Stoking the Fire: How Dying Cells Propagate Inflammatory Signalling through Extracellular Vesicle Trafficking

Abstract
1. Introduction
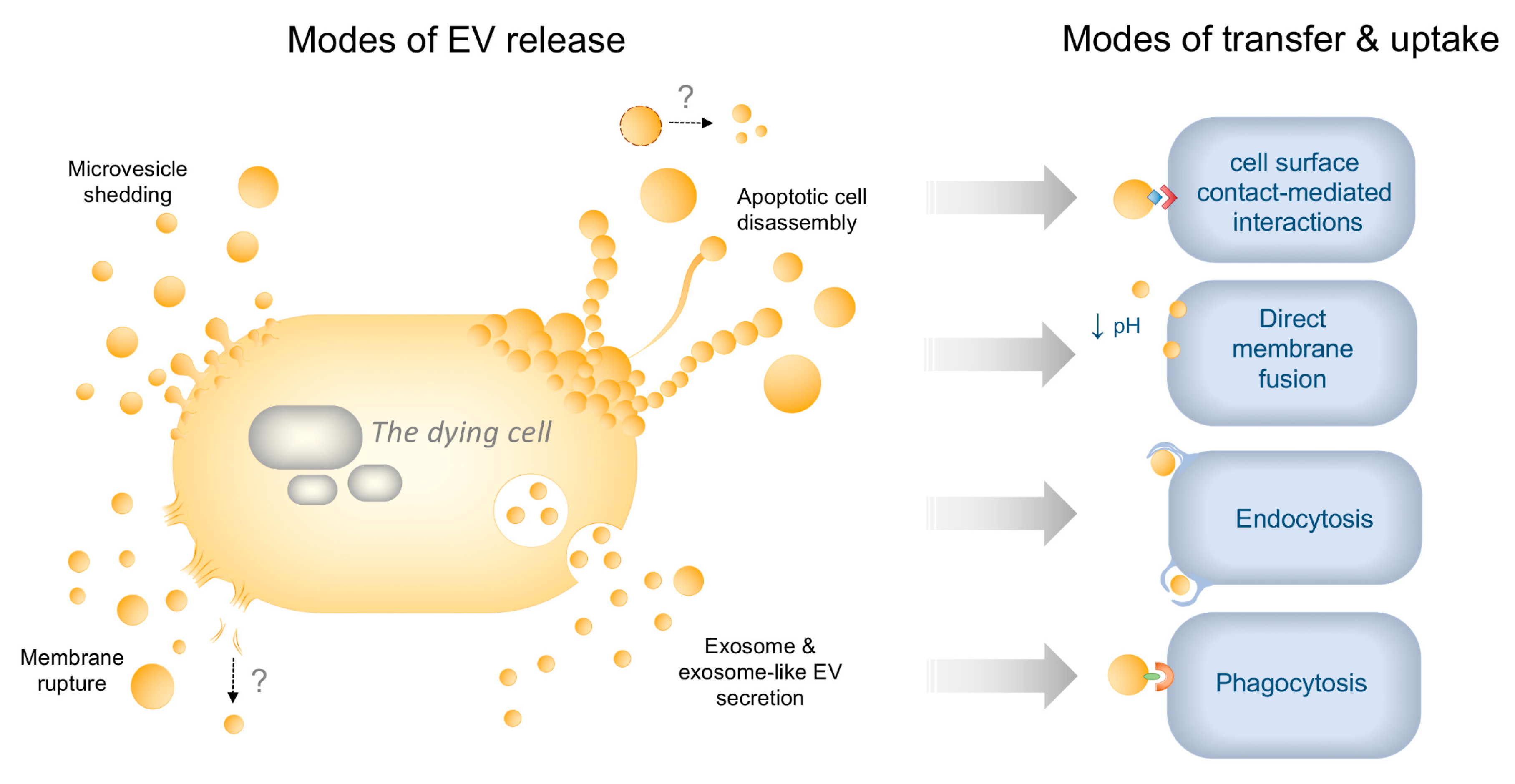
2. EV Diversity in Healthy and Dying Cells—Mechanisms of Biogenesis and Uptake
2.1. Biogenesis and Cargo Sorting
2.1.1. Exosomes
2.1.2. Microvesicles (MVs)
2.1.3. ApoBDs
2.1.4. Alternative Nomenclature and Biogenesis during Cell Death
2.2. Uptake Mechanisms
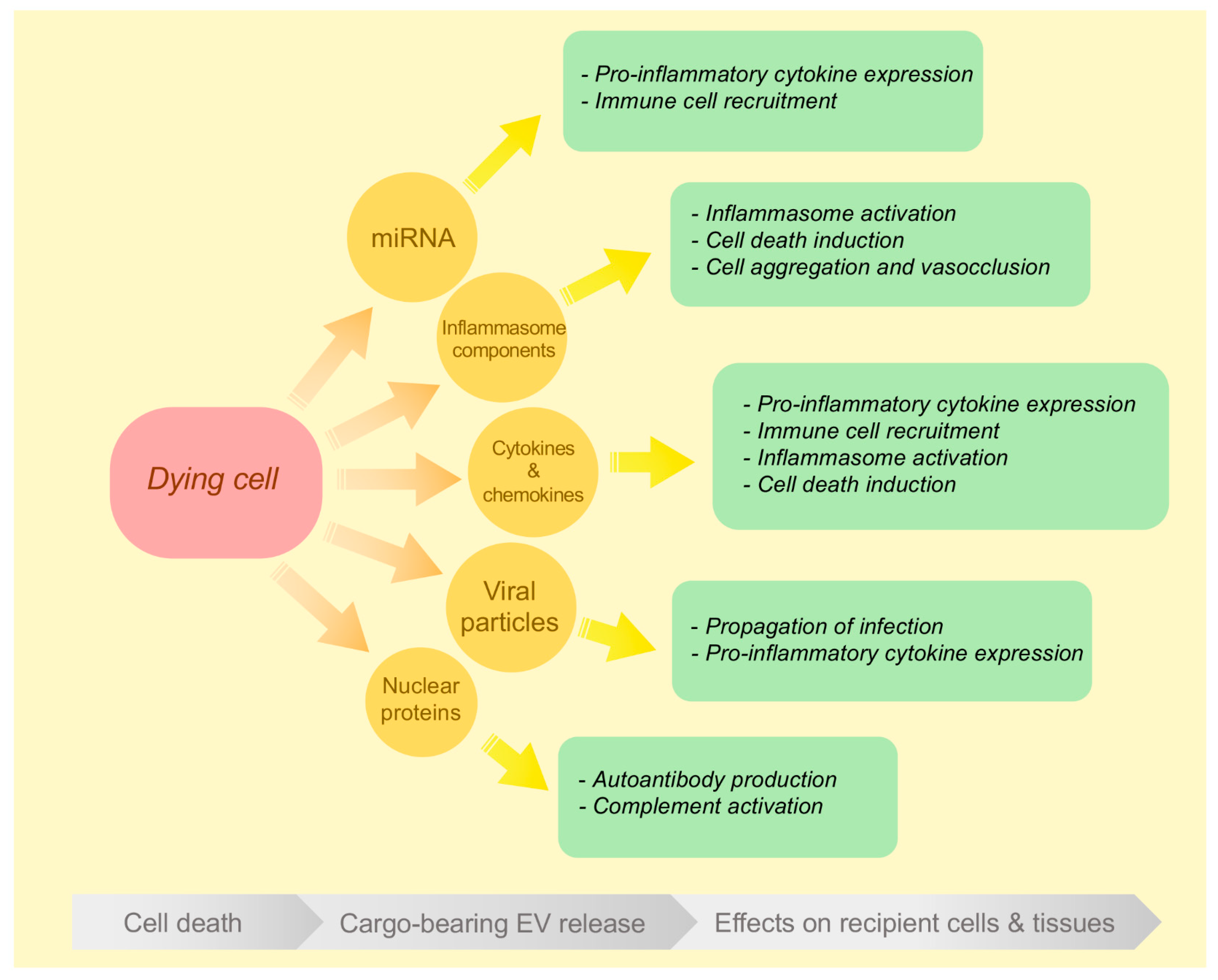
3. Inflammatory EVs Generated during Different Modes of Cell Death
3.1. EVs Released during Apoptosis and Secondary Necrosis
3.2. EVs Released during Primary Necrosis
3.3. EVs Released during Inflammasome Activation and Pyroptosis
3.4. EVs Released during Necroptosis
4. Challenges in Reporting on Dying Cell-Derived EVs
5. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Raposo, G.; Stahl, P.D. Extracellular vesicles: A new communication paradigm? Nat. Rev. Mol. Cell Biol. 2019, 20, 509–510. [Google Scholar] [CrossRef]
- Alcamí, P.; Pereda, A.E. Beyond plasticity: The dynamic impact of electrical synapses on neural circuits. Nat. Rev. Neurosci. 2019, 20, 253–271. [Google Scholar] [CrossRef]
- Dustin, M.L. The immunological synapse. Cancer Immunol. Res. 2014, 2, 1023–1033. [Google Scholar] [CrossRef]
- Matzuk, M.M.; Burns, K.H.; Viveiros, M.M.; Eppig, J.J. Intercellular Communication in the Mammalian Ovary: Oocytes Carry the Conversation. Science 2002, 296, 2178–2180. [Google Scholar] [CrossRef]
- Rawlins, E.L. The building blocks of mammalian lung development. Dev. Dyn. 2010, 240, 463–476. [Google Scholar] [CrossRef]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Salomon, C.; Rice, G.E. Role of Exosomes in Placental Homeostasis and Pregnancy Disorders. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands; Volume 145, pp. 163–179.
- Dai, J.; Su, Y.; Zhong, S.; Cong, L.; Liu, B.; Yang, J.; Tao, Y.; He, Z.; Chen, C.; Jiang, Y. Exosomes: Key players in cancer and potential therapeutic strategy. Signal Transduct. Target. Ther. 2020, 5, 1–10. [Google Scholar] [CrossRef]
- Turpin, D.; Truchetet, M.E.; Faustin, B.; Augusto, J.F.; Contin-Bordes, C.; Brisson, A.R.; Blanco, P.; Duffau, P. Role of extracellular vesicles in autoimmune diseases. Autoimmun. Rev. 2016, 15, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Quek, C.; Hill, A. The role of extracellular vesicles in neurodegenerative diseases. Biochem. Biophys. Res. Commun. 2017, 483, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Sequeiros, T.; Rigau, M.; Chiva, C.; Montes, M.; Garcia-Grau, I.; Garcia, M.; Diaz, S.; Celma, A.; Bijnsdorp, I.; Campos, A.; et al. Targeted proteomics in urinary extracellular vesicles identifies biomarkers for diagnosis and prognosis of prostate cancer. Oncotarget 2016, 8, 4960–4976. [Google Scholar] [CrossRef] [PubMed]
- Kerr, N.; García-Contreras, M.; Abbassi, S.; Mejias, N.H.; DeSousa, B.R.; Ricordi, C.; Dietrich, W.D.; Keane, R.W.; Vaccari, J.P.D.R. Inflammasome Proteins in Serum and Serum-Derived Extracellular Vesicles as Biomarkers of Stroke. Front. Mol. Neurosci. 2018, 11, 309. [Google Scholar] [CrossRef] [PubMed]
- Ostenfeld, M.S.; Jensen, S.G.; Jeppesen, D.K.; Christensen, L.L.; Thorsen, S.B.; Stenvang, J.; Hvam, M.L.; Thomsen, A.; Mouritzen, P.; Rasmussen, M.H.; et al. miRNA profiling of circulating EpCAM(+) extracellular vesicles: Promising biomarkers of colorectal cancer. J. Extracell. Vesicles 2016, 5, 31488. [Google Scholar] [CrossRef] [PubMed]
- Gasecka, A.; Böing, A.N.; Filipiak, K.J.; Nieuwland, R. Platelet extracellular vesicles as biomarkers for arterial thrombosis. Platelets 2016, 28, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Khatri, M.; Richardson, L.A.; Meulia, T. Mesenchymal stem cell-derived extracellular vesicles attenuate influenza virus-induced acute lung injury in a pig model. Stem Cell Res. Ther. 2018, 9, 17. [Google Scholar] [CrossRef]
- Drommelschmidt, K.; Serdar, M.; Bendix, I.; Herz, J.; Bertling, F.; Prager, S.; Keller, M.; Ludwig, A.-K.; Duhan, V.; Radtke, S.; et al. Mesenchymal stem cell-derived extracellular vesicles ameliorate inflammation-induced preterm brain injury. Brain Behav. Immun. 2017, 60, 220–232. [Google Scholar] [CrossRef]
- Barreca, M.M.; Cancemi, P.; Geraci, F. Mesenchymal and Induced Pluripotent Stem Cells-Derived Extracellular Vesicles: The New Frontier for Regenerative Medicine? Cells 2020, 9, 1163. [Google Scholar] [CrossRef]
- Nathan, C.; Ding, A. Nonresolving Inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef]
- Van Hezel, M.E.; Nieuwland, R.; Van Bruggen, R.; Juffermans, N.P. The Ability of Extracellular Vesicles to Induce a Pro-Inflammatory Host Response. Int. J. Mol. Sci. 2017, 18, 1285. [Google Scholar] [CrossRef]
- Hosseinkhani, B.; Akker, N.M.V.D.; Molin, D.G.; Michiels, L. (Sub)populations of extracellular vesicles released by TNF-α—triggered human endothelial cells promote vascular inflammation and monocyte migration. J. Extracell. Vesicles 2020, 9, 1801153. [Google Scholar] [CrossRef]
- Boilard, E.; Nigrovic, P.A.; Larabee, K.; Watts, G.F.; Coblyn, J.S.; Weinblatt, M.E.; Massarotti, E.M.; Remold-O’Donnell, E.; Farndale, R.W.; Ware, J.; et al. Platelets Amplify Inflammation in Arthritis via Collagen-Dependent Microparticle Production. Science 2010, 327, 580–583. [Google Scholar] [CrossRef]
- Poon, I.K.H.; Lucas, C.D.; Rossi, A.G.; Ravichandran, K.S. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Medina, C.B.; Mehrotra, P.; Arandjelovic, S.; Perry, J.S.A.; Guo, Y.; Morioka, S.; Barron, B.; Walk, S.F.; Ghesquière, B.; Krupnick, A.S.; et al. Metabolites released from apoptotic cells act as tissue messengers. Nature 2020, 580, 130–135. [Google Scholar] [CrossRef]
- Brock, C.K.; Wallin, S.T.; Ruiz, O.E.; Samms, K.M.; Mandal, A.; Sumner, E.A.; Eisenhoffer, G.T. Stem cell proliferation is induced by apoptotic bodies from dying cells during epithelial tissue maintenance. Nat. Commun. 2019, 10, 1044. [Google Scholar] [CrossRef] [PubMed]
- Shlomovitz, I.; Erlich, Z.; Speir, M.; Zargarian, S.; Baram, N.; Engler, M.; Edry-Botzer, L.; Munitz, A.; Croker, B.A.; Gerlic, M. Necroptosis directly induces the release of full-length biologically active IL-33 in vitro and in an inflammatory disease model. FEBS J. 2019, 286, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S. Apoptosis and autoimmune diseases. Ann. N. Y. Acad. Sci. 2010, 1209, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Paone, S.; Baxter, A.A.; Hulett, M.D.; Poon, I.K.H. Endothelial cell apoptosis and the role of endothelial cell-derived extracellular vesicles in the progression of atherosclerosis. Cell. Mol. Life Sci. 2018, 76, 1093–1106. [Google Scholar] [CrossRef] [PubMed]
- Withrow, J.; Murphy, C.; Liu, Y.; Hunter, M.; Fulzele, S.; Hamrick, M.W. Extracellular vesicles in the pathogenesis of rheumatoid arthritis and osteoarthritis. Arthritis Res. 2016, 18, 286. [Google Scholar] [CrossRef] [PubMed]
- Meehan, B.; Rak, J.; Di Vizio, D. Oncosomes—large and small: What are they, where they came from? J. Extracell. Vesicles 2016, 5, 33109. [Google Scholar] [CrossRef]
- Baxter, A.A.; Phan, T.K.; Hanssen, E.; Liem, M.; Hulett, M.D.; Mathivanan, S.; Poon, I.K.H. Analysis of extracellular vesicles generated from monocytes under conditions of lytic cell death. Sci. Rep. 2019, 9, 7538. [Google Scholar] [CrossRef]
- Zargarian, S.; Shlomovitz, I.; Erlich, Z.; Hourizadeh, A.; Ofir-Birin, Y.; Croker, B.A.; Regev-Rudzki, N.; Edry-Botzer, L.; Gerlic, M. Phosphatidylserine externalization, “necroptotic bodies” release, and phagocytosis during necroptosis. PLoS Biol. 2017, 15, e2002711. [Google Scholar] [CrossRef]
- Kuwabara, Y.; Ono, K.; Horie, T.; Nishi, H.; Nagao, K.; Kinoshita, M.; Watanabe, S.; Baba, O.; Kojima, Y.; Shizuta, S.; et al. Increased MicroRNA-1 and MicroRNA-133a Levels in Serum of Patients With Cardiovascular Disease Indicate Myocardial Damage. Circ. Cardiovasc. Genet. 2011, 4, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Zuo, P.; Lin, X.; Li, X.; Zhang, Y. Macrophage-derived extracellular vesicles transfer inflammasome components to endothelial cells and induces endothelial injury. FASEB J. 2017, 31, 825.13. [Google Scholar]
- Zhang, Y.; Liu, F.; Yuan, Y.; Jin, C.; Chang, C.; Zhu, Y.; Zhang, X.; Tian, C.; He, F.; Wang, J. Inflammasome-Derived Exosomes Activate NF-κB Signaling in Macrophages. J. Proteome Res. 2016, 16, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Kim, J.M.; Kim, J.; Hur, J.; Park, S.; Kim, K.; Shin, H.J.; Chwae, Y.J. Molecular mechanisms of biogenesis of apoptotic exosome-like vesicles and their roles as damage-associated molecular patterns. Proc. Natl. Acad. Sci. USA 2018, 115, E11721–E11730. [Google Scholar] [CrossRef]
- Yoon, S.; Kovalenko, A.; Bogdanov, K.; Wallach, D. MLKL, the Protein that Mediates Necroptosis, also Regulates Endosomal Trafficking and Extracellular Vesicle Generation. Immunity 2017, 47, 51–65. [Google Scholar] [CrossRef]
- Hong, J.; Bhat, O.M.; Li, G.; Dempsey, S.K.; Zhang, Q.; Ritter, J.K.; Li, W.; Li, P.L. Lysosomal regulation of extracellular vesicle excretion during d-ribose-induced NLRP3 inflammasome activation in podocytes. Biochim. Biophys. Acta Bioenerg. 2019, 1866, 849–860. [Google Scholar] [CrossRef]
- Gong, Y.N.; Guy, C.; Olauson, H.; Becker, J.U.; Yang, M.; Fitzgerald, P.; Linkermann, A.; Green, D.R. ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and its Consequences. Cell 2017, 169, 286–300. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, X.; Wang, G.; Tong, H.S.; Su, L.; Li, X. Proteomic analysis of extracellular vesicles released from heat-stroked hepatocytes reveals promotion of programmed cell death pathway. Biomed. Pharmacother. 2020, 129, 110489. [Google Scholar] [CrossRef]
- Lee, J.Y.; Park, J.K.; Lee, E.Y.; Lee, E.B.; Song, Y.W. Circulating exosomes from patients with systemic lupus erythematosus induce a proinflammatory immune response. Arthritis Res. Ther. 2016, 18, 1–8. [Google Scholar] [CrossRef]
- Nevzorova, T.A.; Evtugina, N.G.; Litvinov, R.I. Cellular Microvesicles in the Blood of Patients with Systemic Lupus Erythematosus. BioNanoSci. 2018, 8, 441–445. [Google Scholar] [CrossRef]
- Nielsen, C.T.; Østergaard, O.; Stener, L.; Iversen, L.V.; Truedsson, L.; Gullstrand, B.; Jacobsen, S.; Heegaard, N.H.H. Increased IgG on cell-derived plasma microparticles in systemic lupus erythematosus is associated with autoantibodies and complement activation. Arthritis Rheum. 2012, 64, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Zirngibl, M.; Fürnrohr, B.G.; Janko, C.; Munoz, L.; Voll, R.E.; Gregory, C.D.; Schett, G.; Herrmann, M. Loading of nuclear autoantigens prototypically recognized by systemic lupus erythematosus sera into late apoptotic vesicles requires intact microtubules and myosin light chain kinase activity. Clin. Exp. Immunol. 2014, 179, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Niessen, A.; Heyder, P.; Krienke, S.; Blank, N.; Tykocinski, L.O.; Lorenz, H.M.; Schiller, M. Apoptotic-cell-derived membrane microparticles and IFN-α induce an inflammatory immune response. J. Cell Sci. 2015, 128, 2443–2453. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Zhang, D.; Zhu, Z.; Cruz, C.S.D.; Jin, Y. Epithelial cell-derived microvesicles activate macrophages and promote inflammation via microvesicle-containing microRNAs. Sci. Rep. 2016, 6, 35250. [Google Scholar] [CrossRef]
- O’Dea, K.P.; Porter, J.R.; Tirlapur, N.; Katbeh, U.; Singh, S.; Handy, J.M.; Takata, M. Circulating Microvesicles are Elevated Acutely following Major Burns Injury and Associated with Clinical Severity. PLoS ONE 2016, 11, e0167801. [Google Scholar] [CrossRef]
- Berda-Haddad, Y.; Robert, S.; Salers, P.; Zekraoui, L.; Farnarier, C.; Dinarello, C.A.; Dignat-George, F.; Kaplanski, G. Sterile inflammation of endothelial cell-derived apoptotic bodies is mediated by interleukin-1. Proc. Natl. Acad. Sci. USA 2011, 108, 20684–20689. [Google Scholar] [CrossRef]
- Atkin-Smith, G.K.; Duan, M.; Zanker, D.J.; Loh, L.; Nguyen, T.H.O.; Koutsakos, M.; Nguyen, T.; Jiang, X.; Carrera, J.; Phan, T.K.; et al. Monocyte apoptotic bodies are vehicles for influenza A virus propagation. Commun. Biol. 2020, 3, 1–14. [Google Scholar] [CrossRef]
- Hirsova, P.; Ibrahim, S.H.; Krishnan, A.; Verma, V.K.; Bronk, S.F.; Werneburg, N.W.; Charlton, M.R.; Shah, V.H.; Malhi, H.; Gores, G.J. Lipid-Induced Signaling Causes Release of Inflammatory Extracellular Vesicles From Hepatocytes. Gastroenterology 2016, 150, 956–967. [Google Scholar] [CrossRef]
- Loyer, X.; Zlatanova, I.; Devue, C.; Yin, M.; Howangyin, K.Y.; Klaihmon, P.; Guerin, C.L.; Kheloufi, M.; Vilar, J.; Zannis, K.; et al. Intra-cardiac release of extracellular vesicles shapes inflammation following myocardial infarction short communication. Circ. Res. 2018, 123, 100–106. [Google Scholar] [CrossRef]
- Couch, Y.; Akbar, N.; Davis, S.; Fischer, R.; Dickens, A.M.; Neuhaus, A.; Burgess, A.I.; Rothwell, P.M.; Buchan, A.M. Inflammatory Stroke Extracellular Vesicles Induce Macrophage Activation. Stroke 2017, 48, 2292–2296. [Google Scholar] [CrossRef]
- Vats, R.; Brzoska, T.; Bennewitz, M.F.; Jimenez, M.A.; Pradhan-Sundd, T.; Tutuncuoglu, E.; Jonassaint, J.; Gutierrez, E.; Watkins, S.C.; Shiva, S.; et al. Platelet Extracellular Vesicles Drive Inflammasome–IL-1β–Dependent Lung Injury in Sickle Cell Disease. Am. J. Respir. Crit. Care Med. 2020, 201, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Kerr, N.A.; Vaccari, J.P.D.R.; Umland, O.; Bullock, R.; Conner, G.E.; Dietrich, W.D.; Keane, R.W. Human Lung Cell Pyroptosis Following Traumatic Brain Injury. Cells 2019, 8, 69. [Google Scholar] [CrossRef]
- Catalano, M.; O’Driscoll, L. Inhibiting extracellular vesicles formation and release: A review of EV inhibitors. J. Extracell. Vesicles 2019, 9, 1703244. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.; Samuel, M.; Kumar, S.; Mathivanan, S. Ticket to a bubble ride: Cargo sorting into exosomes and extracellular vesicles. Biochim.Biophys. Acta Proteins Proteom. 2019, 1867, 140203. [Google Scholar] [CrossRef] [PubMed]
- Tricarico, C.; Clancy, J.; D’Souza-Schorey, C. Biology and biogenesis of shed microvesicles. Small GTPases 2016, 8, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Antonyak, M.A.; Zhang, J.; Cerione, R.A. RhoA triggers a specific signaling pathway that generates transforming microvesicles in cancer cells. Oncogene 2012, 31, 4740–4749. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan-Chari, V.; Clancy, J.; Plou, C.; Romao, M.; Chavrier, P.; Raposo, G.; D’Souza-Schorey, C. ARF6-Regulated Shedding of Tumor Cell-Derived Plasma Membrane Microvesicles. Curr. Biol. 2009, 19, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Tixeira, R.; Phan, T.K.; Caruso, S.; Shi, B.; Atkin-Smith, G.K.; Nedeva, C.; Chow, J.D.Y.; Puthalakath, H.; Hulett, M.D.; Herold, M.J.; et al. ROCK1 but not LIMK1 or PAK2 is a key regulator of apoptotic membrane blebbing and cell disassembly. Cell Death Differ. 2019, 27, 102–116. [Google Scholar] [CrossRef]
- Atkin-Smith, G.K.; Tixeira, R.; Paone, S.; Mathivanan, S.; Collins, C.; Liem, M.; Goodall, K.J.; Ravichandran, K.S.; Hulett, M.D.; Poon, I.K.H. A novel mechanism of generating extracellular vesicles during apoptosis via a beads-on-a-string membrane structure. Nat. Commun. 2015, 6, 7439. [Google Scholar] [CrossRef]
- Atkin-Smith, G.K.; Miles, M.A.; Tixeira, R.; Lay, F.T.; Duan, M.; Hawkins, C.J.; Phan, T.K.; Paone, S.; Mathivanan, S.; Hulett, M.D.; et al. Plexin B2 is a Regulator of Monocyte Apoptotic Cell Disassembly. Cell Rep. 2019, 29, 1821–1831. [Google Scholar] [CrossRef]
- Poon, I.K.H.; Parkes, M.A.F.; Jiang, L.; Atkin-Smith, G.K.; Tixeira, R.; Gregory, C.D.; Ozkocak, D.C.; Rutter, S.F.; Caruso, S.; Santavanond, J.P.; et al. Moving beyond size and phosphatidylserine exposure: Evidence for a diversity of apoptotic cell-derived extracellular vesicles in vitro. J. Extracell. Vesicles 2019, 8, 1608786. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Paone, S.; Caruso, S.; Atkin-Smith, G.K.; Phan, T.K.; Hulett, M.D.; Poon, I.K.H. Determining the contents and cell origins of apoptotic bodies by flow cytometry. Sci. Rep. 2017, 7, 14444. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef]
- Mastoridis, S.; Bertolino, G.M.; Whitehouse, G.; Dazzi, F.; Sanchez-Fueyo, A.; Martinez-Llordella, M. Multiparametric Analysis of Circulating Exosomes and Other Small Extracellular Vesicles by Advanced Imaging Flow Cytometry. Front. Immunol. 2018, 9, 1583. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, L.; Toyofuku, M.; Hynen, A.L.; Kurosawa, M.; Pessi, G.; Petty, N.K.; Osvath, S.R.; Cárcamo-Oyarce, G.; Gloag, E.S.; Shimoni, R.; et al. Explosive cell lysis as a mechanism for the biogenesis of bacterial membrane vesicles and biofilms. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Mathieu, M.; Martín-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Mallat, Z.; Hugel, B.; Ohan, J.; Lesѐche, G.; Freyssinet, J.M.; Tedgui, A. Shed Membrane Microparticles With Procoagulant Potential in Human Atherosclerotic Plaques. Circulation 1999, 99, 348–353. [Google Scholar] [CrossRef]
- Combes, V.; Simon, A.C.; Grau, G.E.; Arnoux, D.; Camoin, L.; Sabatier, F.; Mutin, M.; Sanmarco, M.; Sampol, J.; Dignat-George, F. In vitro generation of endothelial microparticles and possible prothrombotic activity in patients with lupus anticoagulant. J. Clin. Investig. 1999, 104, 93–102. [Google Scholar] [CrossRef]
- Rautou, P.E.; Leroyer, A.; Ramkhelawon, B.; Devue, C.; Duflaut, D.; Vion, A.; Nalbone, G.; Castier, Y.; Leseche, G.; Lehoux, S.; et al. Microparticles From Human Atherosclerotic Plaques Promote Endothelial ICAM-1–Dependent Monocyte Adhesion and Transendothelial Migration. Circ. Res. 2011, 108, 335–343. [Google Scholar] [CrossRef]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; de Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH is a Key Factor for Exosome Traffic in Tumor Cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef]
- Torr, E.E.; Gardner, D.H.; Thomas, L.; Goodall, D.M.; Bielemeier, A.; Willetts, R.; Griffiths, H.R.; Marshall, L.J.; Devitt, A. Apoptotic cell-derived ICAM-3 promotes both macrophage chemoattraction to and tethering of apoptotic cells. Cell Death Differ. 2011, 19, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, A.; Mulya, A.; Lazić, M.; Radhakrishnan, D.; Berk, M.P.; Povero, D.; Gornicka, A.; Feldstein, A.E. Microparticles Release by Adipocytes Act as “Find-Me” Signals to Promote Macrophage Migration. PLoS ONE 2015, 10, e0123110. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, M.; Shah, N.; Zanetti, B.R.; Maugeri, M.; Silvestre, R.N.; Fatima, F.; Neder, L.; Valadi, H. Extracellular Vesicles and Matrix Remodeling Enzymes: The Emerging Roles in Extracellular Matrix Remodeling, Progression of Diseases and Tissue Repair. Cells 2018, 7, 167. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Morioka, S.; Maueröder, C.; Ravichandran, K.S. Living on the Edge: Efferocytosis at the Interface of Homeostasis and Pathology. Immunity 2019, 50, 1149–1162. [Google Scholar] [CrossRef]
- Silva, M.T. Secondary necrosis: The natural outcome of the complete apoptotic program. FEBS Lett. 2010, 584, 4491–4499. [Google Scholar] [CrossRef]
- Gaipl, U.; Munoz, L.; Grossmayer, G.; Lauber, K.; Franz, S.; Sarter, K.; Voll, R.E.; Winkler, T.; Kuhn, A.; Kalden, J.; et al. Clearance deficiency and systemic lupus erythematosus (SLE). J. Autoimmun. 2007, 28, 114–121. [Google Scholar] [CrossRef]
- Cui, S. Formation of Necrotic Cores in the Growth of Tumors: Analytic Results. Acta Math. Sci. 2006, 26, 781–796. [Google Scholar] [CrossRef]
- Hasegawa, H.; Watanabe, T.; Kato, S.; Toshima, T.; Yokoyama, M.; Aida, Y.; Nishiwaki, M.; Kadowaki, S.; Narumi, T.; Honda, Y.; et al. The role of macrophage transcription factor MafB in atherosclerotic plaque stability. Atherosclerosis 2016, 250, 133–143. [Google Scholar] [CrossRef]
- Jiang, L.; Tixeira, R.; Caruso, S.; Atkin-Smith, G.K.; Baxter, A.A.; Paone, S.; Hulett, M.D.; Poon, I.K.H. Monitoring the progression of cell death and the disassembly of dying cells by flow cytometry. Nat. Protoc. 2016, 11, 655–663. [Google Scholar] [CrossRef]
- Jansen, F.; Yang, X.; Baumann, K.; Przybilla, D.; Schmitz, T.; Flender, A.; Paul, K.; Alhusseiny, A.; Nickenig, G.; Werner, N. Endothelial microparticles reduce ICAM-1 expression in a microRNA-222-dependent mechanism. J. Cell. Mol. Med. 2015, 19, 2202–2214. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kasagi, S.; Chia, C.; Zhang, D.; Tu, E.; Wu, R.; Zanvit, P.; Goldberg, N.; Jin, W.; Chen, W. Extracellular Vesicles from Apoptotic Cells Promote TGFβ Production in Macrophages and Suppress Experimental Colitis. Sci. Rep. 2019, 9, 5875. [Google Scholar] [CrossRef] [PubMed]
- Østergaard, O.; Nielsen, C.T.; Iversen, L.V.; Tanassi, J.T.; Knudsen, S.; Jacobsen, S.; Heegaard, N.H.H. Unique protein signature of circulating microparticles in systemic lupus erythematosus. Arthritis Rheum. 2013, 65, 2680–2690. [Google Scholar] [CrossRef]
- Edinger, A.L.; Thompson, C.B. Death by design: Apoptosis, necrosis and autophagy. Curr. Opin. Cell Biol. 2004, 16, 663–669. [Google Scholar] [CrossRef]
- Sachet, M.; Liang, Y.Y.; Oehler, R. The immune response to secondary necrotic cells. Apoptosis 2017, 22, 1189–1204. [Google Scholar] [CrossRef] [PubMed]
- Samson, A.L.; Zhang, Y.; Geoghegan, N.D.; Gavin, X.J.; Davies, K.A.; Mlodzianoski, M.J.; Whitehead, L.; Frank, D.; Garnish, S.E.; FitzGibbon, C.; et al. MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Wang, X.; Zheng, Y.; Jiang, J.; Hu, J. What role does pyroptosis play in microbial infection? J. Cell. Physiol. 2018, 234, 7885–7892. [Google Scholar] [CrossRef] [PubMed]
- Barrington, J.; LeMarchand, E.; Allan, S.M. A brain in flame; do inflammasomes and pyroptosis influence stroke pathology? Brain Pathol. 2017, 27, 205–212. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef]
- Doitsh, G.; Greene, W.C. Dissecting How CD4 T Cells are Lost During HIV Infection. Cell Host Microbe 2016, 19, 280–291. [Google Scholar] [CrossRef]
- Giordano, A.; Murano, I.; Mondini, E.; Perugini, J.; Smorlesi, A.; Severi, I.; Barazzoni, R.; Scherer, P.E.; Cinti, S. Obese adipocytes show ultrastructural features of stressed cells and die of pyroptosis. J. Lipid Res. 2013, 54, 2423–2436. [Google Scholar] [CrossRef] [PubMed]
- Cypryk, W.; Nyman, T.A.; Matikainen, S. From Inflammasome to Exosome—Does Extracellular Vesicle Secretion Constitute an Inflammasome-Dependent Immune Response? Front. Immunol. 2018, 9, 2188. [Google Scholar] [CrossRef] [PubMed]
- González-Juarbe, N.; Gilley, R.P.; Hinojosa, C.A.; Bradley, K.M.; Kamei, A.; Gao, G.; Dube, P.H.; Bergman, M.A.; Orihuela, C.J. Pore-Forming Toxins Induce Macrophage Necroptosis during Acute Bacterial Pneumonia. PLOS Pathog. 2015, 11, e1005337. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Li, H.; Yang, M.; Ren, J.; Huang, Z.; Han, F.; Huang, J.; Ma, J.; Zhang, D.W.; Zhang, Z.; et al. A Role of RIP3-Mediated Macrophage Necrosis in Atherosclerosis Development. Cell Rep. 2013, 3, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Luedde, T. Apoptosis and necroptosis in the liver: A matter of life and death. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 738–752. [Google Scholar] [CrossRef]
- Frank, D.; Vince, J.E. Pyroptosis versus necroptosis: Similarities, differences, and crosstalk. Cell Death Differ. 2018, 26, 99–114. [Google Scholar] [CrossRef]
- Lane, R.E.; Korbie, D.; Hill, M.M.; Trau, M. Extracellular vesicles as circulating cancer biomarkers: Opportunities and challenges. Clin. Transl. Med. 2018, 7, 14. [Google Scholar] [CrossRef]
- Ludwig, N.; Whiteside, T.L.; Reichert, T.E. Challenges in Exosome Isolation and Analysis in Health and Disease. Int. J. Mol. Sci. 2019, 20, 4684. [Google Scholar] [CrossRef]
- Bewicke-Copley, F.; Mulcahy, L.A.; Jacobs, L.A.; Samuel, P.; Akbar, N.; Pink, R.; Carter, D.R.F. Extracellular vesicles released following heat stress induce bystander effect in unstressed populations. J. Extracell. Vesicles 2017, 6, 1340746. [Google Scholar] [CrossRef]
- Benedikter, B.J.; Bouwman, F.G.; Heinzmann, A.C.A.; Vajen, T.; Mariman, E.C.M.; Wouters, E.F.M.; Savelkoul, P.H.M.; Koenen, R.R.; Rohde, G.G.U.; Van Oerle, R.; et al. Proteomic analysis reveals procoagulant properties of cigarette smoke-induced extracellular vesicles. J. Extracell. Vesicles 2019, 8, 1585163. [Google Scholar] [CrossRef]
- Haanen, C.; Vermes, I. Apoptosis: Programmed cell death in fetal development. Eur. J. Obstet. Gynecol. Reprod. Biol. 1996, 64, 129–133. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed]
- Morán, G.A.G.; Parra-Medina, R.; Cardona, A.G.; Quintero-Ronderos, P.; Rodríguez, É.G. Cytokines, chemokines and growth factors. In Autoimmunity: From Bench to Bedside; El Rosario University Press: Bogota, Colombia, 2013. [Google Scholar]
- Delgado-Rizo, V.; Martínez-Guzmán, M.A.; Iñiguez-Gutierrez, L.; García-Orozco, A.; Alvarado-Navarro, A.; Fafutis-Morris, M. Neutrophil Extracellular Traps and its Implications in Inflammation: An Overview. Front. Immunol. 2017, 8, 81. [Google Scholar] [CrossRef]
- Riegman, M.; Sagie, L.; Galed, C.; Levin, T.; Steinberg, N.; Dixon, S.J.; Wiesner, U.; Bradbury, M.S.; Niethammer, P.; Zaritsky, A.; et al. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat. Cell Biol. 2020, 22, 1–7. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| EV Type | Apoptosis | Secondary Necrosis | Primary Necrosis | Pyroptosis | Necroptosis | Reference |
|---|---|---|---|---|---|---|
| Exosomes | - | - | + | + | + | [32,33,34] |
| Exosome-like EVs | + | - | + | - | + | [35,36,37,38,39] |
| Microvesicles | + | - | + | - | - | [40,41,42,43,44,45,46] |
| ApoBDs | + | - | - | - | - | [47,48] |
| Small EVs * | - | + | + | + | - | [12,30,31,49,50,51,52,53] |
| Medium EVs * | - | + | + | + | - | [30] |
| Large EVs * | - | - | + | + | - | [30] |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baxter, A.A. Stoking the Fire: How Dying Cells Propagate Inflammatory Signalling through Extracellular Vesicle Trafficking. Int. J. Mol. Sci. 2020, 21, 7256. https://doi.org/10.3390/ijms21197256
Baxter AA. Stoking the Fire: How Dying Cells Propagate Inflammatory Signalling through Extracellular Vesicle Trafficking. International Journal of Molecular Sciences. 2020; 21(19):7256. https://doi.org/10.3390/ijms21197256
Chicago/Turabian StyleBaxter, Amy A. 2020. "Stoking the Fire: How Dying Cells Propagate Inflammatory Signalling through Extracellular Vesicle Trafficking" International Journal of Molecular Sciences 21, no. 19: 7256. https://doi.org/10.3390/ijms21197256
APA StyleBaxter, A. A. (2020). Stoking the Fire: How Dying Cells Propagate Inflammatory Signalling through Extracellular Vesicle Trafficking. International Journal of Molecular Sciences, 21(19), 7256. https://doi.org/10.3390/ijms21197256