The Effects of Immune System Modulation on Prion Disease Susceptibility and Pathogenesis




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
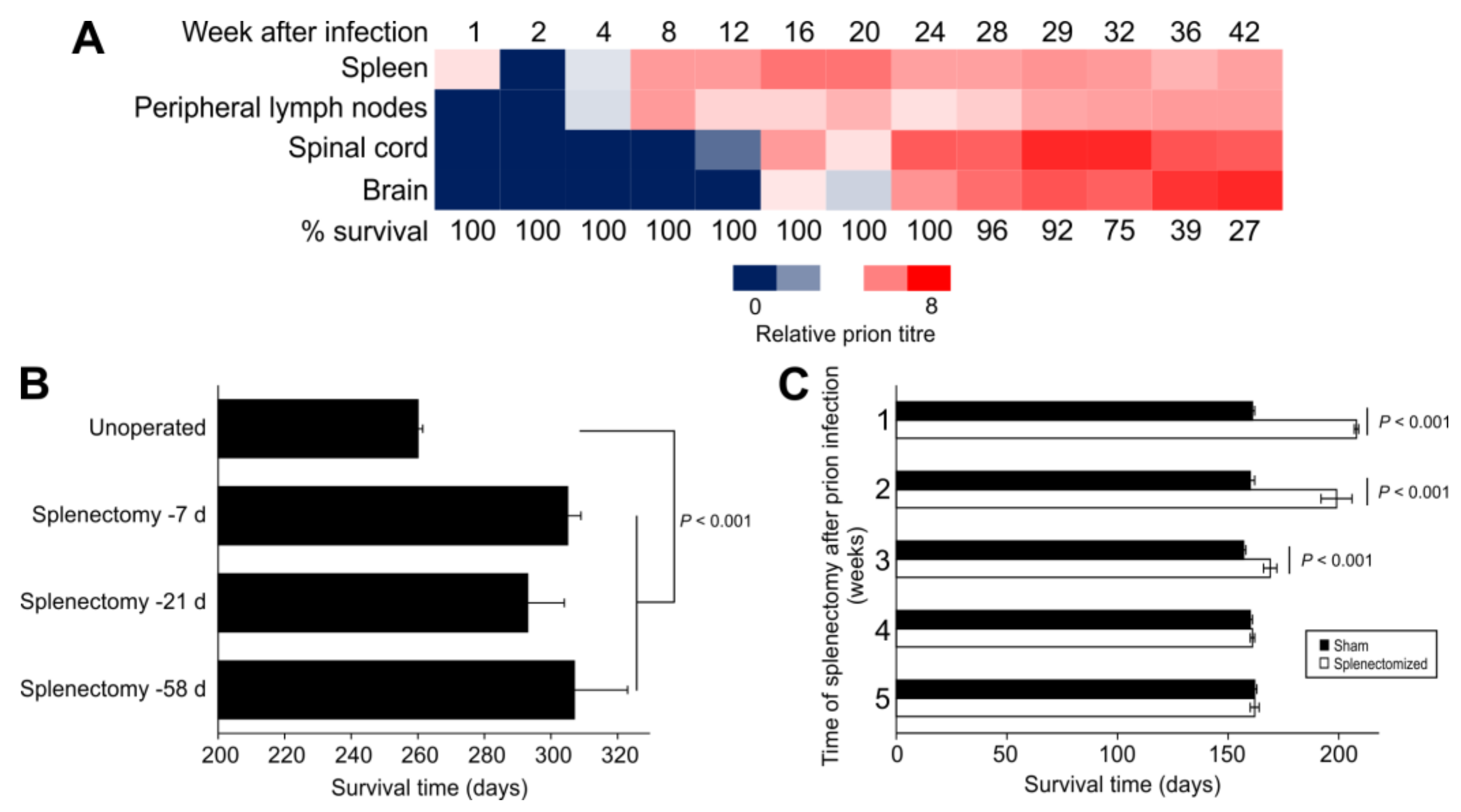
2. Splenectomy before Intraperitoneal Prion Infection Extends Survival Times
3. Immune Stimulation Accelerates, Immunosuppression Delays
4. Major Histocompatibility Complex (MHC)
5. Prions First Replicate upon Follicular Dendritic Cells in SLO
5.1. FDC Trap Prions in a Complement-Dependent Manner
5.2. Ageing Affects FDC and their Ability to Trap Prions
5.3. PrPC Abundance on FDC Affects Disease Susceptibility
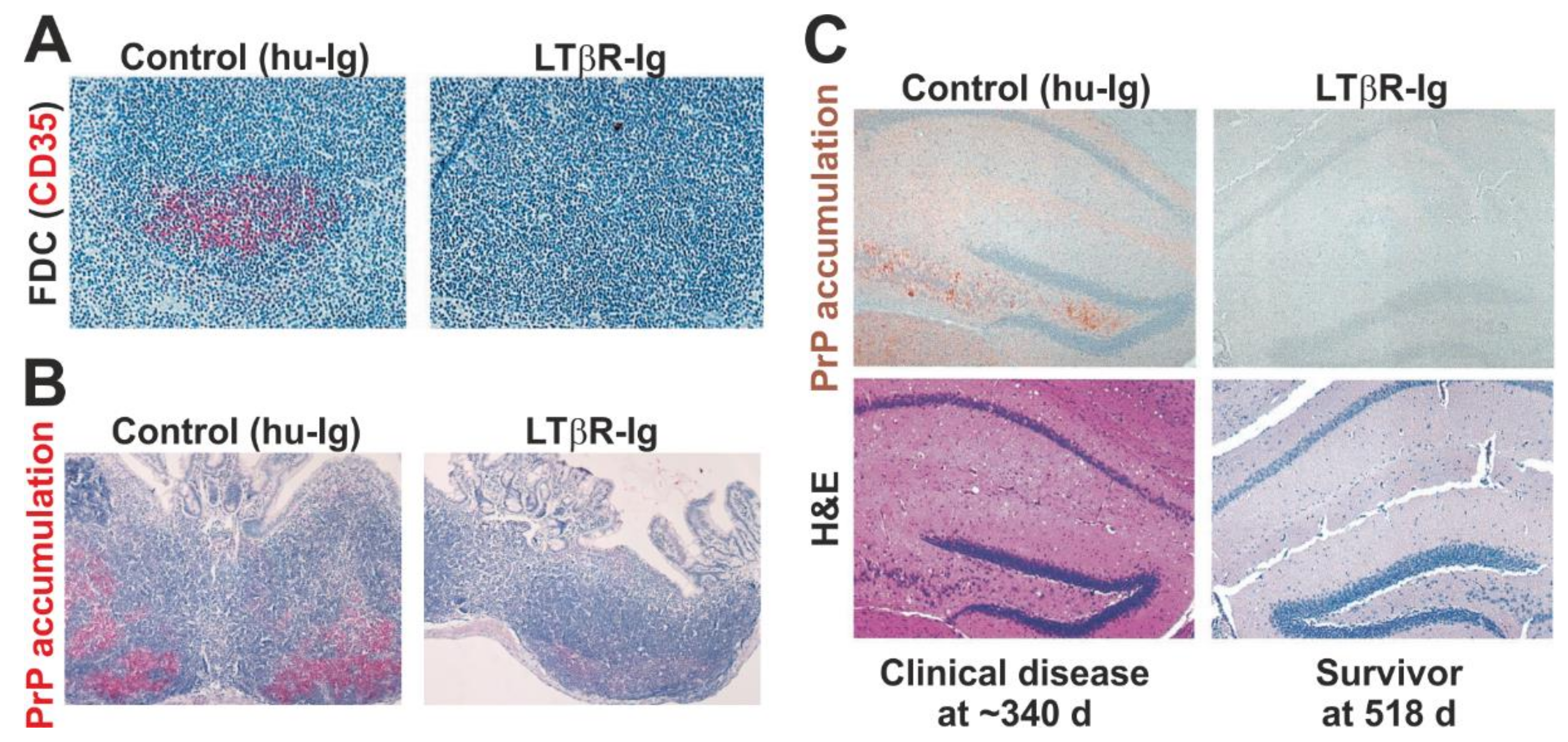
5.4. The Distance between FDC and Nerves Is Rate Limiting
6. Propagation of Prions to FDC in Peyer’s Patches
6.1. M Cells are the Gate Keepers of Prions in the Intestine
6.2. Conventional DC Shuttle Prions to FDC
6.3. Macrophages Can Destroy Prions
7. Chronic Inflammation Can Facilitate Prion Targeting in Non-SLO Tissues
8. Pathogen Co-Infections Can Affect Oral Prion Disease
8.1. Gastrointestinal Helminths
8.2. Pathogenic Bacteria
9. CNS Prion Disease
9.1. The Yin and Yang of the Microglia
9.1.1. Microglia Can Phagocytose and Destroy Prions in the Steady State
9.1.2. Microglia Engulf Apoptotic Bodies
9.1.3. Microglia Can Cause Neurodegeneration
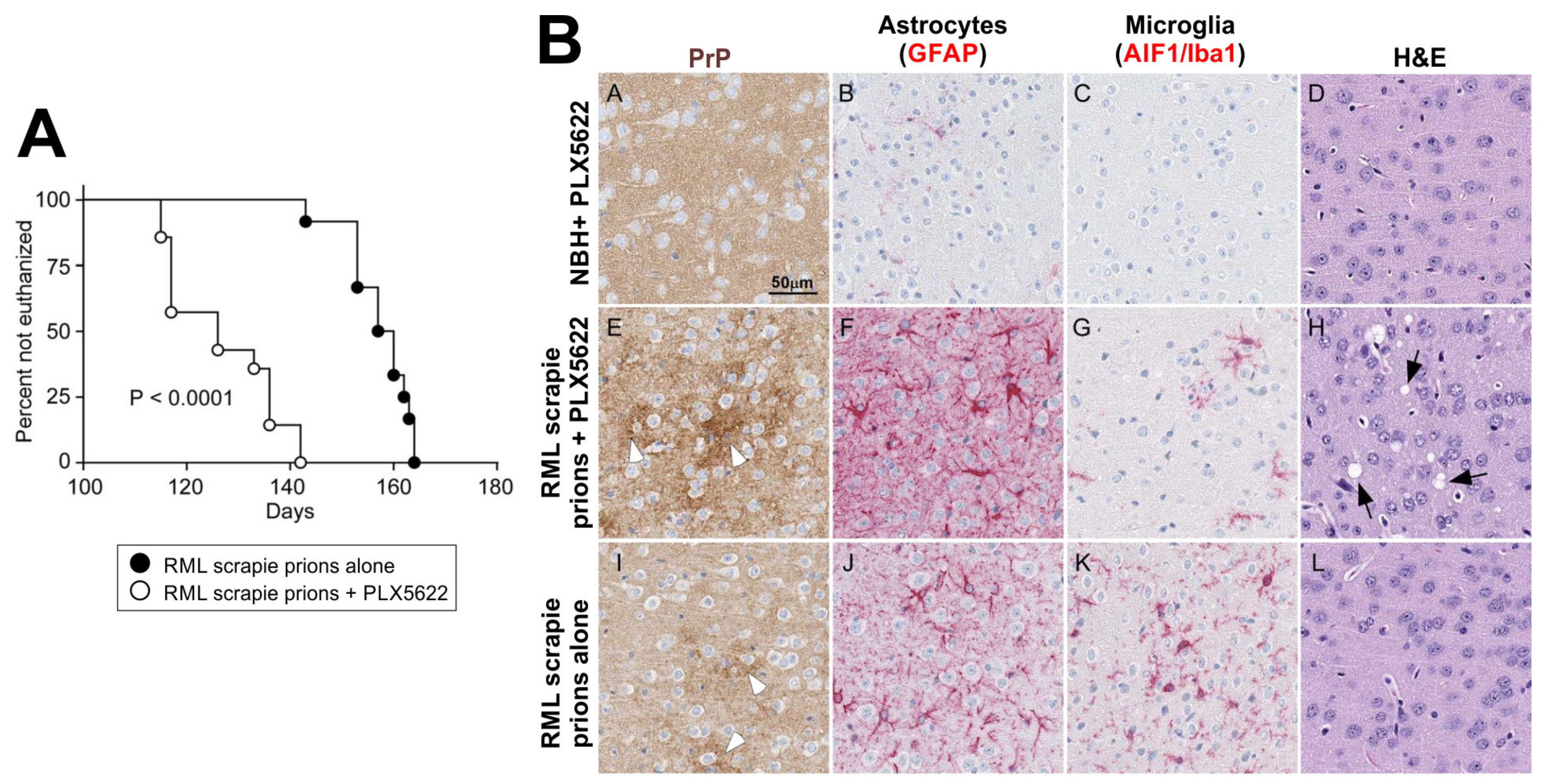
9.1.4. Microglia as Therapeutic Targets
9.1.5. The Commensal Gut Microbiome Constitutively Modulates Microglia Status
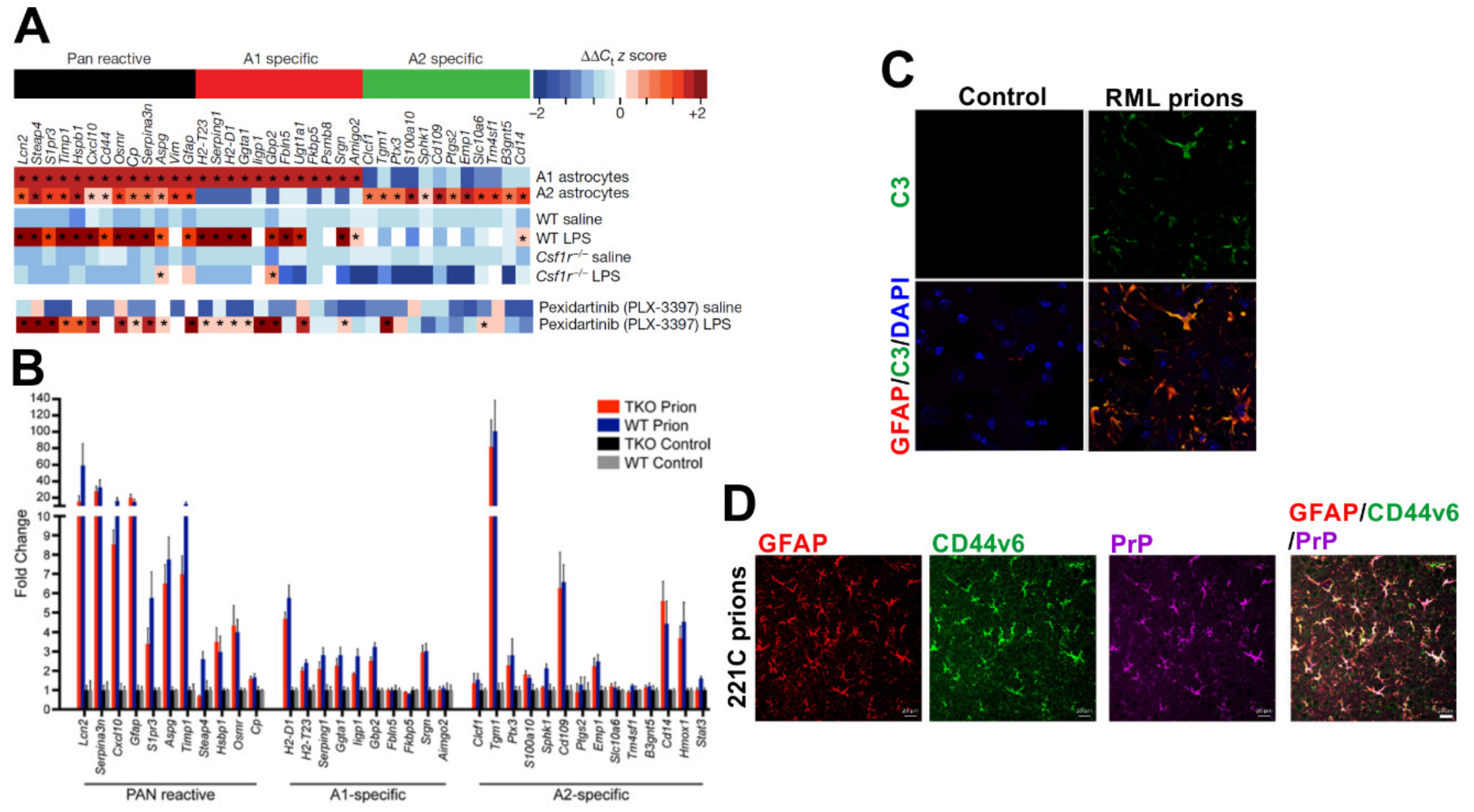
9.2. Reactive Astrocytes: Neuroprotective or Neurotoxic?
9.2.1. Microglia Can Modify the Phenotype of Reactive Astrocytes
9.2.2. Systemic Inflammation and Reactive Astrocyte Activation During Prion Disease
9.3. Pathogen Co-Infection Can Modify CNS Prion Disease
9.3.1. Virus Co-Infections
9.3.2. Gastrointestinal Helminth Parasites
9.4. The Contrasting Effects of Type I Interferons
9.5. COVID-19
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BSE | Bovine spongiform encephalopathy |
| cDC | Conventional dendritic cell |
| CNS | Central nervous system |
| CSF1/R | Colony-stimulating factor 1/receptor |
| FAE | Follicle-associated epithelium |
| FDC | Follicular dendritic cell |
| GALT | Gut-associated lymphoid tissue |
| GSS | Gerstmann–Sträussler–Scheinker disease |
| IC | Intracerebral |
| IFN | Interferon |
| IFNGR1 | Interferon gamma receptor 1 |
| IL | Interleukin |
| IP | Intraperitoneal |
| LPS | Lipopolysaccharide |
| MFGE8 | Milk fat globule epidermal growth factor 8 |
| MHC | Major histocompatibility complex |
| MNP | Mononuclear phagocyte |
| PrPC | Cellular PrP isoform |
| PrPSc | Prion disease-specific PrP isoform |
| sCJD | Sporadic Creutzfeldt–Jakob disease |
| SED | Subepithelial dome |
| SFB | Segmented filamentous bacteria |
| SLO | Secondary lymphoid organ |
| TGF | Transforming growth factor |
| TNF | Tumour necrosis factor |
| UPR | Unfolded protein response |
| vCJD | Variant Creutzfeldt–Jakob disease |
References
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Bolton, D.C.; McKinley, M.P.; Prusiner, S.B. Identification of a protein that purifies with the scrapie prion. Science 1982, 218, 1309–1311. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Küffer, A.; Lakkraju, A.K.; Mogha, A.; Petersen, S.C.; Airich, K.; Doucerain, C.; Marpakwar, R.; Bakirci, P.; Senatore, A.; Monnard, A.; et al. The prion protein is an agonistic ligand of the G protein-coupled receptor Adgrg6. Nature 2016, 536, 464–468. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Groth, D.; Serban, A.; Koehler, R.; Foster, D.; Torchia, M.; Burton, D.; Yang, S.-L.; DeArmond, S.J. Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP antibodies. Proc. Natl. Acad. Sci. USA 1993, 90, 10608–10612. [Google Scholar] [CrossRef]
- Manson, J.C.; Clarke, A.R.; Hooper, M.L.; Aitchison, L.; McConnell, I.; Hope, J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol. Neurobiol. 1994, 8, 121–127. [Google Scholar] [CrossRef]
- Benestad, S.L.; Austbo, L.; Tranulis, M.A.; Espenes, A.; Olsaker, I. Healthy goats naturally devoid of prion protein. Vet. Res. 2012, 43, 87. [Google Scholar] [CrossRef]
- Minikel, E.V.; Karczewski, K.J.; Martin, H.C.; Cummings, B.B.; Whiffin, N.; Rhodes, D.; Alfoldi, J.; Trembath, R.C.; van Heel, D.A.; Daly, M.J.; et al. Evaluating drug targets through human loss-of-function genetic variation. Nature 2020, 581, 459–464. [Google Scholar] [CrossRef]
- Nakato, G.; Hase, K.; Suzuki, M.; Kimura, M.; Ato, M.; Hanazato, M.; Tobiume, M.; Horiuchi, M.; Atarashi, R.; Nishida, N.; et al. Cutting edge: Brucella abortus exploits a cellular prion protein on intestinal M cells as an invasive receptor. J. Immunol. 2012, 189, 1540–1544. [Google Scholar] [CrossRef]
- Chida, J.; Hara, H.; Uchiyama, K.; Takahashi, E.; Miyata, H.; Kosako, H.; Tomioka, Y.; Ito, T.; Horiuchi, H.; Matsuda, H.; et al. Prion protein signaling induces M2 macrophage polarization and protects from lethal influenza infection in mice. PLoS Pathog. 2020, 16, e1008823. [Google Scholar] [CrossRef]
- McCulloch, L.; Brown, K.L.; Mabbott, N.A. Ablation of the cellular prion protein, PrPC, specifcally on follicular dendritic cells has no effect on their maturation or function. Immunology 2013, 138, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.W.; Dong, A.; Bhat, K.S.; Ernst, D.; Hayes, S.F.; Caughey, W. Secondary structure analysis of the scrapie-associated protein PrP 27-30 in water by infrared spectroscopy. Biochemistry 1991, 30, 7672–7680. [Google Scholar] [CrossRef] [PubMed]
- Bradford, B.M.; Piccardo, P.; Ironside, J.; Mabbott, N.A. Human prion diseases and their risk of transmission during anatomical dissection. Clin. Anat. 2014, 27, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.E.; Will, R.G.; Ironside, J.W.; McConnell, I.; Drummond, D.; Suttie, A.; McCardle, L.; Chree, A.; Hope, J.; Birkett, C.; et al. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature 1997, 389, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.F.; Desbruslais, M.; Joiner, S.; Sidle, K.C.L.; Gowland, I.; Collinge, J. The same prion strain causes vCJD and BSE. Nature 1997, 389, 448–450. [Google Scholar] [CrossRef] [PubMed]
- Mabbott, N. How do PrPSc prions spread between and within host species, and within hosts? Pathogens 2017, 6, 60. [Google Scholar] [CrossRef] [PubMed]
- Eklund, C.M.; Kennedy, R.C.; Hadlow, W.J. Pathogenesis of scrapie virus infections in the mouse. J. Infect. Dis. 1967, 117, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.C.; Haig, D.A. Attempts to demonstrate neutralising antibodies in the sera of scrapie-infected animals. Vet. Rec. 1966, 19, 647–649. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.D.; Porter, H.G.; Cox, N.A. Failure to Demonstrate a Humoral Immune Response to Scrapie Infection in Mice. J. Immunol. 1973, 111, 1407–1410. [Google Scholar] [PubMed]
- Tsukamoto, T.; Diringer, H.; Ludwig, H. Absence of autoantibodies against neurofilament proteins in the sera of scarpie infected mice. Tohoku J. Exp. Med. 1985, 4, 483–484. [Google Scholar] [CrossRef]
- Fraser, H.; Dickinson, A.G. Pathogenesis of scrapie in the mouse: The role of the spleen. Nature 1970, 226, 462–463. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, A.G.; Fraser, H.; McConnell, I.; Outram, G.W. Mitogenic Stimulation of the Host Enhances Susceptibility to Scrapie. Nature 1978, 272, 54–55. [Google Scholar] [CrossRef] [PubMed]
- Kimberlin, R.H.; Cunnington, P.G. Reduction fo scrapie incubation time in mice and hamsters by a single infection of methanol extraction residue of BCG. Fems Microbiol. Lett. 1978, 3, 169–172. [Google Scholar] [CrossRef][Green Version]
- Outram, G.W.; Dickinson, A.G.; Fraser, H. Slow encephalopathies, inflammatory responses, and arachis oil. Lancet 1975, 198–200. [Google Scholar] [CrossRef]
- Farquhar, C.F.; Dickinson, A.G. Prolongation of scrapie incubation period by an injection of dextran sulphate 500 within the month before or after infection. J. Gen. Virol. 1986, 67, 463–473. [Google Scholar] [CrossRef]
- Dickinson, A.G.; Meikle, V.M.; Fraser, H. Genetical control of the concentration of ME7 scrapie agent in the brain of mice. J. Comp. Pathol. 1969, 79, 15–22. [Google Scholar] [CrossRef]
- Junt, T.; Scandella, E.; Ludewig, B. Form follows function: Lymphoid tissue microarchitecture in antimicrobial immune defence. Nat. Rev. Immunol. 2008, 8, 763–775. [Google Scholar] [CrossRef]
- Fraser, H.; Dickinson, A.G. Studies of the lymphoreticular system in the pathogenesis of scrapie: The role of spleen and thymus. J. Comp. Pathol. 1978, 88, 563–573. [Google Scholar] [CrossRef]
- Kitamoto, T.; Muramoto, T.; Mohri, S.; Doh-Ura, K.; Tateishi, J. Abnormal Isoform of Prion Protein Accumulates in Follicular Dendritic Cells in Mice with Creutzfeldt-Jakob Disease. J. Virol. 1991, 65, 6292–6295. [Google Scholar] [CrossRef]
- Klein, M.A.; Frigg, R.; Flechsig, E.; Raeber, A.J.; Kalinke, U.; Bluethman, H.; Bootz, F.; Suter, M.; Zinkernagel, R.M.; Aguzzi, A. A crucial role for B cells in neuroinvasive scrapie. Nature 1997, 390, 687–691. [Google Scholar] [CrossRef]
- Kimberlin, R.H.; Walker, C.A. The role of the spleen in the neuroinvasion of scrapie in mice. Virus Res. 1989, 12, 201–211. [Google Scholar] [CrossRef]
- van Keulen, L.J.; Schreuder, B.E.; Vromans, M.E.; Langeveld, J.P.; Smits, M.A. Pathogenesis of natural scrapie in sheep. Arch. Virol. Suppl. 2000, 16, 57–71. [Google Scholar]
- van Keulen, L.J.M.; Vromans, M.E.W.; van Zijderveld, F.G. Ealry and late pathogenesis of natural scrapie infection in sheep. APMIS 2002, 110, 23–32. [Google Scholar] [CrossRef] [PubMed]
- van Keulen, L.J.M.; Bossers, A.; Van Zijderveld, F.G. TSE pathogenesis in cattle and sheep. Vet. Res. 2008, 39, 24. [Google Scholar] [CrossRef]
- Sigurdson, C.J.; Williams, E.S.; Miller, M.W.; Spraker, T.R.; O’Rourke, K.I.; Hoover, E.A. Oral transmission and early lymphoid tropism of chronic wasting disease PrPres in mule deer fawns (Odocoileus hemionus). J. Gen. Virol. 1999, 80, 2757–2764. [Google Scholar] [CrossRef]
- Glaysher, B.R.; Mabbott, N.A. Role of the draining lymph node in scrapie agent transmission from the skin. Immunol. Lett. 2007, 109, 64–71. [Google Scholar] [CrossRef]
- Prinz, M.; Huber, G.; Macpherson, A.J.S.; Heppner, F.L.; Glatzel, M.; Eugster, H.-P.; Wagner, N.; Aguzzi, A. Oral prion infection requires normal numbers of Peyer’s patches but not of enteric lymphocytes. Am. J. Pathol. 2003, 162, 1103–1111. [Google Scholar] [CrossRef]
- Horiuchi, M.; Furuoka, H.; Kitamura, N.; Shinagawa, M. Alymphoplasia mice are resistant to prion infection via oral route. Jpn. J. Vet. Res. 2006, 53, 149–157. [Google Scholar]
- Glaysher, B.R.; Mabbott, N.A. Role of the GALT in scrapie agent neuroinvasion from the intestine. J. Immunol. 2007, 178, 3757–3766. [Google Scholar] [CrossRef]
- Donaldson, D.S.; Else, K.J.; Mabbott, N.A. The gut-associated lymphoid tissues in the small intestine, not the large intestine, play a major role in oral prion disease pathogenesis. J. Virol. 2015, 15, 9532–9547. [Google Scholar] [CrossRef]
- Mabbott, N.A.; MacPherson, G.G. Prions and their lethal journey to the brain. Nat. Rev. Microbiol. 2006, 4, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Heikenwalder, M. Pathogenesis of prion diseases: Current status and future outlook. Nat. Rev. Microbiol. 2006, 2006, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Mabbott, N.A. Prion pathogenesis and secodary lymphoid organs (SLO): Tracking the SLO spread of prions to the brain. Prion 2012, 6, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Nuvolone, M.; Zhu, C. The immunobiology of prion diseases. Nat. Rev. Immunol. 2013, 13, 888–902. [Google Scholar] [CrossRef]
- Mabbott, N.A.; Alibhai, J.D.; Manson, J.C. The role of the immune system in prion infection. In Human Prion Diseases; Pocchiari, M., Manson, J., Eds.; Elsevier BV: San Diego, CA, USA, 2018; pp. 85–110. [Google Scholar] [CrossRef]
- Marsh, R.F. Effect of Vaccina-activated macrophages on scrapie infection in hamsters. In Hamster Immune Responses in Infectious and Oncologic Diseases. Advances in Experimental Medicine and Biology; Wayne Streilein, J., Hart, D.A., Stein-Streilein, J., Duncan, W.R., Billingham, R.E., Eds.; Springer: Boston, MA, USA, 1981; Volume 134, pp. 359–363. [Google Scholar]
- Outram, G.W.; Dickinson, A.G.; Fraser, H. Reduced Susceptibility to Scrapie In Mice After Steroid Administration. Nature 1974, 249, 855–856. [Google Scholar] [CrossRef]
- Sethi, S.; Lipford, G.; Wagner, H.; Kretzschmar, H. Postexposure prophylaxis against prion disease with a stimulator of innate immunity. Lancet 2002, 360, 229–230. [Google Scholar] [CrossRef]
- Heikenwalder, M.; Polymenidou, M.; Junt, T.; Sigurdson, C.; Wagner, H.; Akira, S.; Zinkernagel, R.; Aguzzi, A. Lymphoid follicle destruction and immunosuppression after repeated CpG oligodeoxynucleotide administration. Nat. Med. 2004, 10, 187–192. [Google Scholar] [CrossRef]
- Apanius, V.; Penn, D.; Slev, P.R.; Ruff, L.R.; Potts, W.K. The nature of selection on the major histocompatibility complex. Crit. Rev. Immunol. 1997, 17, 179–224. [Google Scholar] [CrossRef]
- Jackson, G.S.; Beck, J.A.; Navarrete, C.; Brown, J.; Sutton, P.M.; Contreras, M.; Collinge, J. HLA-DQ7 antigen and resistance to variant CJD. Nature 2001, 414, 269–270. [Google Scholar] [CrossRef]
- Pepys, M.B.; Bybee, A.; Booth, D.R.; Bishop, M.T.; Will, R.G.; Little, A.-M.; Prokupek, B.; Madrigal, J.A. MHC typing in variant Creutzfeldt-Jakob disease. Lancet 2003, 361, 487–489. [Google Scholar] [CrossRef]
- Lewicki, H.; Tishon, A.; Homann, D.; Mazarguil, H.; Laval, F.; Asensio, V.C.; Campbell, I.L.; DeArmond, S.; Coon, B.; Teng, C.; et al. T cells infiltrate the brain in murine and human transmissible spongiform encephalopathies. J. Virol. 2003, 77, 3799–3808. [Google Scholar] [CrossRef] [PubMed]
- Kujala, P.; Raymond, C.; Romeijn, M.; Godsave, S.F.; van Kasteren, S.I.; Wille, H.; Prusiner, S.B.; Mabbott, N.A.; Peters, P.J. Prion uptake in the gut: Identification of the first uptake and replication sites. PLoS Pathog. 2011, 7, e1002449. [Google Scholar] [CrossRef] [PubMed]
- Fraser, H.; Brown, K. Peripheral Pathogenesis of Scrapie in Normal and Immunocompromised Mice. Anim. Technol. 1994, 45, 21–22. [Google Scholar]
- Brown, K.L.; Stewart, K.; Ritchie, D.; Mabbott, N.A.; Williams, A.; Fraser, H.; Morrison, W.I.; Bruce, M.E. Scrapie replication in lymphoid tissues depends on PrP-expressing follicular dendritic cells. Nat. Med. 1999, 5, 1308–1312. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, L.; Brown, K.L.; Bradford, B.M.; Hopkins, J.; Bailey, M.; Rajewsky, K.; Manson, J.C.; Mabbott, N.A. Follicular dendritic cell-specific prion protein (PrPC) expression alone is sufficient to sustain prion infection in the spleen. PLoS Pathog. 2011, 7, e1002402. [Google Scholar] [CrossRef] [PubMed]
- Beringue, V.; Herzog, L.; Jaumain, E.; Reine, F.; Sibille, P.; Le Dur, A.; Vilotte, J.-L.; Laude, H. Facilitated cross-species transmission of prions in extraneural tissue. Science 2012, 335, 472–475. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.L.; Mabbott, N.A. Evidence of subclinical prion disease in aged mice following exposure to bovine spongiform encephalopathy. J. Gen. Virol. 2014, 95, 231–243. [Google Scholar] [CrossRef]
- Beringue, V.; Tixador, P.; Andreoletti, O.; Reine, F.; Castille, J.; Laï, T.-L.; Le Dur, A.; Laisne, A.; Herzog, L.; Passet, B.; et al. Host prion protein expression levels impact prion tropism for the spleen. PLoS Pathog. 2020, 16, e1008283. [Google Scholar] [CrossRef]
- Mabbott, N.A.; Young, J.; McConnell, I.; Bruce, M.E. Follicular dendritic cell dedifferentiation by treatment with an inhibitor of the lymphotoxin pathway dramatically reduces scrapie susceptibility. J. Virol. 2003, 77, 6845–6854. [Google Scholar] [CrossRef]
- Victoratos, P.; Lagnel, J.; Tzima, S.; Alimzhanov, M.B.; Rajewsky, K.; Pasparakis, M.; Kollias, G. FDC-specific functions of p55TNFR and IKK2 in the development of FDC networks and of antibody responses. Immunity 2006, 24, 65–77. [Google Scholar] [CrossRef]
- Aguzzi, A.; Kranich, J.; Krautler, N.J. Follicular dendritic cells: Origin, phenotype, and function in health and disease. Trends Immunol. 2014, 35, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Heesters, B.A.; Myers, R.C.; Carroll, M.C. Follicular dendritic cells: Dynamic antigen libraries. Nat. Rev. Immunol. 2014, 14, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Mabbott, N.A.; Bruce, M.E.; Botto, M.; Walport, M.J.; Pepys, M.B. Temporary depletion of complement component C3 or genetic deficiency of C1q significantly delays onset of scrapie. Nat. Med. 2001, 7, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.A.; Kaeser, P.S.; Schwarz, P.; Weyd, H.; Xenarios, I.; Zinkernagel, R.M.; Carroll, M.C.; Verbeek, J.S.; Botto, M.; Walport, M.J.; et al. Complement facilitates early prion pathogenesis. Nat. Med. 2001, 7, 488–492. [Google Scholar] [CrossRef]
- Zabel, M.D.; Heikenwalder, M.; Prinz, M.; Arright, I.; Schwarz, P.; Kranich, J.; Von Teichman, A.; Haas, K.M.; Zeller, N.; Tedder, T.F.; et al. Stromal complement receptor CD21/35 facilitates lymphoid prion colonization and pathogenesis. J. Immunol. 2007, 179, 6144–6152. [Google Scholar] [CrossRef]
- Kane, S.J.; Farley, T.K.; Gordon, E.O.; Estep, J.; Bender, H.R.; Moreno, J.A.; Bartz, J.; Telling, G.C.; Pickering, M.C.; Zabel, M.D. Complement regulatory protein factor H is a soluble prion receptor that potentiates peripheral prion pathogenesis. J. Immunol. 2017, 199, 3821–3827. [Google Scholar] [CrossRef]
- Kane, S.J.; Swanson, E.; Gordon, E.O.; Rocha, S.; Bender, H.R.; Donius, L.R.; Hannan, J.P.; Zabel, M.D. Relative impact of complement receptors CD21/35 (Cr2/1) on scrapie pathogenesis in mice. mSphere 2017, 2, e00493-17. [Google Scholar] [CrossRef]
- Michel, B.; Ferguson, A.; Johnson, T.; Bender, H.; Meyerett-Reid, C.; Pulford, B.; von Teichman, A.; Seelig, D.; Weiss, J.H.; Telling, G.C.; et al. Genetic depletion of complement receptors CD21/35 prevents terminal prion disease in a mouse model of chronic wasting disease. J. Immunol. 2012, 189, 4520–4527. [Google Scholar] [CrossRef]
- Michel, B.; Ferguson, A.; Johnson, T.; Bender, H.; Meyerett-Reid, C.; Wycoff, A.C.; Pulford, B.; Telling, G.C.; Zabel, M.D. Complement protein C3 exacerbates prion disease in a mouse model of chronic wasting disease. Int. Immunol. 2013, 25, 697–702. [Google Scholar] [CrossRef]
- Mabbott, N.A.; Mackay, F.; Minns, F.; Bruce, M.E. Temporary inactivation of follicular dendritic cells delays neuroinvasion of scrapie. Nat. Med. 2000, 6, 719–720. [Google Scholar] [CrossRef]
- Mabbott, N.A.; McGovern, G.; Jeffrey, M.; Bruce, M.E. Temporary blockade of the tumour necrosis factor signaling pathway impedes the spread of scrapie to the brain. J. Virol. 2002, 76, 5131–5139. [Google Scholar] [CrossRef] [PubMed]
- Mohan, J.; Bruce, M.E.; Mabbott, N.A. Follicular dendritic cell dedifferentiation reduces scrapie susceptibility following inoculation via the skin. Immunology 2005, 114, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Montrasio, F.; Frigg, R.; Glatzel, M.; Klein, M.A.; Mackay, F.; Aguzzi, A.; Weissmann, C. Impaired prion replication in spleens of mice lacking functional follicular dendritic cells. Science 2000, 288, 1257–1259. [Google Scholar] [CrossRef]
- Bremer, J.; Heikenwalder, M.; Haybaeck, J.; Tiberi, C.; Krautler, N.J.; Kurrer, M.O.; Aguzzi, A. Repetitive immunization enhances the susceptibility of mice to peripherally administered prions. PLoS ONE 2009, 4, e7160. [Google Scholar] [CrossRef] [PubMed]
- Monflacone, A.P.; Szakal, A.K.; Tew, J.G. Increased leukocyte diversity and responsiveness to B-cell and T-cell mitogens in cell suspensiions prepared by enzymatically dissociating murine lymph nodes. J. Leukoc. Biol. 1986, 39, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.L.; Wathne, G.J.; Sales, J.; Bruce, M.E.; Mabbott, N.A. The effects of host age on follicular dendritic cell status dramatically impair scrapie agent neuroinvasion in aged mice. J. Immunol. 2009, 183, 5199–5207. [Google Scholar] [CrossRef]
- Avrahami, D.; Gabizon, R. Age-related alterations affect the susceptibility of mice to prion infection. Neurobiol. Aging 2011, 32, 2006–2015. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.L.; Gossner, A.; Mok, S.; Mabbott, N.A. The effects of host age on the transport of complement-bound complexes to the spleen and the pathogenesis of intravenous scrapie infection. J. Virol. 2012, 86, 1228–1237. [Google Scholar] [CrossRef]
- Turner, V.M.; Mabbott, N.A. Structural and functional changes to lymph nodes in ageing mice. Immunology 2017, 151, 239–247. [Google Scholar] [CrossRef]
- Turner, V.M.; Mabbott, N.A. Ageing adversely affects the migration and function of marginal zone B cells. Immunology 2017, 151, 349–362. [Google Scholar] [CrossRef]
- Diack, A.B.; Head, M.W.; McCutcheon, S.; Boyle, A.; Knight, R.; Ironside, J.W.; Manson, J.C.; Will, R.G. Variant CJD. 18 years of research and surveillance. Prion 2014, 2014, 286–295. [Google Scholar] [CrossRef] [PubMed]
- St. Rose, S.; Hunter, N.; Matthews, D.; Foster, J.; Chase-Topping, M.E.; Kruuk, L.E.B.; Shaw, D.J.; Rhind, S.M.; Will, R.G.; Woolhouse, M.E.J. Comparative evidence for a link between Peyer’s patch development and susceptibility to transmissible spongiform encephalopathies. Bmc Infect. Dis. 2006, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.C.; Clarke, A.R.; McBride, P.A.; McConnell, I.; Hope, J. PrP gene dosage determines the timing but not the final intensity or distribution of lesions in scrapie pathology. Neurodegeneration 1994, 3, 331–340. [Google Scholar]
- Klein, M.A.; Frigg, R.; Raeber, A.J.; Flechsig, E.; Hegyi, I.; Zinkernagel, R.M.; Weissmann, C.; Aguzzi, A. PrP expression in B lymphocytes is not required for prion neuroinvasion. Nat. Med. 1998, 4, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Ierna, M.I.; Farquhar, C.F.; Outram, G.W.; Bruce, M.E. Resistance of neonatal mice to scrapie is associated with inefficient infection of the immature spleen. J. Virol. 2006, 80, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Lötscher, M.; Recher, M.; Hunzinker, L.; Klein, M.A. Immunologically induced, complement-dependent up-regulation of the prion protein in the mouse spleen: Follicular dendritic cells versus capsule and trabeculae. J. Immunol. 2003, 170, 6040–6047. [Google Scholar]
- Glatzel, M.; Heppner, F.L.; Albers, K.M.; Aguzzi, A. Sympathetic innervation of lymphoreticular organs is rate limiting for prion neuroinvasion. Neuron 2001, 31, 25–34. [Google Scholar] [CrossRef]
- McBride, P.A.; Schulz-Shaeffer, W.J.; Donaldson, M.; Bruce, M.; Diringer, H.; Kretzschmar, H.A.; Beekes, M. Early spread of scrapie from the gastrointestinal tract to the central nervous system involves autonomic fibers of the splanchnic and vagus nerves. J. Virol. 2001, 75, 9320–9327. [Google Scholar] [CrossRef]
- Prinz, M.; Heikenwalder, M.; Junt, T.; Schwarz, P.; Glatzel, M.; Heppner, F.L.; Fu, Y.-X.; Lipp, M.; Aguzzi, A. Positioning of follicular dendritic cells within the spleen controls prion neuroinvasion. Nature 2003, 425, 957–962. [Google Scholar] [CrossRef]
- Mabbott, N.A.; Donaldson, D.S.; Ohno, H.; Williams, I.R.; Mahajan, A. Microfold (M) cells: Important immunosurveillance posts in the intestinal epithelium. Mucosal Immunol. 2013, 6, 666–677. [Google Scholar] [CrossRef]
- Rios, D.; Wood, M.B.; Li, J.; Chassaing, B.; Gewirtz, A.T.; Williams, I.R. Antigen sampling by intestinal M cells is the principal pathway initiating mucosal IgA production to commensal enteric bacteria. Mucosal Immunol. 2016, 9, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Takakura, I.; Miyazawa, K.; Kanaya, T.; Itani, W.; Watanabe, K.; Ohwada, S.; Watanabe, H.; Hondo, T.; Rose, M.T.; Mori, T.; et al. Orally administered prion protein is incorporated by M cells and spreads to lymphoid tissues with macrophages in prion protein knockout mice. Am. J. Pathol. 2011, 179, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, D.S.; Kobayashi, A.; Ohno, H.; Yagita, H.; Williams, I.R.; Mabbott, N.A. M cell depletion blocks oral prion disease pathogenesis. Mucosal Immunol. 2012, 5, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, D.S.; Sehgal, A.; Rios, D.; Williams, I.R.; Mabbott, N.A. Increased abundance of M cells in the gut epithelium dramatically enhances oral prion disease susceptibility. PLoS Pathog. 2016, 12, e1006075. [Google Scholar] [CrossRef]
- Tahoun, A.; Mahajan, S.; Paxton, E.; Malterer, G.; Donaldson, D.S.; Wang, D.; Tan, A.; Gillespie, T.L.; O’Shea, M.; Rose, A.; et al. Salmonella transforms follicle-associated epithelial cells into M cells to promote intestinal invasion. Cell Host Microbe 2012, 12, 645–666. [Google Scholar] [CrossRef]
- Donaldson, D.S.; Pollock, J.; Vohra, P.; Stevens, M.P.; Mabbott, N.A. Microbial stimulation reverses the age-related decline in M cells in aged mice. iScience 2020, 23, 101147. [Google Scholar] [CrossRef]
- Knoop, K.A.; Kumar, N.; Butler, B.R.; Sakthivel, S.K.; Taylor, R.T.; Nochi, T.; Akiba, H.; Yagita, H.; Kiyono, H.; Williams, I.R. RANKL is necessary and sufficient to initiate development of antigen-sampling M cells in the intestinal epithelium. J. Immunol. 2009, 183, 5738–5747. [Google Scholar] [CrossRef]
- Nakato, G.; Fukuda, S.; Hase, K.; Goitsuka, R.; Cooper, M.D.; Ohno, H. New approach for M-cell-specific molecules by screening comprehensive transcriptome analysis. Dna Res. 2009, 16, 227–235. [Google Scholar] [CrossRef]
- Sigurdson, C.J.; Heikenwalder, M.; Manco, G.; Barthel, M.; Schwarz, P.; Stecher, B.; Krautler, N.J.; Hardt, W.-D.; Seifert, B.; MacPherson, A.J.S.; et al. Bacterial colitis increases susceptibility to oral prion pathogenesis. J. Infect. Dis. 2009, 199, 243–252. [Google Scholar] [CrossRef]
- Marshall, A.; Bradford, B.M.; Clarke, A.R.; Manson, J.C.; Mabbott, N.A. Oral prion neuroinvasion occurs independently of PrPC expression in the gut epithelium. J. Virol. 2018. [Google Scholar] [CrossRef]
- Kobayashi, A.; Donaldson, D.S.; Erridge, C.; Kanaya, T.; Williams, I.R.; Ohno, H.; Mahajan, A.; Mabbott, N.A. The functional maturation of M cells is dramatically reduced in the Peyer’s patches of aged mice. Mucosal Immunol. 2013, 6, 1027–1037. [Google Scholar] [CrossRef] [PubMed]
- Bennet, K.M.; Parnell, E.A.; Sanscartier, C.; Parks, S.; Chen, G.; Nair, M.G.; Lo, D.D. Induction of colonic M cells during intestinal inflammation. Am. J. Pathol. 2016, 186, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Wood, M.B.; Rios, D.; Williams, I.R. TNF-a augments RANKL-dependent intestinal M-cell differentiation in enteroid cultures. Am. J. Physiol. Cell Physiol. 2016, 311, C498–C507. [Google Scholar] [CrossRef] [PubMed]
- Krautler, N.J.; Kana, V.; Kranich, J.; Tian, Y.; Perera, D.; Lemm, D.; Schwarz, P.; Armulik, A.; Browning, J.L.; Tallquist, M.; et al. Follicular dendritic cells emerge from ubiquitous perivascular precursors. Cell 2012, 150, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Wykes, M.; Pombo, A.; Jenkins, C.; MacPherson, G.G. Dendritic cells interact directly with Naive B lymphocytes to transfer antigen and initiate class switching in a primary T-dependent response. J. Immunol. 1998, 161, 1313–1319. [Google Scholar] [PubMed]
- Raymond, C.R.; Aucouturier, P.; Mabbott, N.A. In vivo depletion of CD11c+ cells impairs scrapie agent neuroinvasion from the intestine. J. Immunol. 2007, 179, 7758–7766. [Google Scholar] [CrossRef] [PubMed]
- Cordier-Dirikoc, S.; Chabry, J. Temporary depletion of CD11c+ dendritic cells delays lymphoinvasion after intraperitoneal scrapie infection. J. Virol. 2008, 82, 8933–8936. [Google Scholar] [CrossRef]
- Wathne, G.J.; Kissenpfennig, A.; Malissen, B.; Zurzolo, C.; Mabbott, N.A. Determining the role of mononuclear phagocytes in prion neuroinvasion from the skin. J. Leukoc. Biol. 2012, 91, 817–828. [Google Scholar] [CrossRef]
- Bradford, B.M.; Reizis, B.; Mabbott, N.A. Oral prion disease pathogenesis is impeded in the specific absence of CXCR5-expressing dendritic cells. J. Virol. 2017, 91, e00124-17. [Google Scholar] [CrossRef]
- Bradford, B.M.; Sester, D.; Hume, D.A.; Mabbott, N.A. Defining the anatomical localisation of subsets of the murine mononuclear phagocyte system using integrin alpha X (Itgax, CD11c) and colony stimulating factor 1 receptor (Csf1r, CD115) expression fails to discriminate dendritic cells from macrophages. Immunobiology 2011, 216, 1228–1237. [Google Scholar] [CrossRef]
- Bonnardel, J.; Da Silva, C.; Henri, S.; Tamoutounour, S.; Chasson, L.; Mantanana-Sanchis, F.; Gorvel, J.P.; Lelouard, H. Innate and adaptive immune functions of Peyer’s patch-derived monocyte-derived cells. Cell Rep. 2015, 11, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Bonnardel, J.; Da Silva, C.; Wagner, C.; Bonifay, R.; Chasson, L.; Masse, M.; Pollet, E.; Dalod, M.; Gorvel, J.-P.; Lelouard, H. Distribution, location, and transcriptional profile of Peyer’s patch conventional DC subsets at steady state and under TLR7 ligand stimulation. Mucosal Immunol. 2017, 10, 1412–1430. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Kerksiek, K.M.; Brocker, T.; Kretzschmar, H. Role of the CD8+ dendritic cell subset in transmission of prions. J. Virol. 2007, 81, 4877–4880. [Google Scholar] [CrossRef] [PubMed]
- Oldstone, M.B.A.; Race, R.; Thomas, D.; Lewicki, H.; Homman, D.; Smelt, S.; Holz, A.; Koni, P.; Lo, D.; Chesebro, B.; et al. Lymphotoxin-a- and lymphotoxin-b-deficient mice differ in susceptibility to scrapie: Evidence against dendritic cell involvement. J. Virol. 2002, 76, 4357–4363. [Google Scholar] [CrossRef]
- Ano, Y.; Sakudo, A.; Nakayama, H.; Onodera, T. Uptake and dynamics of infectious prion protein in the intestine. Prot. Pept. Lett. 2009, 16, 247–255. [Google Scholar] [CrossRef]
- Carp, R.I.; Callahan, S.M. In vitro interaction of scrapie agent and mouse peritoneal macrophages. Intervirology 1981, 16, 8–13. [Google Scholar] [CrossRef]
- Carp, R.I.; Callahan, S.M. Effect of mouse peritoneal macrophages on scrapie infectivity during extended in vitro incubation. Intervirology 1982, 17, 201–207. [Google Scholar] [CrossRef]
- Beringue, V.; Demoy, M.; Lasmezas, C.I.; Gouritin, B.; Weingarten, C.; Deslys, J.-P.; Andreux, J.P.; Couvreur, P.; Dormont, D. Role of spleen macrophages in the clearance of scrapie agent early in pathogenesis. J. Pathol. 2000, 190, 495–502. [Google Scholar] [CrossRef]
- Maignien, T.; Shakweh, M.; Calvo, P.; Marce, D.; Sales, N.; Fattal, E.; Deslys, J.-P.; Couvreur, P.; Lasmezas, C.I. Role of gut macrophages in mice orally contaminated with scrapie or BSE. Int. J. Pharm. 2005, 298, 293–304. [Google Scholar] [CrossRef]
- Sehgal, A.; Donaldson, D.S.; Pridans, C.; Sauter, K.A.; Hume, D.A.; Mabbott, N.A. The role of CSF1R-dependent macriophages in control of the intestinal stem-cell niche. Nat. Commun. 2018, 9, 1272. [Google Scholar] [CrossRef]
- Gabanyi, I.; Muller, P.A.; Feighery, L.; Oliveira, T.Y.; Costa-Pinto, F.A.; Mucida, D. Neuro-immune interactions drive tissue programming in intestinal macrophages. Cell 2016, 164, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Koscso, B.; Rajani, G.M.; Stevanovic, K.; Berres, M.-L.; Hashimoto, D.; Mortha, A.; Leboeuf, M.; Li, X.-M.; Mucida, D.; et al. Crosstalk between muscularis macrophages and enteric neurones regulates gastrointestinal motility. Cell 2014, 158, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Courth, L.F.; Ostaff, M.J.; Mailander-Sanchez, D.; Malek, N.P.; Stange, E.F.; Wehkamp, J. Crohn’s disease-derived monocytes fail to induce Paneth cell defensins. Proc. Natl. Acad. Sci. USA 2015, 112, 14000–14005. [Google Scholar] [CrossRef] [PubMed]
- Nusse, Y.M.; Savage, A.K.; Marangoni, P.; Rosendahl-Huber, A.K.M.; Landman, T.A.; de Sauvage, F.J.; Locksley, R.M.; Klein, O.D. Parasitic helminths induce fetal-like reversion in the intestinal stem cell niche. Nature 2018, 559, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Seeger, H.; Heikenwalder, M.; Zeller, N.; Kranich, J.; Schwarz, P.; Gaspert, A.; Seifert, B.; Miele, G.; Aguzzi, A. Coincident scrapie infection and nephritis lead to urinary prion excretion. Science 2005, 310, 324–326. [Google Scholar] [CrossRef]
- Ligios, C.; Cancedda, G.M.; Margalith, I.; Santucciu, C.; Madau, L.; Maestrale, C.; Basagni, M.; Saba, M.; Heikenwalder, M. Intraepithelial and interstitial deposition of pathological prion protein in kidneys of scrapie-affected sheep. PLoS ONE 2007, 2, e859. [Google Scholar] [CrossRef]
- Ligios, C.; Sigurdson, C.; Santucciu, C.; Carcassola, G.; Manco, G.; Basagni, M.; Maestrale, C.; Cancedda, M.G.; Madau, L.; Aguzzi, A. PrPSc in mammary glands of sheep affected by scrapie and mastitis. Nat. Med. 2005, 11, 1137–1138. [Google Scholar] [CrossRef]
- Lacroux, C.; Simon, S.; Benenstad, S.L.; Maillet, S.; Mathey, J.; Lugan, S.; Corbiere, F.; Cassard, H.; Costes, P.; Bergonier, D.; et al. Prions in milk from ewes incubating natural scrapie. PLoS Pathog. 2008, 4, e1000238. [Google Scholar] [CrossRef]
- Ligios, C.; Cancedda, M.G.; Carta, A.; Santucciu, C.; Maestrale, C.; Demontis, F.; Saba, M.; Patta, C.; DeMartini, J.C.; Aguzzi, A.; et al. Sheep with scrapie and mastitis transmit infectious prions through the milk. J. Virol. 2011, 85, 1136–1139. [Google Scholar] [CrossRef]
- Heikenwalder, M.; Kurrer, M.O.; Margalith, I.; Kranich, J.; Zeller, N.; Haybaeck, J.; Polymenidou, M.; Matter, M.; Bremer, J.; Jackson, W.S.; et al. Lymphotoxin-dependent prion replication in inflammatory stromal cells of granulomas. Immunity 2008, 29, 998–1008. [Google Scholar] [CrossRef]
- Al-Dybiat, I.; Moudjou, M.; Martin, D.; Reine, F.; Herzog, L.; Truchet, S.; Berthon, P.; Laude, H.; Rezaei, H.; Andreoletti, O.; et al. Prion strain-dependent tropism is maintained between spleen and granuloma and relies on lymphoreticular structures. Sci. Rep. 2019, 9, 14656. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Quintero, A.; Bradford, B.M.; Maizels, R.; Donaldson, D.S.; Mabbott, N.A. Effect of co-infection with a small intestine-restricted helminth pathogen on oral prion disease pathogenesis in mice. Sci. Rep. 2019, 9, 6674. [Google Scholar] [CrossRef] [PubMed]
- Gruner, L.; Elsen, J.M.; Vu Tien Khang, J.; Eychenne, F.; Caritez, J.C.; Jacquiet, P.; Andreoletti, O.; Sarradin, P.; Cortet, J.; Richer, N.; et al. Nematode parasites and scrapie: Experiments in sheep and mice. Parasitol. Res. 2004, 93, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, D.S.; Bradford, B.M.; Artis, D.; Mabbott, N.A. Reciprocal development of lymphoid tissue development in the large intestine by IL-25 and IL-23. Mucosal Immunol. 2015, 8, 582–595. [Google Scholar] [CrossRef]
- Gonzalez, L.; Martin, S.; Siso, S.; Konold, T.; Ortiz-Pelaez, A.; Phelan, L.; Goldmann, W.; Stewart, P.; Saunders, G.; Windl, O.; et al. High prevalence of scrapie in a dairy goat herd: Tissue distribution of disease-associated PrP and effect of PRNP genotype and age. Vet. Res. 2009, 40, 65. [Google Scholar] [CrossRef]
- Thomsen, B.V.; Schneider, D.A.; O’Rourke, K.I.; Gidlewski, T.; McLane, J.; Allen, R.W.; mcIsaac, A.A.; Mitchell, G.B.; Keane, D.P.; Spraker, T.R.; et al. Diagnostic accuracy of rectal mucosa biopsy testing for chronic wasting disease within white-tailed deer (Odocoileus virginianus) herds in North America: Effects of age, sex, polymorphism at PRNP codon 96, and disease progression. J. Vet. Diagn. Intest. 2012, 24, 878–887. [Google Scholar] [CrossRef]
- Kimura, S.; Yamakami-Kimura, M.; Obata, Y.; Hase, K.; Kitamura, H.; Ohno, H.; Iwanaga, T. Visualization of the entire differentiation process of murine M cells: Suppression of their maturation in caecal patches. Mucosal Immunol. 2015, 8, 650–660. [Google Scholar] [CrossRef]
- Pelaseyed, T.; Bergstrom, J.H.; Gustafsson, J.K.; Emund, A.; Birchenough, G.M.H.; Schutte, A.; van der Post, S.; Svensson, F.; Rodriguez-Pineiro, A.M.; Nystrom, E.E.L.; et al. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol. Rev. 2014, 260, 8–20. [Google Scholar] [CrossRef]
- Lai, N.Y.; Musser, M.A.; Pinho-Ribeiro, F.A.; Baral, P.; Jacobson, A.; Potts, D.E.; Chen, Z.; Paik, D.; Soualhi, S.; Yan, Y.; et al. Gut-innervating nociceptor neurons regulate Peyer’s patch microfold cells and SFB levels to mediate Salmonella host defense. Cell 2020, 180, 33–39. [Google Scholar] [CrossRef]
- Woolf, C.J.; Ma, Q. Nociceptors-Noxious stimulus detectors. Neuron 2007, 55, 353–364. [Google Scholar] [CrossRef]
- Sandberg, M.K.; Al-Doujaily, H.; Shaps, A.R.; Clarke, A.R.; Collinge, J. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature 2011, 470, 540–542. [Google Scholar] [CrossRef] [PubMed]
- Betmouni, S.; Perry, V.H.; Gordon, J.L. Evidence for an early inflammatory response in the central- nervous-system of mice with scrapie. Neuroscience 1996, 74, 1–5. [Google Scholar] [CrossRef]
- Friedman-Levi, Y.; Ovadia, H.; Hoftberger, R.; Einstein, O.; Abramsky, O.; Budka, H.; Gabizon, R. Fatal neurological disease in scrapie-infected mice induced for experimental autoimmune encephalomyelitis. J. Virol. 2007, 81, 9942–9949. [Google Scholar] [CrossRef] [PubMed]
- Vincenti, J.E.; Murphy, L.; Grabert, K.; McColl, B.W.; Cancellotti, E.; Freeman, T.C.; Manson, J.C. Defining the microglial response during the time course of chronic neurodegeneration. J. Virol. 2016, 90, 3003–3017. [Google Scholar] [CrossRef]
- Alibhai, J.; Blanco, R.A.; Barria, M.A.; Piccardo, P.; Caughey, B.; Perry, V.H.; Freeman, T.C.; Manson, J.C. Distribution of misfolded prion protein seeding activity alone does not predict regions of neurodegeneration. Plos Biol. 2016, 14, e1002579. [Google Scholar] [CrossRef]
- Sorce, S.; Nuvulone, M.; Russo, G.; Chincisan, A.; Heinzer, D.; Avar, M.; Pfammatter, M.; Schwarz, P.; Delic, M.; Muller, M.; et al. Genome-wide transcriptomics identifies an early preclinical signature of prion infection. PLoS Pathog. 2020, 16, e1008653. [Google Scholar] [CrossRef]
- Alliot, F.; Godin, I.; Pessac, B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Dev. Brain. Res. 1999, 117, 145–152. [Google Scholar] [CrossRef]
- Askew, K.; Li, K.; Olmos-Alonso, A.; Garcia-Moreno, F.; Liang, Y.; Richardson, P.; Tipton, T.; Chapman, M.A.; Riecken, K.; Beccari, S.; et al. Coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain. Cell Rep. 2017, 18, 391–405. [Google Scholar] [CrossRef]
- Gomez-Nicola, D.; Schetters, S.T.T.; Perry, V.H. Differential role of CCR2 in the dynamics of microglia and perivascular macrophages during prion disease. Glia 2014, 62, 1041–1052. [Google Scholar] [CrossRef]
- Gomez-Nicola, D.; Fransen, N.L.; Suzzi, S.; Perry, V.H. Regulation of microglial proliferation during chronic neurodegeneration. J. Neurosci. 2013, 33, 2481–2493. [Google Scholar] [CrossRef]
- Bruce, M.E.; McBride, P.A.; Farquhar, C.F. Precise targeting of the pathology of the sialoglycoprotein, PrP, and vacuolar degeneration in mouse scrapie. Neurosci. Lett. 1989, 102, 1–6. [Google Scholar] [CrossRef]
- Priller, J.; Prinz, M.; Heikenwalder, M.; Zeller, N.; Schwarz, P.; Heppner, F.L.; Aguzzi, A. Early and rapid engraftment of bone marrow-derived microglia in scrapie. J. Neurosci. 2006, 26, 11753–11762. [Google Scholar] [CrossRef] [PubMed]
- Montrasio, F.; Cozzio, A.; Flechsig, E.; Rossi, D.; Klein, M.A.; Rulicke, T.; Raeber, A.J.; Vosshenrich, C.A.J.; Proft, J.; Aguzzi, A.; et al. B-lymphocyte-restricted expression of the prion protein does not enable prion replication in PrP knockout mice. Proc. Natl. Acad. Sci. USA 2001, 98, 4034–4037. [Google Scholar] [CrossRef] [PubMed]
- Raeber, A.J.; Sailer, A.; Hegyi, I.; Klein, M.A.; Rulicke, T.; Fischer, M.; Brandner, S.; Aguzzi, A.; Weissmann, C. Ectopic expression of prion protein (PrP) in T lymphocytes or hepatocytes of PrP knockout mice is insufficient to sustain prion replication. Proc. Natl. Acad. Sci. USA 1999, 96, 3987–3992. [Google Scholar] [CrossRef] [PubMed]
- Hume, D.A.; Caruso, M.; Ferrari-Cestari, M.; Summers, K.M.; Pridans, C.; Irvine, K.M. Phenotypic impacts of CSF1R deficiencies in humans and model organisms. J. Leukoc. Biol. 2020, 107, 205–219. [Google Scholar] [CrossRef]
- Wang, Y.; Szretter, K.J.; Vermi, W.; Gilfillan, S.; Rossini, C.; Cella, M.; Barrow, A.D.; Diamond, M.S.; Colonna, M. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat. Immunol. 2012, 13, 753–760. [Google Scholar] [CrossRef]
- Zhu, C.; Hermann, U.S.; Falsig, J.; Abakumova, I.; Nuvolone, M.; Schwarz, P.; Frauenknecht, K.; Rushing, E.J.; Aguzzi, A. A neuroprotective role for microglia during prion diseaes. J. Exp. Med. 2016, 213, 1047–1059. [Google Scholar] [CrossRef]
- Carroll, J.A.; Race, B.; Williams, K.; Striebel, J.; Chesebro, B. Microglia are critical in host defence against prion disease. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Lei, F.; Cui, N.; Zhou, C.; Chodosh, J.; Vavvas, D.G.; Paschalis, E.I. CSF1R inhibition by a small-molecule inhibitor is not microglia specific; affecting hematopoiesis and the function of macrophages. Proc. Natl. Acad. Sci. USA 2020, in press. [Google Scholar] [CrossRef]
- Kranich, J.; Krautler, N.J.; Falsig, J.; Ballmer, B.; Li, S.; Hutter, G.; Schwarz, P.; Moos, R.; Julius, C.; Miele, G.; et al. Engulfment of cerebral apoptotic bodies controls the course of prion disease in a mouse strain–dependent manner. J. Exp. Med. 2010, 207, 2271–2281. [Google Scholar] [CrossRef]
- Zhu, C.; Herrmann, U.S.; Li, B.; Abakumova, I.; Moos, R.; Schwarz, P.; Rushing, E.J.; Colonna, M.; Aguzzi, A. Triggering receptor expressed on myeloid cells-2 is involved in prion-induced microglial activation but does not contribute to prion pathogenesis in mouse brains. Neurobiol. Aging 2015, 36, 1994–2002. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, F.; Yang, Y.; Chen, J.; Hu, X. SIRP/CD47 signaling in neurological disorders. Brain Res. 2015, 1623, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Nuvulone, M.; Paolucci, M.; Sorce, S.; Kana, V.; Moos, R.; Matozaki, T.; Aguzzi, A. Prion pathogenesis is unaltered in the absence of SIRPa-mediated “dont-eat-me” signaling. PLoS ONE 2017, 12, e0177876. [Google Scholar] [CrossRef]
- Crocker, P.R.; Kelm, S.; Dubois, C.; Martin, B.; McWilliam, A.S.; Shotton, D.M.; Paulson, J.C.; Gordon, S. Purification and properties of sialoadhesin, a sialic acid-binding receptor of murine tissue macrophages. Embo J. 1991, 10, 1661–1669. [Google Scholar] [CrossRef] [PubMed]
- Bogie, J.F.; Boelen, E.; Louagie, E.; Delputte, P.; Elewaut, D.; van Horssen, J.; Hendricks, J.J.; Hellings, N. CD169 is a marker for highly pathogenic phagocytes in multiple sclerosis. Mult. Scler. 2018, 24, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Chang, J.C.-Y.; Baskakov, I.V. Region-specific sialylation pattern of prion strains provides novel insight into prion neurotropism. Int. J. Mol. Sci. 2020, 21, 828. [Google Scholar] [CrossRef] [PubMed]
- Bradford, B.M.; Crocker, P.R.; Mabbott, N.A. Peripheral prion disease pathogenesis is unaltered in the absence of sialoadhesin (Siglec-1/CD169). Immunology 2014, 143, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.; Wilcockson, D.C.; Boche, D.; Perry, V.H. Comparison of inflammatory and acute-phase responses in the brain and peripheral organs of the ME7 model of prion disease. J. Virol. 2005, 79, 5174–5184. [Google Scholar] [CrossRef]
- Fadok, V.A.; Bratton, D.L.; Konowal, A.; Freed, P.W.; Westcott, J.Y.; Henson, P.M. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Investig. 1998, 101, 890–898. [Google Scholar] [CrossRef]
- Nuvolone, M.; Sorce, S.; Schwarz, P.; Aguzzi, A. Prion pathogenesis in the absence of NLRP3/ASC inflammasomes. PLoS ONE 2015, 10, e0117208. [Google Scholar] [CrossRef]
- Julius, C.; Heikenwalder, M.; Schwarz, P.; Marcel, A.; Karin, M.; Prinz, M.; Pasparakis, M.; Aguzzi, A. Prion propagation in mice lacking central nervous system NF-kappaB signalling. J. Gen. Virol. 2008, 89, 1545–1550. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Heikenwalder, M.; Scwarz, P.; Takeda, K.; Akira, S.; Aguzzi, A. Prion pathogenesis in the absence of Toll-like receptor signalling. Embo Rep. 2003, 4, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Guijarro, I.M.; Garcés, M.; Andrés-Benito, P.; Marín, B.; Otero, A.; Barrio, T.; Carmona, M.; Ferrer, I.; Badiola, J.J.; Monzón, M. Assessment of glial activation response in the progress of natural scrapie after chronic dexamethesone treatment. Int. J. Mol. Sci. 2020, 21, 3231. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.; Boche, D.; Perry, V.H. Transforming growth factor b1, the dominant cytokine in murine prion disease: Influence on inflammatory cytokine synthesis and alteration of vascular extracellular matrix. Neuropathol. Appl. Neurobiol. 2002, 28, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.A.; Race, B.; Williams, K.; Striebel, J.; Chesebro, B. RNA-seq and network analysis reveal unique glial gene expression signatures during prion infection. Mol. Brain 2020, 13, 71. [Google Scholar] [CrossRef]
- De Lucia, C.; Rinchon, A.; Olmos-Alsonso, A.; Riecken, K.; Fehse, B.; Boche, D.; Perry, V.H.; Gomez-Nicola, D. Microglia regulate hippocampal neurogenesis during chronic neurodegeneration. BrainBehav. Immun. 2016, 55, 179–190. [Google Scholar] [CrossRef]
- Boche, D.; Cunningham, C.; Gauldie, J.; Perry, V.H. Transforming growth factor-beta 1-mediated neuroprotection against excitotoxic injury in vivo. J. Cereb. Blood Flow Metab. 2003, 23, 1174–1182. [Google Scholar] [CrossRef]
- Srivastava, S.; Katorcha, E.; Makarava, N.; Barrett, J.P.; Loane, D.J.; Baskakov, I.V. Inflammatory response of microglia to prions is controlled by sialylation of PrPSc. Sci. Rep. 2018, 8, 11326. [Google Scholar] [CrossRef]
- Perry, V.H.; Newma, T.A.; Cunningham, C. The impact of systemic infection on the progression of neurodegenerative disease. Nat. Rev. Neurosci. 2003, 4, 103–112. [Google Scholar] [CrossRef]
- Cunningham, C.; Wilcockson, D.C.; Campion, S.; Lunnon, K.; Perry, V.H. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J. Neurosci. 2005, 25, 9275–9284. [Google Scholar] [CrossRef]
- Cunningham, C.; Campion, S.; Lunnon, K.; Murray, C.L.; Woods, J.F.C.; Deacon, R.M.J.; Rawlins, J.N.P.; Perry, V.H. Systemic inflammation induces acute behavioural and cognitive changes and accelerates neurodegenerative disease. Biol. Psychiatry 2009, 65, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Lunnon, K.; Teeling, J.L.; Tutt, A.L.; Cragg, M.S.; Glennie, M.J.; Perry, V.H. Systemic inflammation modulates Fc receptor expression on microglia during chronic neurodegeneration. J. Immunol. 2011, 186, 7215–7224. [Google Scholar] [CrossRef] [PubMed]
- Nakagaki, T.; Ishibashi, D.; Mori, T.; Miyazaki, Y.; Takatsuki, H.; Tange, H.; Tagauchi, Y.; Satoh, K.; Atarashi, R.; Nishida, N. Administration of FK506 from late stage of disease prolongs survival of human prion-inoculated mice. Neurotherapeutics 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.R.; Hoyles, L.; Flint, H.J.; Dumas, M.E. Colonic bacterial metabolites and human health. Curr. Opin. Microbiol. 2013, 16, 246–254. [Google Scholar] [CrossRef]
- Kamada, N.; Kim, Y.G.; Sham, H.P.; Vallance, B.A.; Puente, J.L.; Martens, E.C.; Nunez, G. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science 2012, 336, 1325–1329. [Google Scholar] [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, c.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef]
- Erny, D.; Hrabě de Angelis, A.L.; Prinz, M. Communicating systems in the body: How microbiota and microglia cooperate. Immunology 2017, 150, 7–15. [Google Scholar] [CrossRef]
- Ceppa, F.A.; Izzo, L.; Sardelli, L.; Raimondi, I.; Tunesi, M.; Albani, D.; Giordano, C. Human gut-microbiota interaction in neurodegenerative disorders and current engineered tools for its modeling. Front. Cell. Infect. Microbiol. 2020, 10, 296. [Google Scholar] [CrossRef]
- Erny, D.; Hrabe de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- Fang, P.; Kazmi, S.A.; Jameson, K.G.; Hsiao, E.Y. The microbiome as a modifier of neurodegenerative disease risk. Cell Host Microbe 2020, 28, 201–222. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, E.; Kim, S.-W.; Suda, W.; Kawasumi, M.; Onawa, S.; Taguchi-Atarashi, N.; Morita, H.; Taylor, T.D.; Hattori, M.; Ohno, H. Gut microorganisms act together to exacerbate inflammation in spinal cords. Nature 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Bradford, B.M.; Tetlow, L.; Mabbott, N.A. Prion disease pathogenesis in the absence of the commensal microbiota. J. Gen. Virol. 2017, 98, 1943–1952. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, D.S.; Mabbott, N.A. The influence of the commensal and pathogenic gut microbiota on prion disease pathogenesis. J. Gen. Virol. 2016, 97, 1725–1738. [Google Scholar] [CrossRef]
- De Luigi, A.; Colombo, L.; Diomede, L.; Capobianco, R.; Mangieri, M.; Miccolo, C.; Limido, L.; Forloni, G.; Tagliavini, F.; Salmona, M. The efficacy of tetracyclines in peripheral and intracerebral prion infection. PLoS ONE 2008, 3, e1888. [Google Scholar] [CrossRef]
- Sun, J.; Shigemi, H.; Tanaka, Y.; Yamauchi, T.; Ueda, T.; Iwasaki, H. Tetracyclines downregulate the production of LPS-induced cytokines and chemokines in THP-1 cells via ERK, p38, and nuclear factor kB signaling pathways. Biochem. Biophys. Rep. 2015, 4, 397–404. [Google Scholar] [CrossRef]
- Tagliavini, F.; Forloni, G.; Colombo, L.; Rossi, G.; Girola, L.; Canciani, B.; Angretti, N.; Giampaolo, L.; Pressini, E.; Awan, T.; et al. Tetracycline affects abnormal properties of synthetic PrP peptides and PrPSc in vitro. J. Mol. Biol. 2000, 300, 1309–1322. [Google Scholar] [CrossRef]
- Allen, N.J.; Bennett, M.L.; Foo, L.C.; Wang, G.X.; Chakroborty, C.; Smith, S.J.; Barres, B.A. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 2012, 486, 410–414. [Google Scholar] [CrossRef]
- Kucukdereli, H.; Allen, N.J.; Lee, A.T.; Feng, A.; Ozlu, M.I.; Conaster, L.M.; Chakroborty, C.; Workman, G.; Weaver, M.; Sage, E.H.; et al. Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc. Natl. Acad. Sci. USA 2011, 108, E440–E449. [Google Scholar] [CrossRef]
- Chung, W.-S.; Clarke, L.E.; Wang, G.X.; Stafford, B.K.; Sher, A.; Chakroborty, C.; Joung, J.; Foo, L.C.; Thompson, A.; Chen, C.; et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 2013, 504, 394–400. [Google Scholar] [CrossRef]
- Smith, H.L.; Freeman, O.J.; Butcher, A.J.; Holmqvist, S.; Humoud, I.; Schatzl, T.; Hughes, D.T.; Verity, N.C.; Swinden, D.P.; Hayes, J.; et al. Astrocyte unfolded protein response induces a specific reactivity state that causes non-cell-autonomous neuronal degeneration. Neuron 2020, 105, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Astrocyte reactivity: Subtypes, states, and functions in CNS innate immunity. Trends Immunol. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Damisah, E.C.; Hill, R.A.; Rai, A.; Chen, F.; Rothlin, C.V.; Ghosh, S.; Grutzendler, J. Astrocytes and microglia play orchestrated roles and respect phagocytic territories during neuronal corpse removal in vivo. Sci. Adv. 2020, 6, eaba3239. [Google Scholar] [CrossRef] [PubMed]
- Raeber, A.J.; Race, R.E.; Brandner, S.; Priola, S.A.; Sailer, A.; Bessen, R.A.; Mucke, L.; Manson, J.; Aguzzi, A.; Oldstone, B.A.; et al. Astrocyte-specific expression of hamster prion protein (PrP) renders PrP knockout mice susceptible to hamster scrapie. Embo J. 1997, 16, 6057–6065. [Google Scholar] [CrossRef]
- Moreno, J.A.; Radford, H.; Peretti, D.; Steinert, J.R.; Verity, N.; Martin, M.G.; Halliday, M.; Morgan, J.; Dinsdale, D.; Ortori, C.A.; et al. Sustained translational repression by eIF2aP mediates prion neurodegeneration. Nature 2012, 485, 507–511. [Google Scholar] [CrossRef]
- Kitchen, P.; Salman, M.M.; Halsey, A.M.; Clarke-Bland, C.; MacDonald, J.A.; Ishida, H.; Vogel, H.J.; Almuturi, S.; Logan, A.; Kreida, S.; et al. Targeting aquaporin-4 subcellular localization to treat central nervous system edema. Cell 2020, 181, 7799–7894. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Marsh, S.E.; Stevens, B. Microglia and astrocytes in disease: Dynamic duo or partners in crime? Trends Immunol. 2020, in press. [Google Scholar] [CrossRef]
- Bohlen, C.J.; Bennett, F.C.; Tucker, A.F.; Collins, H.Y.; Mulinyawe, S.B.; Barres, B.A. Diverse requirements for microglial survival, specification and funciton revealed by defined-medium cultures. Neuron 2017, 94, 759–773. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennet, F.L.; Bohlen, C.J.; Schirmer, L.; Bennet, M.L.; Munch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Guttenplan, K.A.; Stafford, B.K.; El-Danaf, R.; Adler, D.I.; Munch, A.E.; Weigel, M.K.; Huberman, A.D.; Liddelow, S.A. Neurotoxic reactive astrocytes drive neuronal death after retinal injury. Cell Rep. 2020, 31, 107776. [Google Scholar] [CrossRef]
- Donaldson, D.S.; Bradford, B.M.; Else, K.J.; Mabbott, N.A. Accelerated onset of CNS prion disease in mice co-infected with a gastrointestinal helminth pathogen during the preclincal phase. Sci. Rep. 2020, 10, 4554. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, K.; Sepulveda-Falla, D.; Rose, I.V.L.; Madore, C.; Muth, C.; Matschke, J.; Butovsky, O.; Liddelow, S.A.; Glatzel, M.; Krasemann, S. Complement 3+-astrocytes are highly abundant in prion diseases, but their abolishment led to an accelerated disease course and early dysregulation of microglia. Acta Neuropath. Commun. 2020, 7, 83. [Google Scholar] [CrossRef] [PubMed]
- Bradford, B.M.; Wijaya, C.A.W.; Mabbott, N.A. Discrimination of prion strain targeting in the central nervous system via reactive astrocyte heterogeneity in CD44 expression. Front. Cell. Neurosci. 2019, 13, 411. [Google Scholar] [CrossRef] [PubMed]
- Mabbott, N.A.; Williams, A.; Farquhar, C.F.; Pasparakis, M.; Kollias, G.; Bruce, M.E. Tumor necrosis factor-alpha-deficient, but not interleukin-6-deficient, mice resist peripheral infection with scrapie. J. Virol. 2000, 74, 3338–3344. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Montrasio, F.; Klein, M.A.; Schwarz, P.; Priller, J.; Odermatt, B.; Pfeffer, K.; Aguzzi, A. Lymph nodal prion replication and neuroinvasion in mice devoid of follicular dendritic cells. Proc. Natl. Acad. Sci. USA 2002, 99, 919–924. [Google Scholar] [CrossRef]
- Rojo, R.; Raper, A.; Ozdemir, D.D.; Lefevre, L.; Grabert, K.; Wollscheid-Lengeling, E.; Bradford, B.; Caruso, M.; Gazova, I.; Sanchez, A.; et al. Deletion of a Csf1r enhancer selectively impacts CSF1R expression and development of tissue macrophage populations. Nat. Commun. 2019, 10, 3215. [Google Scholar] [CrossRef]
- Hennessy, E.; Griffin, E.W.; Cunningham, C. Astrocytes are primed by chronic neurodegeneration to produce exaggerated chemokine and cell infiltration responses to acute stimulation with the cytokines IL-1b and TNF-a. J. Neurosci. 2015, 35, 8411–8422. [Google Scholar] [CrossRef]
- Wheeler, M.A.; Clark, I.C.; Tjon, E.C.; Li, Z.; Zandee, S.E.J.; Couturier, C.P.; Watson, B.R.; Scalisis, G.; Alkwai, S.; Rothhammer, V.; et al. MAFG-driven astrocytes promote CNS inflammation. Nature 2020, 578, 593–599. [Google Scholar] [CrossRef]
- Ehresmann, D.W.; Hogan, R.N. Acceleration of scrapie disease in mice by an adenovirus. Intervirology 1986, 25, 103–110. [Google Scholar] [CrossRef]
- Lins, N.; Mourao, L.; Trévia, N.; Passos, A.; Farias, J.A.; Assunção, J.; Quintairos, A.; Bento-Torres, J.; Sosthene, M.C.K.; Diniz, J.A.P.; et al. Virus infections on prion diseased mice exacerbate inflammatory microglial response. Oxidative Med. Cell. Longev. 2016, 2016, 3974648. [Google Scholar] [CrossRef]
- Muth, C.; Schrock, K.; Madore, C.; Hartmann, K.; Fanek, Z.; Butovsky, O.; Glatzel, M.; Krasemann, S. Activation of microglia by retroviral infection correlates with transient clearance of prions from the brain but does not change incubation time. Brain Pathol. 2017, 27, 590–602. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Neumann, M.; Luepke, J.-P.; Grashorn, J.; Wurr, S.; Stocking, C.; Glatzel, M. Persistent retroviral infection with MoMuLV influences neuropathological signature and phenotype of prion disease. Acta Neuropathol. 2012, 124, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Bancroft, A.J.; Else, K.J.; Grencis, R.K. Low-level infection with Trichuris muris significantly affects the polarization of the CD4 response. Eur. J. Immunol. 1994, 24, 3113–3118. [Google Scholar] [CrossRef] [PubMed]
- Else, K.J.; Finkelman, F.D.; Maliszewski, C.R.; Grencis, R.K. Cytokine-mediated regulation of chronic intestinal helminth infection. J. Exp. Med. 1994, 179, 347–351. [Google Scholar] [CrossRef]
- Hashioka, S.; Klegeris, A.; Schwab, S.; McGeer, P.L. Interferon-g-dependent cytotoxic activation of human astrocytes and astrocytoma cells. Neurobiol. Aging 2009, 30, 1924–1935. [Google Scholar] [CrossRef]
- Walsh, D.T.; Betmouni, S.; Perry, V.H. Absence of detectable IL-1 beta production in murine prion disease: A model of chronic neurodegeneration. J. Neuropathol. Exp. Neurol. 2001, 60, 173–182. [Google Scholar] [CrossRef]
- Halonen, S.K.; Woods, T.A.; McInnerney, K.; Weiss, L.M. Microarray analysis of IFN-gamma response genes in astrocytes. J. Neuroimmunol. 2006, 175, 19–30. [Google Scholar] [CrossRef]
- Lundh, M.; Bugliani, M.; Dahlby, T.; Chou, D.H.C.; Wagner, B.; Ghiasi, S.M.; De Tata, V.; Chen, Z.; Lund, M.N.; Davies, M.J.; et al. The immunoproteasome is induced by cytokines and regulates apoptosis in human islets. J. Endocrinol. 2017, 233, 369–379. [Google Scholar] [CrossRef]
- McFarlin, D.E.; Raff, M.C.; Simpson, E.; Nehlsen, S.H. Scrapie in immunologically deficient mice. Nature 1971, 233, 336. [Google Scholar] [CrossRef]
- Bonney, S.; Seitz, S.; Ryan, C.A.; Jones, K.L.; Clarke, P.; Tyler, K.L.; Siegenthaler, J.A. Gamma interferon alters junctional integrity via Rho kinase resulting in blood-brain barrier leakage in experimental viral encephalitis. mBio 2019, 10, e01675-19. [Google Scholar] [CrossRef]
- Gate, D.; Saligrama, N.; Leventhal, O.; Yang, A.C.; Unger, M.S.; Middeldorp, J.; Chen, K.; Lehallier, B.; Channappa, D.; De Los Santos, M.B.; et al. Clonally-expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 2020, 577, 399–404. [Google Scholar] [CrossRef]
- Owens, T.; Khorooshi, R.; Wlodarczyk, A.; Asgari, N. Interferons in the central nervous system: A few instruments play many tunes. Glia 2014, 62, 339–355. [Google Scholar] [CrossRef] [PubMed]
- Nazmi, A.; Field, R.H.; Griffin, E.W.; Haugh, O.; Hennessy, E.; Cox, D.; Reis, R.; Tortorelli, L.; Murray, C.L.; Lopez-Rodriguea, A.B.; et al. Chronic neurodegeneration induces type I interferon systhesis via STING, shaping microglial phenotype and accelerating disease progression. Glia 2019, 67, 1254–1276. [Google Scholar] [CrossRef] [PubMed]
- Keating, S.E.; Baran, M.; Bowie, A.G. Cytosolic DNA sensors regulating type I interferon induction. Trends Immunol. 2011, 32, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Baker, C.A.; Lu, Z.Y.; Manuelidis, L. Early induction of interferon-responsive mRNAs in Creutzfeldt-Jakob disease. J. Neurovirology 2004, 10, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Field, R.; Campion, S.; Warren, C.; Murray, C.; Cunningham, C. Systemic challenge with the TLR3 agonist poly I:C induces amplified IFNa/b and IL-1b responses in the disease brain and exacerbates chronic neurodegeneration. Brain Behav. Immun. 2010, 24, 996–1007. [Google Scholar] [CrossRef]
- Ishibashi, D.; Atarashi, R.; Fuse, T.; Nagaki, T.; Yamaguchi, N.; Satoh, K.; Honda, K.; Nishida, N. Protective role of interferon regulatory factor 3-mediated signaling against prion infection. J. Virol. 2012, in press. [Google Scholar] [CrossRef]
- Ishibashi, D.; Homma, T.; Nakagaki, T.; Fuse, T.; Sano, K.; Sato, K.; Mori, T.; Atarashi, R.; Nishida, N. Type I interferon protects neurons from prions in in vivo models. Brain 2019, 142, 1035–1050. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-S.; Hu, Y.; Tao, Z.-W.; Pei, Y.-Y.; Yuan, M.-L.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- WHO. COVID-19 situation reports; WHO: Geneva, Switzerland, 2020; Volume 2020. [Google Scholar]
- England, N. COVID-19 Daily Deaths. Available online: https://web.archive.org/web/20200501094237 (accessed on 25 August 2020).
- Atkins, J.L.; Masoli, J.A.H.; Delgado, J.; Pilling, L.C.; Kuo, C.L.; Kuchel, G.A.; Melzer, D. Preexisting comorbidities predicting COVID-19 and mortality in the UK biobank community cohort. J. Gerontol. 2020, in press. [Google Scholar] [CrossRef]
- Williamson, E.J.; Walker, A.J.; Bhaskaran, K.; Bacon, S.; Bates, C.; Morton, C.E.; Curtis, H.J.; Mehrkar, A.; Evans, D.; Inglesby, P.; et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 2020, 584, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford, D.; Perry, V.H. Systemic inflammation and disease progression in Alzheimer disease. Neurology 2009, 73, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Combrinck, M.I.; Perry, V.H.; Cunningham, C. Peripheral infection evokes exaggerated sickness behaviour in pre-clinical murine prion disease. Neuroscience 2002, 112, 7–11. [Google Scholar] [CrossRef]
- Nerius, M.; Doblhammer, G.; Tamguney, G. GI infections are associated with an increased risk of Parkinson’s disease. Gut 2020, 69, 1154–1156. [Google Scholar] [CrossRef]
- Burberry, A.; Wells, M.F.; Limone, F.; Couto, A.; Smith, K.S.; Keaney, J.; Gillet, G.; van Gastel, N.; Wang, J.-Y.; Pietilainen, O.; et al. C9orf72 suppresses sytemic and neural inflammation induced by gut bacteria. Nature 2020, 582, 89–94. [Google Scholar] [CrossRef]
- Young, M.J.; O’Hare, M.; Matiello, M.; Schmahmann, J.D. Creutzfeldt-Jakob disease in a man with COVID-19: SARS-CoV-2-accelerated neurodegeneration? Brain Behav. Immun. 2020, in press. [Google Scholar] [CrossRef]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudual analyses reveal immunological misfiring in severe COVID-19. Nature 2020, in press. [Google Scholar] [CrossRef]
- Hennessy, E.; Gormley, S.; Lopez-Rodriguez, A.B.; Murray, C.; Murray, C.; Cunningham, C. Systemic TNF-a produces acute cognitive dysfunction and exagerated sickness behavior when superimposed upon progressive neurodegeneration. Brain Behav. Immun. 2017, 59, 233–244. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Payan-Gomez, C.; Rodriguez, D.; Amador-Munoz, D.; Ramirez-Clavijo, S. Integrative analysis of global gene expression identifies opposite patterns of reactive astrogliosis in aged human prefrontal cortex. Brain Sci. 2018, 8, 27. [Google Scholar] [CrossRef]
- Bishop, M.T.; Hart, P.; Aitchison, L.; Baybutt, H.N.; Plinston, C.; Thomson, V.; Tuzi, N.L.; Head, M.W.; Ironside, J.W.; Will, R.G.; et al. Predicting susceptibility and incubation time of human-to-human transmission of vCJD. Lancet Neurol. 2006, 5, 393–398. [Google Scholar] [CrossRef]
- Mabbott, N.A. Prospects for safe and effective vaccines against prion diseases. Expert Rev. Vaccines 2015, 14, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Frontzek, K.; Aguzzi, A. Recent developments in antibody therapeutics against prion disease. Emerg. Top. Life Sci. 2020, in press. [Google Scholar] [CrossRef]
- Heppner, F.L.; Musahl, C.; Arrighi, I.; Klein, M.A.; Rulicke, T.; Oesch, B.; Zinkernagel, R.M.; Kalinke, U.; Aguzzi, A. Prevention of scrapie pathogenesis by transgenic expression of anti-prion protein antibodies. Science 2001, 294, 178–182. [Google Scholar] [CrossRef]
- White, A.R.; Enever, P.; Tayebi, M.; Mushens, R.; Lineham, J.; Brandner, S.; Anstee, D.; Collinge, J.; Hawke, S. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature 2003, 422, 80–83. [Google Scholar] [CrossRef]
- University College London Hospitals. Sixth UCLH Patient to Recieve Innovative Drug for CJD. UCL News, 9 October 2019. [Google Scholar]
- Neves, V.; Aires-da-Silva, F.; Corte-Real, S.; Castanho, M.A.R.B. Antibody approaches to treat brain diseases. Trends Biotechnol. 2015, 34, 36–48. [Google Scholar] [CrossRef]
- Pain, C.; Dumont, J.; Dumoulin, M. Camelid single-domain antibody fragments: Uses and prospectives to investigate protein misfolding and aggregation, and to treat diseases associated with these phenomena. Biochimie 2015, 111, 82–106. [Google Scholar] [CrossRef]
- Sonati, T.; Reimann, R.R.; Falsig, J.; Baral, P.K.; O’Connor, T.; Hornemann, S.; Yaganoglu, S.; Li, B.; Herrmann, U.S.; Wieland, B.; et al. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 2013, 501, 102–106. [Google Scholar] [CrossRef]
- Reimann, R.R.; Sonati, T.; Hornemann, S.; Hermann, U.S.; Arand, M.; Hawke, S.; Aguzzi, A. Differential toxicity of antibodies to the prion protein. PLoS Pathog. 2016, 12, e1005401. [Google Scholar] [CrossRef]
- Goñi, F.; Knudsen, E.; Schreiber, F.; Scholtzova, H.; Pankiewicz, J.; Carp, R.; Meeker, H.C.; Rubenstein, R.; Brown, D.R.; Sy, M.S.; et al. Mucosal vaccination delays or prevents prion infection via the oral route. Neuroscience 2005, 133, 413–421. [Google Scholar] [CrossRef]
- Goñi, F.; Mathiason, C.K.; Yim, L.; Wong, K.; Hayes-Klug, J.; Nalls, A.; Peyser, D.; Estevez, V.; Denkers, N.; Xu, J.; et al. Mucosal immunization with an attenuated Salmonella vaccine partially protects white-tailed deer from chronic wasting disease. Vaccine 2015, 33, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Claypool, S.M.; Wagner, J.S.; Mizoguchi, E.; Mizoguchi, A.; Roopenian, D.C.; Lencer, W.I.; Blumberg, R.S. Human neonatal Fc receptor mediates transport of IgG into luminal secretions for delivery of antigens to mucosal dendritic cells. Immunity 2004, 20, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Ano, Y.; Sakudo, A.; Uraki, R.; Sato, Y.; Kono, J.; Sugiura, K.; Yokoyama, T.; Itohara, S.; Nakayama, H.; Yukawa, M.; et al. Enhanced enteric invasion of scrapie agents into the villous columnar epithelium via maternal immunoglobulin. Int. J. Mol. Med. 2010, 26, 845–851. [Google Scholar] [CrossRef]
- Frontzek, K.; Carta, M.; Losa, M.; Epskamp, M.; Meisl, G.; Anane, A.; Brandel, J.-P.; Camenisch, U.; Castilla, J.; Haik, S.; et al. Autoantibodies against the prion protein in individuals with PRNP mutations. Neurology 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Senatore, A.; Frontzek, K.; Emmenegger, M.; Chincisan, A.; Losa, M.; Reimann, R.; Horny, G.; Guo, J.; Fels, S.; Sorce, S.; et al. Protective anti-prion antibodies in human immunoglobulin repertoires. Embo Mol. Med. 2020, e12739, in press. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mabbott, N.A.; Bradford, B.M.; Pal, R.; Young, R.; Donaldson, D.S. The Effects of Immune System Modulation on Prion Disease Susceptibility and Pathogenesis. Int. J. Mol. Sci. 2020, 21, 7299. https://doi.org/10.3390/ijms21197299
Mabbott NA, Bradford BM, Pal R, Young R, Donaldson DS. The Effects of Immune System Modulation on Prion Disease Susceptibility and Pathogenesis. International Journal of Molecular Sciences. 2020; 21(19):7299. https://doi.org/10.3390/ijms21197299
Chicago/Turabian StyleMabbott, Neil A., Barry M. Bradford, Reiss Pal, Rachel Young, and David S. Donaldson. 2020. "The Effects of Immune System Modulation on Prion Disease Susceptibility and Pathogenesis" International Journal of Molecular Sciences 21, no. 19: 7299. https://doi.org/10.3390/ijms21197299
APA StyleMabbott, N. A., Bradford, B. M., Pal, R., Young, R., & Donaldson, D. S. (2020). The Effects of Immune System Modulation on Prion Disease Susceptibility and Pathogenesis. International Journal of Molecular Sciences, 21(19), 7299. https://doi.org/10.3390/ijms21197299