Beyond the Canonical Endocannabinoid System. A Screening of PPAR Ligands as FAAH Inhibitors

 ,
,  , , ,
, , ,
Abstract
1. Introduction
2. Results and Discussion
2.1. Screening Results
2.2. Chemistry
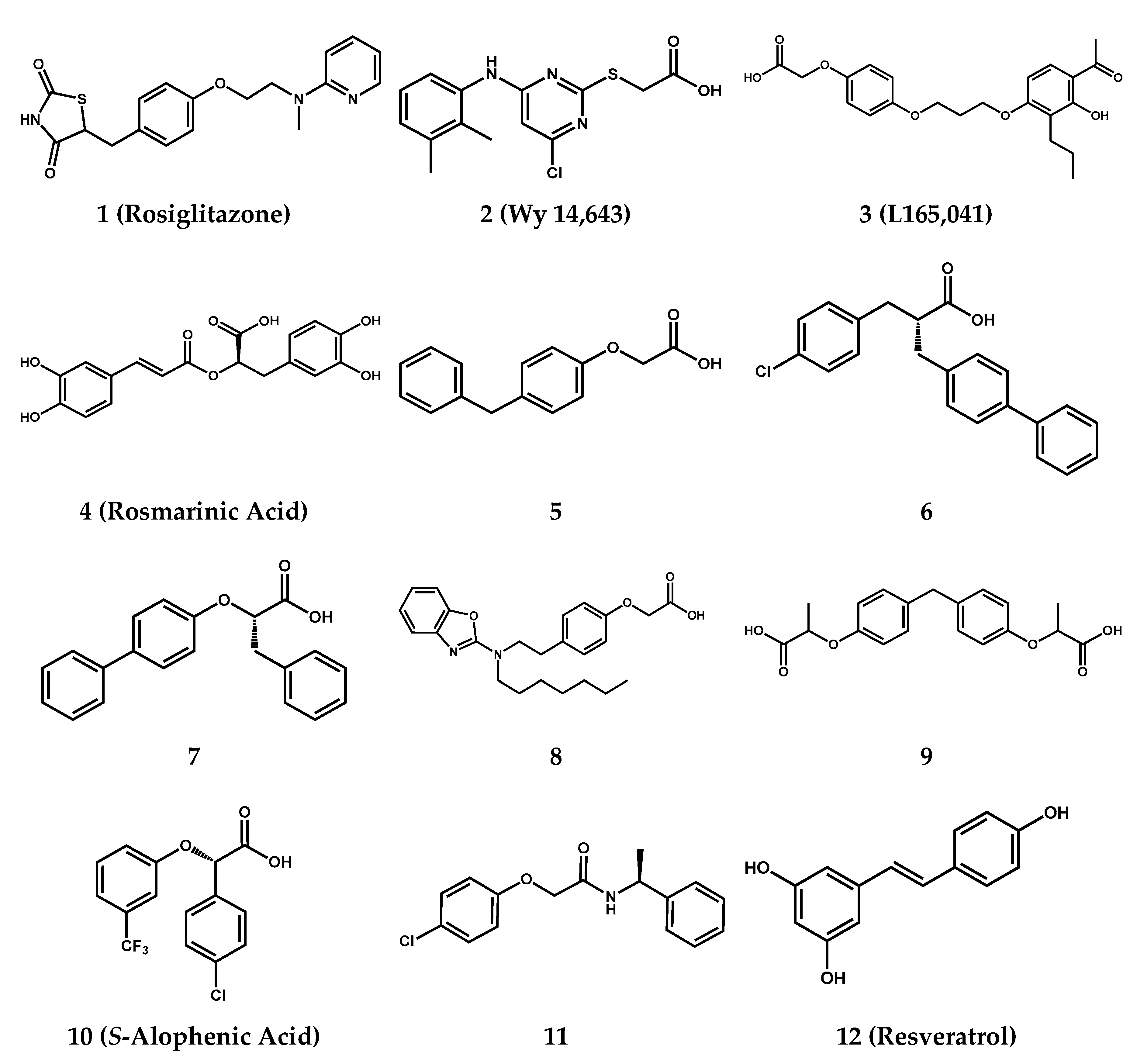
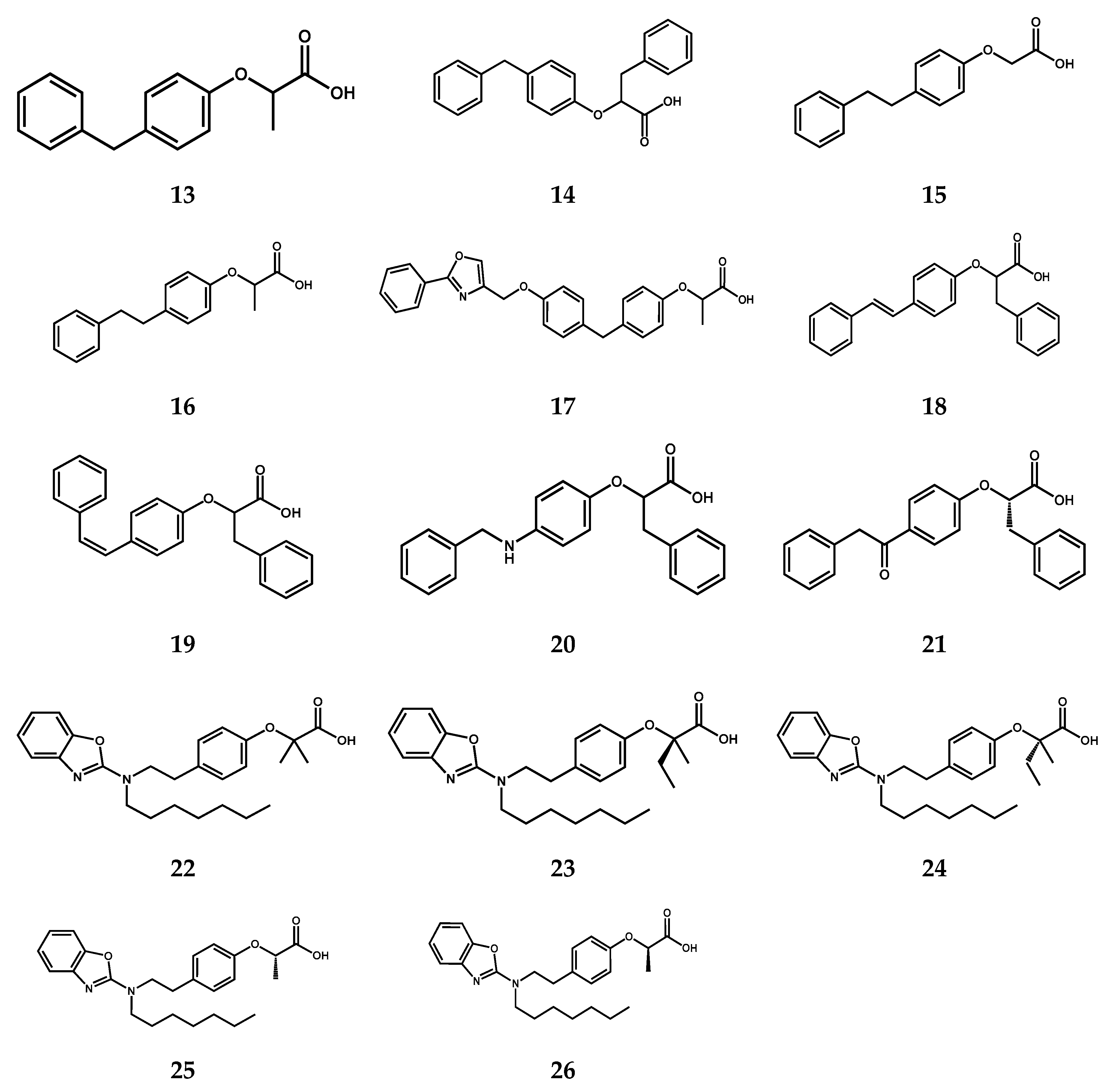
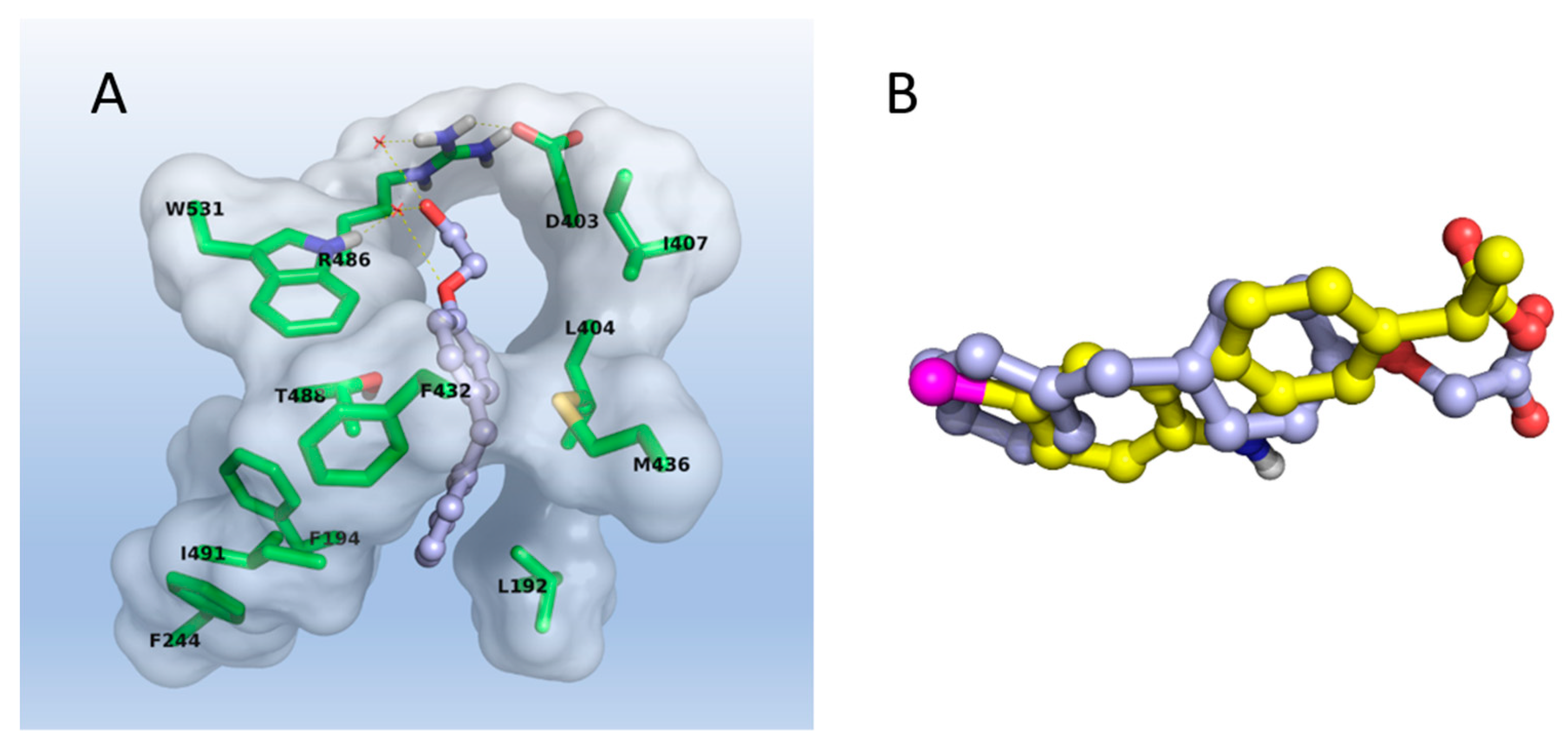
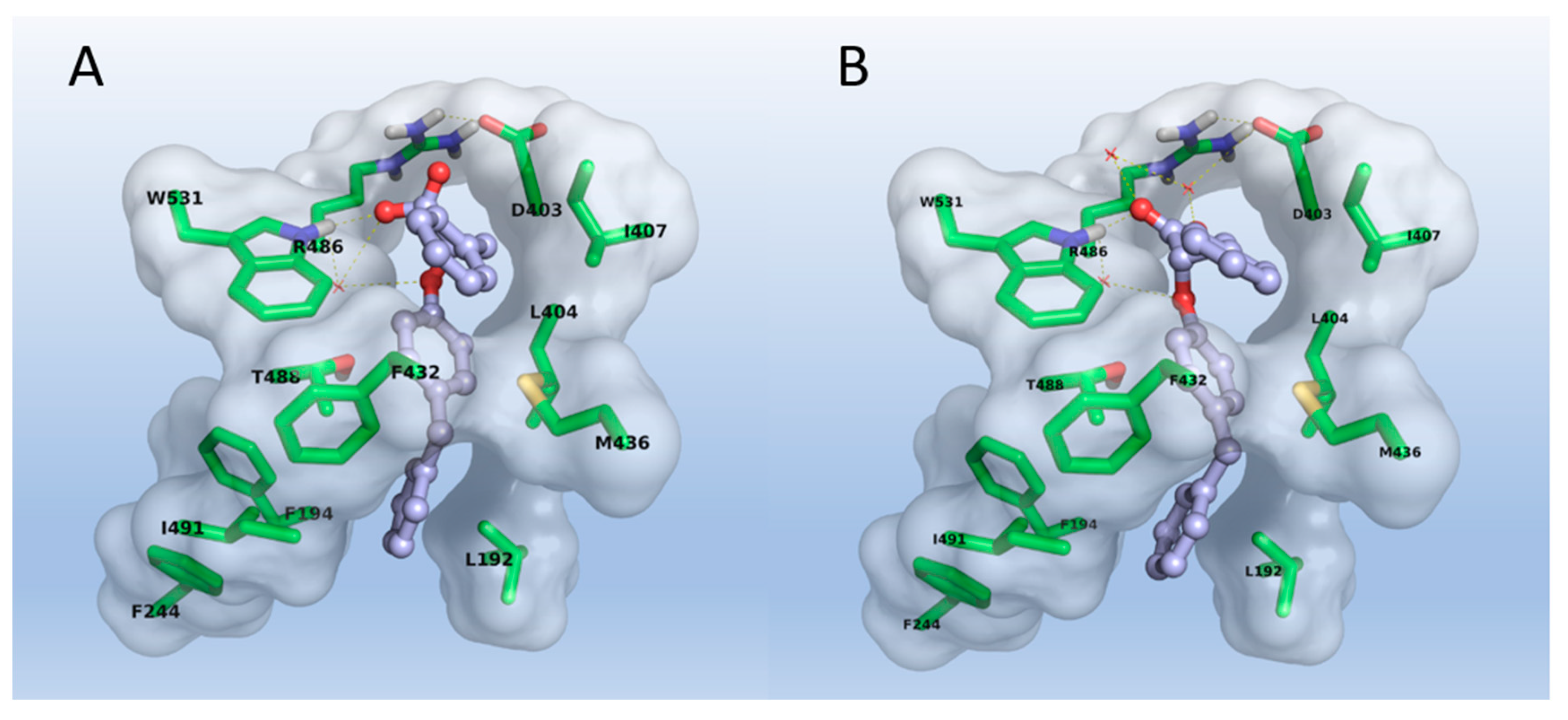
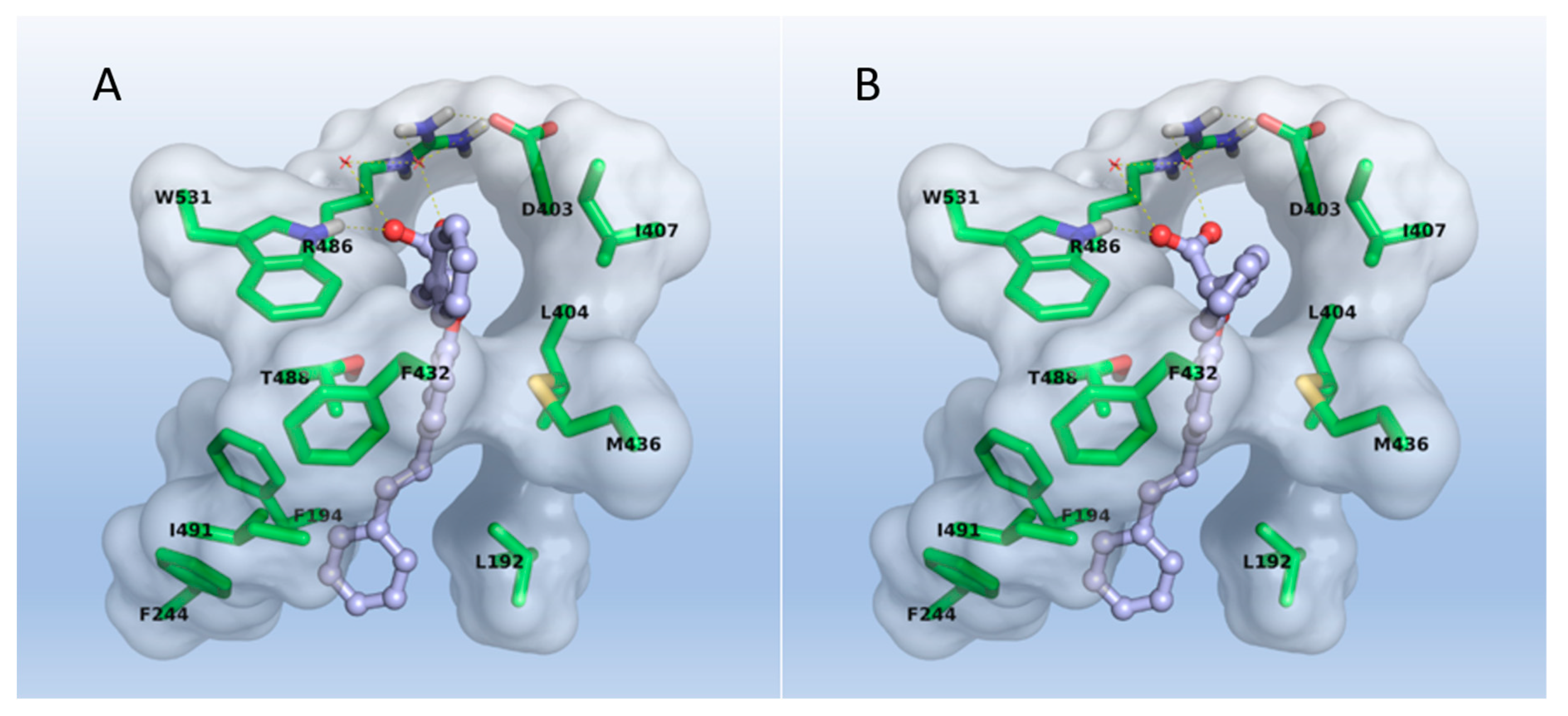
2.3. Molecular Modeling
3. Materials and Methods
3.1. Chemical Methods
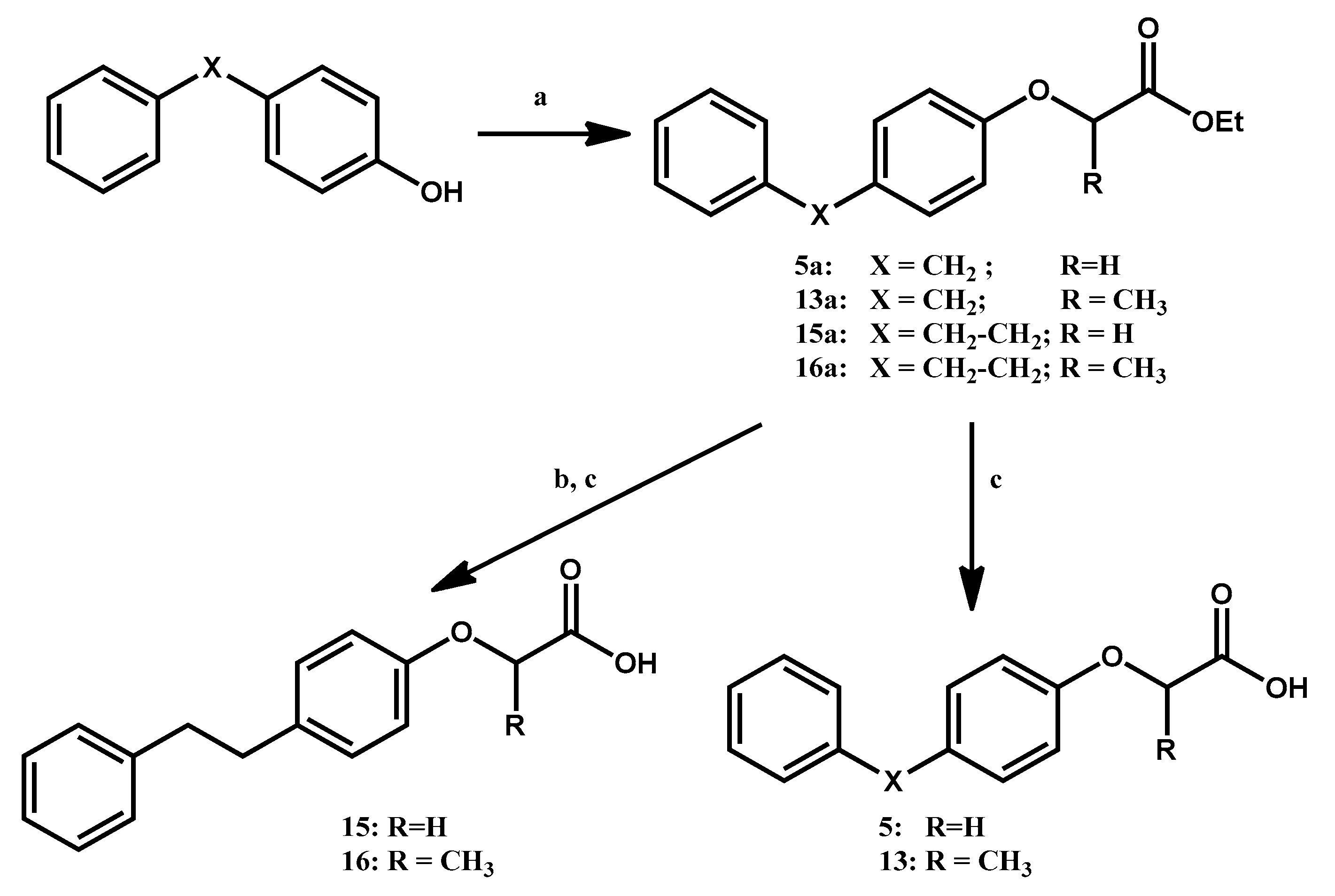
3.1.1. General Procedure for the Preparation of Ethyl Aryloxyacetates 5a, (R,S)-15a and Ethyl 2-Aryloxy-propanoates (R,S)-13a, (R,S)-16a
3.1.2. General Procedure for the Preparation of Ethyl (4-Phenethyl-phenoxy)-acetate 15b and (R,S)-Ethyl 2-(4-Phenethyl-phenoxy)-propanoate 16b
3.1.3. General Procedure for the Preparation of 4-Aryl-phenoxyacetic Acids 5 and 15 and (R,S)-2-(4-Arylphenoxy)-propanoic Acids 13 and 16
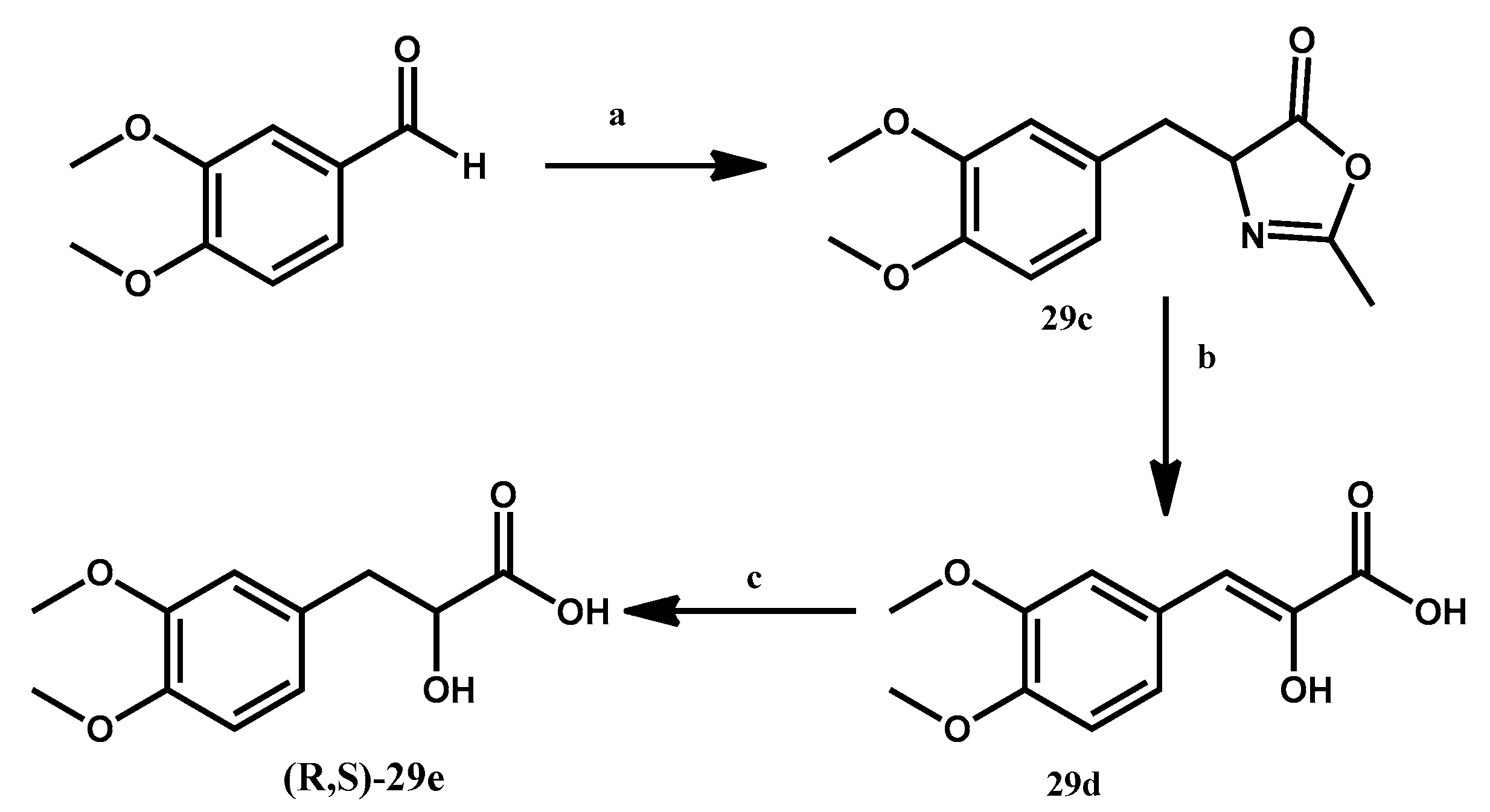
3.1.4. Preparation of 4-[(3,4-Dimethoxyphenyl)methyl-idene]-2-methyl-4,5-dihydro-1,3-oxazol-5-one 29c
3.1.5. Preparation of 3-(3,4-Dimethoxyphenyl)-2-hydroxyacrylic Acid 29d
3.1.6. Preparation of (R,S)-3-(3,4-Dimethoxyphenyl)-2-hydroxypropanoic Acid 29e
3.1.7. General Procedure for the Preparation of Allyl 3-Phenyl-2-hydroxypropanoates (R)-28a, (S)-28a, (R,S)29a
3.1.8. General Procedure for the Preparation of 1-(Allyloxy)-1-oxo-3-phenylpropan-2-yl-2-phenylacrylates
3.1.9. General Procedure for the Preparation of 1-(Allyloxy)-1-oxo-3-phenylpropan-2-yl 2-Arylacrylic Acids
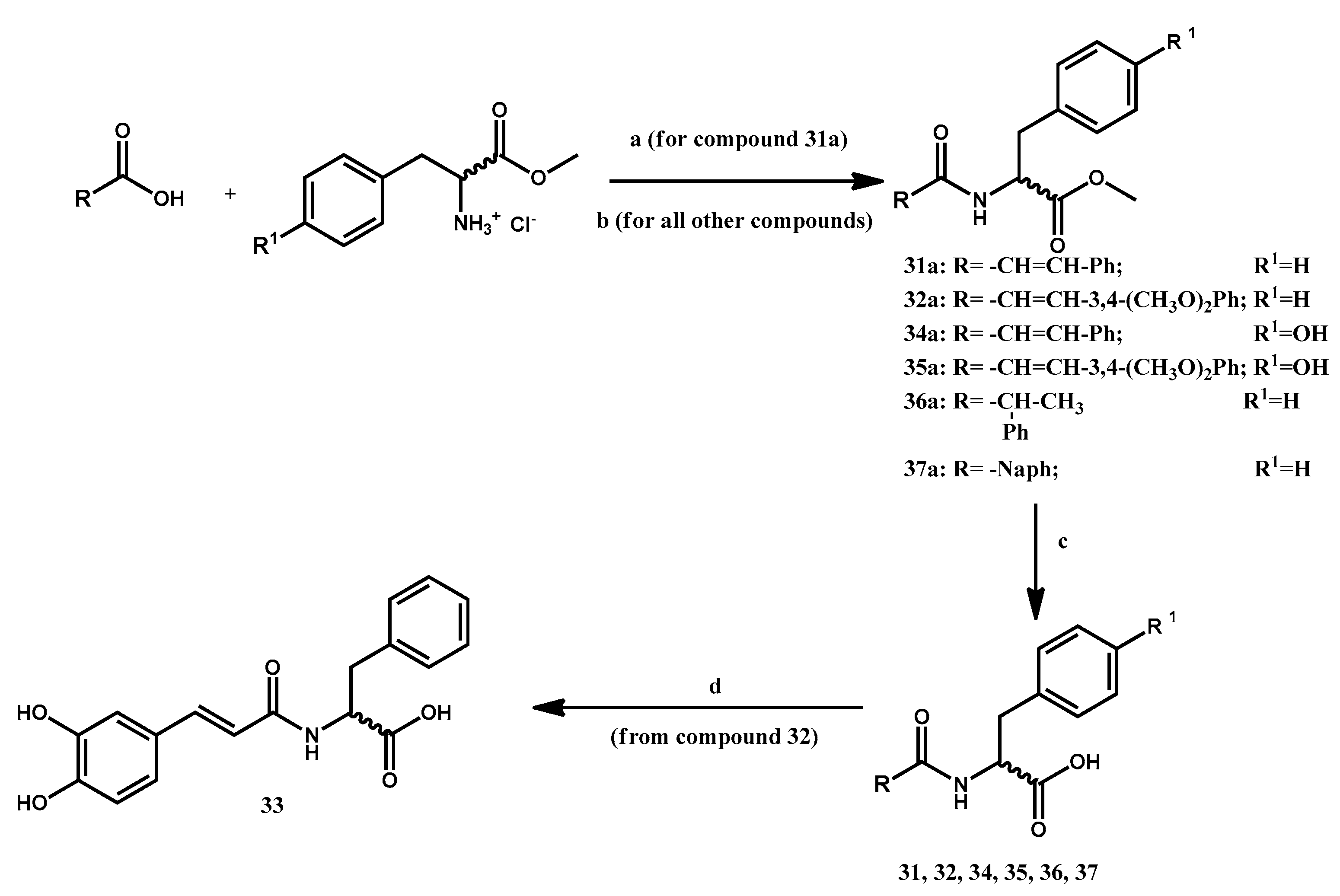
3.1.10. Preparation of Methyl 2-Cinnamamido-3-phenylpropanoate 31a
3.1.11. General Procedure for the Preparation of Methyl 2-(3-Arylacrylamido)-3-arylpropanoates 32a, 34a, 35a
3.1.12. Preparation of Methyl (R,S)-2-phenylpropanoylamido-(S)-3-phenylpropanoate 36a
3.1.13. Preparation of Methyl 2-(2-Naphthamido)-(S)-3-phenylpropanoate 37a
3.1.14. General Procedure for the Preparation of 2-Substituted-3-arylpropanoic Acids 31, 32, 34–37
3.1.15. Preparation of (S)-2-(3-(3,4-Dihydroxyphenyl)acrylamido-3-phenylpropanoic Acid 33
3.2. Biological Methods
3.2.1. Plasmids
3.2.2. Cell Culture and Transfection
3.2.3. FAAH Inhibition Assay
3.3. Molecular Modeling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AEA | Arachidonoylethanolamide, Anandamide |
| CNS | Central Nervous System |
| COX | Cyclooxigenase |
| ECS | Endocannabinoid System |
| ER | Endoplasmic Reticulum |
| GC-MS | Gas Chromatography—Mass Spectrometry |
| HRMS | High Resolution Mass Spectrometry |
| FAAH | Fatty Acid Amide Hydrolase |
| NAAA | N-Acylethanolamine Acid Amidase |
| NAEs | N-Acylethanolamides |
| OEA | Oleoylethanolamide |
| PEA | Palmitoylethanolamide |
| PPARs | Peroxisome Proliferator-Activated Receptors |
| NMR | Nuclear Magnetic Resonance |
| T2DM | Type 2 Diabetes Mellitus |
References
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications--A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Mansour, M. The roles of peroxisome proliferator-activated receptors in the metabolic syndrome. Prog. Mol. Biol. Transl. Sci. 2014, 121, 217–266. [Google Scholar] [CrossRef] [PubMed]
- Laghezza, A.; Montanari, R.; Lavecchia, A.; Piemontese, L.; Pochetti, G.; Iacobazzi, V.; Infantino, V.; Capelli, D.; De Bellis, M.; Liantonio, A.; et al. On the Metabolically Active Form of Metaglidasen: Improved Synthesis and Investigation of Its Peculiar Activity on Peroxisome Proliferator-Activated Receptors and Skeletal Muscles. ChemMedChem 2015, 10, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Pirat, C.; Farce, A.; Lebègue, N.; Renault, N.; Furman, C.; Millet, R.; Yous, S.; Speca, S.; Berthelot, P.; Desreumaux, P.; et al. Targeting Peroxisome Proliferator-Activated Receptors (PPARs): Development of Modulators. J. Med. Chem. 2012, 55, 4027–4061. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, S.E. Cannabinoids go nuclear: Evidence for activation of peroxisome proliferator-activated receptors. Br. J. Pharmacol. 2007, 152, 576–582. [Google Scholar] [CrossRef]
- O’Sullivan, S.E. An update on PPAR activation by cannabinoids. Br. J. Pharmacol. 2016, 173, 1899–1910. [Google Scholar] [CrossRef]
- Sun, Y.; Bennett, A. Cannabinoids: A New Group of Agonists of PPARs. PPAR Res. 2007, 2007, 7. [Google Scholar] [CrossRef]
- Orellana-Serradell, O.; Poblete, C.E.; Sanchez, C.; Castellón, E.A.; Gallegos, I.; Huidobro, C.; Llanos, M.N.; Contreras, H.R. Proapoptotic effect of endocannabinoids in prostate cancer cells. Oncol. Rep. 2015, 33, 1599–1608. [Google Scholar] [CrossRef]
- Ortega, A.; García-Hernández, V.M.; Ruiz-García, E.; Meneses-García, A.; Herrera-Gómez, A.; Aguilar-Ponce, J.L.; Montes-Servín, E.; Prospero-García, O.; Del Angel, S.A. Comparing the effects of endogenous and synthetic cannabinoid receptor agonists on survival of gastric cancer cells. Life Sci. 2016, 165, 56–62. [Google Scholar] [CrossRef]
- Toczek, M.; Malinowska, B. Enhanced endocannabinoid tone as a potential target of pharmacotherapy. Life Sci. 2018, 204, 20–45. [Google Scholar] [CrossRef]
- Petrosino, S.; Di Marzo, V. FAAH and MAGL inhibitors: Therapeutic opportunities from regulating endocannabinoid levels. Curr. Opin. Investig. Drugs (Lond. Engl. 2000) 2010, 11, 51–62. [Google Scholar]
- Chiou, L.-C.; Hu, S.S.-J.; Ho, Y.-C. Targeting the cannabinoid system for pain relief? Acta Anaesthesiol. Taiwanica 2013, 51, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, L.; Loiodice, F.; Piemontese, L.; Tortorella, P.; Laghezza, A. New Approaches to Cancer Therapy: Combining Fatty Acid Amide Hydrolase (FAAH) Inhibition with Peroxisome Proliferator-Activated Receptors (PPARs) Activation. J. Med. Chem. 2019, 62, 10995–11003. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, L.; Laghezza, A.; Loiodice, F.; Tortorella, P.; Piemontese, L. Combining fatty acid amide hydrolase (FAAH) inhibition with peroxisome proliferator-activated receptor (PPAR) activation: A new potential multi-target therapeutic strategy for the treatment of Alzheimer’s disease. Neural Regen. Res. 2020, 15, 67. [Google Scholar] [CrossRef] [PubMed]
- Panlilio, L.V.; Justinova, Z.; Goldberg, S.R. Inhibition of FAAH and activation of PPAR: New approaches to the treatment of cognitive dysfunction and drug addiction. Pharmacol. Ther. 2013, 138, 84–102. [Google Scholar] [CrossRef]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef]
- Alhouayek, M.; Muccioli, G.G. Harnessing the anti-inflammatory potential of palmitoylethanolamide. Drug Discov. Today 2014, 19, 1632–1639. [Google Scholar] [CrossRef]
- Gabrielsson, L.; Gouveia-Figueira, S.; Häggström, J.; Alhouayek, M.; Fowler, C.J. The anti-inflammatory compound palmitoylethanolamide inhibits prostaglandin and hydroxyeicosatetraenoic acid production by a macrophage cell line. Pharmacol. Res. Perspect. 2017, 5. [Google Scholar] [CrossRef]
- Massaro, M.; Scoditti, E.; Pellegrino, M.; Carluccio, M.A.; Calabriso, N.; Wabitsch, M.; Storelli, C.; Wright, M.; De Caterina, R. Therapeutic potential of the dual peroxisome proliferator activated receptor (PPAR)α/γ agonist aleglitazar in attenuating TNF-α-mediated inflammation and insulin resistance in human adipocytes. Pharmacol. Res. 2016, 107, 125–136. [Google Scholar] [CrossRef]
- Fracchiolla, G.; Laghezza, A.; Piemontese, L.; Carbonara, G.; Lavecchia, A.; Tortorella, P.; Crestani, M.; Novellino, E.; Loiodice, F. Synthesis, Biological Evaluation, and Molecular Modeling Investigation of Chiral Phenoxyacetic Acid Analogues with PPARα and PPARγ Agonist Activity. ChemMedChem 2007, 2, 641–654. [Google Scholar] [CrossRef]
- Fracchiolla, G.; Laghezza, A.; Piemontese, L.; Parente, M.; Lavecchia, A.; Pochetti, G.; Montanari, R.; Di Giovanni, C.; Carbonara, G.; Tortorella, P.; et al. Synthesis, biological evaluation and molecular investigation of fluorinated peroxisome proliferator-activated receptors α/γ dual agonists. Bioorg. Med. Chem. 2012, 20, 2141–2151. [Google Scholar] [CrossRef] [PubMed]
- Piemontese, L.; Fracchiolla, G.; Carrieri, A.; Parente, M.; Laghezza, A.; Carbonara, G.; Sblano, S.; Tauro, M.; Gilardi, F.; Tortorella, P.; et al. Design, synthesis and biological evaluation of a class of bioisosteric oximes of the novel dual peroxisome proliferator-activated receptor α/γ ligand LT175. Eur. J. Med. Chem. 2015, 90, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Fracchiolla, G.; Lavecchia, A.; Laghezza, A.; Piemontese, L.; Trisolini, R.; Carbonara, G.; Tortorella, P.; Novellino, E.; Loiodice, F. Synthesis, biological evaluation, and molecular modeling investigation of chiral 2-(4-chloro-phenoxy)-3-phenyl-propanoic acid derivatives with PPARα and PPARγ agonist activity. Bioorganic Med. Chem. 2008, 16, 9498–9510. [Google Scholar] [CrossRef]
- De Vivo, M.; Scarpelli, R.; Cavalli, A.; Migliore, M.; Piomelli, D.; Habrant, D.; Favia, A. Multitarget Faah and Cox Inhibitors and Therapeutical Uses Thereof. U.S. Patent WO2014023643A1, 13 February 2014. [Google Scholar]
- Favia, A.D.; Habrant, D.; Scarpelli, R.; Migliore, M.; Albani, C.; Bertozzi, S.M.; Dionisi, M.; Tarozzo, G.; Piomelli, D.; Cavalli, A.; et al. Identification and Characterization of Carprofen as a Multitarget Fatty Acid Amide Hydrolase/Cyclooxygenase Inhibitor. J. Med. Chem. 2012, 55, 8807–8826. [Google Scholar] [CrossRef] [PubMed]
- Bertolacci, L.; Romeo, E.; Veronesi, M.; Magotti, P.; Albani, C.; Dionisi, M.; Lambruschini, C.; Scarpelli, R.; Cavalli, A.; De Vivo, M.; et al. A Binding Site for Nonsteroidal Anti-inflammatory Drugs in Fatty Acid Amide Hydrolase. J. Am. Chem. Soc. 2013, 135, 22–25. [Google Scholar] [CrossRef]
- Calleri, E.; Pochetti, G.; Dossou, K.S.S.; Laghezza, A.; Montanari, R.; Capelli, D.; Prada, E.; Loiodice, F.; Massolini, G.; Bernier, M.; et al. Resveratrol and its metabolites bind to PPARs. ChemBioChem 2014, 15, 1154–1160. [Google Scholar] [CrossRef]
- Rauf, A.; Imran, M.; Suleria, H.A.R.; Ahmad, B.; Peters, D.G.; Mubarak, M.S. A comprehensive review of the health perspectives of resveratrol. Food Funct. 2017, 8, 4284–4305. [Google Scholar] [CrossRef]
- Porcelli, L.; Gilardi, F.; Laghezza, A.; Piemontese, L.; Mitro, N.; Azzariti, A.; Altieri, F.; Cervoni, L.; Fracchiolla, G.; Giudici, M.; et al. Synthesis, characterization and biological evaluation of ureidofibrate-like derivatives endowed with peroxisome proliferator-activated receptor activity. J. Med. Chem. 2012, 55, 37–54. [Google Scholar] [CrossRef]
- Han, J.; Wang, D.; Ye, L.; Li, P.; Hao, W.; Chen, X.; Ma, J.; Wang, B.; Shang, J.; Li, D.; et al. Rosmarinic acid protects against inflammation and cardiomyocyte apoptosis during myocardial ischemia/reperfusion injury by activating peroxisome proliferator-activated receptor gamma. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef]
- Fracchiolla, G.; Laghezza, A.; Piemontese, L.; Tortorella, P.; Mazza, F.; Montanari, R.; Pochetti, G.; Lavecchia, A.; Novellino, E.; Pierno, S.; et al. New 2-Aryloxy-3-phenyl-propanoic Acids As Peroxisome Proliferator-Activated Receptors α/γ Dual Agonists with Improved Potency and Reduced Adverse Effects on Skeletal Muscle Function. J. Med. Chem. 2009, 52, 6382–6393. [Google Scholar] [CrossRef]
- Piemontese, L.; Cerchia, C.; Laghezza, A.; Ziccardi, P.; Sblano, S.; Tortorella, P.; Iacobazzi, V.; Infantino, V.; Convertini, P.; Dal Piaz, F.; et al. New diphenylmethane derivatives as peroxisome proliferator-activated receptor alpha/gamma dual agonists endowed with anti-proliferative effects and mitochondrial activity. Eur. J. Med. Chem. 2017, 127, 379–397. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Skillman, A.G.; Nicholls, A. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef]
- OpenEye Scientific Software ROCS. Available online: https://www.eyesopen.com/rocs (accessed on 23 September 2020).
- Raspé, E.; Madsen, L.; Lefebvre, A.M.; Leitersdorf, I.; Gelman, L.; Peinado-Onsurbe, J.; Dallongeville, J.; Fruchart, J.C.; Berge, R.; Staels, B. Modulation of rat liver apolipoprotein gene expression and serum lipid levels by tetradecylthioacetic acid (TTA) via PPARalpha activation. J. Lipid Res. 1999, 40, 2099–2110. [Google Scholar]
- OpenEye Scientific Software QUACPAC. Available online: https://www.eyesopen.com/quacpac (accessed on 23 September 2020).
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- El Khoury, L.; Santos-Martins, D.; Sasmal, S.; Eberhardt, J.; Bianco, G.; Ambrosio, F.A.; Solis-Vasquez, L.; Koch, A.; Forli, S.; Mobley, D.L. Comparison of affinity ranking using AutoDock-GPU and MM-GBSA scores for BACE-1 inhibitors in the D3R Grand Challenge 4. J. Comput. Aided. Mol. Des. 2019, 33, 1011–1020. [Google Scholar] [CrossRef]
- Forli, S.; Olson, A.J. A force field with discrete displaceable waters and desolvation entropy for hydrated ligand docking. J. Med. Chem. 2012, 55, 623–638. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | FAAH | PPARα | PPARγ | ||
|---|---|---|---|---|---|
| IC50 (μM) | EC50 (µM) | Emax | EC50 (µM) | Emax | |
| 1 | i | i | 0.04 ± 0.02 | 100 ± 9 | |
| 2 | i | 1.6 ± 0.3 | 100 ± 10 | i | |
| 3 | >100 (40 ± 20% @ 100 μM) | i | i | ||
| 4 | ≈100 (62.05 ± 5.00% @ 100 μM) | i | i | ||
| 5 | 40.2 ± 17.7 | 24 ± 5 | 34 ± 8% | 33 ± 23 | 9 ± 6% |
| 8 [29] | 30.5 ± 12.6 | 0.12 ± 0.16 | 87 ± 7% | 0.83 ± 0.75 | 37 ± 11% |
| 12 | 26.1 ± 11.4 | i | i | ||
| JZL195 | 0.019 ± 0.003 | - | - | ||
| Compound | FAAH | PPARα | PPARγ | ||
|---|---|---|---|---|---|
| IC50 (μM) | EC50 (µM) | Emax | EC50 (µM) | Emax | |
| 13 | >100 (30.0 ± 3.0% @ 100 μM) | 4.6 ± 1.8 | 116 ± 1% | i | |
| 14 [31] | i | 0.0159 ± 0.0003 | 115 ± 27% | 0.58 ± 0.19 | 42 ± 1% |
| 15 | ≈100 (63 ± 14% @ 100 μM) | i | i | ||
| 16 | >100 (32.2 ± 2.8% @100 μM) | 17.6 ± 3 | 116 ± 1% | 24 ± 6 | 18 ± 7% |
| 17 [32] | i | 0.039 ± 0.013 | 47 ± 13 | 0.29 ± 0.08 | 26 ± 3 |
| 18 [31] | 24 ± 2.5 | 1.75 ± 0.12 | 57 ± 4% | 0.72 ± 0.27 | 50 ± 1% |
| 19 [31] | >100 (30.5 ± 3.2% @ 100 μM) | 0.16 ± 0.02 | 107 ± 2% | 1.5 ± 0.4 | 34 ± 1% |
| 20 [31] | >100 (39.2 ± 7.3% @ 100 μM) | 3.6 ± 1.5 | 78 ± 4% | 13 ± 3 | 27 ± 6% |
| 21 [31] | >100 (34.0 ± 4.6% @ 100 μM) | 1.8 ± 1.0 | 106 ± 1% | 9.1 ± 0.9 | 40 ± 2% |
| 22 [29] | >100 (9.9 ± 3.8% @ 100 μM) | 0.030 ± 0.016 | 82 ± 7% | 0.11 ± 0.06 | 50 ± 4% |
| 23 [29] | >100 (12.9 ± 3.3% @ 100 μM) | 0.003 ± 0.001 | 91 ± 3% | 0.07 ± 0.05 | 116 ± 9% |
| 24 [29] | >100 (8.7 ± 1.8% @ 100 μM) | 0.056 ± 0.034 | 75 ± 4% | 0.59 ± 0.11 | 50 ± 5% |
| 25 [29] | ≈100 (45.1 ± 7.3% @ 100 μM) | 0.62 ± 0.26 | 78 ± 4% | 2.3 ± 1.0 | 32 ± 5% |
| 26 [29] | >100 (35 ± 12% @ 100 μM) | 0.025 ± 0.017 | 93 ± 6% | 0.15 ± 0.06 | 59 ± 2% |

| Compound | R | R’ | X |
|---|---|---|---|
| 27 (Clovamide) | OH | OH | NH |
| (R)-28, (S)-28 | H | H | O |
| 29 | OCH3 | H | O |
| (R)-30 | H | OCH3 | O |
| 31 | H | H | NH |
| 32 | OCH3 | H | NH |
| 33 | OH | H | NH |
| 34 | H | 4-OH | NH |
| 35 | OCH3 | 4-OH | NH |
 |  | ||
| 36 | 37 | ||
| Compound | FAAH | PPARα | PPARγ | ||
|---|---|---|---|---|---|
| IC50 (μM) | EC50 (µM) | Emax | EC50 (µM) | Emax | |
| (S)-28 | i | 24 ± 5 | 34 ± 8% | 33 ± 23 | 9 ± 6% |
| 33 | 12.6 ± 3.1 | i | i | ||
| Compound | FEB a | LE b | POP c | CSA d |
|---|---|---|---|---|
| 5 | −7.10 | −0.395 | 45/1000 | 351.02 |
| (R)-14 | −8.97 | −0.359 | 258/1000 | 385.37 |
| (S)-14 | −8.96 | −0.358 | 131/1000 | 436.09 |
| (R)-18 | −9.29 | −0.357 | 256/1000 | 484.92 |
| (S)-18 | −9.37 | −0.360 | 170/1000 | 482.02 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brunetti, L.; Carrieri, A.; Piemontese, L.; Tortorella, P.; Loiodice, F.; Laghezza, A. Beyond the Canonical Endocannabinoid System. A Screening of PPAR Ligands as FAAH Inhibitors. Int. J. Mol. Sci. 2020, 21, 7026. https://doi.org/10.3390/ijms21197026
Brunetti L, Carrieri A, Piemontese L, Tortorella P, Loiodice F, Laghezza A. Beyond the Canonical Endocannabinoid System. A Screening of PPAR Ligands as FAAH Inhibitors. International Journal of Molecular Sciences. 2020; 21(19):7026. https://doi.org/10.3390/ijms21197026
Chicago/Turabian StyleBrunetti, Leonardo, Antonio Carrieri, Luca Piemontese, Paolo Tortorella, Fulvio Loiodice, and Antonio Laghezza. 2020. "Beyond the Canonical Endocannabinoid System. A Screening of PPAR Ligands as FAAH Inhibitors" International Journal of Molecular Sciences 21, no. 19: 7026. https://doi.org/10.3390/ijms21197026
APA StyleBrunetti, L., Carrieri, A., Piemontese, L., Tortorella, P., Loiodice, F., & Laghezza, A. (2020). Beyond the Canonical Endocannabinoid System. A Screening of PPAR Ligands as FAAH Inhibitors. International Journal of Molecular Sciences, 21(19), 7026. https://doi.org/10.3390/ijms21197026