Caffeoyl-Prolyl-Histidine Amide Inhibits Fyn and Alleviates Atopic Dermatitis-Like Phenotypes via Suppression of NF-κB Activation

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
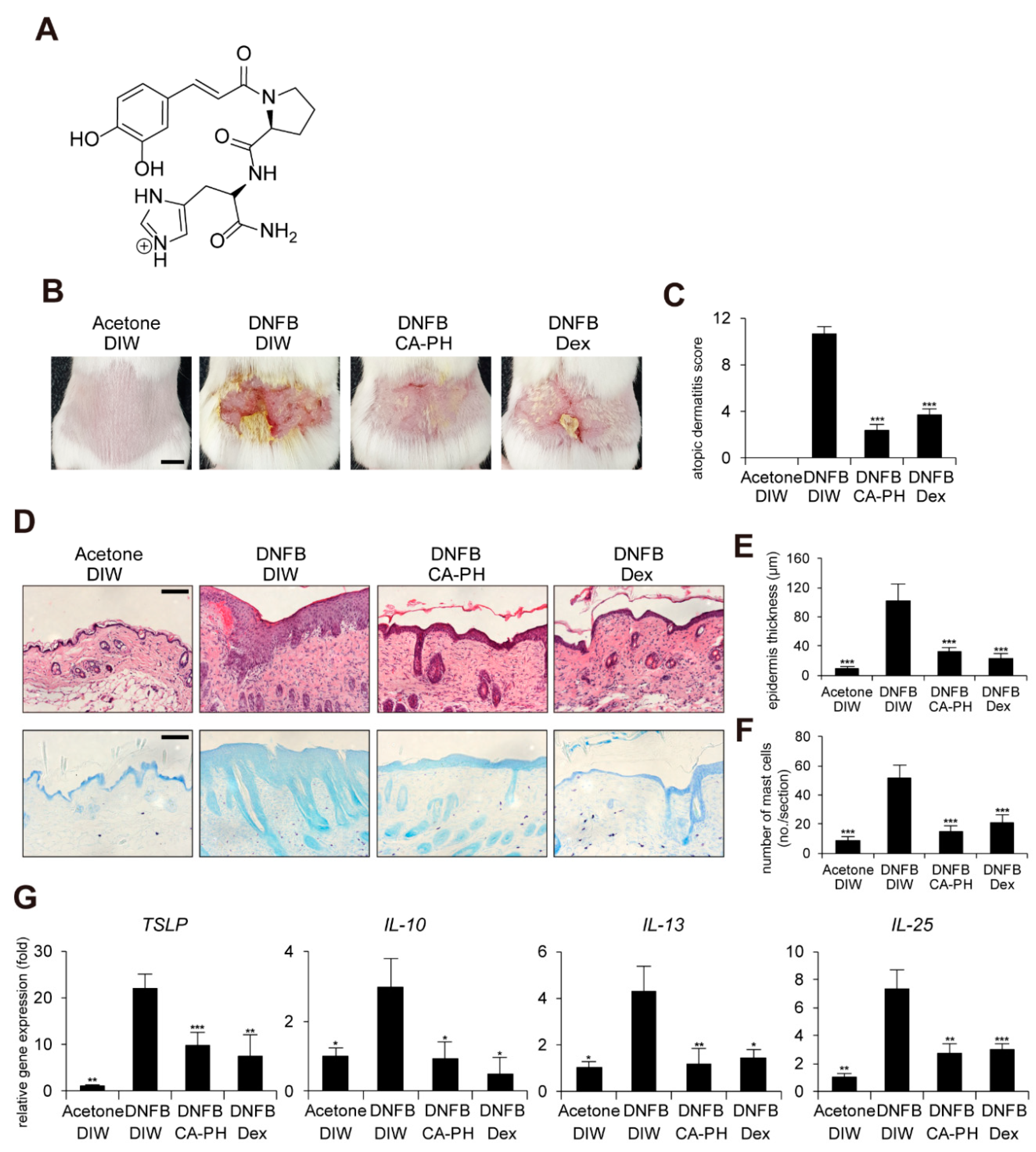
2.1. CA-PH Alleviates AD-Like Phenotypes Induced by DNFB Treatment
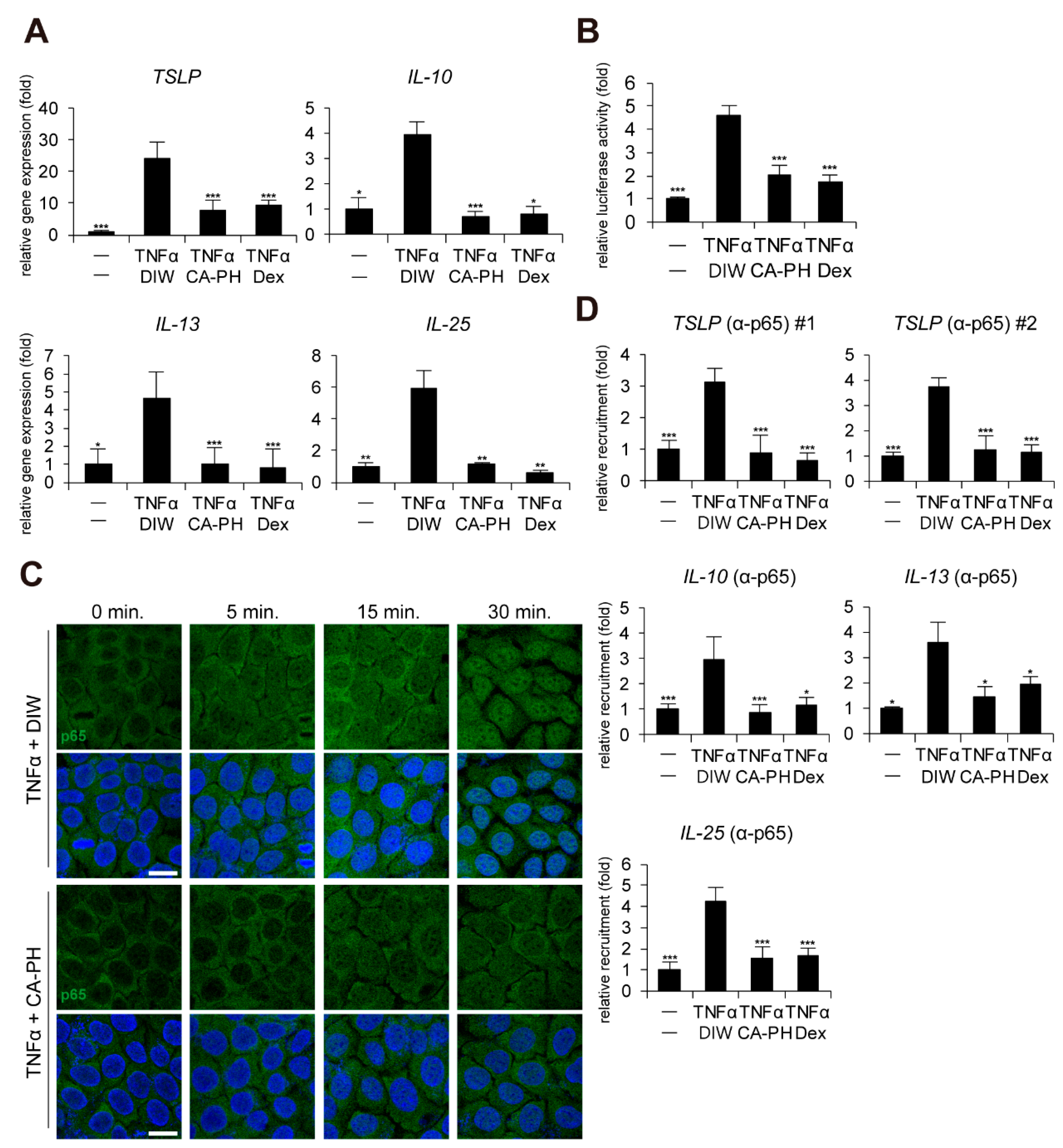
2.2. CA-PH Suppresses Nuclear Translocation of NF-κB
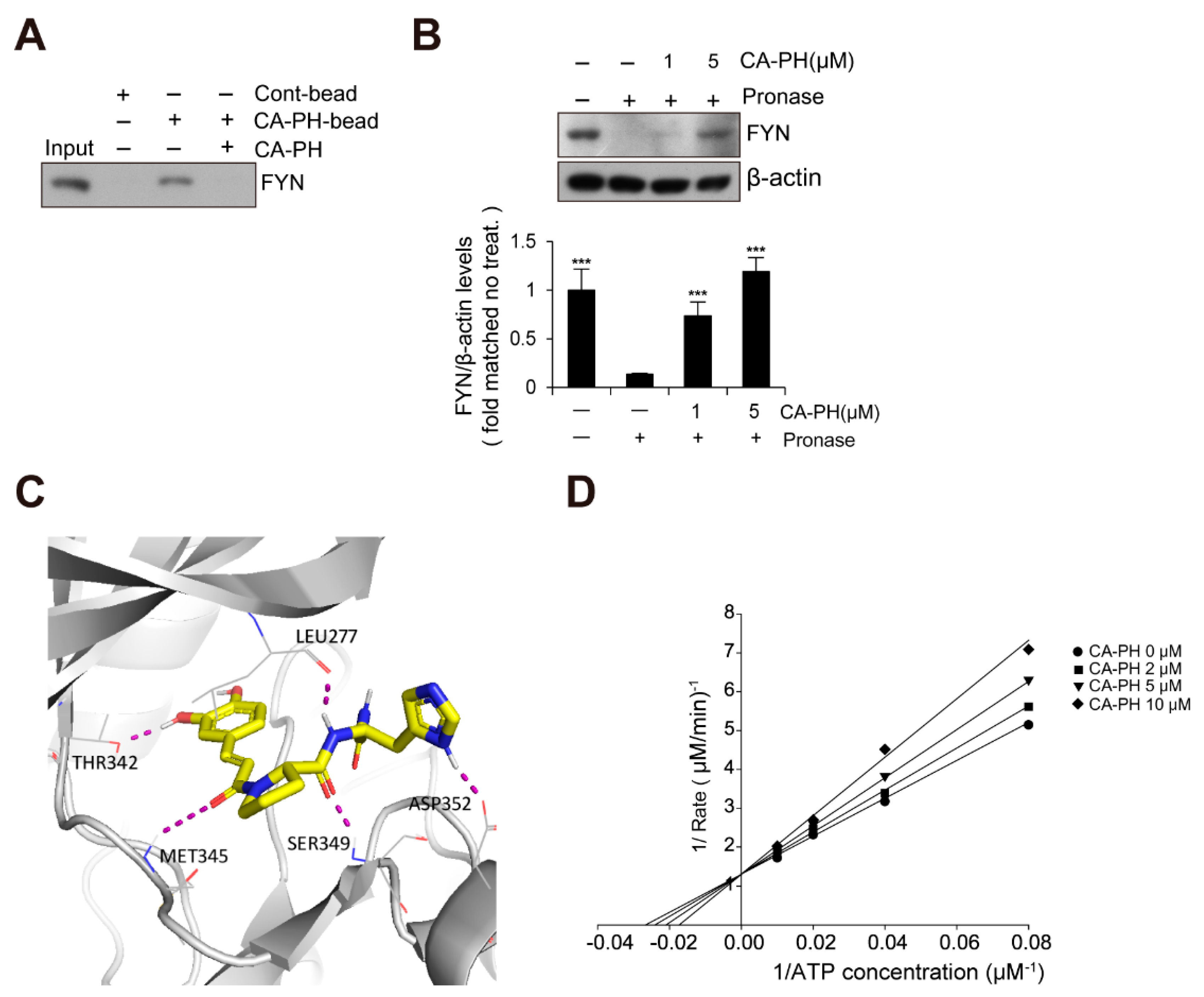
2.3. CA-PH Binds to and Inhibits Fyn
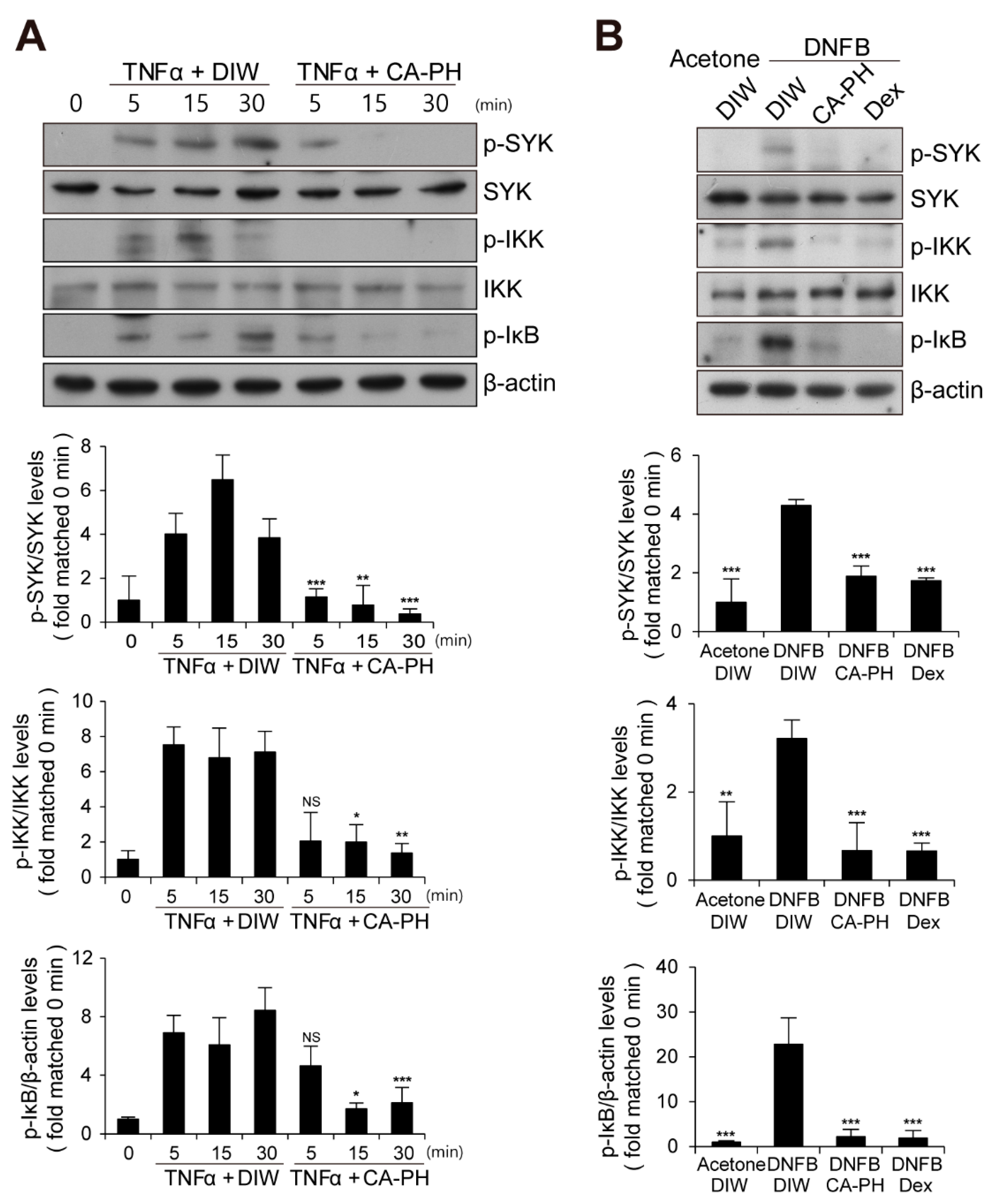
2.4. CA-PH Suppresses NF-κB Signaling Pathway by Inhibition of Fyn Activation
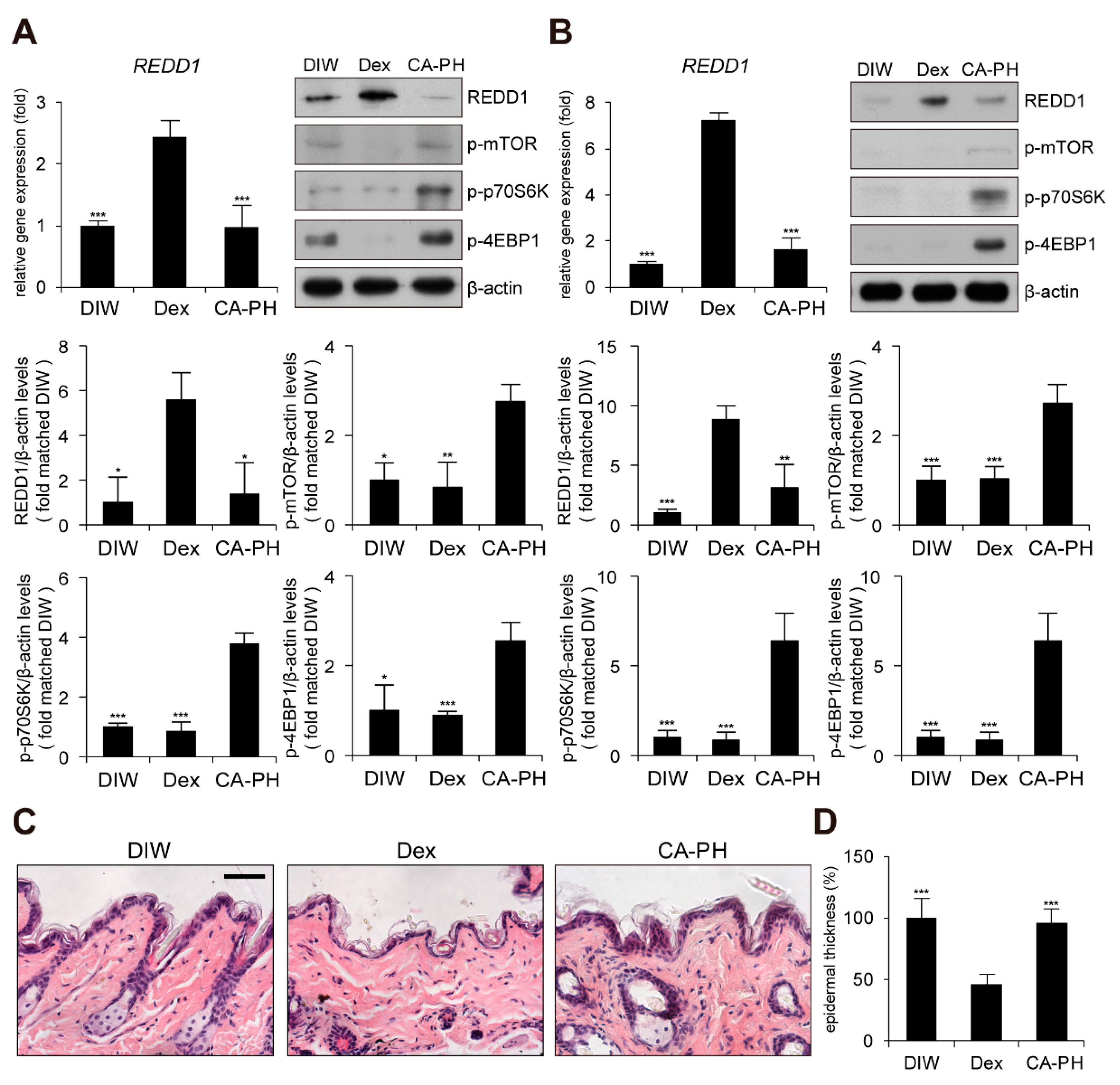
2.5. CA-PH Does not Induce Skin Atrophy
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Mouse AD Model
4.3. Mouse Skin Atrophy Model
4.4. Skin Lesion and AD-Like Phenotype Score
4.5. Quantitative PCR
4.6. Immunocytochemistry and Histology
4.7. Immunoprecipitation and Western Blot Analysis
4.8. Promoter Reporter Assay
4.9. Chromatin Immunoprecipitation (ChIP)
4.10. Preparation of CA-PH Beads
4.11. Pull-Down Assay
4.12. Drug Affinity Responsive Target Stability (DARTS) Assay
4.13. Molecular Docking Prediction
4.14. Kinase Assay and Enzyme Kinetic Study
4.15. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CA-PH | caffeoyl-prolyl-histidine amide |
| AD | atopic dermatitis |
| DNFB | 1-fluoro-2,4-dinitrobenzene |
| NF-κB | nuclear factor kappa-light-chain enhancer of activated B cells |
| SYK | spleen-associated tyrosine kinase |
| JNK | c-JUN N-terminal kinase |
| REDD1 | regulated in development and DNA damage response 1 |
| DARTS | drug affinity responsive target stability |
| ChIP | chromatin immunoprecipitation |
References
- Janbaz, K.; Saeed, S.; Gilani, A. Studies on the protective effects of caffeic acid and quercetin on chemical-induced hepatotoxicity in rodents. Phytomedicine 2004, 11, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Yasui, H.; Sakurai, H. Suppressive Effect of Caffeic Acid and its Derivatives on the Generation of UVA-induced Reactive Oxygen Species in the Skin of Hairless Mice and Pharmacokinetic Analysis on Organ Distribution of Caffeic Acid in ddY Mice. Photochem. Photobiol. 2006, 82, 1668. [Google Scholar] [CrossRef]
- Genaro-Mattos, T.C.; Maurício, Â.Q.; Rettori, D.; Alonso, A.; Hermes-Lima, M. Antioxidant Activity of Caffeic Acid against Iron-Induced Free Radical Generation—A Chemical Approach. PLoS ONE 2015, 10, e0129963. [Google Scholar] [CrossRef]
- Lima, V.N.; Oliveira-Tintino, C.D.; Santos, E.S.; Morais, L.P.; Tintino, S.R.; Freitas, T.S.; Geraldo, Y.S.; Pereira, R.L.; Cruz, R.P.; Menezes, I.R.; et al. Antimicrobial and enhancement of the antibiotic activity by phenolic compounds: Gallic acid, caffeic acid and pyrogallol. Microb. Pathog. 2016, 99, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Hirose, M.; Takesada, Y.; Tanaka, H.; Tamano, S.; Kato, T.; Shirai, T. Carcinogenicity of antioxidants BHA, caffeic acid, sesamol, 4-methoxyphenol and catechol at low doses, either alone or in combination, and modulation of their effects in a rat medium-term multi-organ carcinogenesis model. Carcinogenesis 1998, 19, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Chao, P.-C.; Hsu, C.-C.; Yin, M.-C. Anti-inflammatory and anti-coagulatory activities of caffeic acid and ellagic acid in cardiac tissue of diabetic mice. Nutr. Metab. 2009, 6, 33. [Google Scholar] [CrossRef]
- Jeon, Y.-D.; Kee, J.-Y.; Kim, D.-S.; Han, Y.-H.; Kim, S.-H.; Kim, S.-J.; Um, J.-Y.; Hong, S.-H. Effects of Ixeris dentata water extract and caffeic acid on allergic inflammation in vivo and in vitro. BMC Complement. Altern. Med. 2015, 15, 196. [Google Scholar] [CrossRef]
- Seo, H.-S.; Kwak, S.-Y.; Lee, J.-Y. Antioxidative activities of histidine containing caffeic acid-dipeptides. Bioorganic Med. Chem. Lett. 2010, 20, 4266–4272. [Google Scholar] [CrossRef]
- Kwak, S.-Y.; Lee, H.J.; Yang, J.-K.; Lee, E.J.; Seo, M.; Lee, J.-Y. Antioxidant activity of caffeoyl-prolyl-histidine amide and its effects on PDGF-induced proliferation of vascular smooth muscle cells. Amino Acids 2014, 46, 2777–2785. [Google Scholar] [CrossRef]
- Jang, S.; Ohn, J.; Kim, J.W.; Kang, S.M.; Jeon, D.; Heo, C.Y.; Lee, Y.-S.; Kwon, O.; Kim, K.H. Caffeoyl–Pro–His amide relieve DNCB-Induced Atopic Dermatitis-Like phenotypes in BALB/c mice. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Parsons, S.J.; Parsons, J.T. Src family kinases, key regulators of signal transduction. Oncogene 2004, 23, 7906–7909. [Google Scholar] [CrossRef] [PubMed]
- Okada, M. Regulation of the Src Family Kinases by Csk. Int. J. Boil. Sci. 2012, 8, 1385–1397. [Google Scholar] [CrossRef]
- Comba, A.; Dunn, P.J.; Argento, A.E.; Kadiyala, P.; Ventosa, M.; Patel, P.; Zamler, D.B.; Nunez, F.J.; Zhao, L.; Castro, M.G.; et al. Fyn tyrosine kinase, a downstream target of receptor tyrosine kinases, modulates antiglioma immune responses. Neuro-Oncol. 2020, 22, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Groves, T.; Smiley, P.; Cooke, M.P.; Forbush, K.; Perlmutter, R.M.; Guidos, C.J. Fyn Can Partially Substitute for Lck in T Lymphocyte Development. Immunity 1996, 5, 417–428. [Google Scholar] [CrossRef]
- White, R.; Krämer-Albers, E.-M. Axon-glia interaction and membrane traffic in myelin formation. Front. Cell. Neurosci. 2014, 7, 284. [Google Scholar] [CrossRef] [PubMed]
- Resh, M.D. Fyn, a Src family tyrosine kinase. Int. J. Biochem. Cell Boil. 1998, 30, 1159–1162. [Google Scholar] [CrossRef]
- Tang, X.; Feng, Y.; Ye, K. Src-family tyrosine kinase fyn phosphorylates phosphatidylinositol 3-kinase enhancer-activating Akt, preventing its apoptotic cleavage and promoting cell survival. Cell Death Differ. 2006, 14, 368–377. [Google Scholar] [CrossRef]
- Davidson, D.; Fournel, M.; Veillette, A. Oncogenic activation of p59fyn tyrosine protein kinase by mutation of its carboxyl-terminal site of tyrosine phosphorylation, tyrosine 528. J. Boil. Chem. 1994, 269, 10956–10963. [Google Scholar]
- Lee, G.; Thangavel, R.; Sharma, V.M.; Litersky, J.M.; Bhaskar, K.; Fang, S.M.; Do, L.H.; Andreadis, A.; Van Hoesen, G.; Ksiezak-Reding, H. Phosphorylation of Tau by Fyn: Implications for Alzheimer’s Disease. J. Neurosci. 2004, 24, 2304–2312. [Google Scholar] [CrossRef]
- Yamada, E.; Okada, S.; Bastie, C.C.; Vatish, M.; Nakajima, Y.; Shibusawa, R.; Ozawa, A.; Pessin, J.E.; Yamada, M. Fyn phosphorylates AMPK to inhibit AMPK activity and AMP-dependent activation of autophagy. Oncotarget 2016, 7, 74612–74629. [Google Scholar] [CrossRef]
- Calautti, E.; Grossi, M.; Mammucari, C.; Aoyama, Y.; Pirro, M.; Ono, Y.; Li, J.; Dotto, G.P. Fyn tyrosine kinase is a downstream mediator of Rho/PRK2 function in keratinocyte cell–cell adhesion. J. Cell Boil. 2002, 156, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Cabodi, S.; Calautti, E.; Talora, C.; Kuroki, T.; Stein, P.L.; Dotto, G. A PKC-η/Fyn-Dependent Pathway Leading to Keratinocyte Growth Arrest and Differentiation. Mol. Cell 2000, 6, 1121–1129. [Google Scholar] [CrossRef]
- Tu, C.-L.; Chang, W.; Bikle, D.D. The Calcium-Sensing Receptor-Dependent Regulation of Cell–Cell Adhesion and Keratinocyte Differentiation Requires Rho and Filamin, A. J. Investig. Dermatol. 2011, 131, 1119–1128. [Google Scholar] [CrossRef]
- Calautti, E.; Cabodi, S.; Stein, P.L.; Hatzfeld, M.; Kedersha, N.; Dotto, G.P. Tyrosine Phosphorylation and Src Family Kinases Control Keratinocyte Cell–Cell Adhesion. J. Cell Boil. 1998, 141, 1449–1465. [Google Scholar] [CrossRef] [PubMed]
- Fenton, S.E.; Denning, M.F. FYNagling divergent adhesive functions for Fyn in keratinocytes. Exp. Dermatol. 2014, 24, 81–85. [Google Scholar] [CrossRef]
- Ayli, E.E.; Li, W.; Brown, T.T.; Witkiewicz, A.; Elenitsas, R.; Seykora, J.T. Activation of Src-family tyrosine kinases in hyperproliferative epidermal disorders. J. Cutan. Pathol. 2008, 35, 273–277. [Google Scholar] [CrossRef]
- Zhao, L.; Li, W.; Marshall, C.; Griffin, T.; Hanson, M.; Hick, R.; Dentchev, T.; Williams, E.; Werth, A.; Miller, C.; et al. Srcasm Inhibits Fyn-Induced Cutaneous Carcinogenesis with Modulation of Notch1 and p53. Cancer Res. 2009, 69, 9439–9447. [Google Scholar] [CrossRef]
- Hwang, M.K.; Kang, N.J.; Heo, Y.-S.; Lee, K.W.; Lee, H.J. Fyn kinase is a direct molecular target of delphinidin for the inhibition of cyclooxygenase-2 expression induced by tumor necrosis factor-α. Biochem. Pharmacol. 2009, 77, 1213–1222. [Google Scholar] [CrossRef]
- Lim, T.-G.; Kwon, J.Y.; Kim, J.; Song, N.R.; Lee, K.M.; Heo, Y.-S.; Lee, H.J.; Lee, K. Cyanidin-3-glucoside suppresses B[a]PDE-induced cyclooxygenase-2 expression by directly inhibiting Fyn kinase activity. Biochem. Pharmacol. 2011, 82, 167–174. [Google Scholar] [CrossRef]
- Park, Y.H.; Kim, K.; Kim, H.S.; Lee, D.; Lee, M.B.; Min, K.Y.; Jo, M.G.; Lee, J.E.; Kim, Y.M.; Choi, W.S. WZ3146 inhibits mast cell Lyn and Fyn to reduce IgE-mediated allergic responses in vitro and in vivo. Toxicol. Appl. Pharmacol. 2019, 383, 114763. [Google Scholar] [CrossRef]
- Lee, S.-H.; Shin, H.J.; Kim, N.-Y.; Shim, D.-W.; Kim, T.-J.; Ye, S.-K.; Won, H.-S.; Koppula, S.; Kang, T.-B.; Lee, K.-H. Streptochlorin Suppresses Allergic Dermatitis and Mast Cell Activation via Regulation of Lyn/Fyn and Syk Signaling Pathways in Cellular and Mouse Models. PLoS ONE 2013, 8, e74194. [Google Scholar] [CrossRef] [PubMed]
- Albanesi, C. Keratinocytes in allergic skin diseases. Curr. Opin. Allergy Clin. Immunol. 2010, 10, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Na, J.; Lee, K.; Na, W.; Shin, J.-Y.; Lee, M.-J.; Yune, T.Y.; Lee, H.K.; Jung, H.-S.; Kim, W.S.; Ju, B.-G. Histone H3K27 Demethylase JMJD3 in Cooperation with NF-κB Regulates Keratinocyte Wound Healing. J. Investig. Dermatol. 2016, 136, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Shin, J.Y.; Kim, M.-J.; Na, J.; Ju, B.-G. Activation of Aryl Hydrocarbon Receptor Negatively Regulates Thymic Stromal Lymphopoietin Gene Expression via Protein Kinase Cδ-p300–NF-κB Pathway in Keratinocytes under Inflammatory Conditions. J. Investig. Dermatol. 2019, 139, 1098–1109. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Han, S.-B.; Ham, H.J.; Park, J.H.; Lee, J.S.; Hwang, D.Y.; Jung, Y.S.; Yoon, D.-Y.; Hong, J.T. IL-32γ suppressed atopic dermatitis through inhibition of miR-205 expression via inactivation of nuclear factor-kappa B. J. Allergy Clin. Immunol. 2020, 146, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Kang, N.J.; Lee, K.W.; Shin, B.J.; Jung, S.K.; Hwang, M.K.; Bode, A.M.; Heo, Y.-S.; Lee, H.J.; Dong, Z. Caffeic acid, a phenolic phytochemical in coffee, directly inhibits Fyn kinase activity and UVB-induced COX-2 expression. Carcinogenesis 2008, 30, 321–330. [Google Scholar] [CrossRef]
- Tintori, C.; La Sala, G.; Vignaroli, G.; Botta, L.; Fallacara, A.L.; Falchi, F.; Radi, M.; Zamperini, C.; Dreassi, E.; Iacono, L.D.; et al. Studies on the ATP Binding Site of Fyn Kinase for the Identification of New Inhibitors and Their Evaluation as Potential Agents against Tauopathies and Tumors. J. Med. Chem. 2015, 58, 4590–4609. [Google Scholar] [CrossRef]
- Sultan, M.M.; Kiss, G.; Pande, V.S. Towards simple kinetic models of functional dynamics for a kinase subfamily. Nat. Chem. 2018, 10, 903–909. [Google Scholar] [CrossRef]
- Cotter, D.G.; Schairer, D.; Eichenfield, L.F. Emerging therapies for atopic dermatitis: JAK inhibitors. J. Am. Acad. Dermatol. 2018, 78, S53–S62. [Google Scholar] [CrossRef]
- Shreberk-Hassidim, R.; Ramot, Y.; Zlotogorski, A. Janus kinase inhibitors in dermatology: A systematic review. J. Am. Acad. Dermatol. 2017, 76, 745–753. [Google Scholar] [CrossRef]
- Pavel, A.B.; Song, T.; Kim, H.J.; Del Duca, E.; Krueger, J.G.; Dubin, C.; Peng, X.; Xu, H.; Zhang, N.; Estrada, Y.D.; et al. Oral Janus kinase/SYK inhibition (ASN002) suppresses inflammation and improves epidermal barrier markers in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2019, 144, 1011–1024. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, R.; Maari, C.; Forman, S.; Bhatia, N.; Lee, M.; Fowler, J.; Tyring, S.; Pariser, D.; Sofen, H.; Dhawan, S.; et al. The oral Janus kinase/spleen tyrosine kinase inhibitor ASN 002 demonstrates efficacy and improves associated systemic inflammation in patients with moderate-to-severe atopic dermatitis: Results from a randomized double-blind placebo-controlled study. Br. J. Dermatol. 2019, 181, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Larson, M.; Sherman, M.A.; Amar, F.; Nuvolone, M.; Schneider, J.A.; Bennett, D.A.; Aguzzi, A.; Lesne, S. The complex PrP(c)-Fyn couples human oligomeric Aβ with pathological tau changes in Alzheimer’s disease. J. Neurosci. 2012, 32, 16857–716871a. [Google Scholar] [CrossRef] [PubMed]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef]
- Panicker, N.; Saminathan, H.; Jin, H.; Neal, M.; Harischandra, D.; Gordon, R.; Kanthasamy, K.; Lawana, V.; Sarkar, S.; Luo, J.; et al. Fyn Kinase Regulates Microglial Neuroinflammatory Responses in Cell Culture and Animal Models of Parkinson’s Disease. J. Neurosci. 2015, 35, 10058–10077. [Google Scholar] [CrossRef]
- Lu, Y.; Piao, D.; Zhang, H.; Li, X.; Chao, G.H.; Park, S.J.; Chang, Y.-C.; Kim, C.-H.; Murakami, M.; Jung, S.-H.; et al. Saucerneol F inhibits tumor necrosis factor-α and IL-6 production by suppressing Fyn-mediated pathways in FcεRI-mediated mast cells. Food Chem. Toxicol. 2013, 59, 696–702. [Google Scholar] [CrossRef]
- Lu, Y.; Hwang, S.-L.; Son, J.K.; Chang, H.W. Manassantin B isolated from Saururus chinensis inhibits cyclooxygenase-2-dependent prostaglandin D2 generation by blocking Fyn-mediated nuclear factor-kappaB and mitogen activated protein kinase pathways in bone marrow derived-mast cells. Boil. Pharm. Bull. 2013, 36, 1370–1374. [Google Scholar] [CrossRef][Green Version]
- Zhang, J.; Billingsley, M.L.; Kincaid, R.L.; Siraganian, R.P. Phosphorylation of Syk Activation Loop Tyrosines Is Essential for Syk Function. J. Boil. Chem. 2000, 275, 35442–35447. [Google Scholar] [CrossRef]
- Wu, N.-L.; Huang, D.-Y.; Tsou, H.-N.; Lin, Y.-C.; Lin, W.-W. Syk Mediates IL−17-Induced CCL20 Expression by Targeting Act1-Dependent K63-Linked Ubiquitination of TRAF6. J. Investig. Dermatol. 2015, 135, 490–498. [Google Scholar] [CrossRef]
- Bukong, T.N.; Iracheta-Vellve, A.; Saha, B.; Ambade, A.; Satishchandran, A.; Gyongyosi, B.; Lowe, P.; Catalano, N.; Kodys, K.; Szabo, G. Inhibition of spleen tyrosine kinase activation ameliorates inflammation, cell death, and steatosis in alcoholic liver disease. Hepatology 2016, 64, 1057–1071. [Google Scholar] [CrossRef]
- Woodbury, R.; Kligman, A.M. The hairless mouse model for assaying the atrophogenicity of topical corticosteroids. Acta Derm. Venereol. 1992, 72, 403–406. [Google Scholar] [PubMed]
- Schoepe, S.; Schäcke, H.; May, E.; Asadullah, K. Glucocorticoid therapy-induced skin atrophy. Exp. Dermatol. 2006, 15, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, C.; Averbeck, M.; Diedenhofen, N.; Willenberg, A.; Anderegg, U.; Sleeman, J.P.; Simon, J.C. Dermal Hyaluronan Is Rapidly Reduced by Topical Treatment with Glucocorticoids. J. Investig. Dermatol. 2010, 130, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Barnes, L.; Kaya, G.; Rollason, V. Topical Corticosteroid-Induced Skin Atrophy: A Comprehensive Review. Drug Saf. 2015, 38, 493–509. [Google Scholar] [CrossRef]
- Baida, G.; Bhalla, P.; Kirsanov, K.I.; Lesovaya, E.A.; Yakubovskaya, M.; Yuen, K.; Guo, S.; Lavker, R.M.; Readhead, B.; Dudley, J.T.; et al. REDD 1 functions at the crossroads between the therapeutic and adverse effects of topical glucocorticoids. EMBO Mol. Med. 2014, 7, 42–58. [Google Scholar] [CrossRef]
- Gamaro, G.D.; Suyenaga, E.; Borsoi, M.; Lermen, J.; Pereira, P.; Ardenghi, P. Effect of Rosmarinic and Caffeic Acids on Inflammatory and Nociception Process in Rats. ISRN Pharmacol. 2011, 2011, 1–6. [Google Scholar] [CrossRef]
- Gülçin, I. Antioxidant activity of caffeic acid (3,4-dihydroxycinnamic acid). Toxicology 2006, 217, 213–220. [Google Scholar] [CrossRef]
- Romano, V.; De Beer, T.A.P.; Schwede, T. A computational protocol to evaluate the effects of protein mutants in the kinase gatekeeper position on the binding of ATP substrate analogues. BMC Res. Notes 2017, 10, 104. [Google Scholar] [CrossRef]
- Azam, M.; Seeliger, M.; Gray, N.S.; Kuriyan, J.; Daley, G.Q. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat. Struct. Mol. Boil. 2008, 15, 1109–1118. [Google Scholar] [CrossRef]
- Alaimo, P.J.; Knight, Z.; Shokat, K.M. Targeting the gatekeeper residue in phosphoinositide 3-kinases. Bioorganic Med. Chem. 2005, 13, 2825–2836. [Google Scholar] [CrossRef]
- Tou, W.I.; Chen, Y.-C. In Silico Investigation of Potential Src Kinase Ligands from Traditional Chinese Medicine. PLoS ONE 2012, 7, e33728. [Google Scholar] [CrossRef]
- Renert-Yuval, Y.; Guttman-Yassky, E. New treatments for atopic dermatitis targeting beyond IL-4/IL-13 cytokines. Ann. Allergy Asthma Immunol. 2019, 124, 28–35. [Google Scholar] [CrossRef]
- Lee, D.E.; Clark, A.K.; Tran, K.A.; Shi, V.Y. New and emerging targeted systemic therapies: A new era for atopic dermatitis. J. Dermatol. Treat. 2017, 29, 364–374. [Google Scholar] [CrossRef]
- Szilveszter, K.P.; Németh, T.; Mócsai, A. Tyrosine Kinases in Autoimmune and Inflammatory Skin Diseases. Front. Immunol. 2019, 10, 1862. [Google Scholar] [CrossRef]
- Lee, D.; Park, Y.H.; Lee, J.E.; Kim, H.S.; Min, K.Y.; Jo, M.G.; Kim, H.S.; Choi, W.S.; Kim, Y.M. Dasatinib Inhibits Lyn and Fyn Src-Family Kinases in Mast Cells to Suppress Type I Hypersensitivity in Mice. Biomol. Ther. 2020. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, A.-R.; Kim, H.S.; Kim, H.W.; Park, Y.H.; You, J.S.; Park, Y.-M.; Her, E.; Kim, H.S.; Kim, Y.M.; et al. Rhamnus davurica leaf extract inhibits Fyn activation by antigen in mast cells for anti-allergic activity. BMC Complement. Altern. Med. 2015, 15, 80. [Google Scholar] [CrossRef]
- Ko, H.M.; Lee, S.H.; Kim, K.C.; Joo, S.H.; Choi, W.S.; Shin, C.Y. The Role of TLR4 and Fyn Interaction on Lipopolysaccharide-Stimulated PAI-1 Expression in Astrocytes. Mol. Neurobiol. 2014, 52, 8–25. [Google Scholar] [CrossRef]
- Lee, C.; Low, C.Y.B.; Wong, S.Y.; Lai, M.K.P.; Tan, M.G. Selective induction of alternatively spliced FynT isoform by TNF facilitates persistent inflammatory responses in astrocytes. Sci. Rep. 2017, 7, 43651. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Sano, T.; Nabetani, T.; Asano, Y.; Hirabayashi, Y. GPRC5B Activates Obesity-Associated Inflammatory Signaling in Adipocytes. Sci. Signal 2012, 5, ra85. [Google Scholar] [CrossRef]
- Jung, S.K.; Lee, K.W.; Byun, S.; Kang, N.J.; Lim, S.H.; Heo, Y.-S.; Bode, A.M.; Bowden, G.; Lee, H.J.; Dong, Z. Myricetin Suppresses UVB-Induced Skin Cancer by Targeting Fyn. Cancer Res. 2008, 68, 6021–6029. [Google Scholar] [CrossRef]
- Oh, J.; Yu, T.; Choi, S.J.; Yang, Y.; Baek, H.S.; An, S.A.; Kwon, L.K.; Kim, J.; Rho, H.S.; Shin, S.S.; et al. Syk/Src Pathway-Targeted Inhibition of Skin Inflammatory Responses by Carnosic Acid. Mediat. Inflamm. 2012, 2012, 1–13. [Google Scholar] [CrossRef]
- Németh, T.; Virtic, O.; Sitaru, C.; Mócsai, A. The Syk Tyrosine Kinase Is Required for Skin Inflammation in an In Vivo Mouse Model of Epidermolysis Bullosa Acquisita. J. Investig. Dermatol. 2017, 137, 2131–2139. [Google Scholar] [CrossRef]
- Samavedam, U.K.; Mitschker, N.; Kasprick, A.; Bieber, K.; Schmidt, E.; Laskay, T.; Recke, A.; Goletz, S.; Vidarsson, G.; Schulze, F.S.; et al. Whole-Genome Expression Profiling in Skin Reveals SYK As a Key Regulator of Inflammation in Experimental Epidermolysis Bullosa Acquisita. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Alzahrani, K.S.; Nadeem, A.; Ahmad, S.F.; Al-Harbi, N.O.; Ibrahim, K.E.; El-Sherbeeny, A.M.; Alhoshani, A.; Alshammari, M.A.; Alotaibi, M.R.; Al-Harbi, M.M. Inhibition of spleen tyrosine kinase attenuates psoriasis-like inflammation in mice through blockade of dendritic cell-Th17 inflammation axis. Biomed. Pharmacother. 2019, 111, 347–358. [Google Scholar] [CrossRef]
- Ye, J.; Piao, H.; Jiang, J.; Jin, G.; Zheng, M.; Yang, J.; Jin, X.; Sun, T.; Choi, Y.H.; Li, L.; et al. Polydatin inhibits mast cell-mediated allergic inflammation by targeting PI3K/Akt, MAPK, NF-κB and Nrf2/HO-1 pathways. Sci. Rep. 2017, 7, 11895. [Google Scholar] [CrossRef]
- Kim, W.-J.; Cha, H.-S.; Lee, M.-H.; Kim, S.-Y.; Kim, S.H.; Kim, T.-J. Effects of Cymbidium Root Ethanol Extract on Atopic Dermatitis. Evidence Based Complement. Altern. Med. 2016, 2016, 1–10. [Google Scholar] [CrossRef]
- Han, H.-M.; Kim, S.-J.; Kim, J.-S.; Kim, B.H.; Lee, H.W.; Lee, Y.T.; Kang, K.-H. Ameliorative effects of Artemisia argyi Folium extract on 2,4-dinitrochlorobenzene-induced atopic dermatitis-like lesions in BALB/c mice. Mol. Med. Rep. 2016, 14, 3206–3214. [Google Scholar] [CrossRef]
- Lesovaya, E.A.; Agarwal, S.; Readhead, B.; Vinokour, E.; Baida, G.; Bhalla, P.; Kirsanov, K.; Yakubovskaya, M.; Platanias, L.C.; Dudley, J.T.; et al. Rapamycin Modulates Glucocorticoid Receptor Function, Blocks Atrophogene REDD1, and Protects Skin from Steroid Atrophy. J. Investig. Dermatol. 2018, 138, 1935–1944. [Google Scholar] [CrossRef]
- Agarwal, S.; Mirzoeva, S.; Readhead, B.; Dudley, J.T.; Budunova, I. PI3K inhibitors protect against glucocorticoid-induced skin atrophy. EBioMedicine 2019, 41, 526–537. [Google Scholar] [CrossRef]
- Han, N.-R.; Moon, P.-D.; Kim, H.-M.; Jeong, H.-J. Tryptanthrin ameliorates atopic dermatitis through down-regulation of TSLP. Arch. Biochem. Biophys. 2014, 542, 14–20. [Google Scholar] [CrossRef]
- Hua, G.; Zein, N.; Paulen, L.; Chambon, P. The glucocorticoid receptor agonistic modulators CpdX and CpdX-D3 do not generate the debilitating effects of synthetic glucocorticoids. Proc. Natl. Acad. Sci. USA 2019, 116, 14200–14209. [Google Scholar] [CrossRef]
- Leung, N.Y.; Hirsch, R.L.; Schneider, L.; Moody, C.; Takaoka, R.; Li, S.H.; Meyerson, L.A.; Mariam, S.G.; Goldstein, G.; Hanifin, J.M. Thymopentin therapy reduces the clinical severity of atopic dermatitis. J. Allergy Clin. Immunol. 1990, 85, 927–933. [Google Scholar] [CrossRef]
- Lee, K.; Na, W.; Lee, J.Y.; Na, J.; Cho, H.; Wu, H.; Yune, T.Y.; Kim, W.-S.; Ju, B.-G. Molecular mechanism of Jmjd3-mediated interleukin-6 gene regulation in endothelial cells underlying spinal cord injury. J. Neurochem. 2012, 122, 272–282. [Google Scholar] [CrossRef]
- Pai, M.Y.; Lomenick, B.; Hwang, H.; Schiestl, R.; McBride, W.; Loo, J.A.; Huang, J. Drug affinity responsive target stability (DARTS) for small-molecule target identification. Breast Cancer 2015, 1263, 287–298. [Google Scholar] [CrossRef]
- Burnham, K.P.; Anderson, D.R. Multimodel inference: Understanding AIC and BIC in model selection. Sociological methods & research 2004, 33, 261–304. [Google Scholar]
- Burlingham, B.T.; Widlanski, T.S. An Intuitive Look at the Relationship of Ki and IC50: A More General Use for the Dixon Plot. J. Chem. Educ. 2003, 80, 214. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, H.; Shin, J.Y.; Lee, K.; Lee, S.-J.; Chong, H.-J.; Jeong, H.; Jeon, Y.-E.; Shin, D.-S.; Jang, S.; Kim, K.H.; et al. Caffeoyl-Prolyl-Histidine Amide Inhibits Fyn and Alleviates Atopic Dermatitis-Like Phenotypes via Suppression of NF-κB Activation. Int. J. Mol. Sci. 2020, 21, 7160. https://doi.org/10.3390/ijms21197160
Jeong H, Shin JY, Lee K, Lee S-J, Chong H-J, Jeong H, Jeon Y-E, Shin D-S, Jang S, Kim KH, et al. Caffeoyl-Prolyl-Histidine Amide Inhibits Fyn and Alleviates Atopic Dermatitis-Like Phenotypes via Suppression of NF-κB Activation. International Journal of Molecular Sciences. 2020; 21(19):7160. https://doi.org/10.3390/ijms21197160
Chicago/Turabian StyleJeong, Hayan, Jee Youn Shin, Kwanghyun Lee, Su-Jin Lee, Hyo-Jin Chong, Hyeri Jeong, Young-Eun Jeon, Dong-Sik Shin, Sunhyae Jang, Kyu Han Kim, and et al. 2020. "Caffeoyl-Prolyl-Histidine Amide Inhibits Fyn and Alleviates Atopic Dermatitis-Like Phenotypes via Suppression of NF-κB Activation" International Journal of Molecular Sciences 21, no. 19: 7160. https://doi.org/10.3390/ijms21197160
APA StyleJeong, H., Shin, J. Y., Lee, K., Lee, S.-J., Chong, H.-J., Jeong, H., Jeon, Y.-E., Shin, D.-S., Jang, S., Kim, K. H., Kim, S.-I., Lee, Y.-S., & Ju, B.-G. (2020). Caffeoyl-Prolyl-Histidine Amide Inhibits Fyn and Alleviates Atopic Dermatitis-Like Phenotypes via Suppression of NF-κB Activation. International Journal of Molecular Sciences, 21(19), 7160. https://doi.org/10.3390/ijms21197160