Mucolipidoses Overview: Past, Present, and Future


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
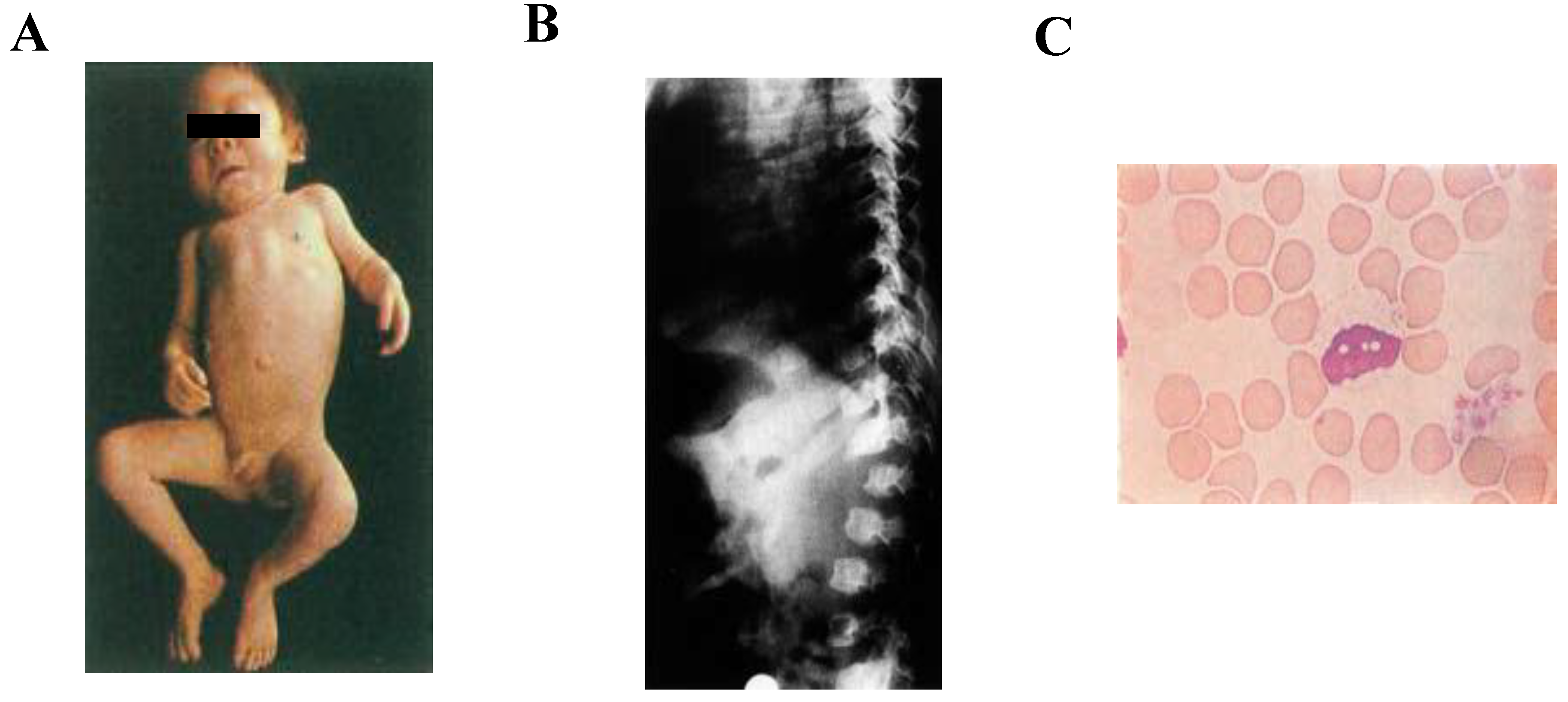
2. Diagnosis of ML II/III Patients
2.1. Clinical Diagnosis
2.2. Genetic Diagnosis
2.3. Biochemical Diagnosis
2.4. Storage Materials
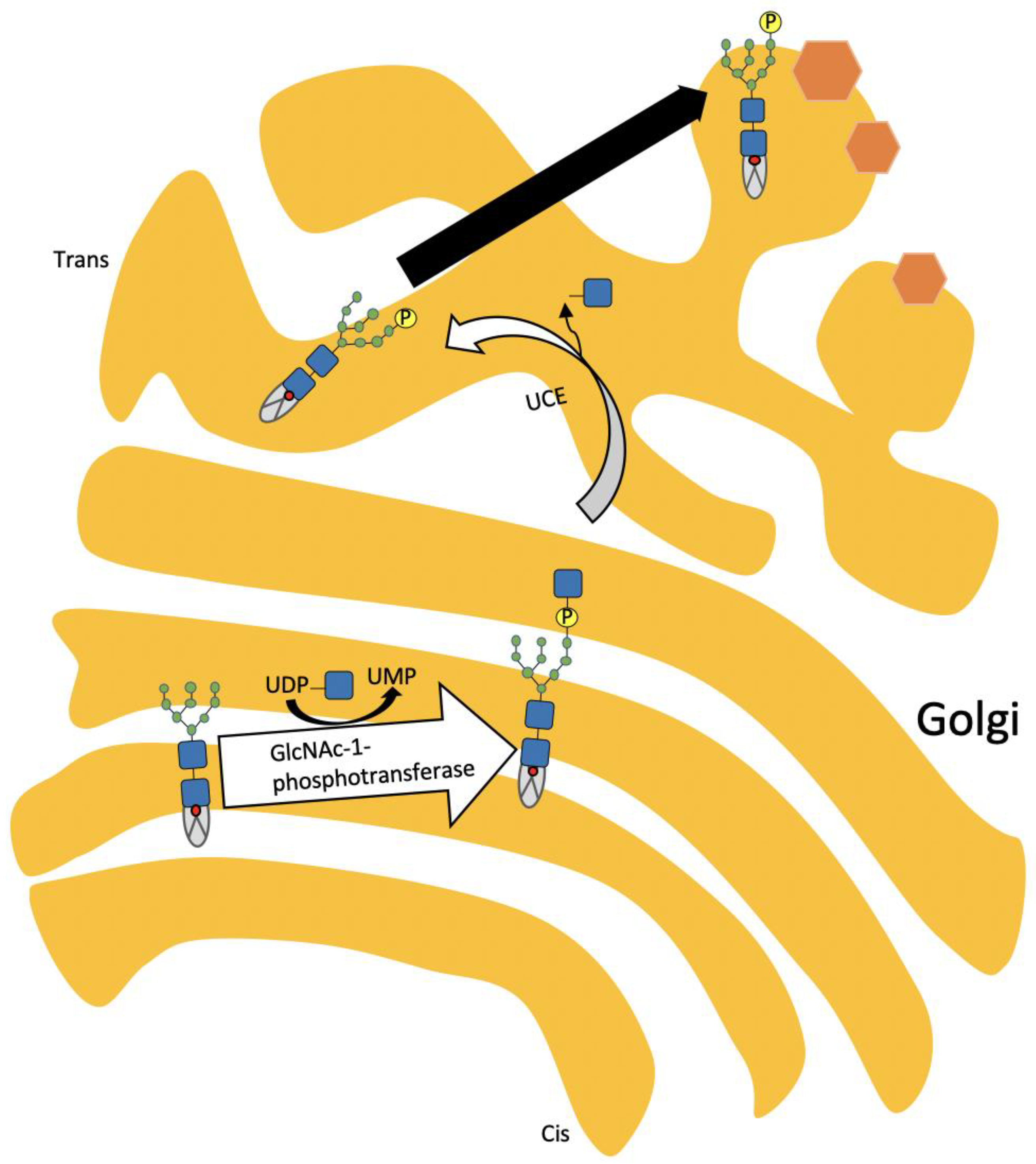
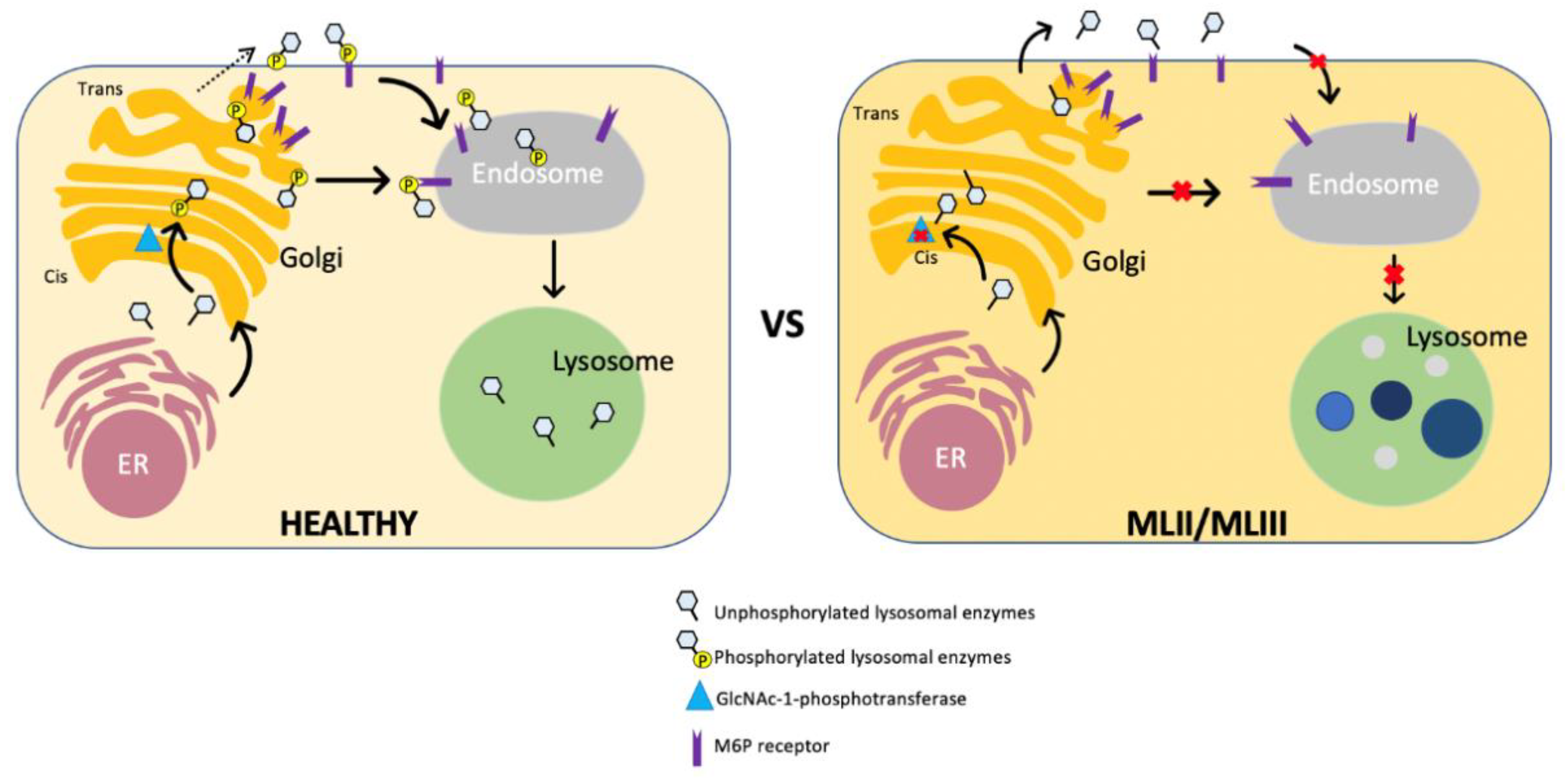
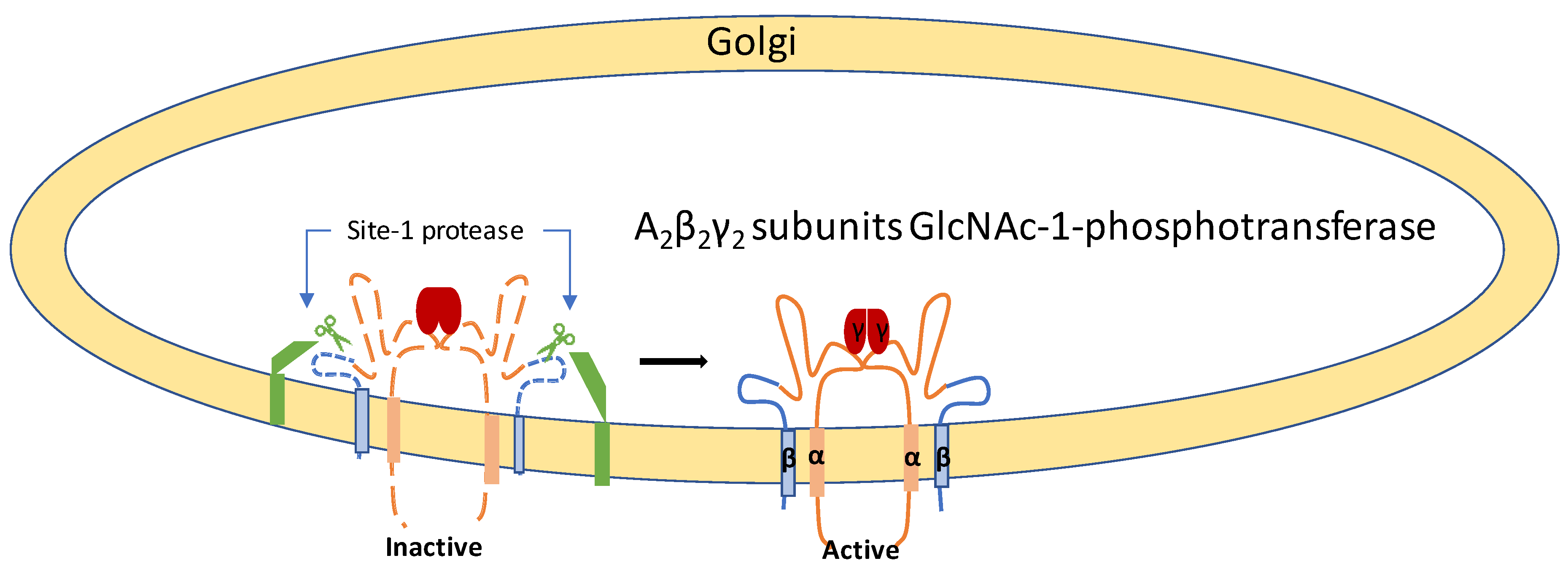
3. Pathophysiology of ML
4. Animal and Zebrafish Models of ML
4.1. Animal Models
4.1.1. Feline Model
4.1.2. Mouse Models
4.2. Zebrafish Models
5. Therapy and Management of ML Patients
5.1. Supportive Therapy
5.2. Hematopoietic Stem Cell Transplantation (HSCT)
5.3. Future Therapies
5.3.1. Enzyme Replacement Therapy (ERT)
5.3.2. Gene Therapy
5.3.3. Pharmacological Chaperon
5.3.4. Antisense Oligonucleotides
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated Virus |
| BMT | Bone marrow transplantation |
| CSF | Cerebrospinal fluid |
| CS | Chondroitin sulfate |
| DS | Dermatan sulfate |
| ELISA | Enzyme-linked immunosorbent assay |
| ERT | Enzyme Replacement Therapy |
| FGFs | Fibroblast growth factors |
| GAGs | Glycosaminoglycans |
| GlcNAc | N-acetylglucosamine |
| GNPTAB | N-acetylglucosamine-1-phosphate transferase subunits α and β |
| GNPTG | N-acetylglucosamine-1-phosphate transferase subunit gamma |
| HGMD | The Human Gene Mutation Database |
| HSCT | Hematopoietic Stem Cell Transplantation |
| HS | Heparan sulfate |
| KS | Keratan sulfate |
| LAL | Lysosomal acid lipase |
| LC-MS/MS | Liquid chromatography-tandem mass spectrometry |
| LSDs | Lysosomal storage diseases |
| ML | Mucolipidoses |
| MPS | Mucopolysaccharidoses |
| M6P | Mannose 6-phosphate |
| TRAP | Tartrate-resistant acid phosphatase |
| UDP | Uridine-diphosphate |
| UMP | Uridine-monophosphate |
References
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of lysosomal storage disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef] [PubMed]
- National Institute of Neurological Disorders and Stroke. Mucolipidoses Fact Sheet. September 2015, NIH Publication No. 15-4899. Available online: https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Mucolipidoses-Fact-Sheet (accessed on 29 July 2020).
- Leroy, J.G.; Demars, R.I. Mutant enzymatic and cytological phenotypes in cultured human fibroblasts. Science 1967, 157, 804–806. [Google Scholar] [CrossRef] [PubMed]
- Cathey, S.S.; Kudo, M.; Tiede, S.; Raas-Rothschild, A.; Braulke, T.; Beck, M.; Taylor, H.A.; Canfield, W.M.; Leroy, J.G.; Neufeld, E.F.; et al. Molecular order in mucolipidosis II and III nomenclature. Am. J. Med. Genet. Part A 2008, 146A, 512–513. [Google Scholar] [CrossRef] [PubMed]
- Spranger, J.W.; Wiedemann, H.R. The genetic mucolipidoses. Diagnosis and differential diagnosis. Humangenetik 1970, 9, 113–139. [Google Scholar] [PubMed]
- Schiff, M.; Maire, I.; Bertrand, Y.; Cochat, P.; Guffon, N. Long-term follow-up of metachronous marrow-kidney transplantation in severe type II sialidosis: What does success mean? Nephrol. Dial. Transpl. 2005, 20, 2563–2565. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Coutinho, M.F.; Dalal, A.B.; Mohamed Nurul Jain, S.J.; Prata, M.J.; Alves, S. Prenatal skeletal dysplasia phenotype in severe MLII alpha/beta with novel GNPTAB mutation. Gene 2014, 542, 266–268. [Google Scholar] [CrossRef] [PubMed]
- Alegra, T.; Koppe, T.; Acosta, A.; Sarno, M.; Burin, M.; Kessler, R.G.; Sperb-Ludwig, F.; Cury, G.; Baldo, G.; Matte, U.; et al. Pitfalls in the prenatal diagnosis of mucolipidosis II alpha/beta: A case report. Meta Gene 2014, 2, 403–406. [Google Scholar] [CrossRef]
- Costain, G.; Inbar-Feigenberg, M.; Saleh, M.; Yaniv-Salem, S.; Ryan, G.; Morgen, E.; Goh, E.S.; Nishimura, G.; Chitayat, D. Challenges in Diagnosing Rare Genetic Causes of Common In Utero Presentations: Report of Two Patients with Mucolipidosis Type II (I-Cell Disease). J. Pediatr. Genet. 2018, 7, 134–137. [Google Scholar] [CrossRef]
- Velho, R.V.; Harms, F.L.; Danyukova, T.; Ludwig, N.F.; Friez, M.J.; Cathey, S.S.; Filocamo, M.; Tappino, B.; Gunes, N.; Tuysuz, B.; et al. The lysosomal storage disorders mucolipidosis type II, type III alpha/beta, and type III gamma: Update on GNPTAB and GNPTG mutations. Hum. Mutat. 2019, 40, 842–864. [Google Scholar]
- Heo, J.S.; Choi, K.Y.; Sohn, S.H.; Kim, C.; Kim, Y.J.; Shin, S.H.; Lee, J.M.; Lee, J.; Sohn, J.A.; Lim, B.C.; et al. A case of mucolipidosis II presenting with prenatal skeletal dysplasia and severe secondary hyperparathyroidism at birth. Korean J. Pediatr. 2012, 55, 438–444. [Google Scholar] [CrossRef]
- Yang, M.; Cho, S.Y.; Park, H.D.; Choi, R.; Kim, Y.E.; Kim, J.; Lee, S.Y.; Ki, C.S.; Kim, J.W.; Sohn, Y.B.; et al. Clinical, biochemical and molecular characterization of Korean patients with mucolipidosis II/III and successful prenatal diagnosis. Orphanet J. Rare Dis. 2017, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Yuksel, A.; Kayserili, H.; Gungor, F. Short femurs detected at 25 and 31 weeks of gestation diagnosed as Leroy I-cell disease in the postnatal period: A report of two cases. Fetal Diagn. Ther. 2007, 22, 198–202. [Google Scholar] [CrossRef]
- Sprigz, R.A.; Doughty, R.A.; Spackman, T.J.; Murane, M.J.; Coates, P.M.; Koldovsky, O.; Zackai, E.H. Neonatal presentation of I-cell disease. J. Pediatr. 1978, 93, 954–958. [Google Scholar] [CrossRef]
- Nishimura, F.; Naruishi, H.; Naruishi, K.; Yamada, T.; Sasaki, J.; Peters, C.; Uchiyama, Y.; Murayama, Y. Cathepsin-L, a key molecule in the pathogenesis of drug-induced and I-cell disease-mediated gingival overgrowth: A study with cathepsin-L-deficient mice. Am. J. Pathol. 2002, 161, 2047–2052. [Google Scholar] [CrossRef]
- Nogami, H.; Oohira, A.; Suzuki, F.; Tsuda, K. Cartilage of I-cell disease. Pediatr. Res. 1981, 15, 330–334. [Google Scholar] [CrossRef]
- Wooten, W.I., 3rd; Muhlebach, M.S.; Muenzer, J.; Loughlin, C.E.; Vaughn, B.V. Progression of Polysomnographic Abnormalities in Mucolipidosis II (I-Cell Disease). J. Clin. Sleep Med. 2016, 12, 1695–1696. [Google Scholar] [CrossRef]
- Tabone, L.; Caillaud, C.; Amaddeo, A.; Khirani, S.; Michot, C.; Couloigner, V.; Brassier, A.; Cormier-Daire, V.; Baujat, G.; Fauroux, B. Sleep-disordered breathing in children with mucolipidosis. Am. J. Med. Genet. Part A 2019, 179, 1196–1204. [Google Scholar] [CrossRef]
- Mallen, J.; Highstein, M.; Smith, L.; Cheng, J. Airway management considerations in children with I-cell disease. Int. J. Pediatr. Otorhinolaryngol. 2015, 79, 760–762. [Google Scholar] [CrossRef]
- Cathey, S.S.; Leroy, J.G.; Wood, T.; Eaves, K.; Simensen, R.J.; Kudo, M.; Stevenson, R.E.; Friez, M.J. Phenotype and genotype in mucolipidoses II and III alpha/beta: A study of 61 probands. J. Med. Genet. 2010, 47, 38–48. [Google Scholar] [CrossRef]
- Coutinho, M.F.; Santos Lda, S.; Girisha, K.M.; Satyamoorthy, K.; Lacerda, L.; Prata, M.J.; Alves, S. Mucolipidosis type II alpha/beta with a homozygous missense mutation in the GNPTAB gene. Am. J. Med. Genet. Part A 2012, 158A, 1225–1228. [Google Scholar] [CrossRef]
- Raas-Rothschild, A.; Pohl, S.; Braulke, T. Multiple enzyme deficiencies: Defects in transport: Mucolipidosis II alpha/beta; mucolipidosis III alpha/beta and mucolipidosis III gamma. Lysosomal Storage Dis. A Pract. Guide 2012, 121–126. [Google Scholar]
- Spranger, J.; Brill, P.; Poznanski, A. Bone Dysplasias. An Atlas of Genetic Disorders of Skeletal Development; Oxford University Press: Oxford, UK, 2002. [Google Scholar]
- Wood, K.A.; Zambrano, R.M.; Cheek, B.J.; Arcement, C.; Haymon, M.; Steinkampf, J.; Sampath, S.; Hyland, J.C.; Lacassie, Y. Neonatal mucolipidosis type II alpha/beta due to compound heterozygosity for a known and novel GNPTAB mutation, and a concomitant heterozygous change in SERPINF1 inherited from the mother. Clin. Case Rep. 2017, 5, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Leroy, J.G.; Sillence, D.; Wood, T.; Barnes, J.; Lebel, R.R.; Friez, M.J.; Stevenson, R.E.; Steet, R.; Cathey, S.S. A novel intermediate mucolipidosis II/IIIalphabeta caused by GNPTAB mutation in the cytosolic N-terminal domain. Eur. J. Hum. Genet. EJHG 2014, 22, 594–601. [Google Scholar] [CrossRef][Green Version]
- Liu, S.; Zhang, W.; Shi, H.; Yao, F.; Wei, M.; Qiu, Z. Mutation Analysis of 16 Mucolipidosis II and III Alpha/Beta Chinese Children Revealed Genotype-Phenotype Correlations. PLoS ONE 2016, 11, e0163204. [Google Scholar] [CrossRef] [PubMed]
- Oussoren, E.; van Eerd, D.; Murphy, E.; Lachmann, R.; van der Meijden, J.C.; Hoefsloot, L.H.; Verdijk, R.; Ruijter, G.J.G.; Maas, M.; Hollak, C.E.M.; et al. Mucolipidosis type III, a series of adult patients. J. Inherit. Metab. Dis. 2018, 41, 839–848. [Google Scholar] [CrossRef]
- Pohl, S.; Encarnacao, M.; Castrichini, M.; Muller-Loennies, S.; Muschol, N.; Braulke, T. Loss of N-acetylglucosamine-1-phosphotransferase gamma subunit due to intronic mutation in GNPTG causes mucolipidosis type III gamma: Implications for molecular and cellular diagnostics. Am. J. Med. Genet. Part A 2010, 152A, 124–132. [Google Scholar] [CrossRef]
- Raas-Rothschild, A.; Bargal, R.; Goldman, O.; Ben-Asher, E.; Groener, J.E.; Toutain, A.; Stemmer, E.; Ben-Neriah, Z.; Flusser, H.; Beemer, F.A.; et al. Genomic organisation of the UDP-N-acetylglucosamine-1-phosphotransferase gamma subunit (GNPTAG) and its mutations in mucolipidosis III. J. Med. Genet. 2004, 41, e52. [Google Scholar] [CrossRef]
- Tuysuz, B.; Kasapcopur, O.; Alkaya, D.U.; Sahin, S.; Sozeri, B.; Yesil, G. Mucolipidosis type III gamma: Three novel mutation and genotype-phenotype study in eleven patients. Gene 2018, 642, 398–407. [Google Scholar] [CrossRef]
- Velho, R.V.; Ludwig, N.F.; Alegra, T.; Sperb-Ludwig, F.; Guarany, N.R.; Matte, U.; Schwartz, I.V. Enigmatic in vivo GlcNAc-1-phosphotransferase (GNPTG) transcript correction to wild type in two mucolipidosis III gamma siblings homozygous for nonsense mutations. J. Hum. Genet. 2016, 61, 555–560. [Google Scholar] [CrossRef]
- Hasilik, A.; Waheed, A.; von Figura, K. Enzymatic phosphorylation of lysosomal enzymes in the presence of UDP-N-acetylglucosamine. Absence of the activity in I-cell fibroblasts. Biochem. Biophys. Res. Commun. 1981, 98, 761–767. [Google Scholar] [CrossRef]
- Reitman, M.L.; Kornfeld, S. UDP-N-acetylglucosamine:glycoprotein N-acetylglucosamine-1-phosphotransferase. Proposed enzyme for the phosphorylation of the high mannose oligosaccharide units of lysosomal enzymes. J. Biol. Chem. 1981, 256, 4275–4281. [Google Scholar]
- Reitman, M.L.; Kornfeld, S. Lysosomal enzyme targeting. N-Acetylglucosaminylphosphotransferase selectively phosphorylates native lysosomal enzymes. J. Biol. Chem. 1981, 256, 11977–11980. [Google Scholar] [PubMed]
- Rohrer, J.; Kornfeld, R. Lysosomal hydrolase mannose 6-phosphate uncovering enzyme resides in the trans-Golgi network. Mol. Biol. Cell 2001, 12, 1623–1631. [Google Scholar] [CrossRef] [PubMed]
- Pohl, S.; Marschner, K.; Storch, S.; Braulke, T. Glycosylation- and phosphorylation-dependent intracellular transport of lysosomal hydrolases. Biol. Chem. 2009, 390, 521–527. [Google Scholar] [CrossRef]
- Varki, A.; Sherman, W.; Kornfeld, S. Demonstration of the enzymatic mechanisms of alpha-N-acetyl-D-glucosamine-1-phosphodiester N-acetylglucosaminidase (formerly called alpha-N-acetylglucosaminylphosphodiesterase) and lysosomal alpha-N-acetylglucosaminidase. Arch. Biochem. Biophys. 1983, 222, 145–149. [Google Scholar] [CrossRef]
- Varki, A.; Kornfeld, S. Identification of a rat liver alpha-N-acetylglucosaminyl phosphodiesterase capable of removing ”blocking” alpha-N-acetylglucosamine residues from phosphorylated high mannose oligosaccharides of lysosomal enzymes. J. Biol. Chem. 1980, 255, 8398–8401. [Google Scholar]
- Braulke, T.; Bonifacino, J.S. Sorting of lysosomal proteins. Biochim. Biophys. Acta 2009, 1793, 605–614. [Google Scholar] [CrossRef]
- Bao, M.; Booth, J.L.; Elmendorf, B.J.; Canfield, W.M. Bovine UDP-N-acetylglucosamine:lysosomal-enzyme N-acetylglucosamine-1-phosphotransferase. I. Purification and subunit structure. J. Biol. Chem. 1996, 271, 31437–31445. [Google Scholar] [CrossRef]
- Tiede, S.; Storch, S.; Lubke, T.; Henrissat, B.; Bargal, R.; Raas-Rothschild, A.; Braulke, T. Mucolipidosis II is caused by mutations in GNPTA encoding the alpha/beta GlcNAc-1-phosphotransferase. Nat. Med. 2005, 11, 1109–1112. [Google Scholar] [CrossRef]
- Kudo, M.; Brem, M.S.; Canfield, W.M. Mucolipidosis II (I-cell disease) and mucolipidosis IIIA (classical pseudo-hurler polydystrophy) are caused by mutations in the GlcNAc-phosphotransferase alpha/beta -subunits precursor gene. Am. J. Hum. Genet. 2006, 78, 451–463. [Google Scholar] [CrossRef]
- Kudo, M.; Bao, M.; D’Souza, A.; Ying, F.; Pan, H.; Roe, B.A.; Canfield, W.M. The alpha- and beta-subunits of the human UDP-N-acetylglucosamine:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase [corrected] are encoded by a single cDNA. J. Biol. Chem. 2005, 280, 36141–36149. [Google Scholar] [CrossRef] [PubMed]
- Marschner, K.; Kollmann, K.; Schweizer, M.; Braulke, T.; Pohl, S. A key enzyme in the biogenesis of lysosomes is a protease that regulates cholesterol metabolism. Science 2011, 333, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Raas-Rothschild, A.; Cormier-Daire, V.; Bao, M.; Genin, E.; Salomon, R.; Brewer, K.; Zeigler, M.; Mandel, H.; Toth, S.; Roe, B.; et al. Molecular basis of variant pseudo-hurler polydystrophy (mucolipidosis IIIC). J. Clin. Investig. 2000, 105, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Muller-Loennies, S.; Galliciotti, G.; Kollmann, K.; Glatzel, M.; Braulke, T. A novel single-chain antibody fragment for detection of mannose 6-phosphate-containing proteins: Application in mucolipidosis type II patients and mice. Am. J. Pathol. 2010, 177, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Pohl, S.; Braulke, T.; Müller-Loennies, S. A Novel Mannose 6-phosphate Specific Antibody Fragment for Diagnosis of Mucolipidosis type II and III. In Anticarbohydrate Antibodies; Springer: Berlin/Heidelberg, Germany, 2012; pp. 307–325. [Google Scholar]
- Sperb-Ludwig, F.; Alegra, T.; Velho, R.V.; Ludwig, N.; Kim, C.A.; Kok, F.; Kitajima, J.P.; van Meel, E.; Kornfeld, S.; Burin, M.G.; et al. Exome sequencing for mucolipidosis III: Detection of a novel GNPTAB gene mutation in a patient with a very mild phenotype. Mol. Genet. Metab. Rep. 2015, 2, 34–37. [Google Scholar] [CrossRef]
- Steet, R.A.; Hullin, R.; Kudo, M.; Martinelli, M.; Bosshard, N.U.; Schaffner, T.; Kornfeld, S.; Steinmann, B. A splicing mutation in the alpha/beta GlcNAc-1-phosphotransferase gene results in an adult onset form of mucolipidosis III associated with sensory neuropathy and cardiomyopathy. Am. J. Med. Genet. Part A 2005, 132A, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Tiede, S.; Cantz, M.; Raas-Rothschild, A.; Muschol, N.; Burger, F.; Ullrich, K.; Braulke, T. A novel mutation in UDP-N-acetylglucosamine-1-phosphotransferase gamma subunit (GNPTAG) in two siblings with mucolipidosis type III alters a used glycosylation site. Hum. Mutat. 2004, 24, 535. [Google Scholar] [CrossRef]
- Tiede, S.; Muschol, N.; Reutter, G.; Cantz, M.; Ullrich, K.; Braulke, T. Missense mutations in N-acetylglucosamine-1-phosphotransferase alpha/beta subunit gene in a patient with mucolipidosis III and a mild clinical phenotype. Am. J. Med. Genet. Part A 2005, 137A, 235–240. [Google Scholar] [CrossRef]
- Qian, Y.; van Meel, E.; Flanagan-Steet, H.; Yox, A.; Steet, R.; Kornfeld, S. Analysis of mucolipidosis II/III GNPTAB missense mutations identifies domains of UDP-GlcNAc:lysosomal enzyme GlcNAc-1-phosphotransferase involved in catalytic function and lysosomal enzyme recognition. J. Biol. Chem. 2015, 290, 3045–3056. [Google Scholar] [CrossRef]
- Qian, Y.; Flanagan-Steet, H.; van Meel, E.; Steet, R.; Kornfeld, S.A. The DMAP interaction domain of UDP-GlcNAc:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase is a substrate recognition module. Proc. Natl. Acad. Sci. USA 2013, 110, 10246–10251. [Google Scholar] [CrossRef]
- van Meel, E.; Qian, Y.; Kornfeld, S.A. Mislocalization of phosphotransferase as a cause of mucolipidosis III alphabeta. Proc. Natl. Acad. Sci. USA 2014, 111, 3532–3537. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.C.; Zhang, L.; Wu, H.C.; Tsui, K.M. A Maximum A Posteriori Probability and Time-Varying Approach for Inferring Gene Regulatory Networks from Time Course Gene Microarray Data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2015, 12, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Otomo, T.; Muramatsu, T.; Yorifuji, T.; Okuyama, T.; Nakabayashi, H.; Fukao, T.; Ohura, T.; Yoshino, M.; Tanaka, A.; Okamoto, N.; et al. Mucolipidosis II and III alpha/beta: Mutation analysis of 40 Japanese patients showed genotype-phenotype correlation. J. Hum. Genet. 2009, 54, 145–151. [Google Scholar] [CrossRef]
- Plante, M.; Claveau, S.; Lepage, P.; Lavoie, E.M.; Brunet, S.; Roquis, D.; Morin, C.; Vezina, H.; Laprise, C. Mucolipidosis II: A single causal mutation in the N-acetylglucosamine-1-phosphotransferase gene (GNPTAB) in a French Canadian founder population. Clin. Genet. 2008, 73, 236–244. [Google Scholar] [CrossRef]
- Paik, K.H.; Song, S.M.; Ki, C.S.; Yu, H.W.; Kim, J.S.; Min, K.H.; Chang, S.H.; Yoo, E.J.; Lee, I.J.; Kwan, E.K.; et al. Identification of mutations in the GNPTA (MGC4170) gene coding for GlcNAc-phosphotransferase alpha/beta subunits in Korean patients with mucolipidosis type II or type IIIA. Hum. Mutat. 2005, 26, 308–314. [Google Scholar] [CrossRef]
- Raza, M.H.; Domingues, C.E.; Webster, R.; Sainz, E.; Paris, E.; Rahn, R.; Gutierrez, J.; Chow, H.M.; Mundorff, J.; Kang, C.S.; et al. Mucolipidosis types II and III and non-syndromic stuttering are associated with different variants in the same genes. Eur. J. Hum. Genet. EJHG 2016, 24, 529–534. [Google Scholar] [CrossRef]
- Wiesmann, U.; Vassella, F.; Herschkowitz, N. “I-cell” disease: Leakage of lysosomal enzymes into extracellular fluids. N. Engl. J. Med. 1971, 285, 1090–1091. [Google Scholar]
- Leroy, J.G.; Ho, M.W.; MacBrinn, M.C.; Zielke, K.; Jacob, J.; O’Brien, J.S. I-cell disease: Biochemical studies. Pediatr. Res. 1972, 6, 752–757. [Google Scholar] [CrossRef]
- Bitter, T.; Muir, H.M. A modified uronic acid carbazole reaction. Anal. Biochem. 1962, 4, 330–334. [Google Scholar] [CrossRef]
- Boas, N.F. Method for the determination of hexosamines in tissues. J. Biol. Chem. 1953, 204, 553–563. [Google Scholar]
- Reitman, M.L.; Varki, A.; Kornfeld, S. Fibroblasts from patients with I-cell disease and pseudo-Hurler polydystrophy are deficient in uridine 5’-diphosphate-N-acetylglucosamine: Glycoprotein N-acetylglucosaminylphosphotransferase activity. J. Clin. Investig. 1981, 67, 1574–1579. [Google Scholar] [CrossRef] [PubMed]
- Whelan, D.T.; Chang, P.L.; Cockshott, P.W. Mucolipidosis II. The clinical, radiological and biochemical features in three cases. Clin. Genet. 1983, 24, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Tames, I.; Gracia, A.; Aladro, A.; Vieito, X.; Gonzalez, F.A.; Chabas, A. Mucolipidosis II or ”I-cell diseas” in the newborn infant. 2 new cases. Esp. Pediatr. 1987, 27, 297–302. [Google Scholar]
- Singh, A.; Prasad, R.; Gupta, A.K.; Sharma, A.; Alves, S.; Coutinho, M.F.; Kapoor, S.; Mishra, O.P. I Cell Disease (Mucolipidosis II Alpha/Beta): From Screening to Molecular Diagnosis. Indian J. Pediatr. 2017, 84, 144–146. [Google Scholar] [CrossRef]
- Leroy, J.G.; Spranger, J.W.; Feingold, M.; Opitz, J.M.; Crocker, A.C. I-cell disease: A clinical picture. J. Pediatr. 1971, 79, 360–365. [Google Scholar] [CrossRef]
- Coppa, G.V.; Maiorana, A.; Gabrielli, O.; Sani, S. Glycosaminoglycans from urine and tissues in mucolipidosis II (I-cell disease). Clin. Chim. Acta Int. J. Clin. Chem. 1979, 95, 135–137. [Google Scholar] [CrossRef]
- Tomatsu, S.; Gutierrez, M.A.; Ishimaru, T.; Pena, O.M.; Montano, A.M.; Maeda, H.; Velez-Castrillon, S.; Nishioka, T.; Fachel, A.A.; Cooper, A.; et al. Heparan sulfate levels in mucopolysaccharidoses and mucolipidoses. J. Inherit. Metab. Dis. 2005, 28, 743–757. [Google Scholar] [CrossRef]
- Tomatsu, S.; Okamura, K.; Maeda, H.; Taketani, T.; Castrillon, S.V.; Gutierrez, M.A.; Nishioka, T.; Fachel, A.A.; Orii, K.O.; Grubb, J.H.; et al. Keratan sulphate levels in mucopolysaccharidoses and mucolipidoses. J. Inherit. Metab. Dis. 2005, 28, 187–202. [Google Scholar] [CrossRef]
- Langereis, E.J.; Wagemans, T.; Kulik, W.; Lefeber, D.J.; van Lenthe, H.; Oussoren, E.; van der Ploeg, A.T.; Ruijter, G.J.; Wevers, R.A.; Wijburg, F.A.; et al. A Multiplex Assay for the Diagnosis of Mucopolysaccharidoses and Mucolipidoses. PLoS ONE 2015, 10, e0138622. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montano, A.M.; Oguma, T.; Dung, V.C.; Oikawa, H.; Gutierrez, M.L.; Yamaguchi, S.; Suzuki, Y.; Fukushi, M.; Barrera, L.A.; et al. Validation of disaccharide compositions derived from dermatan sulfate and heparan sulfate in mucopolysaccharidoses and mucolipidoses II and III by tandem mass spectrometry. Mol. Genet. Metab. 2010, 99, 124–131. [Google Scholar] [CrossRef]
- Tomatsu, S.; Shimada, T.; Mason, R.W.; Montano, A.M.; Kelly, J.; LaMarr, W.A.; Kubaski, F.; Giugliani, R.; Guha, A.; Yasuda, E.; et al. Establishment of glycosaminoglycan assays for mucopolysaccharidoses. Metabolites 2014, 4, 655–679. [Google Scholar] [CrossRef] [PubMed]
- Otomo, T.; Higaki, K.; Nanba, E.; Ozono, K.; Sakai, N. Lysosomal storage causes cellular dysfunction in mucolipidosis II skin fibroblasts. J. Biol. Chem. 2011, 286, 35283–35290. [Google Scholar] [CrossRef]
- Kawashima, I.; Ohsawa, M.; Fukushige, T.; Nagayama, Y.; Niida, Y.; Kotani, M.; Tajima, Y.; Kanekura, T.; Kanzaki, T.; Sakuraba, H. Cytochemical analysis of storage materials in cultured skin fibroblasts from patients with I-cell disease. Clin. Chim. Acta Int. J. Clin. Chem. 2007, 378, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, A.; Niida, Y.; Kuroda, M.; Imi-Hashida, Y.; Toma, T.; Yachie, A. B-cell-specific accumulation of inclusion bodies loaded with HLA class II molecules in patients with mucolipidosis II (I-cell disease). Pediatr. Res. 2019, 86, 85–91. [Google Scholar] [CrossRef]
- Khan, S.A.; Nelson, M.S.; Pan, C.; Gaffney, P.M.; Gupta, P. Endogenous heparan sulfate and heparin modulate bone morphogenetic protein-4 signaling and activity. Am. J. Physiol. Cell Physiol. 2008, 294, C1387–C1397. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Nelson, M.S.; Reyes, M.; Koodie, L.; Brazil, J.J.; Stephenson, E.J.; Zhao, R.C.; Peters, C.; Selleck, S.B.; Stringer, S.E.; et al. Functional abnormalities of heparan sulfate in mucopolysaccharidosis-I are associated with defective biologic activity of FGF-2 on human multipotent progenitor cells. Blood 2005, 106, 1956–1964. [Google Scholar] [CrossRef]
- Kollmann, K.; Pestka, J.M.; Kuhn, S.C.; Schone, E.; Schweizer, M.; Karkmann, K.; Otomo, T.; Catala-Lehnen, P.; Failla, A.V.; Marshall, R.P.; et al. Decreased bone formation and increased osteoclastogenesis cause bone loss in mucolipidosis II. EMBO Mol. Med. 2013, 5, 1871–1886. [Google Scholar] [CrossRef]
- Van Meel, E.; Boonen, M.; Zhao, H.; Oorschot, V.; Ross, F.P.; Kornfeld, S.; Klumperman, J. Disruption of the Man-6-P targeting pathway in mice impairs osteoclast secretory lysosome biogenesis. Traffic 2011, 12, 912–924. [Google Scholar] [CrossRef]
- Vanier, M.T.; Rodriguez-Lafrasse, C.; Rousson, R.; Gazzah, N.; Juge, M.C.; Pentchev, P.G.; Revol, A.; Louisot, P. Type C Niemann-Pick disease: Spectrum of phenotypic variation in disruption of intracellular LDL-derived cholesterol processing. Biochim. Biophys. Acta 1991, 1096, 328–337. [Google Scholar] [CrossRef]
- Vanier, M.T. Complex lipid trafficking in Niemann-Pick disease type C. J. Inherit. Metab. Dis. 2015, 38, 187–199. [Google Scholar] [CrossRef]
- Willenborg, M.; Schmidt, C.K.; Braun, P.; Landgrebe, J.; von Figura, K.; Saftig, P.; Eskelinen, E.L. Mannose 6-phosphate receptors, Niemann-Pick C2 protein, and lysosomal cholesterol accumulation. J. Lipid Res. 2005, 46, 2559–2569. [Google Scholar] [CrossRef]
- Furuchi, T.; Aikawa, K.; Arai, H.; Inoue, K. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, blocks lysosomal cholesterol trafficking in macrophages. J. Biol. Chem. 1993, 268, 27345–27348. [Google Scholar]
- Pittman, R.C.; Williams, J.C.; Miller, A.L.; Steinberg, D. Acid acylhydrolase deficiency in I-cell disease and pseudo-Hurler polydystrophy. Biochim. Biophys. Acta 1979, 575, 399–409. [Google Scholar] [CrossRef]
- Kollmann, K.; Damme, M.; Markmann, S.; Morelle, W.; Schweizer, M.; Hermans-Borgmeyer, I.; Rochert, A.K.; Pohl, S.; Lubke, T.; Michalski, J.C.; et al. Lysosomal dysfunction causes neurodegeneration in mucolipidosis II ‘knock-in’ mice. Brain 2012, 135, 2661–2675. [Google Scholar] [CrossRef] [PubMed]
- Tondeur, M.; Vamos-Hurwitz, E.; Mockel-Pohl, S.; Dereume, J.P.; Cremer, N.; Loeb, H. Clinical, biochemical, and ultrastructural studies in a case of chondrodystrophy presenting the I-cell phenotype in tissue culture. J. Pediatr. 1971, 79, 366–378. [Google Scholar] [CrossRef]
- Owada, M.; Neufeld, E.F. Is there a mechanism for introducing acid hydrolases into liver lysosomes that is independent of mannose 6-phosphate recognition? Evidence from I-cell disease. Biochem. Biophys. Res. Commun. 1982, 105, 814–820. [Google Scholar] [CrossRef]
- Waheed, A.; Pohlmann, R.; Hasilik, A.; von Figura, K.; van Elsen, A.; Leroy, J.G. Deficiency of UDP-N-acetylglucosamine:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase in organs of I-cell patients. Biochem. Biophys. Res. Commun. 1982, 105, 1052–1058. [Google Scholar] [CrossRef]
- Koster, A.; von Figura, K.; Pohlmann, R. Mistargeting of lysosomal enzymes in M(r) 46,000 mannose 6-phosphate receptor-deficient mice is compensated by carbohydrate-specific endocytotic receptors. Eur. J. Biochem. 1994, 224, 685–689. [Google Scholar] [CrossRef]
- Idol, R.A.; Wozniak, D.F.; Fujiwara, H.; Yuede, C.M.; Ory, D.S.; Kornfeld, S.; Vogel, P. Neurologic abnormalities in mouse models of the lysosomal storage disorders mucolipidosis II and mucolipidosis III gamma. PLoS ONE 2014, 9, e109768. [Google Scholar] [CrossRef]
- Paton, L.; Bitoun, E.; Kenyon, J.; Priestman, D.A.; Oliver, P.L.; Edwards, B.; Platt, F.M.; Davies, K.E. A novel mouse model of a patient mucolipidosis II mutation recapitulates disease pathology. J. Biol. Chem. 2014, 289, 26709–26721. [Google Scholar] [CrossRef]
- Hubler, M.; Haskins, M.E.; Arnold, S.; Kaser-Hotz, B.; Bosshard, N.U.; Briner, J.; Spycher, M.A.; Gitzelmann, R.; Sommerlade, H.J.; von Figura, K. Mucolipidosis type II in a domestic shorthair cat. J. Small Anim. Pr. 1996, 37, 435–441. [Google Scholar] [CrossRef]
- Mazrier, H.; Van Hoeven, M.; Wang, P.; Knox, V.W.; Aguirre, G.D.; Holt, E.; Wiemelt, S.P.; Sleeper, M.M.; Hubler, M.; Haskins, M.E.; et al. Inheritance, biochemical abnormalities, and clinical features of feline mucolipidosis II: The first animal model of human I-cell disease. J. Hered. 2003, 94, 363–373. [Google Scholar] [CrossRef]
- Wang, P.; Mazrier, H.; Caverly Rae, J.; Raj, K.; Giger, U. A GNPTAB nonsense variant is associated with feline mucolipidosis II (I-cell disease). BMC Vet. Res. 2018, 14, 416. [Google Scholar] [CrossRef]
- Gelfman, C.M.; Vogel, P.; Issa, T.M.; Turner, C.A.; Lee, W.S.; Kornfeld, S.; Rice, D.S. Mice lacking alpha/beta subunits of GlcNAc-1-phosphotransferase exhibit growth retardation, retinal degeneration, and secretory cell lesions. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5221–5228. [Google Scholar] [CrossRef]
- Otomo, T.; Schweizer, M.; Kollmann, K.; Schumacher, V.; Muschol, N.; Tolosa, E.; Mittrucker, H.W.; Braulke, T. Mannose 6 phosphorylation of lysosomal enzymes controls B cell functions. J. Cell Biol. 2015, 208, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.R.; Jin, D.K.; Cho, S.Y.; Park, S.W.; Przybylska, M.; Yew, N.S.; Cheng, S.H.; Kim, J.S.; Kwak, M.J.; Kim, S.J.; et al. AAV8-mediated expression of N-acetylglucosamine-1-phosphate transferase attenuates bone loss in a mouse model of mucolipidosis II. Mol. Genet. Metab. 2016, 117, 447–455. [Google Scholar] [CrossRef]
- Lee, W.S.; Payne, B.J.; Gelfman, C.M.; Vogel, P.; Kornfeld, S. Murine UDP-GlcNAc:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase lacking the gamma-subunit retains substantial activity toward acid hydrolases. J. Biol. Chem. 2007, 282, 27198–27203. [Google Scholar] [CrossRef]
- Vogel, P.; Payne, B.J.; Read, R.; Lee, W.S.; Gelfman, C.M.; Kornfeld, S. Comparative pathology of murine mucolipidosis types II and IIIC. Vet. Pathol. 2009, 46, 313–324. [Google Scholar] [CrossRef]
- Boonen, M.; Vogel, P.; Platt, K.A.; Dahms, N.; Kornfeld, S. Mice lacking mannose 6-phosphate uncovering enzyme activity have a milder phenotype than mice deficient for N-acetylglucosamine-1-phosphotransferase activity. Mol. Biol. Cell 2009, 20, 4381–4389. [Google Scholar] [CrossRef]
- Flanagan-Steet, H.; Sias, C.; Steet, R. Altered chondrocyte differentiation and extracellular matrix homeostasis in a zebrafish model for mucolipidosis II. Am. J. Pathol. 2009, 175, 2063–2075. [Google Scholar] [CrossRef]
- Petrey, A.C.; Flanagan-Steet, H.; Johnson, S.; Fan, X.; De la Rosa, M.; Haskins, M.E.; Nairn, A.V.; Moremen, K.W.; Steet, R. Excessive activity of cathepsin K is associated with cartilage defects in a zebrafish model of mucolipidosis II. Dis. Model. Mech. 2012, 5, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Flanagan-Steet, H.; Matheny, C.; Petrey, A.; Parker, J.; Steet, R. Enzyme-specific differences in mannose phosphorylation between GlcNAc-1-phosphotransferase alphabeta and gamma subunit deficient zebrafish support cathepsin proteases as early mediators of mucolipidosis pathology. Biochim. Biophys. Acta 2016, 1860, 1845–1853. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Flanagan-Steet, H.; Aarnio, M.; Kwan, B.; Guihard, P.; Petrey, A.; Haskins, M.; Blanchard, F.; Steet, R. Cathepsin-Mediated Alterations in TGFss-Related Signaling Underlie Disrupted Cartilage and Bone Maturation Associated With Impaired Lysosomal Targeting. J. Bone Miner. Res. 2016, 31, 535–548. [Google Scholar] [CrossRef]
- Flanagan-Steet, H.; Christian, C.; Lu, P.N.; Aarnio-Peterson, M.; Sanman, L.; Archer-Hartmann, S.; Azadi, P.; Bogyo, M.; Steet, R.A. TGF-ss Regulates Cathepsin Activation during Normal and Pathogenic Development. Cell Rep. 2018, 22, 2964–2977. [Google Scholar] [CrossRef]
- Raas-Rothschild, A.; Spiegel, R. Mucolipidosis III Gamma. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Takanobu, O. Mucolipidoses: Clinical features, biochemistry, diagnosis, genetics, and treatment. In Mucopolysaccharidoses Update; Tomatsu, S., Lavery, C., Giugliani, R., Harmatz, P., Scarpa, M., Wegrzyn, G., Tadao, O., Eds.; Nova Science Publishers Inc.: New York, NY, USA, 2018; Volume 1, pp. 351–373. [Google Scholar]
- Lund, T.C.; Cathey, S.S.; Miller, W.P.; Eapen, M.; Andreansky, M.; Dvorak, C.C.; Davis, J.H.; Dalal, J.D.; Devine, S.M.; Eames, G.M.; et al. Outcomes after hematopoietic stem cell transplantation for children with I-cell disease. Biol. Blood Marrow Transpl. 2014, 20, 1847–1851. [Google Scholar] [CrossRef] [PubMed]
- Grewal, S.; Shapiro, E.; Braunlin, E.; Charnas, L.; Krivit, W.; Orchard, P.; Peters, C. Continued neurocognitive development and prevention of cardiopulmonary complications after successful BMT for I-cell disease: A long-term follow-up report. Bone Marrow Transpl. 2003, 32, 957–960. [Google Scholar] [CrossRef]
- Naumchik, B.M.; Gupta, A.; Flanagan-Steet, H.; Steet, R.A.; Cathey, S.S.; Orchard, P.J.; Lund, T.C. The Role of Hematopoietic Cell Transplant in the Glycoprotein Diseases. Cells 2020, 9, 1141. [Google Scholar] [CrossRef]
- Wyatt, K.; Henley, W.; Anderson, L.; Anderson, R.; Nikolaou, V.; Stein, K.; Klinger, L.; Hughes, D.; Waldek, S.; Lachmann, R.; et al. The effectiveness and cost-effectiveness of enzyme and substrate replacement therapies: A longitudinal cohort study of people with lysosomal storage disorders. Health Technol. Assess. 2012, 16, 1–543. [Google Scholar] [CrossRef]
- Hickman, S.; Neufeld, E.F. A hypothesis for I-cell disease: Defective hydrolases that do not enter lysosomes. Biochem. Biophys. Res. Commun. 1972, 49, 992–999. [Google Scholar] [CrossRef]
- Parenti, G.; Andria, G.; Valenzano, K.J. Pharmacological Chaperone Therapy: Preclinical Development, Clinical Translation, and Prospects for the Treatment of Lysosomal Storage Disorders. Mol. Ther. 2015, 23, 1138–1148. [Google Scholar] [CrossRef]
- Akiyama, T.; Ishida, J.; Nakagawa, S.; Ogawara, H.; Watanabe, S.; Itoh, N.; Shibuya, M.; Fukami, Y. Genistein, a specific inhibitor of tyrosine-specific protein kinases. J. Biol. Chem. 1987, 262, 5592–5595. [Google Scholar] [PubMed]
- Piotrowska, E.; Jakobkiewicz-Banecka, J.; Baranska, S.; Tylki-Szymanska, A.; Czartoryska, B.; Wegrzyn, A.; Wegrzyn, G. Genistein-mediated inhibition of glycosaminoglycan synthesis as a basis for gene expression-targeted isoflavone therapy for mucopolysaccharidoses. Eur. J. Hum. Genet. EJHG 2006, 14, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Malinowska, M.; Wilkinson, F.L.; Bennett, W.; Langford-Smith, K.J.; O’Leary, H.A.; Jakobkiewicz-Banecka, J.; Wynn, R.; Wraith, J.E.; Wegrzyn, G.; Bigger, B.W. Genistein reduces lysosomal storage in peripheral tissues of mucopolysaccharide IIIB mice. Mol. Genet. Metab. 2009, 98, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Marucha, J.; Tylki-Szymanska, A.; Jakobkiewicz-Banecka, J.; Piotrowska, E.; Kloska, A.; Czartoryska, B.; Wegrzyn, G. Improvement in the range of joint motion in seven patients with mucopolysaccharidosis type II during experimental gene expression-targeted isoflavone therapy (GET IT). Am. J. Med. Genet. Part A 2011, 155A, 2257–2262. [Google Scholar] [CrossRef] [PubMed]
- Kingma, S.D.; Wagemans, T.; L, I.J.; Wijburg, F.A.; van Vlies, N. Genistein increases glycosaminoglycan levels in mucopolysaccharidosis type I cell models. J. Inherit. Metab. Dis. 2014, 37, 813–821. [Google Scholar] [CrossRef]
- Kingma, S.D.; Wagemans, T.; L, I.J.; Seppen, J.; Gijbels, M.J.; Wijburg, F.A.; van Vlies, N. Adverse Effects of Genistein in a Mucopolysaccharidosis Type I Mouse Model. JIMD Rep. 2015, 23, 77–83. [Google Scholar] [PubMed]
- Otomo, T.; Hossain, M.A.; Ozono, K.; Sakai, N. Genistein reduces heparan sulfate accumulation in human mucolipidosis II skin fibroblasts. Mol. Genet. Metab. 2012, 105, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Matos, L.; Vilela, R.; Rocha, M.; Santos, J.I.; Coutinho, M.F.; Gaspar, P.; Prata, M.J.; Alves, S. Development of an Antisense Oligonucleotide-Mediated Exon Skipping Therapeutic Strategy for Mucolipidosis II: Validation at RNA Level. Hum. Gene Ther. 2020, 31, 775–783. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, S.A.; Tomatsu, S.C. Mucolipidoses Overview: Past, Present, and Future. Int. J. Mol. Sci. 2020, 21, 6812. https://doi.org/10.3390/ijms21186812
Khan SA, Tomatsu SC. Mucolipidoses Overview: Past, Present, and Future. International Journal of Molecular Sciences. 2020; 21(18):6812. https://doi.org/10.3390/ijms21186812
Chicago/Turabian StyleKhan, Shaukat A., and Saori C. Tomatsu. 2020. "Mucolipidoses Overview: Past, Present, and Future" International Journal of Molecular Sciences 21, no. 18: 6812. https://doi.org/10.3390/ijms21186812
APA StyleKhan, S. A., & Tomatsu, S. C. (2020). Mucolipidoses Overview: Past, Present, and Future. International Journal of Molecular Sciences, 21(18), 6812. https://doi.org/10.3390/ijms21186812