Finding New Molecular Targets of Familiar Natural Products Using In Silico Target Prediction

 , , , , , , ,
, , , , , , ,  , , ,
, , , 
Abstract
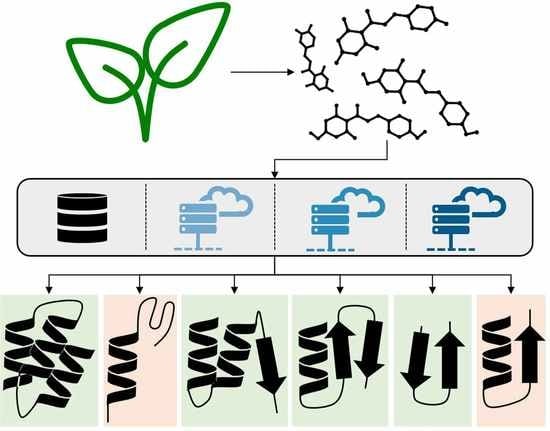
1. Introduction
2. Results
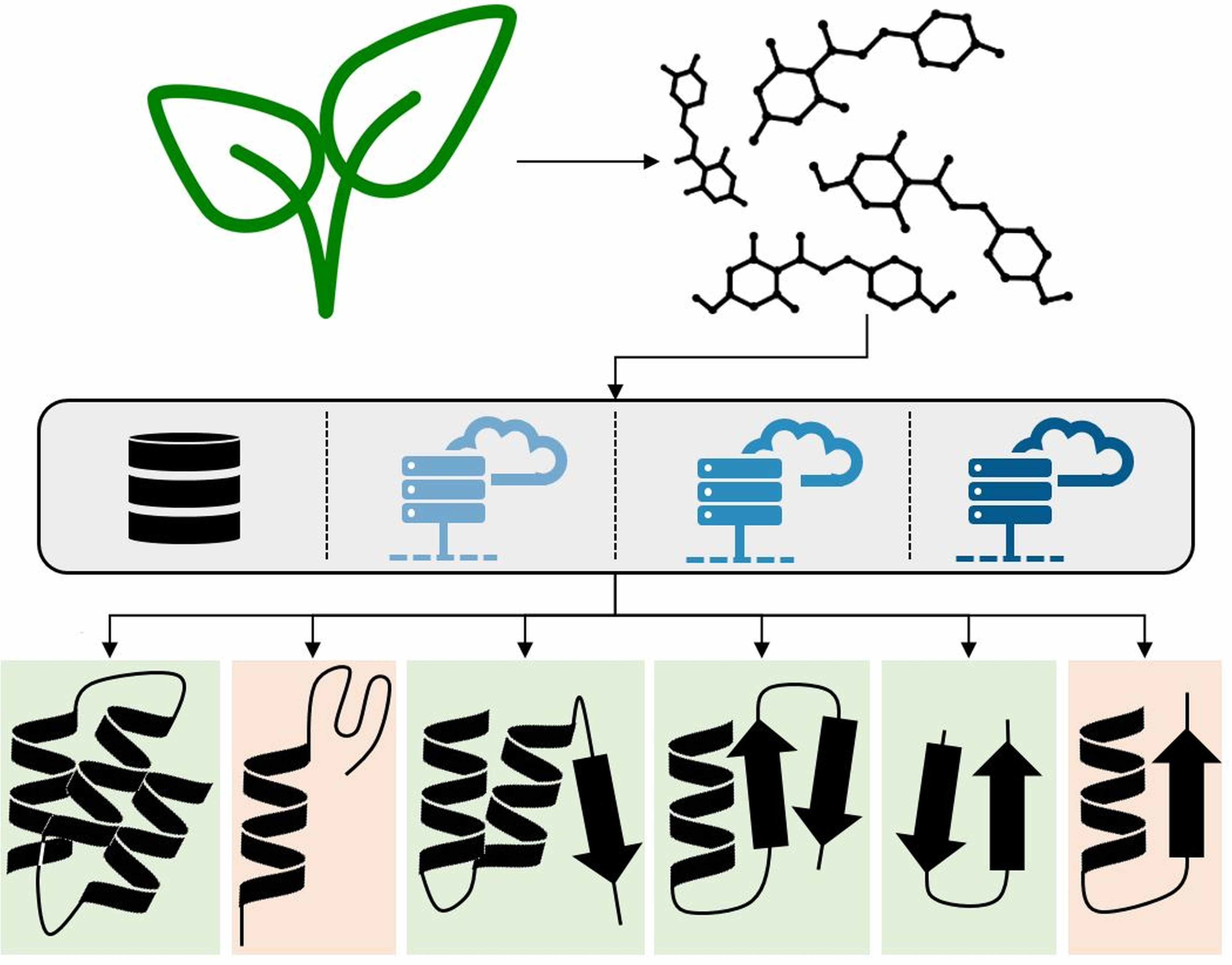
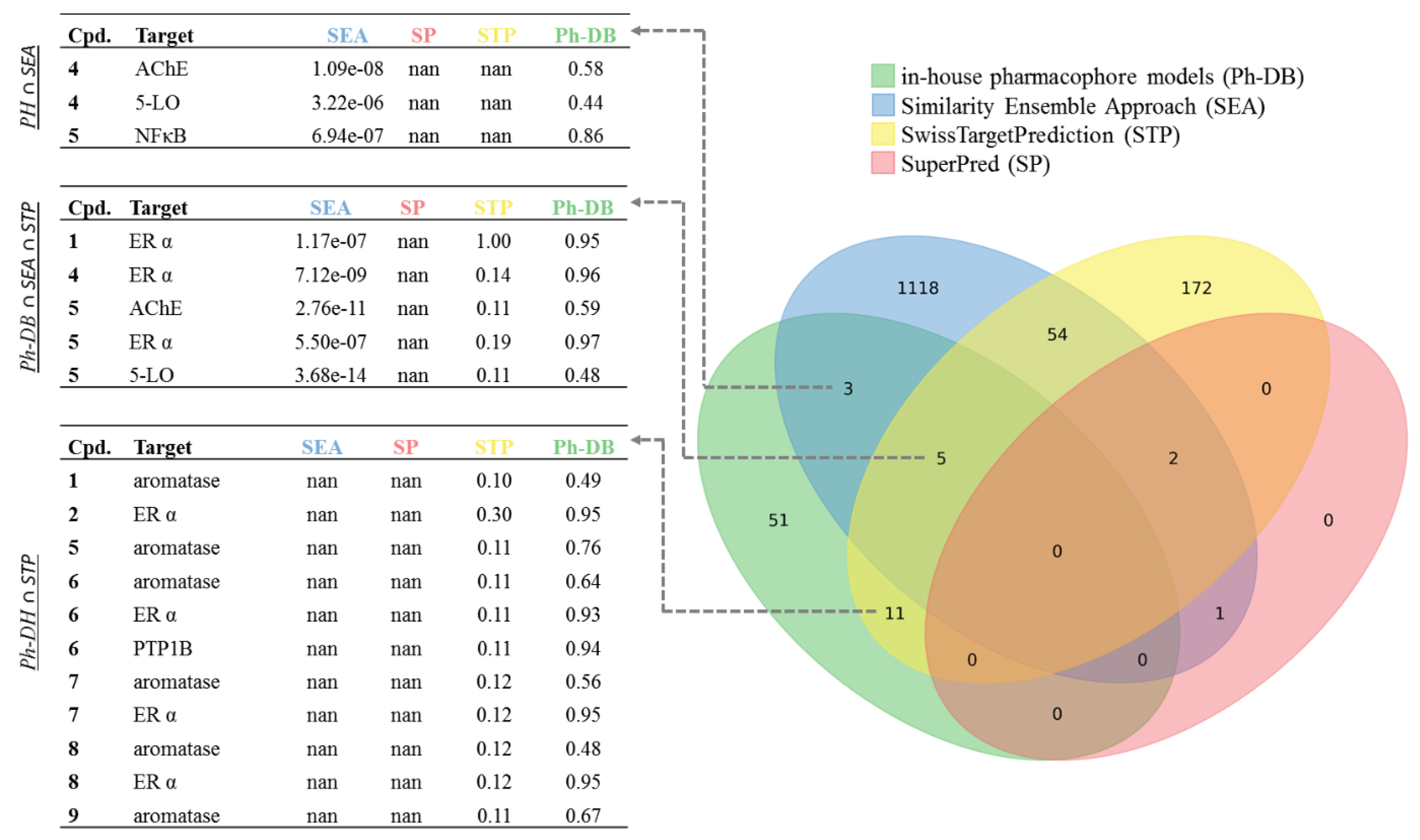
2.1. Data Basis, Curation, and Technical Setup of In Silico Predictions
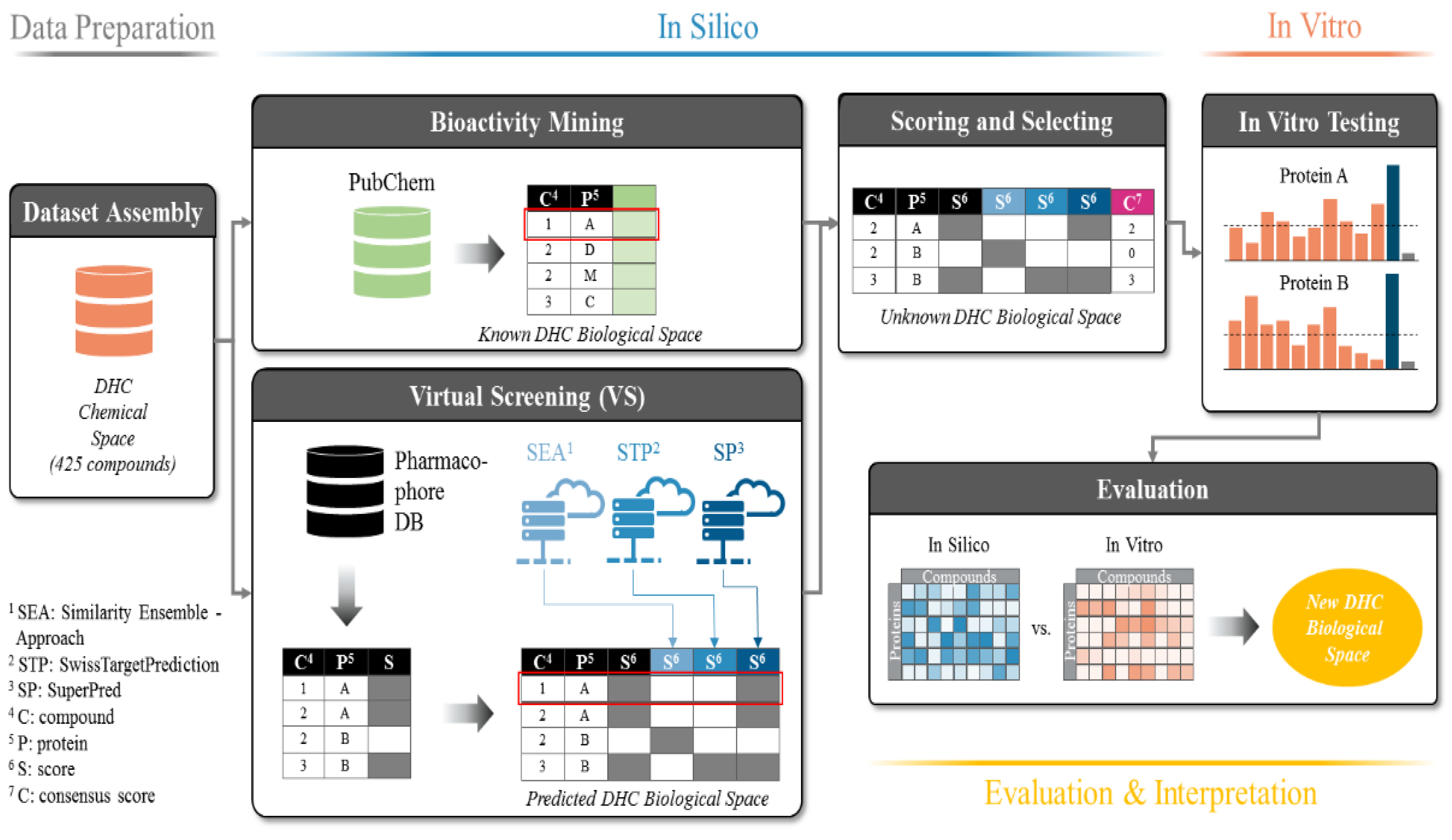
2.2. Predicted and Unknown DHC Biological Space
2.3. New DHC Biological Space
3. Discussion
4. Materials and Methods
4.1. Dataset Assembly
4.2. Bioactivity Mining
4.3. Pharmacophore-based Parallel Virtual Screening
4.4. Target Prediction with Publicly Available Tools
4.5. Biochemical Assays
4.6. Materials
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-LO | 5-lipoxygenase |
| AA | arachidonic acid |
| AChE | acetylcholinesterase |
| AKR1C3 | aldo-keto reductase 1C3 |
| BLAST | Basic Local Alignment Search Tool |
| COX-1 | cyclooxygenase 1 |
| CS | consensus score |
| DHC | dihydrochalcone |
| ER α | estrogen receptor α |
| HSD2 | hydroxysteroid dehydrogenase |
| NF-κB | nuclear factor κB |
| PC | positive control |
| Ph-DB | in-house pharmacophore model database |
| PTP1B | protein-tyrosine phosphatase B1 |
| SEA | Similarity Ensemble Approach |
| SGLT2 | sodium/glucose co-transporter 2 |
| SP | SuperPred |
| STP | SwissTargetPrediction |
| VS | virtual screening |
References
- Campbell, I.B.; Macdonald, S.J.F.; Procopiou, P.A. Medicinal chemistry in drug discovery in big pharma: Past, present and future. Drug Discov. Today 2018, 23, 219–234. [Google Scholar] [CrossRef]
- Chen, C.; Huang, H.; Wu, C.H. Protein bioinformatics databases and resources. In Fundamentals of Protein Bioinformatics; Chen, C., Huang, H., Wu, C.H., Eds.; Humana Press: New York, NY, USA, 2017; Volume 1558, pp. 3–39. [Google Scholar]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W.; Barker, E.L. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Aronson, J.K. Old drugs—New uses. Br. J. Clin. Pharmacol. 2007, 64, 563–565. [Google Scholar] [CrossRef]
- Hurle, M.R.; Yang, L.; Xie, Q.; Rajpal, D.K.; Sanseau, P.; Agarwal, P. Computational drug repositioning: From data to therapeutics. Clin. Pharmacol. Ther. 2013, 93, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.J.; Setola, V.; Irwin, J.J.; Laggner, C.; Abbas, A.I.; Hufeisen, S.J.; Jensen, N.H.; Kuijer, M.B.; Matos, R.C.; Tran, T.B.; et al. Predicting new molecular targets for known drugs. Nature 2009, 462, 175. [Google Scholar] [CrossRef]
- Rush, T.S.; Grant, J.A.; Mosyak, L.; Nicholls, A. A shape-based 3-D scaffold hopping method and its application to a bacterial protein−protein interaction. J. Med. Chem. 2005, 48, 1489–1495. [Google Scholar] [CrossRef] [PubMed]
- Steindl, T.; Schuster, D.; Laggner, C.; Langer, T. Parallel Screening: A novel concept in pharmacophore based modeling and virtual screening. J. Chem. Inf. Model. 2006, 45, 716–724. [Google Scholar] [CrossRef]
- Schuster, D. 3D pharmacophores as tools for activity profiling. Drug Discov. Today Technol. 2010, 7, e205–e211. [Google Scholar] [CrossRef] [PubMed]
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Pujadas, G.; Garcia-Vallve, S. Tools for in silico target fishing. Methods 2015, 71, 98–103. [Google Scholar] [CrossRef]
- Huang, Y.-W.; Pineau, I.; Chang, H.-J.; Azzi, A.; Bellemare, V.r.; Laberge, S.; Lin, S.-X. Critical residues for the specificity of cofactors and substrates in human estrogenic 17β-hydroxysteroid dehydrogenase 1: Variants designed from the three-dimensional structure of the enzyme. Mol. Endocrinol. 2001, 15, 2010–2020. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sydow, D.; Burggraaff, L.; Szengel, A.; van Vlijmen, H.W.T.; Ijzerman, A.P.; van Westen, G.J.P.; Volkamer, A. Advances and challenges in computational target prediction. J. Chem. Inf. Model. 2019, 59, 1728–1742. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Koehn, F.E.; Carter, G.T. The evolving role of natural products in drug discovery. Nat. Rev. Drug Discov. 2005, 4, 206–220. [Google Scholar] [CrossRef]
- Harvey, A.L. Natural products in drug discovery. Drug Discov. Today 2008, 13, 894–901. [Google Scholar] [CrossRef]
- Koeberle, A.; Werz, O. Multi-target approach for natural products in inflammation. Drug Discov. Today 2014, 19, 1871–1882. [Google Scholar] [CrossRef]
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on natural products for drug design. Nat. Chem. 2016, 8, 531. [Google Scholar] [CrossRef]
- Clemons, P.A.; Bodycombe, N.E.; Carrinski, H.A.; Wilson, J.A.; Shamji, A.F.; Wagner, B.K.; Koehler, A.N.; Schreiber, S.L. Small molecules of different origins have distinct distributions of structural complexity that correlate with protein-binding profiles. Proc. Natl. Acad. Sci. USA 2010, 107, 18787. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2018, 47, D1102–D1109. [Google Scholar] [CrossRef]
- Rivière, C. Chapter 7—dihydrochalcones: Occurrence in the plant kingdom, chemistry and biological activities. In Studies in Natural Products Chemistry; Atta ur, R., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 51, pp. 253–381. [Google Scholar]
- Meng, W.; Ellsworth, B.A.; Nirschl, A.A.; McCann, P.J.; Patel, M.; Girotra, R.N.; Wu, G.; Sher, P.M.; Morrison, E.P.; Biller, S.A.; et al. Discovery of dapagliflozin: A potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. J. Med. Chem. 2008, 51, 1145–1149. [Google Scholar] [CrossRef]
- Uthman, L.; Baartscheer, A.; Schumacher, C.A.; Fiolet, J.W.T.; Kuschma, M.C.; Hollmann, M.W.; Coronel, R.; Weber, N.C.; Zuurbier, C.J. Direct cardiac actions of sodium glucose cotransporter 2 inhibitors target pathogenic mechanisms underlying heart failure in diabetic patients. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Orlikova, B.; Schnekenburger, M.; Zloh, M.; Golais, F.; Diederich, M.; Tasdemir, D. Natural chalcones as dual inhibitors of HDACs and NF-κB. Oncol. Rep. 2012, 28, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Dodds, E.C.; Lawson, W.; Dale, H.H. Molecular structure in relation to oestrogenic activity. Compounds without a phenanthrene nucleus. Proc. R. Soc. B 1938, 125, 222–232. [Google Scholar] [CrossRef]
- Hert, J.; Keiser, M.J.; Irwin, J.J.; Oprea, T.I.; Shoichet, B.K. Quantifying the relationships among drug classes. J. Chem. Inf. Model. 2008, 48, 755–765. [Google Scholar] [CrossRef][Green Version]
- Jalencas, X.; Mestres, J. On the origins of drug polypharmacology. MedChemComm 2013, 4, 80–87. [Google Scholar] [CrossRef]
- Matsuura, K.; Shiraishi, H.; Hara, A.; Sato, K.; Deyashiki, Y.; Ninomiya, M.; Sakai, S. Identification of a principal mRNA species for human 3α-hydroxysteroid dehydrogenase isoform (AKR1C3) that exhibits high prostaglandin D2 11-ketoreductase activity. J. Biochem. 1998, 124, 940–946. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef]
- Dunkel, M.; Günther, S.; Ahmed, J.; Wittig, B.; Preissner, R. SuperPred: Drug classification and target prediction. Nucleic Acids Res. 2008, 36, W55–W59. [Google Scholar] [CrossRef]
- Gfeller, D.; Michielin, O.; Zoete, V. Shaping the interaction landscape of bioactive molecules. Bioinformatics 2013, 29, 3073–3079. [Google Scholar] [CrossRef]
- Wolber, G.; Dornhofer, A.A.; Langer, T. Efficient overlay of small organic molecules using 3D pharmacophores. J. Comput. Aided Mol. Des. 2006, 20, 773–788. [Google Scholar] [CrossRef] [PubMed]
- Möller, G.; Deluca, D.; Gege, C.; Rosinus, A.; Kowalik, D.; Peters, O.; Droescher, P.; Elger, W.; Adamski, J.; Hillisch, A. Structure-based design, synthesis and in vitro characterization of potent 17β-hydroxysteroid dehydrogenase type 1 inhibitors based on 2-substitutions of estrone and D-homo-estrone. Bioorg. Med. Chem. Lett. 2009, 19, 6740–6744. [Google Scholar] [CrossRef] [PubMed]
- Schuster, D.; Kowalik, D.; Kirchmair, J.; Laggner, C.; Markt, P.; Aebischer-Gumy, C.; Ströhle, F.; Möller, G.; Wolber, G.; Wilckens, T.; et al. Identification of chemically diverse, novel Inhibitors of 17 beta hydroxysteroid dehydrogenase type 3 and 5 pharmacophore-based virtual screening. J. Steroid Biochem. Mol. Biol. 2011, 125, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Lounkine, E.; Keiser, M.J.; Whitebread, S.; Mikhailov, D.; Hamon, J.; Jenkins, J.L.; Lavan, P.; Weber, E.; Doak, A.K.; Côté, S.; et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature 2012, 486, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Rollinger, J.M. Accessing target information by virtual parallel screening—The impact on natural product research. Phytochem. Lett. 2009, 2, 53–58. [Google Scholar] [CrossRef]
- Rollinger, J.M.; Schuster, D.; Danzl, B.; Schwaiger, S.; Markt, P.; Schmidtke, M.; Gertsch, J.; Raduner, S.; Wolber, G.; Langer, T.; et al. In silico target fishing for rationalized ligand discovery exemplified on constituents of Ruta graveolens. Planta Med. 2009, 75, 195–204. [Google Scholar] [CrossRef]
- Reker, D.; Perna, A.M.; Rodrigues, T.; Schneider, P.; Reutlinger, M.; Mönch, B.; Koeberle, A.; Lamers, C.; Gabler, M.; Steinmetz, H.; et al. Revealing the macromolecular targets of complex natural products. Nat. Chem. 2014, 6, 1072–1078. [Google Scholar] [CrossRef]
- Rodrigues, T.; Reker, D.; Kunze, J.; Schneider, P.; Schneider, G. Revealing the macromolecular targets of fragment-like natural products. Angew. Chem., Int. Ed. 2015, 54, 10516–10520. [Google Scholar] [CrossRef]
- Mayr, F.; Vieider, C.; Temml, V.; Stuppner, H.; Schuster, D. Open-access activity prediction tools for natural products. Case study: hERG blockers. In Progress in the Chemistry of Organic Natural Products 110: Cheminformatics in Natural Product Research; Kinghorn, A.D., Falk, H., Gibbons, S., Kobayashi, J.i., Asakawa, Y., Liu, J.-K., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 177–238. [Google Scholar] [CrossRef]
- Mayr, F.; Sturm, S.; Ganzera, M.; Waltenberger, B.; Martens, S.; Schwaiger, S.; Schuster, D.; Stuppner, H. Mushroom tyrosinase-based enzyme inhibition assays are not suitable for bioactivity-guided fractionation of extracts. J. Nat. Prod. 2019, 82, 136–147. [Google Scholar] [CrossRef]
- Young, S.M.; Bologa, C.; Prossnitz, E.R.; Oprea, T.I.; Sklar, L.A.; Edwards, B.S. High-throughput screening with HyperCyt® flow cytometry to detect small molecule formylpeptide receptor ligands. J. Biomol. Screen. 2005, 10, 374–382. [Google Scholar] [CrossRef]
- Doman, T.N.; McGovern, S.L.; Witherbee, B.J.; Kasten, T.P.; Kurumbail, R.; Stallings, W.C.; Connolly, D.T.; Shoichet, B.K. Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1B. J. Med. Chem. 2002, 45, 2213–2221. [Google Scholar] [CrossRef]
- Polgár, T.; Baki, A.; Szendrei, G.I.; Keserűu, G.M. Comparative virtual and experimental high-throughput screening for glycogen synthase kinase-3β inhibitors. J. Med. Chem. 2005, 48, 7946–7959. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.S.; Simeonov, A.; Jadhav, A.; Eidam, O.; Mott, B.T.; Keiser, M.J.; McKerrow, J.H.; Maloney, D.J.; Irwin, J.J.; Shoichet, B.K. Complementarity between a docking and a high-throughput screen in discovering new cruzain inhibitors. J. Med. Chem. 2010, 53, 4891–4905. [Google Scholar] [CrossRef] [PubMed]
- Le Bail, J.-C.; Pouget, C.; Fagnere, C.; Basly, J.-P.; Chulia, A.-J.; Habrioux, G. Chalcones are potent inhibitors of aromatase and 17β-hydroxysteroid dehydrogenase activities. Life Sci. 2001, 68, 751–761. [Google Scholar] [CrossRef]
- Ripphausen, P.; Nisius, B.; Peltason, L.; Bajorath, J. Quo vadis virtual Screening? A comprehensive survey of prospective applications. J. Med. Chem. 2010, 53, 8461–8467. [Google Scholar] [CrossRef]
- Dutka, M.; Bobiński, R.; Ulman-Włodarz, I.; Hajduga, M.; Bujok, J.; Pająk, C.; Ćwiertnia, M. Various aspects of inflammation in heart failure. Heart Fail. Rev. 2019, 25, 537–548. [Google Scholar] [CrossRef]
- Xu, C.; Wang, W.; Zhong, J.; Lei, F.; Xu, N.; Zhang, Y.; Xie, W. Canagliflozin exerts anti-inflammatory effects by inhibiting intracellular glucose metabolism and promoting autophagy in immune cells. Biochem. Pharmacol. (Amst. Neth.) 2018, 152, 45–59. [Google Scholar] [CrossRef]
- Iannantuoni, F.; M de Marañon, A.; Diaz-Morales, N.; Falcon, R.; Bañuls, C.; Abad-Jimenez, Z.; Victor, V.M.; Hernandez-Mijares, A.; Rovira-Llopis, S. The SGLT2 inhibitor empagliflozin ameliorates the inflammatory profile in type 2 diabetic patients and promotes an antioxidant response in leukocytes. J. Clin. Med. 2019, 8, 1814. [Google Scholar] [CrossRef]
- Hattori, S. Anti-inflammatory effects of empagliflozin in patients with type 2 diabetes and insulin resistance. Diabetol. Metab. Syndr. 2018, 10, 93. [Google Scholar] [CrossRef]
- Margiotti, K.; Kim, E.; Pearce, C.L.; Spera, E.; Novelli, G.; Reichardt, J.K.V. Association of the G289S single nucleotide polymorphism in the HSD17B3 gene with prostate cancer in italian men. Prostate 2002, 53, 65–68. [Google Scholar] [CrossRef]
- Vicker, N.; Sharland, C.M.; Heaton, W.B.; Gonzalez, A.M.R.; Bailey, H.V.; Smith, A.; Springall, J.S.; Day, J.M.; Tutill, H.J.; Reed, M.J.; et al. The design of novel 17β-hydroxysteroid dehydrogenase type 3 inhibitors. Mol. Cell. Endocrinol. 2009, 301, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Neuwirt, H.; Bouchal, J.; Kharaishvili, G.; Ploner, C.; Jöhrer, K.; Pitterl, F.; Weber, A.; Klocker, H.; Eder, I.E. Cancer-associated fibroblasts promote prostate tumor growth and progression through upregulation of cholesterol and steroid biosynthesis. Cell Commun. Signal. 2020, 18, 11. [Google Scholar] [CrossRef] [PubMed]
- Gaucher, M.; Dugé de Bernonville, T.; Lohou, D.; Guyot, S.; Guillemette, T.; Brisset, M.-N.; Dat, J.F. Histolocalization and physico-chemical characterization of dihydrochalcones: Insight into the role of apple major flavonoids. Phytochemistry 2013, 90, 78–89. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2018, 47, D506–D515. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2017, 46, D649–D655. [Google Scholar] [CrossRef]
- McKinney, W. Data structures for statistical computing in python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2020; Volume 445, pp. 51–56. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Gfeller, D.; Grosdidier, A.; Wirth, M.; Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: A web server for target prediction of bioactive small molecules. Nucleic Acids Res. 2014, 42, W32–W38. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef]
- Nickel, J.; Gohlke, B.-O.; Erehman, J.; Banerjee, P.; Rong, W.W.; Goede, A.; Dunkel, M.; Preissner, R. SuperPred: Update on drug classification and target prediction. Nucleic Acids Res. 2014, 42, W26–W31. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef] [PubMed]
- Günther, S.; Kuhn, M.; Dunkel, M.; Campillos, M.; Senger, C.; Petsalaki, E.; Ahmed, J.; Urdiales, E.G.; Gewiess, A.; Jensen, L.J.; et al. SuperTarget and Matador: Resources for exploring drug-target relationships. Nucleic Acids Res. 2008, 36, D919–D922. [Google Scholar] [CrossRef] [PubMed]
- Hecker, N.; Ahmed, J.; von Eichborn, J.; Dunkel, M.; Macha, K.; Eckert, A.; Gilson, M.K.; Bourne, P.E.; Preissner, R. SuperTarget goes quantitative: Update on drug–target interactions. Nucleic Acids Res. 2012, 40, D1113–D1117. [Google Scholar] [CrossRef]
- Armstrong, S.M.; Morris, G.M.; Finn, P.W.; Sharma, R.; Moretti, L.; Cooper, R.I.; Richards, W.G. ElectroShape: Fast molecular similarity calculations incorporating shape, chirality and electrostatics. J. Comput. Aid. Mol. Des. 2010, 24, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.V.; Kempná, P.; Hofer, G.; Mullis, P.E.; Flück, C.E. Modulation of human CYP19A1 activity by mutant NADPH P450 oxidoreductase. Mol. Endocrinol. 2007, 21, 2579–2595. [Google Scholar] [CrossRef] [PubMed]
- Lephart, E.D.; Simpson, E.R. Assay of aromatase activity. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1991; Volume 206, pp. 477–483. [Google Scholar]
- Wetzel, M.; Marchais-Oberwinkler, S.; Perspicace, E.; Möller, G.; Adamski, J.; Hartmann, R.W. Introduction of an electron withdrawing group on the hydroxyphenylnaphthol scaffold improves the potency of 17β-hydroxysteroid dehydrogenase type 2 (17β-HSD2) inhibitors. J. Med. Chem. 2011, 54, 7547–7557. [Google Scholar] [CrossRef]
- Schaible, A.M.; Filosa, R.; Temml, V.; Krauth, V.; Matteis, M.; Peduto, A.; Bruno, F.; Luderer, S.; Roviezzo, F.; Di Mola, A.; et al. Elucidation of the molecular mechanism and the efficacy in vivo of a novel 1,4-benzoquinone that inhibits 5-lipoxygenase. Br. J. Pharmacol. 2014, 171, 2399–2412. [Google Scholar] [CrossRef] [PubMed]
- Koeberle, A.; Siemoneit, U.; Bühring, U.; Northoff, H.; Laufer, S.; Albrecht, W.; Werz, O. Licofelone suppresses prostaglandin E2 formation by interference with the inducible microsomal prostaglandin E2 synthase-1. J. Pharmacol. Exp. Ther. 2008, 326, 975. [Google Scholar] [CrossRef] [PubMed]
- Kratschmar, D.V.; Vuorinen, A.; Da Cunha, T.; Wolber, G.; Classen-Houben, D.; Doblhoff, O.; Schuster, D.; Odermatt, A. Characterization of activity and binding mode of glycyrrhetinic acid derivatives inhibiting 11β-hydroxysteroid dehydrogenase type 2. J. Steroid Biochem. Mol. Biol. 2011, 125, 129–142. [Google Scholar] [CrossRef]
- Udhane, S.S.; Parween, S.; Kagawa, N.; Pandey, A.V. Altered CYP19A1 and CYP3A4 activities due to mutations A115V, T142A, Q153R and P284L in the human P450 oxidoreductase. Front. Pharmacol. 2017, 8, 580. [Google Scholar] [CrossRef] [PubMed]
- Samandari, E.; Kempná, P.; Nuoffer, J.-M.; Hofer, G.; Mullis, P.E.; Flück, C.E. Human adrenal corticocarcinoma NCI-H295R cells produce more androgens than NCI-H295A cells and differ in 3β-hydroxysteroid dehydrogenase type 2 and 17,20 lyase activities. J. Endocrinol. 2007, 195, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Morisseau, C.; Beetham, J.K.; Pinot, F.; Debernard, S.; Newman, J.W.; Hammock, B.D. Cress and potato soluble epoxide hydrolases: Purification, biochemical characterization, and comparison to mammalian enzymes. Arch. Biochem. Biophys. 2000, 378, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Wixtrom, R.N.; Silva, M.H.; Hammock, B.D. Affinity purification of cytosolic epoxide hydrolase using derivatized epoxy-activated sepharose gels. Anal. Biochem. 1988, 169, 71–80. [Google Scholar] [CrossRef]
- Waltenberger, B.; Garscha, U.; Temml, V.; Liers, J.; Werz, O.; Schuster, D.; Stuppner, H. Discovery of potent soluble Epoxide hydrolase (sEH) Inhibitors by pharmacophore-based virtual screening. J. Chem. Inf. Model. 2016, 56, 747–762. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
| No. | Name | R1 | R2 | R3 | R4 |
| 1 | phloretin | OH | H | OH | OH |
| 2 | 3-OH-phloretin | OH | OH | OH | OH |
| 3 | 2′,6′-dihydroxy-4′-methoxy DHC | H | H | OMe | OH |
| 4 | asebogenin | OH | H | OMe | OH |
| 5 | calomelanen | OMe | H | OMe | OH |
| 6 | sieboldin | OH | OH | O-Glc 1 | OH |
| 7 | phloridzin | OH | H | OH | O-Glc 1 |
| 8 | trilobatin | OH | H | O-Glc 1 | OH |
| 9 | phloretin-2′-xyloglucoside | OH | H | OH | O-Rut 2 |
| 10 | neohesperidin DHC | OMe | OH | O-Neo 3 | OH |
| Candidate Target | Selection Criterion I 1 | Selection Criterion II 2 | Selection Criterion III 3 | Selection Criterion IV 4 | Selected |
|---|---|---|---|---|---|
| 17β HSD2 | n.a. 5 | 5th (CS = 3) 12th (CS = 2) | 1-ER α/β | Yes | Yes |
| 17β HSD3 | n.a. 5 | 4th (CS = 3) 7th (CS = 2) | 1-ER α/β | Yes | Yes |
| 5-LO | 4 (CS = 2) 5 (CS = 3) | 3rd (CS = 3) 3rd (CS = 3) | 1-PGDH | Yes | Yes |
| AChE | 4 (CS = 2) 5 (CS = 3) | 1st (CS = 3) 10th (CS = 2) | n.a. 5 | No | No |
| AKR1C3 | n.a. 5 | 6th (CS = 2) | 1–AKR1B10 | Yes | Yes |
| Aromatase | 1 (CS = 2) 5 (CS = 2) 6 (CS = 2) 7 (CS = 2) 8 (CS = 2) 9 (CS = 2) | 1st (CS = 2) | 1-aromatase 1-ER α/β 1-several CYPs | Yes | Yes |
| COX-1 | n.a. 5 | 4th (CS = 3) | 1-PGDH | Yes | Yes |
| ERα | 1 (CS = 3) 2 (CS = 2) 4 (CS = 3) 5 (CS = 3) 6 (CS = 2) 7 (CS = 2) 8 (CS = 2) | 2nd (CS = 3) 2nd (CS = 2) | 1-ER α/β | Yes | No |
| ERβ | n.a. 5 | 5th (CS = 3) 6th (CS = 2) | 1-ER α/β | Yes | No |
| NF-κB | n.a. 5 | 8th (CS = 2) | 1-NF-κB 5-NF-κB | No | No |
| PPARγ | n.a. 5 | 9th (CS = 2) | 1-PPARγ | No | No |
| PTP1B | 6 (CS = 2) | 10th (CS = 2) | n.a. | Yes | No |
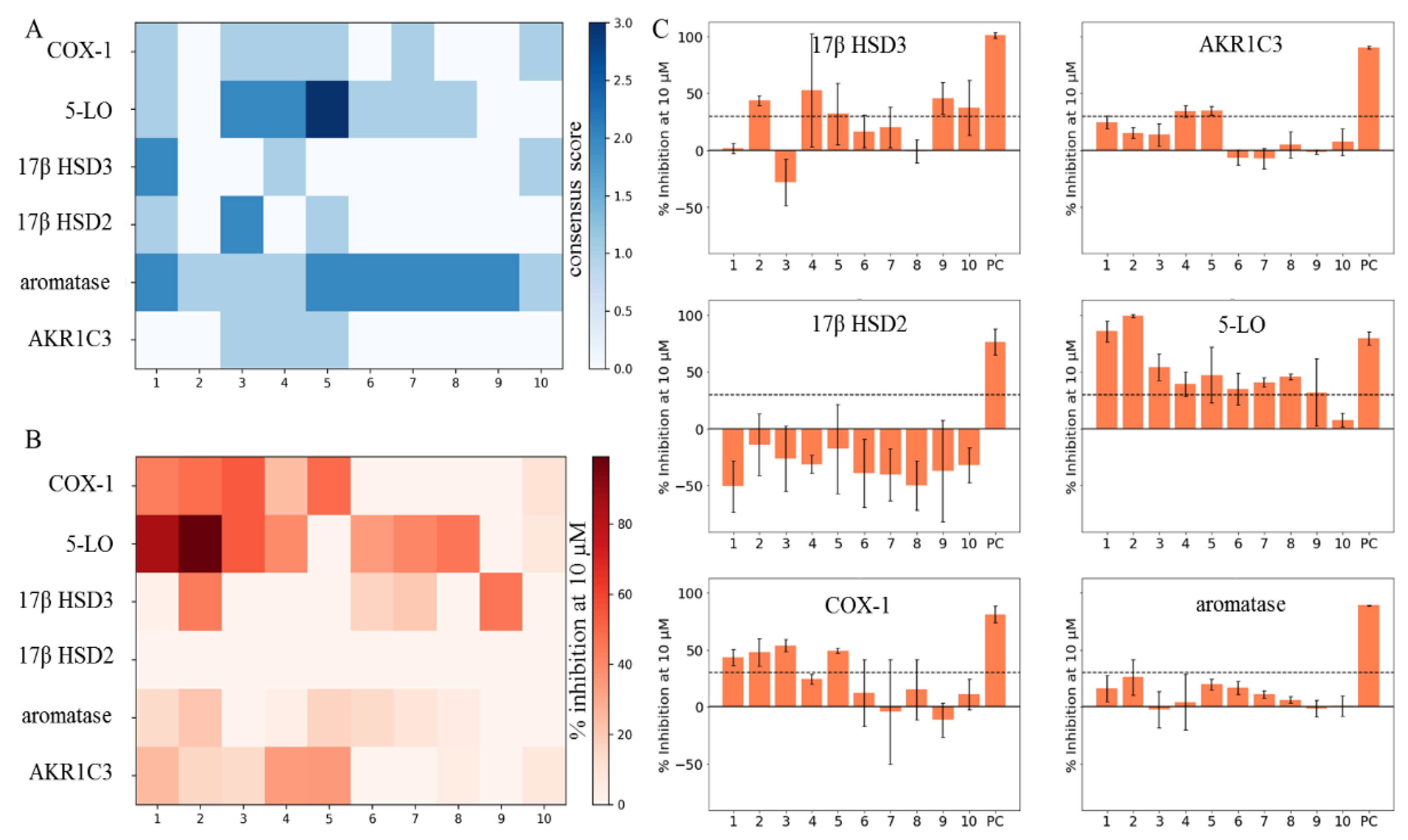
| Compound | Aromatase | 17β HSD2 | 17β HSD3 | AKR1C3 | 5-LO | COX-1 |
|---|---|---|---|---|---|---|
| 1 | 13.8 ± 2.0 | −50.7 ± 22.7 | 1.7 ± 4.5 | 24.8 ± 5.9 | 85.4 ± 9.3 | 43.5 ± 7.2 |
| 2 | 21.1 ± 11.7 | −14.1 ± 27.1 | 43.8 ± 4.7 | 15.5 ± 4.9 | 99.2 ± 1.2 | 48.1 ± 12.0 |
| 3 | −1.0 ± 12.0 | −26.3 ± 28.8 | −28.0 ± 20.5 | 13.8 ± 10.0 | 54.1 ± 11.6 | 53.9 ± 5.3 |
| 4 | 3.5 ± 19.0 | −31.2 ± 8.1 | 52.7 ± 49.6 | 34.4 ± 5.1 | 39.2 ± 11.1 | 24.4 ± 4.2 |
| 5 | 17.0 ± 2.0 | −17.6 ± 39.0 | 32.1 ± 27.2 | 35.2 ± 3.8 | 47.2 ± 24.2 | 49.5 ± 1.9 |
| 6 | 13.8 ± 4.4 | −39 ± 29.9 | 16.7 ± 14.5 | −6.1 ± 6.8 | 34.8 ± 14.2 | 12.34 ± 29.0 |
| 7 | 9.4 ± 3.5 | −40.5 ± 22.9 | 20.2 ± 17.7 | −7.2 ± 8.9 | 40.8 ± 4.1 | −4.2 ± 45.6 |
| 8 | 5.9 ± 3.5 | −49.8 ± 21.4 | −0.6 ± 10.0 | 5.3 ± 11.5 | 45.5 ± 2.7 | 15.3 ± 26.4 |
| 9 | 0.67 ± 4.0 | −37.1 ± 44.3 | 45.8 ± 14.2 | −1.3 ± 1.9 | 31.8 ± 29.4 | −11.4 ± 15.1 |
| 10 | 0 ± 5.8 | −32.2 ± 15.4 | 37.5 ± 23.9 | 7.4 ± 11.8 | 7.7 ± 6.0 | 11.1 ± 13.2 |
| PC | 70.2 ± 0.5 * | 76.1 ± 11.4 † | 101.2 ± 2.4 ‡ | 90.5 ± 1.2 § | 79.26 ± 5.95 ¶ | 81.3 ± 7.5 # |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mayr, F.; Möller, G.; Garscha, U.; Fischer, J.; Rodríguez Castaño, P.; Inderbinen, S.G.; Temml, V.; Waltenberger, B.; Schwaiger, S.; Hartmann, R.W.; et al. Finding New Molecular Targets of Familiar Natural Products Using In Silico Target Prediction. Int. J. Mol. Sci. 2020, 21, 7102. https://doi.org/10.3390/ijms21197102
Mayr F, Möller G, Garscha U, Fischer J, Rodríguez Castaño P, Inderbinen SG, Temml V, Waltenberger B, Schwaiger S, Hartmann RW, et al. Finding New Molecular Targets of Familiar Natural Products Using In Silico Target Prediction. International Journal of Molecular Sciences. 2020; 21(19):7102. https://doi.org/10.3390/ijms21197102
Chicago/Turabian StyleMayr, Fabian, Gabriele Möller, Ulrike Garscha, Jana Fischer, Patricia Rodríguez Castaño, Silvia G. Inderbinen, Veronika Temml, Birgit Waltenberger, Stefan Schwaiger, Rolf W. Hartmann, and et al. 2020. "Finding New Molecular Targets of Familiar Natural Products Using In Silico Target Prediction" International Journal of Molecular Sciences 21, no. 19: 7102. https://doi.org/10.3390/ijms21197102
APA StyleMayr, F., Möller, G., Garscha, U., Fischer, J., Rodríguez Castaño, P., Inderbinen, S. G., Temml, V., Waltenberger, B., Schwaiger, S., Hartmann, R. W., Gege, C., Martens, S., Odermatt, A., Pandey, A. V., Werz, O., Adamski, J., Stuppner, H., & Schuster, D. (2020). Finding New Molecular Targets of Familiar Natural Products Using In Silico Target Prediction. International Journal of Molecular Sciences, 21(19), 7102. https://doi.org/10.3390/ijms21197102