Aging Cell 2008, 7, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Pertusa, M.; García-Matas, S.; Rodríguez-Farré, E.; Sanfeliu, C.; Cristòfol, R. Astrocytes aged in vitro show a decreased neuroprotective capacity. J. Neurochem. 2007, 101, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Dykes-Hoberg, M.; Pardo, C.A.; Bristol, L.A.; Jin, L.; Kuncl, R.W.; Kanai, Y.; Hediger, M.A.; Wang, Y.; Schielke, J.P.; et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 1996, 16, 675–686. [Google Scholar] [CrossRef]
- Han, X.; Zhang, T.; Liu, H.; Mi, Y.; Gou, X. Astrocyte senescence and Alzheimer’s disease: A review. Front. Aging Neurosci. 2020, 12, 148. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.-R.; Yang, Y.; Zhou, W.; Zhang, Y.-X. Expression of VGLUTs contributes to degeneration and acquisition of learning and memory. Neurobiol. Learn. Mem. 2011, 95, 361–375. [Google Scholar] [CrossRef]
- Waller, S.B.; Ingram, N.K.; Reynolds, M.A.; London, E.D. Age and strain comparisons of neurotransmitter synthetic enzyme activities in the mouse. J. Neurochem. 1983, 41, 1421–1428. [Google Scholar] [CrossRef]
- Hascup, E.; Broderick, S.O.; Russell, M.K.; Fang, Y.; Bartke, A.; Boger, H.A.; Hascup, K. Diet-induced insulin resistance elevates hippocampal glutamate as well as VGLUT1 and GFAP expression in AβPP/PS1 mice. J. Neurochem. 2019, 148, 219–237. [Google Scholar] [CrossRef]
- Kitamura, Y.; Zhao, X.-H.; Ohnuki, T.; Takei, M.; Nomura, Y. Age-related changes in transmitter glutamate and NMDA receptor/channels in the brain of senescence-accelerated mouse. Neurosci. Lett. 1992, 137, 169–172. [Google Scholar] [CrossRef]
- Griñán-Ferré, C.; Corpas, R.; Puigoriol-Illamola, D.; Palomera-Avalos, V.; Sanfeliu, C.; Pallás, M. Understanding epigenetics in the neurodegeneration of Alzheimer’s disease: SAMP8 mouse model. J. Alzheimer’s Dis. 2018, 62, 943–963. [Google Scholar] [CrossRef]
- Morley, J.E.; Armbrecht, H.J.; Farr, S.A.; Kumar, V.B. The senescence accelerated mouse (SAMP8) as a model for oxidative stress and Alzheimer’s disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2012, 1822, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, H.; Chellappan, D.K.; Sengupta, P.; Pandey, M.; Gorain, B. Adenosine receptors in modulation of central nervous system disorders. Curr. Pharm. Des. 2019, 25, 2808–2827. [Google Scholar] [CrossRef] [PubMed]
- Poole, R.L.; Braak, D.; Gould, T.J. Concentration-and age-dependent effects of chronic caffeine on contextual fear conditioning in C57BL/6J mice. Behav. Brain Res. 2015, 298, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, J.; Rocha, A.S.; Nunes, F.; Costa, M.S.; Schein, V.; Kazlauckas, V.; Kalinine, E.; Souza, D.O.; Cunha, R.A.; Porciúncula, L.O. Caffeine consumption prevents memory impairment, neuronal damage, and adenosine A2A receptors upregulation in the hippocampus of a rat model of sporadic dementia. J. Alzheimer’s Dis. 2013, 34, 509–518. [Google Scholar] [CrossRef]
- Temido-Ferreira, M.; Ferreira, D.G.; Batalha, V.L.; Marques-Morgado, I.; Coelho, J.E.; Pereira, P.; Gomes, R.; Pinto, A.; Carvalho, S.; Canas, P.M.; et al. Age-related shift in LTD is dependent on neuronal adenosine A2A receptors interplay with mGluR5 and NMDA receptors. Mol. Psychiatry 2018, 1, 1876–1900. [Google Scholar] [CrossRef]
- Deckert, J.; Abel, F.; Künig, G.; Hartmann, J.; Senitz, D.; Maier, H.; Ransmayr, G.; Riederer, P. Loss of human hippocampal adenosine A1 receptors in dementia: Evidence for lack of specificity. Neurosci. Lett. 1998, 244, 1–4. [Google Scholar] [CrossRef]
- Kalaria, R.; Sromek, S.; Wilcox, B.; Unnerstall, J. Hippocampal adenosine A1 receptors are decreased in Alzheimer’s disease. Neurosci. Lett. 1990, 118, 257–260. [Google Scholar] [CrossRef]
- Ciruela, F.; Ecasado, V.; Rodrigues, R.J.; Lujan, R.; Burgueño, J.; Canals, M.; Borycz, J.; Rebola, N.; Goldberg, S.R.; Mallol, J.; et al. Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1–A2A receptor heteromers. J. Neurosci. 2006, 26, 2080–2087. [Google Scholar] [CrossRef]
- Nedeljkovic, N. Complex regulation of ecto-5’-nucleotidase/CD73 and A2AR-mediated adenosine signaling at neurovascular unit: A link between acute and chronic neuroinflammation. Pharmacol. Res. 2019, 144, 99–115. [Google Scholar] [CrossRef]
- Rebola, N.; Lujan, R.; Cunha, R.A.; Mulle, C. Adenosine A2A receptors are essential for long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses. Neuron 2008, 57, 121–134. [Google Scholar] [CrossRef]
- Augusto, E.; Matos, M.; Sévigny, J.; El-Tayeb, A.; Bynoe, M.S.; Müller, C.E.; Cunha, R.A.; Chen, J.-F. Ecto-5’-nucleotidase (CD73)-mediated formation of adenosine is critical for the striatal adenosine A2A receptor functions. J. Neurosci. 2013, 33, 11390–11399. [Google Scholar] [CrossRef] [PubMed]
- León, D.; Albasanz, J.L.; Ruíz, M.A.; Fernández, M.; Martín, M. Adenosine A1 receptor down-regulation in mothers and fetal brain after caffeine and theophylline treatments to pregnant rats. J. Neurochem. 2002, 82, 625–634. [Google Scholar] [CrossRef] [PubMed]
- León-Navarro, D.A.; Albasanz, J.L.; Martín, M.; Martín, M. Hyperthermia-induced seizures alter adenosine A1and A2Areceptors and 5′-nucleotidase activity in rat cerebral cortex. J. Neurochem. 2015, 134, 395–404. [Google Scholar] [CrossRef] [PubMed]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Melgar, A.; Albasanz, J.L.; Pallàs, M.; Martín, M. Adenosine Metabolism in the Cerebral Cortex from Several Mice Models during Aging. Int. J. Mol. Sci. 2020, 21, 7300. https://doi.org/10.3390/ijms21197300
Sánchez-Melgar A, Albasanz JL, Pallàs M, Martín M. Adenosine Metabolism in the Cerebral Cortex from Several Mice Models during Aging. International Journal of Molecular Sciences. 2020; 21(19):7300. https://doi.org/10.3390/ijms21197300
Chicago/Turabian StyleSánchez-Melgar, Alejandro, José Luis Albasanz, Mercè Pallàs, and Mairena Martín. 2020. "Adenosine Metabolism in the Cerebral Cortex from Several Mice Models during Aging" International Journal of Molecular Sciences 21, no. 19: 7300. https://doi.org/10.3390/ijms21197300
APA StyleSánchez-Melgar, A., Albasanz, J. L., Pallàs, M., & Martín, M. (2020). Adenosine Metabolism in the Cerebral Cortex from Several Mice Models during Aging. International Journal of Molecular Sciences, 21(19), 7300. https://doi.org/10.3390/ijms21197300