Metabolism of Glycosphingolipids and Their Role in the Pathophysiology of Lysosomal Storage Disorders

Abstract
1. Introduction.
2. Glycosphingolipids
2.1. Glycosphingolipid Structures
2.2. Gangliosides
2.3. Isolation and Analysis of Glycosphingolipids
2.4. Functions of Glycosphingolipids
2.5. Microbial Interactions with Glycosphingolipids
3. Biosynthesis of Glycosphingolipids
3.1. Biosynthesis of Simple Glycosphingolipids
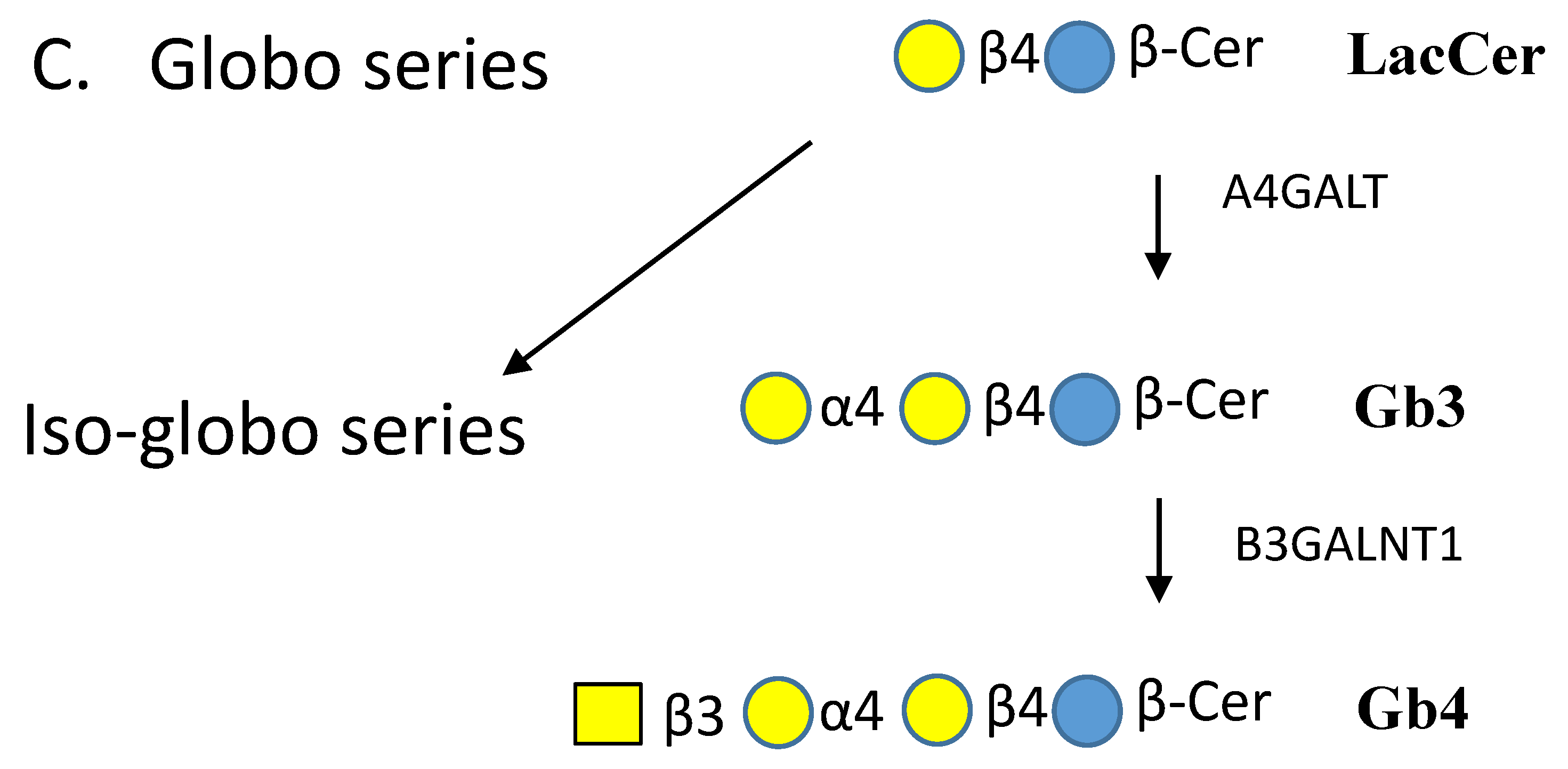
3.2. Glycosyltransferases that Extend Glycan Chains of Glycosphingolipids
3.3. Genetic Defects of Glycosphingolipid Biosynthesis
4. Degradation of Glycosphingolipids
5. Lysosomal Storage Disorders
5.1. Gaucher and Krabbe Disease
5.2. GM1-Gangliosidosis
5.3. GM2-Gangliosidosis: Tay–Sachs and Sandhoff Diseases
5.4. Other Lysosomal Storage Diseases in Brief
6. Pathophysiology of Lysosomal Storage Diseases
6.1. Consequences of Intracellular Aggregation: Lysosomal Membrane Permeability
6.2. Inclusion Body Formation
6.3. Cell–Cell Transmission of Pathology
6.4. Disruption of Autophagy
6.5. Alzheimer’s Disease
6.6. Parkinson’s Disease
7. Therapeutic Approaches for Lysosomal Storage Diseases
8. Conclusions
Funding
Conflicts of Interest
Abbreviations
| Aβ | amyloid β |
| AD | Alzheimer’s disease |
| ALS | autophagic-lysosomal system |
| BBB | Blood-brain barrier |
| CAZy | Carbohydrate Active Enzymes |
| Cer | ceramide |
| CNS | central nervous system |
| EGFR | epidermal growth factor receptor |
| ER | endoplasmic reticulum |
| ERT | enzyme replacement therapy |
| Fuc | fucose |
| Gal | galactose |
| GalNAc | N-acetylgalactosamine |
| GD | Gaucher disease |
| GDNF | glial derived neurotropic factor |
| GH | glycosyl hydrolase |
| Glc | glucose |
| GlcCer | glucosylβ-ceramide |
| GlcNAc | N-acetylglucosamine |
| GLTP | Glycolipid transfer protein |
| GPI | glycosylphosphatidylinositol |
| GSL | glycosphingolipid |
| GT | glycosyltransferase |
| HPLC | high pressure liquid chromatography |
| HPAEC | high performance anion exchange chromatography |
| LacCer | lactosyl-ceramide |
| LC | liquid chromatography |
| LMP | lysosomal membrane permeability |
| LRP | laminin receptor protein |
| LSD | lysosomal storage disease |
| Lyso-GSL | GSL lacking the fatty acid moiety |
| Man-6-P | mannose-6-phosphate |
| MS | mass spectrometry |
| Neu5Ac | 5-N-acetylneuraminic acid |
| Neu5Gc | 5-N-glycolylneuraminic acid |
| NMR | nuclear magnetic resonance |
| PD | Parkinson’s disease |
| PPCA | Cathepsin A |
| SAP | saposin |
| SD | Sandhoff disease |
| SNCA | α-synuclein |
| TNF | tumor necrosis factor |
| TSD | Tay–Sachs disease |
| UPS | ubiquitin-proteasome system |
References
- Russo, D.; Capolupo, L.; Loomba, J.S.; Sticco, L.; D’Angelo, G. Glycosphingolipid metabolism in cell fate specification. J. Cell Sci. 2018, 131, jcs219204. [Google Scholar] [CrossRef] [PubMed]
- Ledeen, R.; Wu, G. Gangliosides of the nervous system. Methods Mol. Biol. 2018, 1804, 19–55. [Google Scholar] [PubMed]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar]
- Honke, K. Biosynthesis and biological function of sulfoglycolipids. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2013, 89, 129–138. [Google Scholar] [CrossRef]
- Sonnino, S.; Chiricozzi, E.; Grassi, S.; Mauri, L.; Prioni, S.; Prinetti, A. Gangliosides in membrane organization. Prog. Mol. Biol. Transl. Sci. 2018, 156, 83–120. [Google Scholar]
- Jennemann, R.; Kaden, S.; Sandhoff, R.; Nordström, V.; Wang, S.; Volz, M.; Robine, S.; Amen, N.; Rothermel, U.; Wiegandt, H.; et al. Glycosphingolipids are essential for intestinal endocytic function. J. Biol. Chem. 2012, 287, 32598–32616. [Google Scholar] [CrossRef]
- Tettamanti, G.; Bonali, F.; Marchesini, S.; Zambotti, V. A new procedure for the extraction, purification and fractionation of brain gangliosides. BBA-Lipid Lipid Metab. 1973, 296, 160–170. [Google Scholar] [CrossRef]
- Ando, T.; Li, S.C.; Ito, M.; Li, Y.T. Facile method for the preparation of lyso-GM1 and lyso-GM2. J. Chromatogr. A 2005, 1078, 193–195. [Google Scholar] [CrossRef]
- Groux-Degroote, S.; Guerardel, Y.; Delannoy, P. Gangliosides: Structures, Biosynthesis, Analysis, and Roles in Cancer. ChemBioChem 2017, 18, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Kolter, T.; Sandhoff, K. Sphingolipid metabolism disease. Biochim. Biophys. Acta (BBA)-Biomembr. 2006, 1758, 2057–2079. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.K.; Tsai, Y.T.; Ariga, T.; Yanagisawa, M. Structures, biosynthesis, and functions of gangliosides–an overview. J. Oleo Sci. 2011, 60, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Vorwerk, C.K. Ganglioside patterns in human spinal cord. Spinal Cord 2001, 39, 628–632. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vajn, K.; Viljetić, B.; Degmečić, I.V.; Schnaar, R.L.; Heffer, M. Differential Distribution of Major Brain Gangliosides in the Adult Mouse Central Nervous System. PLoS ONE 2013, 8, e75720. [Google Scholar] [CrossRef] [PubMed]
- Walia, J.S.; Altaleb, N.; Bello, A.; Kruck, C.; LaFave, M.C.; Varshney, G.K.; Burgess, S.M.; Chowdhury, B.; Hurlbut, D.; Hemming, R.; et al. Long-term correction of Sandhoff disease following intravenous delivery of rAAV9 to mouse neonates. Mol. Ther. 2015, 23, 414–422. [Google Scholar] [CrossRef]
- Indellicato, R.; Parini, R.; Domenighini, R.; Malagolini, N.; Iascone, M.; Gasperini, S.; Masera, N.; dall’Olio, F.; Trinchera, M. Total loss of GM3 synthase activity by a normally processed enzyme in a novel variant and in all ST3GAL5 variants reported to cause a distinct congenital disorder of glycosylation. Glycobiology 2019, 29, 229–241. [Google Scholar] [CrossRef]
- Lawson, C.A.; Martin, D.R. Animal models of GM2 gangliosidosis: Utility and limitations. Appl. Clin. Genet. 2016, 9, 111–120. [Google Scholar] [CrossRef]
- Phaneuf, D.; Wakamatsu, N.; Huang, J.Q.; Borowski, A.; Peterson, A.C.; Fortunato, S.R.; Ritter, G.; Igdoura, S.A.; Morales, C.R.; Benoit, G.; et al. Dramatically different phenotypes in mouse models of human Tay-Sachs and Sandhoff diseases. Hum. Mol. Genet. 1996, 5, 1–14. [Google Scholar] [CrossRef]
- Cong, P.-X.; Gao, R.; Xue, C.-H.; Li, Z.-J.; Zhang, H.-W.; Khan, M.N.; Xue, Y.; Sugawara, T.; Xu, J. Molecular species analysis of monosialogangliosides from sea urchin Strongylocentrotus nudus by RPLC-ESI-MS/MS. Food Chem. 2015, 166, 473–478. [Google Scholar] [CrossRef]
- Huang, Q.; Zhou, X.; Liu, D.; Xin, B.; Cechner, K.; Wang, H.; Zhou, A. A new liquid chromatography/tandem mass spectrometry method for quantification of gangliosides in human plasma. Anal. Biochem. 2014, 455, 26–34. [Google Scholar] [CrossRef]
- Busch, C.M.; Desai, A.V.; Moorthy, G.S.; Fox, E.; Balis, F.M. A validated HPLC-MS/MS method for estimating the concentration of the ganglioside, GD2, in human plasma or serum. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1102–1103, 60–65. [Google Scholar] [CrossRef]
- Suzuki, A.; Suzuki, M.; Ito, E.; Nitta, T.; Inokuchi, J. Mass spectrometry of gangliosides. Methods Mol. Biol. 2018, 1804, 207–221. [Google Scholar] [PubMed]
- Caron, M.; Joubert-Caron, R.; Cartier, J.R.; Chadli, A.; Bladier, D. Study of lectin-ganglioside interactions by high-performance liquid affinity chromatography. J. Chromatogr. A 1993, 646, 327–333. [Google Scholar] [CrossRef]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Jennemann, R.; Sandhoff, R.; Wang, S.; Kiss, E.; Gretz, N.; Zuliani, C.; Martin-Villalba, A.; Jäger, R.; Schorle, H.; Kenzelmann, M.; et al. Cell-specific deletion of glucosylceramide synthase in brain leads to severe neural defects after birth. Proc. Natl. Acad. Sci. USA 2005, 102, 12459–12464. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, B.; Sehl, C.; Wilker, B.; Kamler, M.; Gulbins, E.; Becker, K.A. Glucosylceramide Critically Contributes to the Host Defense of Cystic Fibrosis Lungs. Cell. Physiol. Biochem. 2017, 41, 1208–1218. [Google Scholar] [CrossRef]
- Ngamukote, S.; Yanagisawa, M.; Ariga, T.; Ando, S.; Yu, R.K. Developmental changes of glycosphingolipids and expression of glycogenes in mouse brains. J. Neurochem. 2007, 103, 2327–2341. [Google Scholar] [CrossRef]
- Loberto, N.; Tebon, M.; Lampronti, I.; Marchetti, N.; Aureli, M.; Bassi, R.; Giri, M.G.; Bezzerri, V.; Lovato, V.; Cantù, C.; et al. GBA2-encoded β-glucosidase activity is involved in the inflammatory response to Pseudomonas aeruginosa. PLoS ONE 2014, 9, e104763. [Google Scholar] [CrossRef]
- Qiu, L.; Liu, Z.; Wu, C.; Chen, W.; Chen, Y.; Zhang, B.; Li, J.; Liu, H.; Huang, N.; Jiang, Z.; et al. C6-ceramide induces salivary adenoid cystic carcinoma cell apoptosis via IP3R-activated UPR and UPR-independent pathways. Biochem. Biophys. Res. Commun. 2020, 525, 997–1003. [Google Scholar] [CrossRef]
- Medler, T.R.; Petrusca, D.N.; Lee, P.J.; Hubbard, W.C.; Berdyshev, E.V.; Skirball, J.; Kamocki, K.; Schuchman, E.; Tuder, R.M.; Petrache, I. Apoptotic sphingolipid signaling by ceramides in lung endothelial cells. Am. J. Respir. Cell Mol. Biol. 2008, 38, 639–646. [Google Scholar] [CrossRef]
- Mirkin, B.L.; Clark, S.H.; Zhang, C. Inhibition of human neuroblastoma cell proliferation and EGF receptor phosphorylation by gangliosides GM1, GM3, GD1A and GT1B. Cell Proliferat. 2002, 35, 105–115. [Google Scholar] [CrossRef]
- Zhuo, D.; Guan, F. Ganglioside GM1 promotes contact inhibition of growth by regulating the localization of epidermal growth factor receptor from glycosphingolipid-enriched microdomain to caveolae. Cell Proliferat. 2019, 52, e12639. [Google Scholar] [CrossRef] [PubMed]
- Kabayama, K.; Sato, T.; Saito, K.; Loberto, N.; Prinetti, A.; Sonnino, S.; Kinjo, M.; Igarashi, Y.; Inokuchi, J. Dissociation of the insulin receptor and caveolin-1 complex by ganglioside GM3 in the state of insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 13678–13683. [Google Scholar] [CrossRef] [PubMed]
- Inokuchi, J. Membrane microdomains and insulin resistance. FEBS Lett. 2010, 584, 1864–1871. [Google Scholar] [CrossRef] [PubMed]
- Sonnino, S.; Prinetti, A. The role of sphingolipids in neuronal plasticity of the brain. J. Neurochem. 2016, 137, 485–488. [Google Scholar] [CrossRef]
- Ledeen, R.W.; Wu, G.; André, S.; Bleich, D.; Huet, G.; Kaltner, H.; Kopitz, J.; Gabius, H.J. Beyond glycoproteins as galectin counterreceptors: Tumor-effector T cell growth control via ganglioside GM1. Ann. N. Y. Acad. Sci. 2012, 1253, 206–221. [Google Scholar] [CrossRef]
- Newburn, E.N.; Duchemin, A.M.; Neff, N.H.; Hadjiconstantinou, M. GM1 ganglioside enhances Ret signaling in striatum. J. Neurochem. 2014, 130, 541–554. [Google Scholar] [CrossRef]
- Furukawa, K.; Ohmi, Y.; Ohkawa, Y.; Bhuiyan, R.H.; Zhang, P.; Tajima, O.; Hashimoto, N.; Hamamura, K.; Furukawa, K. New era of research on cancer-associated glycosphingolipids. Cancer Sci. 2019, 110, 1544–1551. [Google Scholar] [CrossRef]
- Mishra, S.K.; Gao, Y.G.; Zou, X.; Stephenson, D.J.; Malinina, L.; Hinchcliffe, E.H.; Chalfant, C.E.; Brown, R.E. Emerging roles for human glycolipid transfer protein superfamily members in the regulation of autophagy, inflammation, and cell death. Prog. Lipid Res. 2020, 78, 101031. [Google Scholar] [CrossRef]
- You, J.; O’Hara, S.D.; Velupillai, P.; Castle, S.; Levery, S.; Garcea, R.L.; Benjamin, T. Ganglioside and Non-ganglioside Mediated Host Responses to the Mouse Polyomavirus. PLOS Pathog. 2015, 11, e1005175. [Google Scholar] [CrossRef]
- Sakai, T.; Nishimura, S.I.; Naito, T.; Saito, M. Influenza A virus hemagglutinin and neuraminidase act as novel motile machinery. Sci. Rep. 2017, 7, 45043. [Google Scholar] [CrossRef]
- Perez-Zsolt, D.; Erkizia, I.; Pino, M.; García-Gallo, M.; Martin, M.T.; Benet, S.; Chojnacki, J.; Fernández-Figueras, M.T.; Guerrero, D.; Urrea, V. Anti-Siglec-1 antibodies block Ebola viral uptake and decrease cytoplasmic viral entry. Nat. Microbiol. 2019, 4, 1558–1570. [Google Scholar] [CrossRef] [PubMed]
- Blixt, O.; Collins, B.E.; van den Nieuwenhof, I.M.; Crocker, P.R.; Paulson, J.C. Sialoside Specificity of the Siglec Family Assessed Using Novel Multivalent probes. Identification of potent inhibitors of myelin-associated glycoprotein. J. Biol. Chem. 2003, 278, 31007–31019. [Google Scholar] [CrossRef] [PubMed]
- Puryear, W.B.; Yu, X.; Ramirez, N.P.; Reinhard, B.M.; Gummuluru, S. HIV-1 incorporation of host-cell-derived glycosphingolipid GM3 allows for capture by mature dendritic cells. Proc. Natl. Acad. Sci. USA 2012, 109, 7475–7480. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Miller, C.; Patel, H.V.; Hatch, S.C.; Archer, J.; Ramirez, N.G.; Gummuluru, S. Virus particle release from glycosphingolipid-enriched microdomains is essential for dendritic cell-mediated capture and transfer of HIV-1 and henipavirus. J. Virol. 2014, 88, 8813–8825. [Google Scholar] [CrossRef]
- Izquierdo-Useros, N.; Lorizate, M.; McLaren, P.J.; Telenti, A.; Kräusslich, H.G.; Martinez-Picado, J. HIV-1 capture and transmission by dendritic cells: The role of viral glycolipids and the cellular receptor Siglec-1. PLoS Pathog. 2014, 10, e1004146. [Google Scholar] [CrossRef]
- Hazlett, L.D.; Masinick, S.; Barrett, R.; Rosol, K. Evidence for asialo GM1 as a corneal glycolipid receptor for Pseudomonas aeruginosa adhesion. Infect. Immun. 1993, 61, 5164–5173. [Google Scholar] [CrossRef]
- Babazadeh, A.; Mohseni Afshar, Z.; Javanian, M.; Mohammadnia-Afrouzi, M.; Karkhah, A.; Masrour-Roudsari, J.; Sabbagh, P.; Koppolu, V.; Vasigala, V.K.; Ebrahimpour, S. Influenza Vaccination and Guillain-Barré Syndrome: Reality or Fear. J. Transl. Int. Med. 2019, 7, 137–142. [Google Scholar] [CrossRef]
- Nachamkin, I.; Shadomy, S.V.; Moran, A.P.; Cox, N.; Fitzgerald, C.; Ung, H.; Corcoran, A.T.; Iskander, J.K.; Schonberger, L.B.; Chen, R.T. Anti-ganglioside antibody induction by swine (A/NJ/1976/H1N1) and other influenza vaccines: Insights into vaccine-associated Guillain-Barré syndrome. J. Infect. Dis. 2008, 198, 226–233. [Google Scholar] [CrossRef]
- Blanco, L.P.; DiRita, V.J. Bacterial-associated cholera toxin and GM1 binding are required for transcytosis of classical biotype Vibrio cholerae through an in vitro M cell model system. Cell. Microbiol. 2006, 8, 982–998. [Google Scholar] [CrossRef]
- Tsukamoto, K.; Kohda, T.; Mukamoto, M.; Takeuchi, K.; Ihara, H.; Saito, M.; Kozaki, S. Binding of Clostridium botulinum type C and D neurotoxins to ganglioside and phospholipid. Novel insights into the receptor for clostridial neurotoxins. J. Biol. Chem. 2005, 280, 35164–35171. [Google Scholar] [CrossRef]
- Lingwood, C. Verotoxin Receptor-Based Pathology and Therapies. Front. Cell. Infect. Microbiol. 2020, 10, 123. [Google Scholar] [CrossRef] [PubMed]
- Gamage, S.D.; McGannon, C.M.; Weiss, A.A. Escherichia coli serogroup O107/O117 lipopolysaccharide binds and neutralizes Shiga toxin 2. J. Bacteriol. 2004, 186, 5506–5512. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Raj, S.; Nazemidashtarjandi, S.; Kim, J.; Joffe, L.; Zhang, X.; Singh, A.; Mor, V.; Desmarini, D.; Djordjevic, J.; Raleigh, D.P.; et al. Changes in glucosylceramide structure affect virulence and membrane biophysical properties of Cryptococcus neoformans. Biochim. Biophys. Acta Biomembr. 2017, 1859, 2224–2233. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.K.; Stephenson, D.J.; Chalfant, C.E.; Brown, R.E. Upregulation of human glycolipid transfer protein (GLTP) induces necroptosis in colon carcinoma cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 158–167. [Google Scholar] [CrossRef]
- Malinina, L.; Malakhova, M.L.; Teplov, A.; Brown, R.E.; Patel, D.J. Structural basis for glycosphingolipid transfer specificity. Nature 2004, 430, 1048–1053. [Google Scholar] [CrossRef]
- Sandhoff, R.; Sandhoff, K. Emerging concepts of ganglioside metabolism. FEBS Lett. 2018, 592, 3835–3864. [Google Scholar] [CrossRef]
- Brockhausen, I. Crossroads between bacterial and mammalian glycosyltransferases. Front. Immunol. 2014, 5, 492. [Google Scholar] [CrossRef]
- Hirschberg, C.B. My journey in the discovery of nucleotide sugar transporters of the Golgi apparatus. J. Biol. Chem. 2018, 293, 12653–12662. [Google Scholar] [CrossRef]
- Liu, L.; Xu, Y.X.; Hirschberg, C.B. The role of nucleotide sugar transporters in development of eukaryotes. Semin. Cell Dev. Biol. 2010, 21, 600–608. [Google Scholar] [CrossRef][Green Version]
- Dauber, A.; Ercan, A.; Lee, J.; James, P.; Jacobs, P.P.; Ashline, D.J.; Wang, S.R.; Miller, T.; Hirschhorn, J.N.; Nigrovic, P.A.; et al. Congenital disorder of fucosylation type 2c (LADII) presenting with short stature and developmental delay with minimal adhesion defect. Hum. Mol Genet. 2014, 23, 2880–2887. [Google Scholar] [CrossRef]
- Mühlenhoff, M.; Frosch, M.; Gerardy-Schahn, R. Nuclear localization signal of murine CMP-Neu5Ac synthetase includes residues required for both nuclear targeting and enzymatic activity. J. Biol. Chem. 2002, 277, 19688–19696. [Google Scholar]
- Hayashi, Y.; Nemoto-Sasaki, Y.; Matsumoto, N.; Hama, K.; Tanikawa, T.; Oka, S.; Saeki, T.; Kumasaka, T.; Koizumi, T.; Arai, S.; et al. Complex formation of sphingomyelin synthase 1 with glucosylceramide synthase increases sphingomyelin and decreases glucosylceramide levels. J. Biol. Chem. 2018, 293, 17505–17522. [Google Scholar] [CrossRef] [PubMed]
- Seko, A.; Yamashita, K. Activation of beta1,3-N-acetylglucosaminyltransferase-2 (beta3Gn-T2) by beta3Gn-T8. Possible involvement of beta3Gn-T8 in increasing poly-N-acetyllactosamine chains in differentiated HL-60 cells. J. Biol. Chem. 2008, 283, 33094–33100. [Google Scholar] [CrossRef]
- Olshefski, R.S.; Ladisch, S. Glucosylceramide synthase inhibition enhances vincristine-induced cytotoxicity. Int. J. Cancer 2001, 93, 131–138. [Google Scholar] [CrossRef]
- Yamaji, T.; Hanada, K. Sphingolipid metabolism and interorganellar transport: Localization of sphingolipid enzymes and lipid transfer proteins. Traffic 2015, 16, 101–122. [Google Scholar] [CrossRef] [PubMed]
- Meech, R.; Hu, D.G.; McKinnon, R.A.; Mubarokah, S.N.; Haines, A.Z.; Nair, P.C.; Rowland, A.; Mackenzie, P.I. The UDP-Glycosyltransferase (UGT) Superfamily: New Members, New Functions, and Novel Paradigms. Physiol. Rev. 2019, 99, 1153–1222. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, T.; Satake, H.; Nishie, T.; Okino, N.; Hatta, T.; Otani, H.; Naruse, C.; Suzuki, H.; Sugihara, K.; Kamimura, E.; et al. Lactosylceramide synthases encoded by B4galt5 and 6 genes are pivotal for neuronal generation and myelin formation in mice. PLoS Genet. 2018, 14, e1007545. [Google Scholar] [CrossRef] [PubMed]
- Nishie, T.; Hikimochi, Y.; Zama, K.; Fukusumi, Y.; Ito, M.; Yokoyama, H.; Naruse, C.; Ito, M.; Asano, M. Beta4-galactosyltransferase-5 is a lactosylceramide synthase essential for mouse extra-embryonic development. Glycobiology 2010, 20, 1311–1322. [Google Scholar] [CrossRef]
- Togayachi, A.; Akashima, T.; Ookubo, R.; Kudo, T.; Nishihara, S.; Iwasaki, H.; Natsume, A.; Mio, H.; Inokuchi, J.; Irimura, T.; et al. Molecular cloning and characterization of UDP-GlcNAc:lactosylceramide beta 1,3-N-acetylglucosaminyltransferase (beta 3Gn-T5), an essential enzyme for the expression of HNK-1 and Lewis X epitopes on glycolipids. J. Biol. Chem. 2001, 276, 22032–22040. [Google Scholar] [CrossRef]
- Giraudo, C.G.; Maccioni, H.J. Ganglioside glycosyltransferases organize in distinct multienzyme complexes in CHO-K1 cells. J. Biol. Chem. 2003, 278, 40262–40271. [Google Scholar] [CrossRef]
- Datta, A.K.; Paulson, J.C. Sialyl motifs of sialyltransferases. Indian J. Biochem. Biophys. 1997, 34, 157–165. [Google Scholar] [PubMed]
- Trinchera, M.; Parini, R.; Indellicato, R.; Domenighini, R.; Dall’Olio, F. Diseases of ganglioside biosynthesis: An expanding group of congenital disorders of glycosylation. Mol. Genet. Metab. 2018, 124, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Max, S.R.; Maclaren, N.K.; Brady, R.O.; Bradley, R.M.; Rennels, M.B.; Tanaka, J.; Garcia, J.H.; Cornblath, M. GM3 (Hematoside) Sphingolipodystrophy. N. Engl. J. Med. 1974, 291, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Indellicato, R.; Domenighini, R.; Malagolini, N.; Cereda, A.; Mamoli, D.; Pezzani, L.; Iascone, M.; Dall’olio, F.; Trinchera, M. A novel nonsense and inactivating variant of ST3GAL3 in two infant siblings suffering severe epilepsy and expressing circulating CA19.9. Glycobiology 2020, 30, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Edvardson, S.; Baumann, A.M.; Mühlenhoff, M.; Stephan, O.; Kuss, A.W.; Shaag, A.; He, L.; Zenvirt, S.; Tanzi, R.; Gerardy-Schahn, R.; et al. West syndrome caused by ST3Gal-III deficiency. Epilepsia 2013, 54, e24–e27. [Google Scholar] [CrossRef] [PubMed]
- van Diepen, L.; Buettner, F.; Hoffmann, D.; Thiesler, C.T.; von Bohlen Und Halbach, O.; von Bohlen Und Halbach, V.; Jensen, L.R.; Steinemann, D.; Edvardson, S.; Elpeleg, O.; et al. A patient-specific induced pluripotent stem cell model for West syndrome caused by ST3GAL3 deficiency. Eur. J. Hum. Genet. 2018, 26, 1773–1783. [Google Scholar] [CrossRef]
- Hu, H.; Eggers, K.; Chen, W.; Garshasbi, M.; Motazacker, M.M.; Wrogemann, K.; Kahrizi, K.; Tzschach, A.; Hosseini, M.; Bahman, I.; et al. ST3GAL3 mutations impair the development of higher cognitive functions. Am. J. Hum. Genet. 2011, 89, 407–414. [Google Scholar] [CrossRef]
- Harlalka, G.V.; Lehman, A.; Chioza, B.; Baple, E.L.; Maroofian, R.; Cross, H.; Sreekantan-Nair, A.; Priestman, D.A.; Al-Turki, S.; McEntagart, M.E.; et al. Mutations in B4GALNT1 (GM2 synthase) underlie a new disorder of ganglioside biosynthesis. Brain 2013, 136 Pt 12, 3618–3624. [Google Scholar] [CrossRef]
- Chiricozzi, E.; Mauri, L.; Lunghi, G.; Di Biase, E.; Fazzari, M.; Maggioni, M.; Valsecchi, M.; Prioni, S.; Loberto, N.; Pomè, D.Y.; et al. Parkinson’s disease recovery by GM1 oligosaccharide treatment in the B4galnt1+/- mouse model. Sci. Rep. 2019, 9, 19330. [Google Scholar] [CrossRef]
- Bhuiyan, R.H.; Ohmi, Y.; Ohkawa, Y.; Zhang, P.; Takano, M.; Hashimoto, N.; Okajima, T.; Furukawa, K.; Furukawa, K. Loss of Enzyme Activity in Mutated B4GALNT1 Gene Products in Patients with Hereditary Spastic Paraplegia Results in Relatively Mild Neurological Disorders: Similarity with Phenotypes of B4galnt1 Knockout Mice. Neuroscience 2019, 397, 94–106. [Google Scholar] [CrossRef]
- Wu, G.; Lu, Z.H.; Kulkarni, N.; Amin, R.; Ledeen, R.W. Mice lacking major brain gangliosides develop parkinsonism. Neurochem. Res. 2011, 36, 1706–1714. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Lu, Z.H.; Seo, J.H.; Alselehdar, S.K.; DeFrees, S.; Ledeen, R.W. Mice deficient in GM1 manifest both motor and non-motor symptoms of Parkinson’s disease; successful treatment with synthetic GM1 ganglioside. Exp. Neurol. 2020, 329, 113284. [Google Scholar] [CrossRef] [PubMed]
- Boukhris, A.; Schule, R.; Loureiro, J.L.; Lourenço, C.M.; Mundwiller, E.; Gonzalez, M.A.; Charles, P.; Gauthier, J.; Rekik, I.; Acosta Lebrigio, R.F.; et al. Alteration of ganglioside biosynthesis responsible for complex hereditary spastic paraplegia. Am. J. Hum. Genet. 2013, 93, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Kornfeld, S. A fascination with sugars. Mol. Biol. Cell 2010, 21, 3773–3775. [Google Scholar] [CrossRef] [PubMed]
- Aureli, M.; Bassi, R.; Loberto, N.; Regis, S.; Prinetti, A.; Chigorno, V.; Aerts, J.M.; Boot, R.G.; Filocamo, M.; Sonnino, S. Cell surface associated glycohydrolases in normal and Gaucher disease fibroblasts. J. Inherit. Metab. Dis. 2012, 35, 1081–1091. [Google Scholar] [CrossRef]
- Bonten, E.J.; Annunziata, I.; d’Azzo, A. Lysosomal multienzyme complex: Pros and cons of working together. Cell. Mol. Life Sci. 2014, 71, 2017–2032. [Google Scholar] [CrossRef] [PubMed]
- Sandhoff, K.; Kolter, T. Processing of sphingolipid activator proteins and the topology of lysosomal digestion. Acta Biochim. Pol. 1998, 45, 373–384. [Google Scholar] [CrossRef]
- Ohto, U.; Usui, K.; Ochi, T.; Yuki, K.; Satow, Y.; Shimizu, T. Crystal structure of human β-galactosidase: Structural basis of Gm1 gangliosidosis and morquio B diseases. J. Biol. Chem. 2012, 287, 1801–1812. [Google Scholar] [CrossRef]
- Garman, S.C.; Garboczi, D.N. The molecular defect leading to Fabry disease: Structure of human alpha-galactosidase. J. Mol. Biol. 2004, 337, 319–335. [Google Scholar] [CrossRef]
- Clark, N.E.; Garman, S.C. The 1.9 Å structure of human alpha-N-acetylgalactosaminidase: The molecular basis of Schindler and Kanzaki diseases. J. Mol. Biol. 2009, 393, 435–447. [Google Scholar] [CrossRef]
- Akiyama, H.; Ide, M.; Nagatsuka, Y.; Sayano, T.; Nakanishi, E.; Uemura, N.; Yuyama, K.; Yamaguchi, Y.; Kamiguchi, H.; Takahashi, R.; et al. Glucocerebrosidases catalyze a transgalactosylation reaction that yields a newly-identified brain sterol metabolite, galactosylated cholesterol. J. Biol. Chem. 2020, 295, 5257–5277. [Google Scholar] [CrossRef] [PubMed]
- Brumshtein, B.; Wormald, M.R.; Silman, I.; Futerman, A.H.; Sussman, J.L. Structural comparison of differently glycosylated forms of acid-beta-glucosidase, the defective enzyme in Gaucher disease. Acta Crystallogr. D Biol. Crystallogr. 2006, 62 Pt 12, 1458–1465. [Google Scholar] [CrossRef]
- Martin, E.; Schüle, R.; Smets, K.; Rastetter, A.; Boukhris, A.; Loureiro, J.L.; Gonzalez, M.A.; Mundwiller, E.; Deconinck, T.; Wessner, M.; et al. Loss of function of glucocerebrosidase GBA2 is responsible for motor neuron defects in hereditary spastic paraplegia. Am. J. Hum. Genet. 2013, 92, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Gebai, A.; Gorelik, A.; Li, Z.; Illes, K.; Nagar, B. Structural basis for the activation of acid ceramidase. Nat. Commun. 2018, 9, 1621. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.P.S.; Amintas, S.; Levade, T.; Medin, J.A. Acid ceramidase deficiency: Farber disease and SMA-PME. Orphanet J. Rare Dis. 2018, 13, 121. [Google Scholar] [CrossRef]
- Sandhoff, K.; Harzer, K. Gangliosides and Gangliosidoses: Principles of Molecular and Metabolic Pathogenesis. J. Neurosci. 2013, 33, 10195–10208. [Google Scholar] [CrossRef] [PubMed]
- Sandhoff, R.; Schulze, H.; Sandhoff, K. Ganglioside metabolism in health and disease. In Progress in Molecular Biology and Translational Science; Chapter 1; Academic Press: Cambridge, MA, USA, 2018; Volome 156, pp. 1–62. [Google Scholar]
- Mehta, A. Epidemiology and natural history of Gaucher’s disease. Eur. J. Intern. Med. 2006, 17, S2–S5. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Gahl, W.A. Lysosomal storage diseases. Transl. Sci. Rare Dis. 2017, 2, 1–71. [Google Scholar] [CrossRef]
- Blumenreich, S.; Barav, O.B.; Jenkins, B.J.; Futerman, A.H. Lysosomal Storage Disorders Shed Light on Lysosomal Dysfunction in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 4966. [Google Scholar] [CrossRef]
- Westbroek, W.; Nguyen, M.; Siebert, M.; Lindstrom, T.; Burnett, R.A.; Aflaki, E.; Jung, O.; Tamargo, R.; Rodriguez-Gil, J.L.; Acosta, W.; et al. A new glucocerebrosidase-deficient neuronal cell model provides a tool to probe pathophysiology and therapeutics for Gaucher disease. Dis. Model. Mech. 2016, 9, 769–778. [Google Scholar] [CrossRef]
- Mistry, P.K.; Lopez, G.; Schiffmann, R.; Barton, N.W.; Weinreb, N.J.; Sidransky, E. Gaucher disease: Progress and ongoing challenges. Mol. Genet. Metab. 2017, 120, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Debs, R.; Froissart, R.; Aubourg, P.; Papeix, C.; Douillard, C.; Degos, B.; Fontaine, B.; Audoin, B.; Lacour, A.; Said, G.; et al. Krabbe disease in adults: Phenotypic and genotypic update from a series of 11 cases and a review. J. Inherit. Metab. Dis. 2013, 36, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Allewelt, H.; Taskindoust, M.; Troy, J.; Page, K.; Wood, S.; Parikh, S.; Prasad, V.K.; Kurtzberg, J. Long-Term Functional Outcomes after Hematopoietic Stem Cell Transplant for Early Infantile Krabbe Disease. Biol. Blood Marrow Transplant. 2018, 24, 2233–2238. [Google Scholar] [CrossRef] [PubMed]
- Caciotti, A.; Garman, S.C.; Rivera-Colón, Y.; Procopio, E.; Catarzi, S.; Ferri, L.; Guido, C.; Martelli, P.; Parini, R.; Antuzzi, D.; et al. GM1 gangliosidosis and Morquio B disease: An update on genetic alterations and clinical findings. Biochim. Biophys. Acta 2011, 1812, 782–790. [Google Scholar] [CrossRef]
- Luu, A.R.; Wong, H.; Agrawal, V.; Wise, N.; Handyside, B.; Lo, M.J.; Pacheco, G.; Felix, J.B.; Giaramita, A.; d’Azzo, A.; et al. Lysosome-targeted β-galactosidase negatively regulates neuraminidase 1 (NEU1) and promotes NEU1 deficiency in GM1 gangliosidosis. J. Biol. Chem. 2020, jbc.RA119.010794. [Google Scholar] [CrossRef]
- Sano, R.; Annunziata, I.; Patterson, A.; Moshiach, S.; Gomero, E.; Opferman, J.; Forte, M.; d’Azzo, A. GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca(2+)-dependent mitochondrial apoptosis. Mol. Cell 2009, 36, 500–511. [Google Scholar] [CrossRef]
- Condori, J.; Acosta, W.; Ayala, J.; Katta, V.; Flory, A.; Martin, R.; Radin, J.; Cramer, C.L.; Radin, D.N. Enzyme replacement for GM1-gangliosidosis: Uptake, lysosomal activation, and cellular disease correction using a novel β-galactosidase:RTB lectin fusion. Mol. Genet. Metab. 2016, 117, 199–209. [Google Scholar] [CrossRef]
- Latour, Y.L.; Yoon, R.; Thomas, S.E.; Grant, C.; Li, C.; Sena-Esteves, M.; Allende, M.L.; Proia, R.L.; Tifft, C.J. Human GLB1 knockout cerebral organoids: A model system for testing AAV9-mediated GLB1 gene therapy for reducing GM1 ganglioside storage in GM1 gangliosidosis. Mol. Genet. Metab. Rep. 2019, 21, 100513. [Google Scholar] [CrossRef]
- Breiden, B.; Sandhoff, K. Mechanism of Secondary Ganglioside and Lipid Accumulation in Lysosomal Disease. Int. J. Mol. Sci. 2020, 21, 2566. [Google Scholar] [CrossRef]
- Mahuran, D.J. Biochemical consequences of mutations causing the GM2 gangliosidoses. Biochim. Biophys. Acta 1999, 1455, 105–138. [Google Scholar] [CrossRef]
- Anheuser, S.; Breiden, B.; Sandhoff, K. Membrane lipids and their degradation compounds control GM2 catabolism at intralysosomal luminal vesicles. J. Lipid Res. 2019, 60, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Tse, R.; Mahuran, D.J. Direct determination of the substrate specificity of the alpha-active site in heterodimeric beta-hexosaminidase A. Biochemistry 1996, 35, 3963–3969. [Google Scholar] [CrossRef] [PubMed]
- Cachon-Gonzalez, M.B.; Zaccariotto, E.; Cox, T.M. Genetics and Therapies for GM2 Gangliosidosis. Curr. Gene Ther. 2018, 18, 68–89. [Google Scholar] [CrossRef]
- Mark, B.L.; Mahuran, D.J.; Cherney, M.M.; Zhao, D.; Knapp, S.; James, M.N.G. Crystal Structure of Human β-Hexosaminidase B: Understanding the Molecular Basis of Sandhoff and Tay–Sachs Disease. J. Mol. Biol. 2003, 327, 1093. [Google Scholar] [CrossRef]
- Sango, K.; Yamanaka, S.; Hoffmann, A.; Okuda, Y.; Grinberg, A.; Westphal, H.; McDonald, M.P.; Crawley, J.N.; Sandhoff, K.; Suzuki, K.; et al. Mouse models of Tay-Sachs and Sandhoff diseases differ in neurologic phenotype and ganglioside metabolism. Nat. Genet. 1995, 11, 170–176. [Google Scholar] [CrossRef]
- Asfaw, B.; Ledvinová, J.; Dobrovolńy, R.; Bakker, H.D.; Desnick, R.J.; van Diggelen, O.P.; de Jong, J.G.; Kanzaki, T.; Chabas, A.; Maire, I.; et al. Defects in degradation of blood group A and B glycosphingolipids in Schindler and Fabry diseases. J. Lipid Res. 2002, 43, 1096–1104. [Google Scholar] [CrossRef]
- Ledvinová, J.; Poupetová, H.; Hanácková, A.; Písacka, M.; Elleder, M. Blood group B glycosphingolipids in alpha-galactosidase deficiency (Fabry disease): Influence of secretor status. Biochim. Biophys. Acta 1997, 1345, 180–187. [Google Scholar] [CrossRef]
- Lukatela, G.; Krauss, N.; Theis, K.; Selmer, T.; Gieselmann, V.; von Figura, K.; Saenger, W. Crystal structure of human arylsulfatase A: The aldehyde function and the metal ion at the active site suggest a novel mechanism for sulfate ester hydrolysis. Biochemistry 1998, 37, 3654–3664. [Google Scholar] [CrossRef]
- Lamichhane, A.; Rocha Cabrero, F. Metachromatic Leukodystrophy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Mitsiakos, G.; Gialamprinou, D.; Chouchou, P.; Chatziioannidis, I.; Karagianni, P. Identification of a homozygous deletion of the NEU1 gene in a patient with type II sialidosis presenting isolated fetal ascites and central nervous system hypoplasia. Hippokratia 2019, 23, 169–171. [Google Scholar]
- Beckmann, N.; Becker, K.A.; Kadow, S.; Schumacher, F.; Kramer, M.; Kühn, C.; Schulz-Schaeffer, W.J.; Edwards, M.J.; Kleuser, B.; Gulbins, E.; et al. Acid Sphingomyelinase Deficiency Ameliorates Farber Disease. Int. J. Mol. Sci. 2019, 20, 6253. [Google Scholar] [CrossRef]
- Dantuma, N.P.; Bott, L.C. The ubiquitin-proteasome system in neurodegenerative diseases: Precipitating factor, yet part of the solution. Front. Mol. Neurosci. 2014, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Fraldi, A.; Jahreiss, L.; Spampanato, C.; Venturi, C.; Medina, D. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2008, 17, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Pereira, V.G.; Gazarini, M.L.; Rodrigues, L.C.; Da Silva, F.H.; Han, S.W.; Martins, A.M.; Tersariol, I.L.S.; D’Almeida, V. Evidence of lysosomal membrane permeabilization in mucopolysaccharidosis type I: Rupture of calcium and proton homeostasis. J. Cell. Physiol. 2010, 223, 335–342. [Google Scholar] [CrossRef]
- Micsenyi, M.C.; Sikora, J.; Stephney, G.; Dobrenis, K.; Walkley, S.U. Lysosomal membrane permeability stimulates protein aggregate formation in neurons of a lysosomal disease. J. Neurosci. 2013, 33, 10815–10827. [Google Scholar] [CrossRef]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef]
- d’Azzo, A.; Tessitore, A.; Sano, R. Gangliosides as apoptotic signals in ER stress response. Cell Death Differ. 2006, 13, 404–414. [Google Scholar] [CrossRef]
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018, 9, 1097. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef]
- Haider, A.; Spurling, B.C.; Sánchez-Manso, J.C. Lewy Body Dementia. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Yu, A.; Shibata, Y.; Shah, B.; Calamini, B.; Lo, D.C.; Morimoto, R.I. Protein aggregation can inhibit clathrin-mediated endocytosis by chaperone competition. Proc. Natl. Acad. Sci. USA 2014, 111, E1481–E1490. [Google Scholar] [CrossRef]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, I.; Patterson, A.; Helton, D.; Hu, H.; Moshiach, S.; Gomero, E.; Nixon, R.; d’Azzo, A. Lysosomal NEU1 deficiency affects amyloid precursor protein levels and amyloid-β secretion via deregulated lysosomal exocytosis. Nat. Commun. 2013, 4, 2734. [Google Scholar] [CrossRef]
- Hoffmann, A.C.; Minakaki, G.; Menges, S.; Salvi, R.; Savitskiy, S.; Kazman, A.; Vicente Miranda, H.; Mielenz, D.; Klucken, J.; Winkler, J.; et al. Extracellular aggregated alpha synuclein primarily triggers lysosomal dysfunction in neural cells prevented by trehalose. Sci. Rep. 2019, 9, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Da, B.; Dias, C.; Jovanovic, K.; Gonsalves, D.; Moodley, K.; Reusch, U.; Knackmuss, S.; Weinberg, M.S.; Little, M.; Weiss, S.T.F. The 37kDa/67kDa Laminin Receptor acts as a receptor for Ab 42 internalization. Sci. Rep. 2014, 4, 5556. [Google Scholar]
- Rauch, J.N.; Luna, G.; Guzman, E.; Audouard, M.; Challis, C.; Sibih, Y.E.; Leshuk, C.; Hernandez, I.; Wegmann, S.; Hyman, B.T.; et al. LRP1 is a master regulator of tau uptake and spread. Nature 2020, 580, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, A.P.; Puertollano, R.; Raben, N.; Slaugenhaupt, S.; Walkley, S.U.; Ballabio, A. Autophagy in lysosomal storage disorders. Autophagy 2012, 8, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, T.; Roth, A.G.; Petersen, N.H.; Mahalka, A.K.; Olsen, O.D.; Moilanen, I.; Zylicz, A.; Knudsen, J.; Sandhoff, K.; Arenz, C.; et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature 2010, 463, 549–553. [Google Scholar] [CrossRef]
- Gabandé-Rodríguez, E.; Boya, P.; Labrador, V.; Dotti, C.G.; Ledesma, M.D. High sphingomyelin levels induce lysosomal damage and autophagy dysfunction in Niemann Pick disease type A. Cell Death Differ. 2014, 21, 864–875. [Google Scholar] [CrossRef]
- Ivanova, M. Altered Sphingolipids Metabolism Damaged Mitochondrial Functions: Lessons Learned from Gaucher and Fabry Diseases. J. Clin. Med. 2020, 9, 1116. [Google Scholar] [CrossRef]
- Kajihara, R.; Numakawa, T.; Odaka, H.; Yaginuma, Y.; Fusaki, N.; Okumiya, T.; Furuya, H.; Inui, S.; Era, T. Novel Drug Candidates Improve Ganglioside Accumulation and Neural Dysfunction in GM1 Gangliosidosis Models with Autophagy Activation. Stem Cell Rep. 2020, 14, 909–923. [Google Scholar] [CrossRef]
- Ariga, T.; McDonald, M.P.; Yu, R.K. Role of ganglioside metabolism in the pathogenesis of Alzheimer’s disease–a review. J. Lipid Res. 2008, 49, 1157–1175. [Google Scholar] [CrossRef] [PubMed]
- Fukami, Y.; Ariga, T.; Yamada, M.; Yuki, N. Brain Gangliosides in Alzheimer’s Disease: Increased Expression of Cholinergic Neuron-Specific Gangliosides. Curr. Alzheimer Res. 2017, 14, 586–591. [Google Scholar] [CrossRef]
- Matsubara, T.; Yasumori, H.; Ito, K.; Shimoaka, T.; Hasegawa, T.; Sato, T. Generation of parallel β-sheets of Ab on ganglioside cluster-1-Amyloid β fibrils assembled on ganglioside-enriched membranes contain both parallel β-sheets and turns. J. Biol. Chem. 2018, 293, 14146–14154. [Google Scholar] [CrossRef] [PubMed]
- Ledeen, R.W.; Wu, G. Gangliosides of the Nuclear Membrane: A Crucial Locus of Cytoprotective Modulation. J. Cell. Biochem. 2006, 97, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Perissinotto, F.; Rondelli, V.; Parisse, P.; Tomena, N.; Zunino, A.; Almasy, L.; Merkel, D.G.; Bottyan, L.; Sajti, S.; Casalis, L. GM1 Ganglioside role in the interaction of Alpha-synuclein with lipid membranes: Morphology and structure. Biophys. Chem. 2019, 255, 106272. [Google Scholar] [CrossRef]
- Fantini, J.; Yahi, N. The driving force of α-synuclein insertion and amyloid channel formation in the plasma membrane of neural cells: Key role of ganglioside- and cholesterol-binding domains. Adv. Exp. Med. Biol. 2013, 991, 15–26. [Google Scholar]
- Saavedra, L.; Mohamed, A.; Ma, V.; Kar, S.; De Chaves, E.P. Internalization of β-amyloid peptide by primary neurons in the absence of apolipoprotein E. J. Biol. Chem. 2007, 282, 35722–35732. [Google Scholar] [CrossRef]
- Park, J.Y.; Kim, K.S.; Lee, S.B.; Ryu, J.S.; Chung, K.C.; Choo, Y.K.; Jou, I.; Kim, J.; Park, S.M. On the mechanism of internalization of α-synuclein into microglia: Roles of ganglioside GM1 and lipid raft. J. Neurochem. 2009, 110, 400–411. [Google Scholar] [CrossRef]
- Zondler, L.; Kostka, M.; Garidel, P.; Heinzelmann, U.; Hengerer, B.; Mayer, B.; Weishaupt, J.H.; Gillardon, F.; Danzer, K.M. Proteasome impairment by α-synuclein. PLoS ONE 2017, 12, e0184040. [Google Scholar] [CrossRef]
- Fantini, J.; Yahi, N.; Garmy, N. Cholesterol accelerates the binding of Alzheimer’s β-amyloid peptide to ganglioside GM1 through a universal hydrogen-bond-dependent sterol tuning of glycolipid conformation. Front. Physiol. 2013, 4, 120. [Google Scholar] [CrossRef]
- Smith, B.R.; Santos, M.B.; Marshall, M.S.; Cantuti-Castelvetri, L.; Lopez-Rosas, A.; Li, G.; van Breemen, R.; Claycomb, K.I.; Gallea, J.I.; Celej, M.S.; et al. Neuronal inclusions of α-synuclein contribute to the pathogenesis of Krabbe disease. J. Pathol. 2014, 232, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Manzoni, C. The LRRK2-macroautophagy axis and its relevance to Parkinson’s disease. Biochem. Soc. Trans. 2017, 45, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Ysselstein, D.; Nguyen, M.; Young, T.J.; Severino, A.; Schwake, M.; Merchant, K.; Krainc, D. LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson’s disease patients. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Zunke, F.; Tsunemi, T.; Toker, N.J.; Jeon, S.; Burbulla, L.F.; Patnaik, S.; Sidransky, E.; Marugan, J.J.; Sue, C.M.; et al. Activation of β-Glucocerebrosidase Reduces Pathological α-Synuclein and Restores Lysosomal Function in Parkinson’s Patient Midbrain Neurons. J. Neurosci. 2016, 36, 7693–7706. [Google Scholar] [CrossRef]
- De Franceschi, G.; Frare, E.; Pivato, M.; Relini, A.; Penco, A.; Greggio, E.; Bubacco, L.; Fontana, A.; de Laureto, P.P. Structural and morphological characterization of aggregated species of α-synuclein induced by docosahexaenoic acid. J. Biol. Chem. 2011, 286, 22262–22274. [Google Scholar] [CrossRef]
- Martinez, Z.; Zhu, M.; Han, S.; Fink, A.L. GM1 specifically interacts with α-synuclein and inhibits fibrillation. Biochemistry 2007, 46, 1868–1877. [Google Scholar] [CrossRef]
- Ledeen, R.W.; Wu, G. Gangliosides, α-Synuclein, and Parkinson’s Disease. Prog. Mol. Biol. Transl. Sci. 2018, 156, 435–454. [Google Scholar]
- Galvagnion, C.; Brown, J.W.; Ouberai, M.M.; Flagmeier, P.; Vendruscolo, M.; Buell, A.K.; Sparr, E.; Dobson, C.M. Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of α-synuclein. Proc. Natl. Acad. Sci. USA 2016, 113, 7065–7070. [Google Scholar] [CrossRef]
- Schneider, J.S. Altered expression of genes involved in ganglioside biosynthesis in substantia nigra neurons in Parkinson’s disease. PLoS ONE 2018, 13, e0199189. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Geisler, F.H.; Schneider, J.S.; Li, P.A.; Fiumelli, H.; Sipione, S. Gangliosides: Treatment Avenues in Neurodegenerative Disease. Front. Neurol. 2019, 10, 859. [Google Scholar] [CrossRef]
- Schneider, J.S.; Gollomp, S.M.; Sendek, S.; Colcher, A.; Cambi, F.; Du, W. A randomized, controlled, delayed start trial of GM1 ganglioside in treated Parkinson’s disease patients. J. Neurol. Sci. 2013, 324, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Galvagnion, C.; Buell, A.K.; Meisl, G.; Michaels, T.C.; Vendruscolo, M.; Knowles, T.P.; Dobson, C.M. Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation. Nat. Chem. Biol. 2015, 11, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Tsigelny, I.F.; Sharikov, Y.; Wrasidlo, W.; Gonzalez, T.; Desplats, P.A.; Crews, L.; Spencer, B.; Masliah, E. Role of α-synuclein penetration into the membrane in the mechanisms of oligomer pore formation. FEBS J. 2012, 279, 1000–1013. [Google Scholar] [CrossRef] [PubMed]
- Su, P.; Khaledi, H.; Waggoner, C.; Gelb, M.H. Detection of GM1-Gangliosidosis in Newborn Dried Blood Spots by Enzyme and Biomarker Assays Using Tandem Mass Spectrometry. J. Inherit. Metab. Dis. 2020. [Google Scholar] [CrossRef]
- Tropak, M.B.; Yonekawa, S.; Karumuthil-Melethil, S.; Thompson, P.; Wakarchuk, W.; Gray, S.J.; Walia, J.S.; Mark, B.L.; Mahuran, D. Construction of a hybrid β-hexosaminidase subunit capable of forming stable homodimers that hydrolyze GM2 ganglioside in vivo. Mol. Ther.-Methods Clin. Dev. 2016, 3, 15057. [Google Scholar] [CrossRef]
- Ullman, J.C.; Arguello, A.; Getz, J.A.; Bhalla, A.; Mahon, C.S.; Wang, J.; Giese, T.; Bedard, C.; Kim, D.J.; Blumenfeld, J.R.; et al. Brain delivery and activity of a lysosomal enzyme using a blood-brain barrier transport vehicle in mice. Sci. Transl. Med. 2020, 12, eaay1163. [Google Scholar] [CrossRef]
- Ivanova, M.M.; Changsila, E.; Iaonou, C.; Goker-Alpan, O. Impaired autophagic and mitochondrial functions are partially restored by ERT in Gaucher and Fabry diseases. PLoS ONE 2019, 14, e0210617. [Google Scholar] [CrossRef]
- Chiricozzi, E.; Niemir, N.; Aureli, M.; Magini, A.; Loberto, N.; Prinetti, A.; Bassi, R.; Polchi, A.; Emiliani, C.; Caillaud, C.; et al. Chaperone Therapy for GM2 Gangliosidosis: Effects of Pyrimethamine on β-Hexosaminidase Activity in Sandhoff Fibroblasts. Mol. Neurobiol. 2014, 50, 159–167. [Google Scholar] [CrossRef]
- Bateman, K.S.; Cherney, M.M.; Mahuran, D.J.; Tropak, M.; James, N.G. Crystal structure of β-hexosaminidase B in complex with pyrimethamine, a potential pharmacological chaperone. J. Med. Chem. 2011, 54, 1421–1429. [Google Scholar] [CrossRef]
- Marshall, J.; Nietupski, J.B.; Park, H.; Cao, J.; Bangari, D.S.; Silvescu, C.; Wilper, T.; Randall, K.; Tietz, D.; Wang, B.; et al. Substrate Reduction Therapy for Sandhoff Disease through Inhibition of Glucosylceramide Synthase Activity. Mol. Ther. 2019, 27, 1495–1506. [Google Scholar] [CrossRef]
- Cox, T.M.; Drelichman, G.; Cravo, R.; Balwani, M.; Burrow, T.A.; Martins, A.M.; Lukina, E.; Rosenbloom, B.; Goker-Alpan, O.; Watman, N.; et al. Eliglustat maintains long- term clinical stability in patients with Gaucher disease type 1 stabilized on enzyme therapy. Blood 2017, 129, 2375–2383. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A.; De Palma, M.; Quattrini, A.; Del Carro, U.; Amadio, S.; Visigalli, I.; Sessa, M.; Fasano, S.; Brambilla, R.; Marchesini, S.; et al. Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J. Clin. Investig. 2004, 113, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A.; Capotondo, A.; Fasano, S.; del Carro, U.; Marchesini, S.; Azuma, H.; Malaguti, M.C.; Amadio, S.; Brambilla, R.; Grompe, M.; et al. Gene therapy of metachromatic leukodystrophy reverses neurological damage and deficits in mice. J. Clin. Investig. 2006, 116, 3070–3082. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013, 34, 1233158. [Google Scholar] [CrossRef]
- Gentner, B.; Visigalli, I.; Hiramatsu, H.; Lechman, E.; Ungari, S.; Giustacchini, A.; Schira, G.; Amendola, M.; Quattrini, A.; Martino, S.; et al. Identification of hematopoietic stem cell-specific miRNAs enables gene therapy of globoid cell leukodystrophy. Sci. Transl. Med. 2010, 2, 58ra84. [Google Scholar] [CrossRef]
- Gomez-Ospina, N.; Scharenberg, S.G.; Mostrel, N.; Bak, R.O.; Mantri, S.; Quadros, R.M.; Gurumurthy, C.B.; Lee, C.; Bao, G.; Suarez, C.J.; et al. Human genome-edited hematopoietic stem cells phenotypically correct Mucopolysaccharidosis type I. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Medin, J.A.; Khan, A.; Huang, J.; Barber, D.; Anthony Rupar, C.; Auray-Blais, C.; Fraser, G.; Fowler, D.H.; Keating, A.; West, M.L.; et al. FACTs Fabry gene therapy clinical trial: Two-year data. Mol. Genet. Metab. 2019, 126, S99. [Google Scholar] [CrossRef]
- Köhn, A.F.; Grigull, L.; du Moulin, M.; Kabisch, S.; Ammer, L.; Rudolph, C.; Muschol, N.M. Hematopoietic stem cell transplantation in mucopolysaccharidosis type IIIA: A case description and comparison with a genotype-matched control group. Mol. Genet. Metab. Rep. 2020, 23, 100578. [Google Scholar] [CrossRef]
- Weismann, C.M.; Ferreira, J.; Keeler, A.M.; Su, Q.; Qui, L.; Shaffer, S.A.; Xu, Z.; Gao, G.; Sena-Esteves, M. Systemic AAV9 gene transfer in adult GM1 gangliosidosis mice reduces lysosomal storage in CNS and extends lifespan. Hum. Mol. Genet. 2015, 24, 4353–4364. [Google Scholar] [CrossRef]
- Akli, S.; Guidotti, J.E.; Vigne, E.; Perricaudet, M.; Sandhoff, K.; Kahn, A.; Poenaru, L. Restoration of hexosaminidase A activity in human Tay-Sachs fibroblasts via adenoviral vector-mediated gene transfer. Gene Ther. 1996, 3, 769–774. [Google Scholar]
- Guidotti, J.E.; Mignon, A.; Haase, G.; Caillaud, C.; McDonell, N.; Kahn, A.; Poenaru, L. Adenoviral gene therapy of the Tay-Sachs disease in hexosaminidase A-deficient knock-out mice. Hum. Mol Genet. 1999, 8, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Osmon, K.J.; Woodley, E.; Thompson, P.; Ong, K.; Karumuthil-Melethil, S.; Keimel, J.G.; Mark, B.L.; Mahuran, D.; Gray, S.J.; Walia, J.S. Systemic Gene Transfer of a Hexosaminidase Variant Using an scAAV9.47 Vector Corrects GM2-Gangliosidosis in Sandhoff Mice. Hum. Gene Ther. 2016, 27, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Woodley, E.; Osmon, K.; Thompson, P.; Richmond, C.; Chen, Z.; Gray, S.J.; Walia, J.S. Efficacy of a Bicistronic Vector for Correction of Sandhoff Disease in a Mouse Model. Mol. Ther.-Methods Clin. Dev. 2019, 12, 47–57. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme Names | Uniprot No. | CAZy Family |
|---|---|---|
| Cer β-Glc-transferase, GlcCer synthase, UGCG | Q16739 | GT21 |
| Cer β-Gal-transferase, GalCer synthase CGT, UGT8 | Q16880 | GT1 |
| Cerebroside sulfotransferase, Gal3ST1, CST | Q99999 | - |
| CerGlc β1,4Gal-transferase, B4GALT6, LacCer synthase | Q9UBX8 | GT7 |
| GalNAc β1,3Gal-transferase, B3GALT4, GM2 synthase | O96024 | GT31 |
| GlcNAc β1,3Gal-transferase, B3GALT5 | Q9Y2C3 | GT31 |
| Gal α1,4Gal-transferase, A4GALT, Gb3 synthase | Q9NPC4 | GT32 |
| LacCer β1,4GalNAc-transferase, B4GALNT1, GD2 synthase | Q00973 | GT12 |
| Gal β1,3GalNAc-transferase, B3GALNT1, Gb4 synthase | O75752 | GT31 |
| Gal β1,3GlcNAc-transferase, B3GNT5 | Q9BYG0 | GT31 |
| Gal α2,3Sialyltransferase, ST3Gal I | Q11201 | GT29 |
| ST3Gal II | Q16842 | GT29 |
| ST3Gal III | Q11203 | GT29 |
| ST3Gal IV | Q11206 | GT29 |
| GM3 synthase, ST3Gal V | Q9UNP4 | GT29 |
| ST3Gal VI | Q9Y274 | GT29 |
| GalNAc α2,6sialyltransferase, ST6GALNAC V | Q9BVH7 | GT29 |
| Sialyl α2,8sialyltransferase, ST8Sia I | Q92185 | GT29 |
| ST8Sia III | O43173 | GT29 |
| ST8Sia V | O15466 | GT29 |
| Name of Disease | Gene | Enzyme | Affected GSL | MIM Phenotype No. |
|---|---|---|---|---|
| Spastic Paraplegia (Type 26) | B4GALNT1 | β1,4GalNAc-transferase | GM2, GM1, etc. | 609195 |
| West Syndrome | ST3GAL III | α2,3Sialyltransferase 3 | GM1, GD1a, etc. | 615006, 611090 |
| Amish Infantile Epilepsy | ST3GAL V | α2,3Sialyltransferase 9 (GM3 synthase) | LacCer | 609056 |
| Enzyme Names | Uniprot No. | CAZy GH/clan | Activator |
|---|---|---|---|
| Glucoceramidase, GCase, GBA1 | P04062 | GH30 / GH-A | SapC |
| β-Galactoceramidase, GALC, Gal-Cer β-galactosidase | P54803 | GH59 / GH-A | SapA |
| Arylsulfatase A, ARSA | P15289 | - | SapB |
| α-Galactosidase A, GLA | P06280 | GH27 / GH-D | SapB |
| β-Galactosidase, GLB1, BGAL | P16278 | GH35 / GH-A | SapB |
| β-Hexosaminidase HEXA | P06865 | GH20 / GH-K | GM2-AP |
| β-Hexosaminidase HEXB | P07686 | GH20 / GH-K | GM2-AP |
| α-N-Acetylgalactosaminidase NAGA | P17050 | GH27 / GH-D | - |
| Sialidase NEU1 | Q99519 | GH33 / GH-E | - |
| Sialidase NEU2 | Q9Y3R4 | GH33 / GH-E | - |
| Sialidase NEU3 | Q9UQ49 | GH33 / GH-E | - |
| Sialidase NEU4 | Q8WWR8 | GH33 / GH-E | - |
| Acid ceramidase, ASAH1 | Q13510 | - | SapD |
| Name of Disease | Gene | Enzyme | Affected GSL | MIM Phenotype No. |
|---|---|---|---|---|
| GM1 Gangliosidosis | BGAL | β-galactosidase | GM1 | Type I: 230500 Type II: 230600 Type III: 230650 |
| GM2 Gangliosidosis Variant B: Tay-Sachs Disease | HEXA | β-hexosaminidase A/S (HexA/S) (α subunit) | GM2 | 272800 |
| GM2 Gangliosidosis Variant 0: Sandhoff Disease | HEXB | β-hexosaminidase B (HexB) (β subunit) | GM2 | 268800 |
| GM2 Gangliosidosis Variant AB: GM2-AP Deficiency | GM2A | GM2-activator protein (GM2-AP) | GM2 | 272750 |
| Fabry Disease | GLA | α-galactosidase A | Globosides, Blood group B | 301500 |
| Gaucher Disease | GBA1 | β-glucoceramidase I (GCase I) | GlcCer | Type I: 230800 Type II: 230900 Type III: 231000 |
| Krabbe Disease | GALC | β-galactoceramidase SapA | GalCer | 245200 606463 |
| Metachromatic Leukodystrophy | ARSA | Arylsulfatase A | Sulfatide | 250100 |
| Saposin deficiency | PSAP | Prosaposin (Saposin precursor protein) | GSLs | 611721 |
| Galactosialidosis, PPCA Deficiency | CTSA | β-galactosidase Sialidase I (NEU1) Cathepsin A | GM1 | 256540 |
| Sialidosis | NEU1 | Sialidase I (NEU1) | Sialylated GSLs | 256550 |
| Niemann-Pick Disease | SMPD1 | Acid sphingomyelinase | Sphingosine | Type A: 257200 Type B: 607616 |
| Schindler Disease | NAGA | α-N-acetylgalactosaminidase B (α-NAGAL) | Lac-Cer, Blood group A | 609241 |
| Farber Disease | ASAH1 | Acid ceramidase | Ceramides | 228000 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryckman, A.E.; Brockhausen, I.; Walia, J.S. Metabolism of Glycosphingolipids and Their Role in the Pathophysiology of Lysosomal Storage Disorders. Int. J. Mol. Sci. 2020, 21, 6881. https://doi.org/10.3390/ijms21186881
Ryckman AE, Brockhausen I, Walia JS. Metabolism of Glycosphingolipids and Their Role in the Pathophysiology of Lysosomal Storage Disorders. International Journal of Molecular Sciences. 2020; 21(18):6881. https://doi.org/10.3390/ijms21186881
Chicago/Turabian StyleRyckman, Alex E., Inka Brockhausen, and Jagdeep S. Walia. 2020. "Metabolism of Glycosphingolipids and Their Role in the Pathophysiology of Lysosomal Storage Disorders" International Journal of Molecular Sciences 21, no. 18: 6881. https://doi.org/10.3390/ijms21186881
APA StyleRyckman, A. E., Brockhausen, I., & Walia, J. S. (2020). Metabolism of Glycosphingolipids and Their Role in the Pathophysiology of Lysosomal Storage Disorders. International Journal of Molecular Sciences, 21(18), 6881. https://doi.org/10.3390/ijms21186881