Toll-Like Receptor as a Potential Biomarker in Renal Diseases

 , , ,
, , ,  , ,
, ,  , ,
, ,
Abstract
1. Introduction
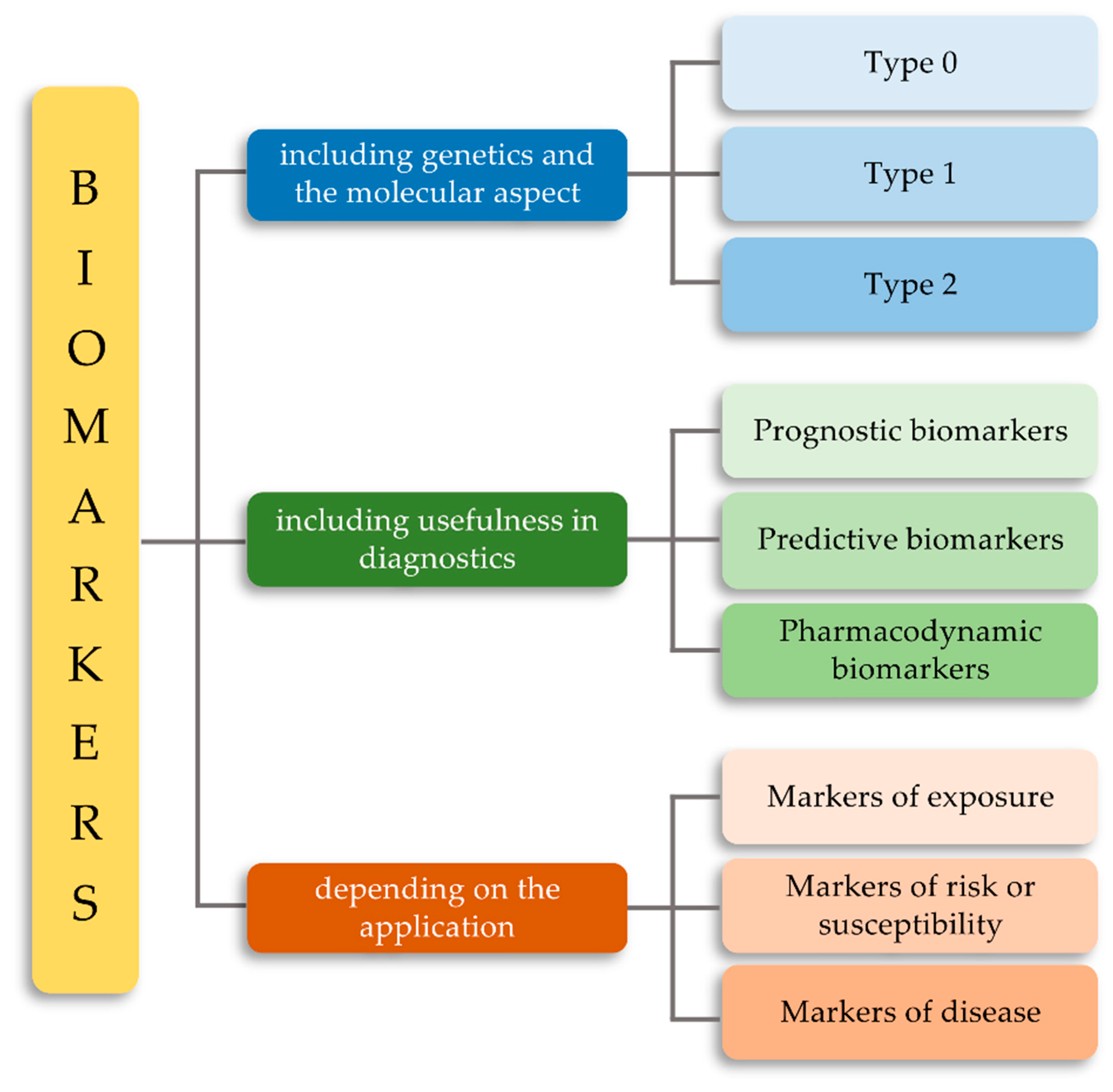
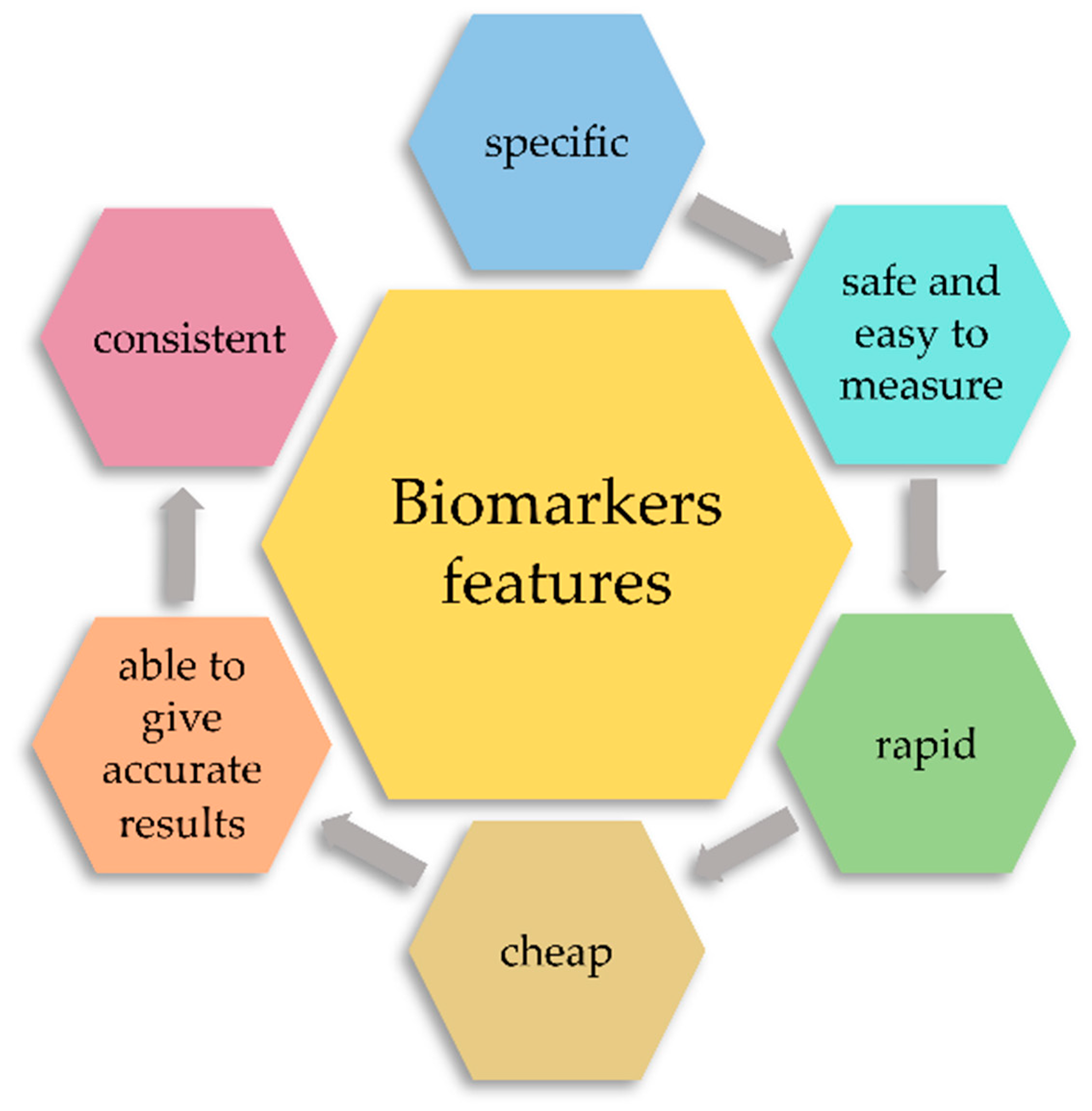
2. Classification of Biomarkers
3. Characteristic of the TLR Receptors
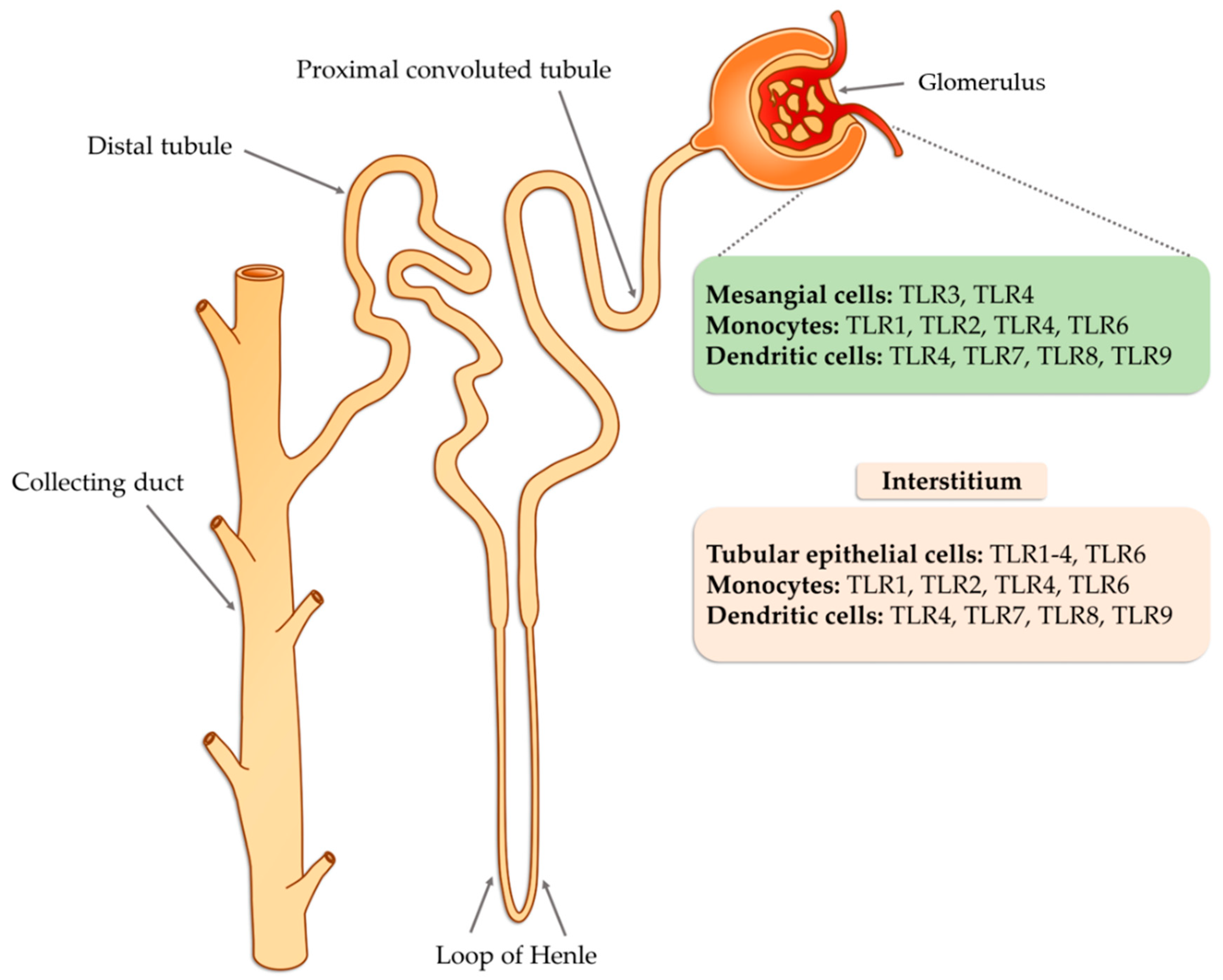
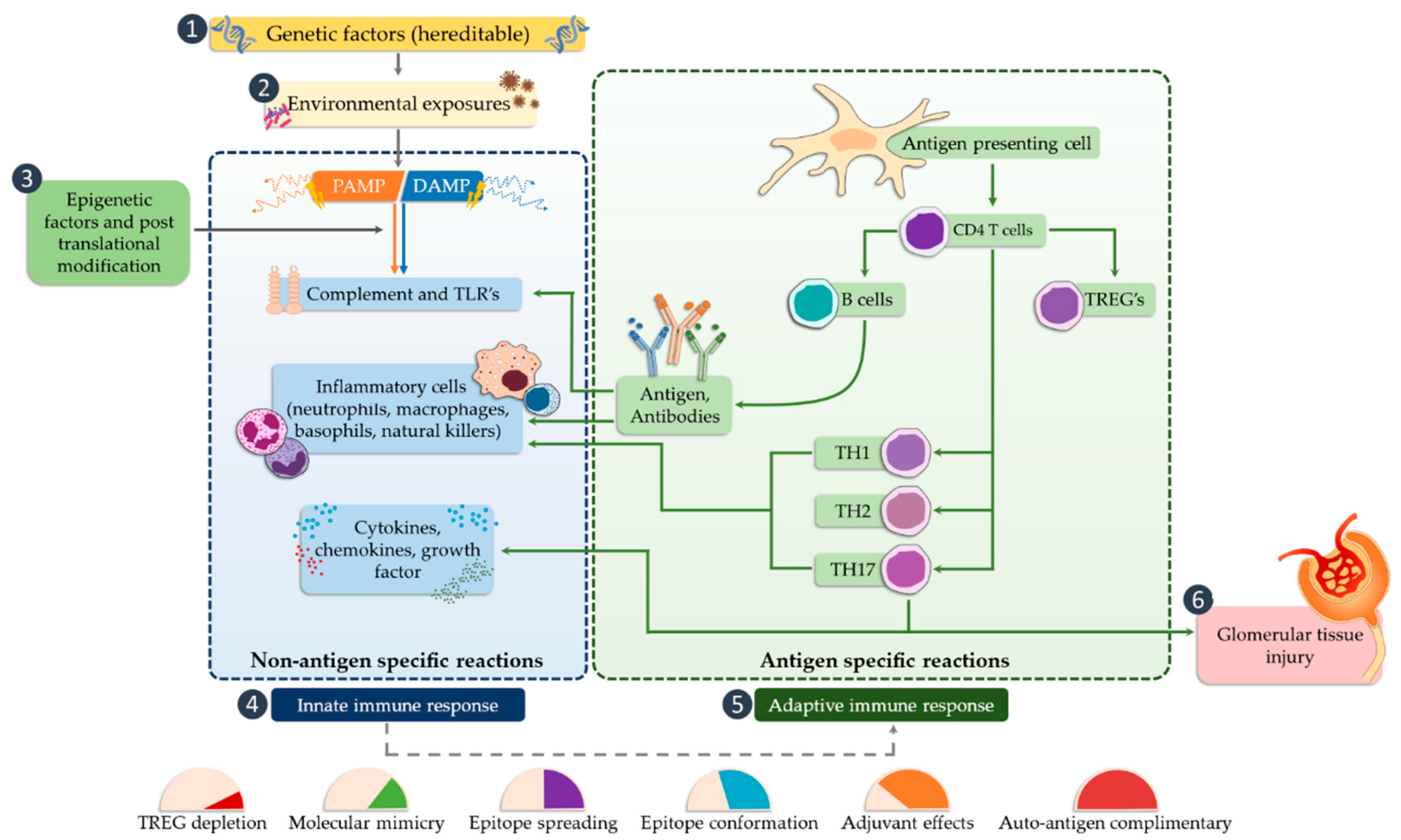
4. The Role of TLRs in Glomerulonephritis
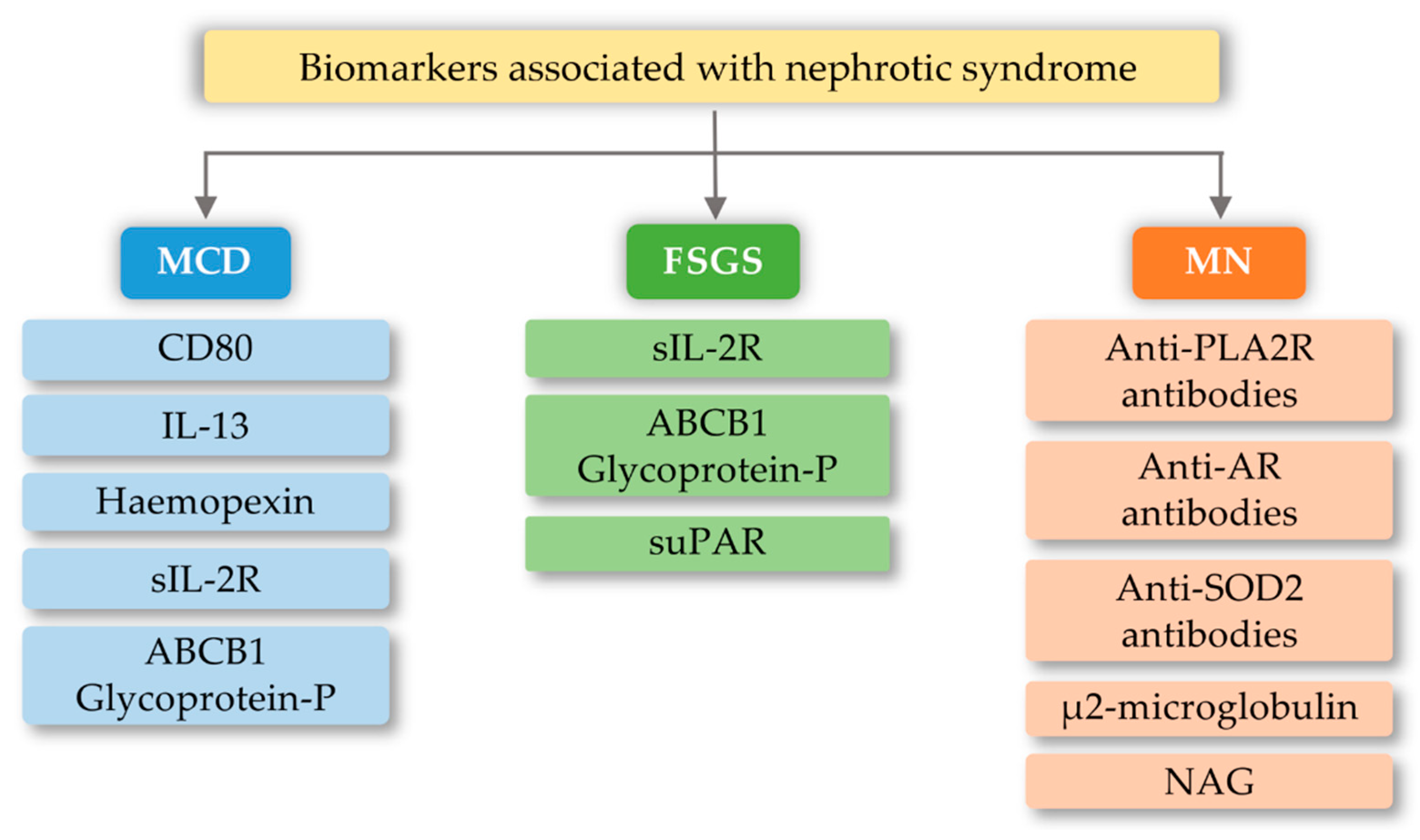
5. Biomarkers and Importance of the TLRs in Selected Glomerular Diseases
6. The Role of TLRs in Primary Non-Proliferative Nephropathies
6.1. Focal Segmental Glomerulosclerosis (FSGS)
6.2. Minimal Change Disease (MCD)
6.3. Membranous Nephropathy (MN)
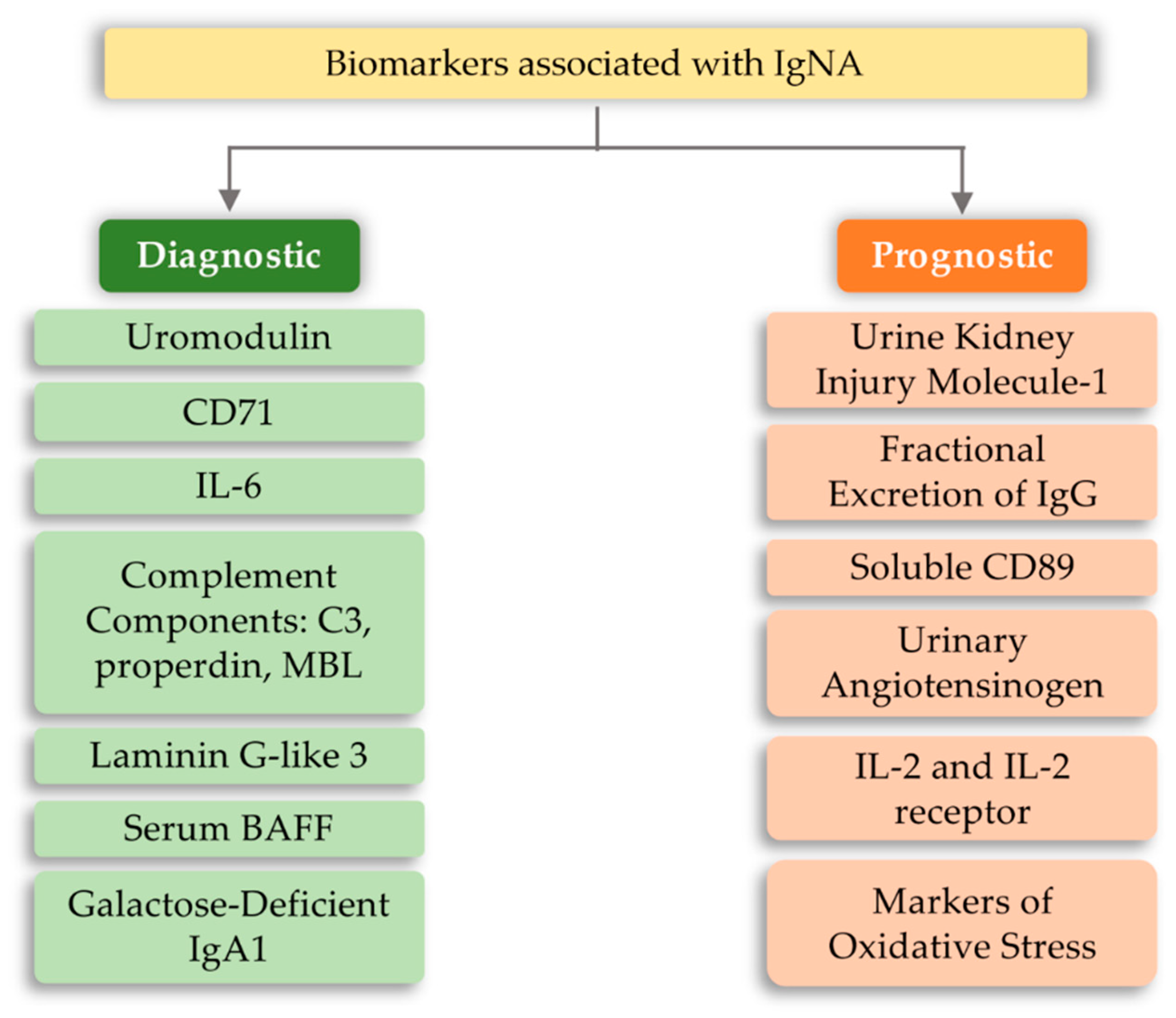
6.4. IgA Nephropathy (IgAN)
7. The Role of TLR Receptors in Secondary Nephropathies
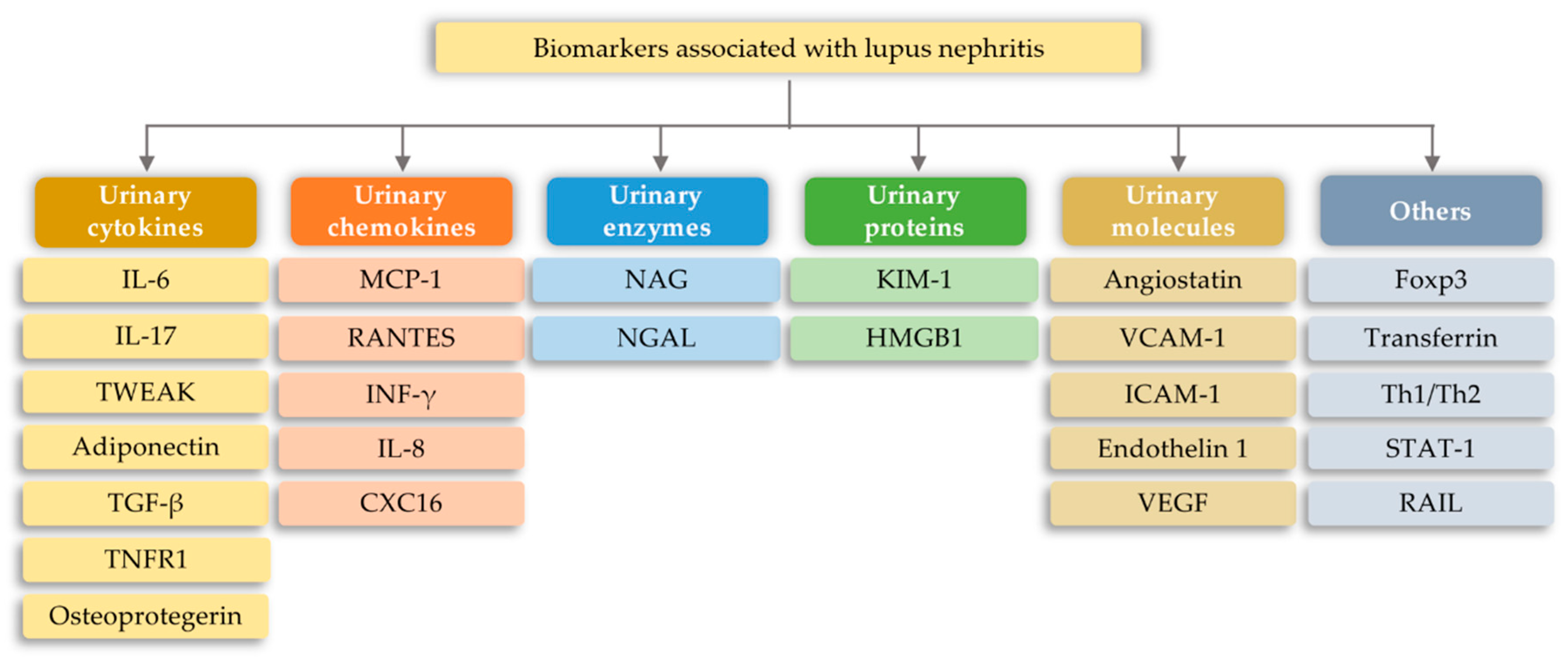
7.1. Lupus Nephritis
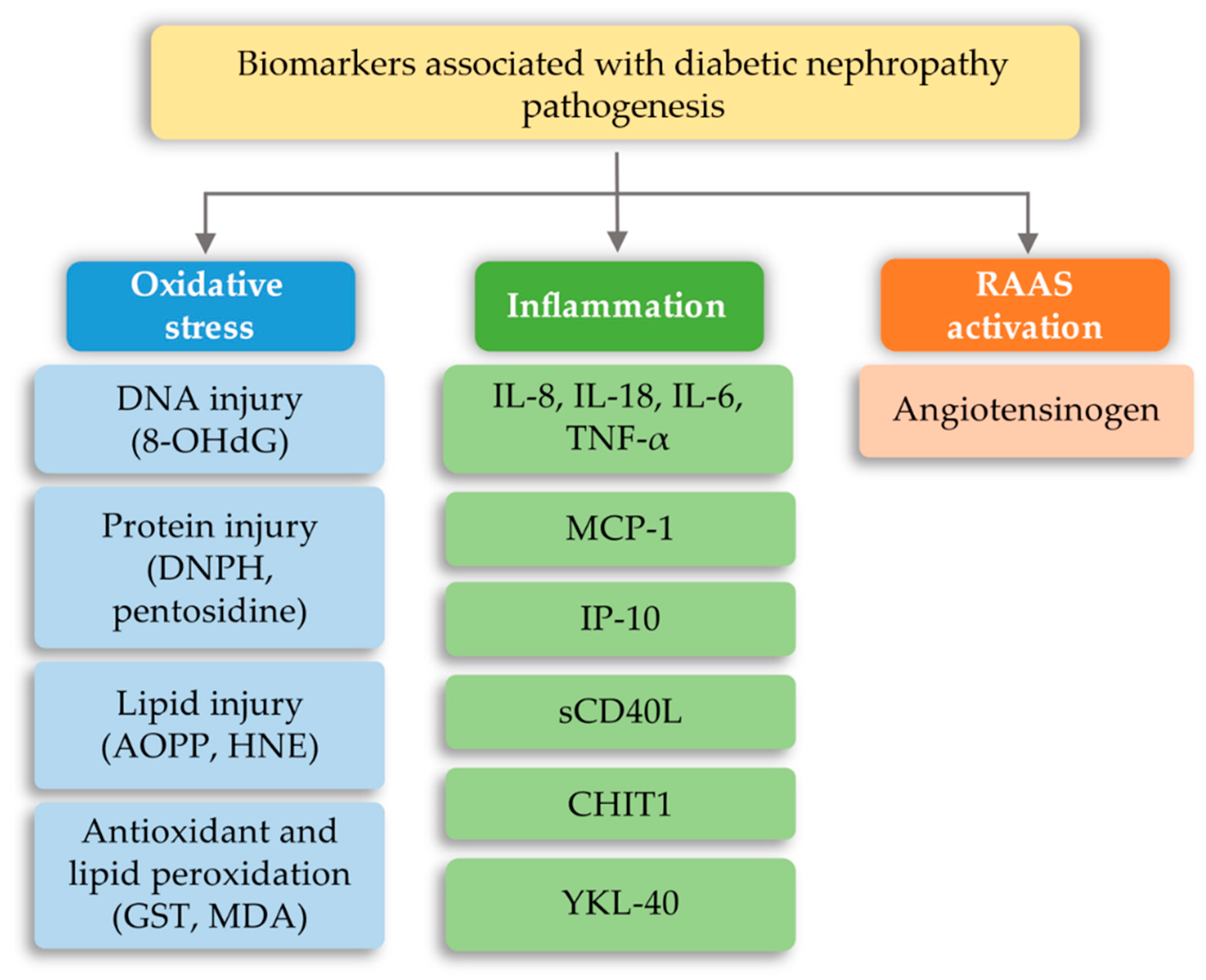
7.2. Diabetic Nephropathy (DN)
7.3. Acute Kidney Injury (AKI) to Chronic Kidney Disease (CKD) Development
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 8-OHdG | 8-hydroxy-2’-deoxyguanine |
| ABCB1 Glycoprotein-P | ATP-binding cassette subfamily B member 1 Glycoprotein-P |
| AKI | Acute kidney injury |
| AOPP | Advanced oxidation protein product |
| AR | Androgen Receptor |
| BAFF | B-cell activation factor |
| CD80 | Cluster of differentiation 80 |
| CHIT1 | Chitotriosidase; DNPH-2,4-dinitrophenylhydrazine |
| CKD | Chronic kidney disease |
| CXC16 | C-X-C motif chemokine 16 |
| DAMP | Damage-associated molecular patterns |
| DN | Diabetic nephropathy |
| FOXP3 | Forkhead box protein P3 |
| FSGS | Focal segmental glomerulosclerosis |
| GST | Glutathione s-transferase |
| HMGB1 | High mobility group box 1 |
| HNE | 4-hydroxy-nonenal |
| HSP70 | Heat shock proteins 70 |
| ICAM | Intercellular Adhesion Molecule 1 |
| IFN-α | Interferon alpha |
| IFN-γ | Interferon gamma |
| IgAN | IgA nephropathy |
| IL-17 | Interleukin 13 |
| IL-13 | Interleukin 13 |
| IL-2 | Interleukin 2 |
| IL-6 | Interleukin 6 |
| IL-7 | Interleukin 7 |
| IL-8 | Interleukin 8 |
| IP-10 | Interferon-inducible protein-10 |
| KIM-1 | Urinary kidney injury molecule-1 |
| LPS | Lipopolysaccharides |
| LRRs | Leucine-rich tandem repeats |
| MCN | Minimal change nephropathy |
| MCP-1 | Monocyte chemoattractant protein-1 |
| MDA | Malondialdehyde |
| MN | Membranous nephropathy |
| NAG | N-Acetyl-β-D Glucosaminidase |
| NAG | N-Acetyl-β-D Glucosaminidase |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NGAL | Neutrophil gelatinase-associated lipocalin |
| NK cells | Natural killer cells |
| PAMP | Pathogen-associated molecular patterns |
| PKC | Protein kinase C |
| PLA2R | M-type phospholipase A2 receptor |
| PRRs | Pattern recognition receptors |
| RAAS | Renin-angiotensin-aldosterone system |
| RAIL | Renal Activity Index for Lupus |
| RANTES | Regulated upon Activation, Normal T cell Expressed, and Secreted |
| ROS | Reactive oxygen species |
| sCD40L | Soluble CD40 ligand |
| sIL-2R | Soluble IL-2 receptor |
| SLE | Systemic lupus erythematosus |
| SOD2 | manganese superoxide-dismutase 2 |
| STAT-1 | Signal transducer and activator of transcription 1 |
| suPAR | Soluble urokinase-type plasminogen activator receptor |
| TGF-β1 | Transforming growth factor beta 1 |
| TLR | Toll-like receptor |
| TNF-a | Tumor necrosis factor alpha |
| TNFR1 | Tumor necrosis factor receptor 1 |
| TWEAK | Urinary TNF-like weak inducer of apoptosis |
| VCAM | Vascular cell adhesion molecule 1 |
| VEGF | Vascular Endothelial Growth Factor |
| YKL-40 | Cartilage glycoprotein 40 |
References
- World Health Organization; International Programme on Chemical Safety. Biomarkers in Risk Assessment: Validity and Validation; World Health Organization: Geneva, Switzerland, 2001; ISBN 978-92-4-157222-4. [Google Scholar]
- Fuentes-Arderiu, X. What is a biomarker? It’s time for a renewed definition. Clin. Chem. Lab. Med. 2013, 51, 1689–1690. [Google Scholar] [CrossRef] [PubMed]
- Goerlich, N.; Brand, H.A.; Langhans, V.; Tesch, S.; Schachtner, T.; Koch, B.; Paliege, A.; Schneider, W.; Grützkau, A.; Reinke, P.; et al. Kidney transplant monitoring by urinary flow cytometry: Biomarker combination of T cells, renal tubular epithelial cells, and podocalyxin-positive cells detects rejection. Sci. Rep. 2020, 10, 796. [Google Scholar] [CrossRef] [PubMed]
- Kasurinen, A.; Hagström, J.; Laitinen, A.; Kokkola, A.; Böckelman, C.; Haglund, C. Evaluation of toll-like receptors as prognostic biomarkers in gastric cancer: High tissue TLR5 predicts a better outcome. Sci. Rep. 2019, 9, 12553. [Google Scholar] [CrossRef] [PubMed]
- Darrabie, M.D.; Cheeseman, J.; Limkakeng, A.T.; Borawski, J.; Sullenger, B.A.; Elster, E.A.; Kirk, A.D.; Lee, J. Toll-like receptor activation as a biomarker in traumatically injured patients. J. Surg. Res. 2018, 231, 270–277. [Google Scholar] [CrossRef]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Dobrică, E.-C.; Găman, M.-A.; Cozma, M.-A.; Bratu, O.G.; Pantea Stoian, A.; Diaconu, C.C. Polypharmacy in Type 2 Diabetes Mellitus: Insights from an Internal Medicine Department. Medicina 2019, 55, 436. [Google Scholar] [CrossRef]
- O’Shaughnessy, M.M.; Hogan, S.L.; Thompson, B.D.; Coppo, R.; Fogo, A.B.; Jennette, J.C. Glomerular disease frequencies by race, sex and region: Results from the International Kidney Biopsy Survey. Nephrol. Dial. Transplant. 2018, 33, 661–669. [Google Scholar] [CrossRef]
- Naylor, S. Biomarkers: Current perspectives and future prospects. Expert Rev. Mol. Diagn. 2003, 3, 525–529. [Google Scholar] [CrossRef]
- Sahu, P.; Pinkalwar, N.; Dubey, R.D.; Paroha, S.; Chatterjee, S.; Chatterjee, T. Biomarkers: An Emerging Tool for Diagnosis of a Disease and Drug Development. Asian J. Res. Pharm. Sci. 2011, 1, 9–16. [Google Scholar]
- Drucker, E.; Krapfenbauer, K. Pitfalls and limitations in translation from biomarker discovery to clinical utility in predictive and personalised medicine. EPMA J. 2013, 4, 7. [Google Scholar] [CrossRef]
- Mayeux, R. Biomarkers: Potential Uses and Limitations. NeuroRx 2004, 1, 182–188. [Google Scholar] [CrossRef] [PubMed]
- FDA. About Biomarkers and Qualification. Available online: https://www.fda.gov/drugs/cder-biomarker-qualification-program/about-biomarkers-and-qualification (accessed on 10 August 2020).
- Levin, A.; Tonelli, M.; Bonventre, J.; Coresh, J.; Donner, J.-A.; Fogo, A.B.; Fox, C.S.; Gansevoort, R.T.; Heerspink, H.J.L.; Jardine, M.; et al. Global kidney health 2017 and beyond: A roadmap for closing gaps in care, research, and policy. Lancet 2017, 390, 1888–1917. [Google Scholar] [CrossRef]
- Devarapu, S.K.; Anders, H. Toll-like receptors in lupus nephritis. J. Biomed. Sci. 2018, 25, 35. [Google Scholar] [CrossRef]
- Luyckx, V.A.; Tonelli, M.; Stanifer, J.W. The global burden of kidney disease and the sustainable development goals. Bull. World Health Organ. 2018, 96, 414–422D. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.C.; Cheung, C.K.; Barratt, J. New insights into the pathogenesis of IgA nephropathy. Pediatr. Nephrol. 2018, 33, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Zheng, F. Immune Cells and Inflammation in Diabetic Nephropathy. J. Diabetes Res. 2016, 2016, 1841690. [Google Scholar] [CrossRef]
- Mohammad Hosseini, A.; Majidi, J.; Baradaran, B.; Yousefi, M. Toll-Like Receptors in the Pathogenesis of Autoimmune Diseases. Adv. Pharm. Bull. 2015, 5, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Imig, J.D.; Ryan, M.J. Immune and Inflammatory Role in Renal Disease. Compr. Physiol. 2013, 3, 957–976. [Google Scholar] [CrossRef]
- Berger, S.P.; Daha, M.R. Complement in glomerular injury. Semin. Immunopathol. 2007, 29, 375–384. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Lawson, B.R. Toll-like receptors and kidney diseases. Inflamm. Allergy Drug Targets 2009, 8, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xue, G.; Li, S.; Fu, Y.; Yin, J.; Zhang, R.; Li, J. Effect of Intermittent and Mild Cold Stimulation on the Immune Function of Bursa in Broilers. Animals 2020, 10, 1275. [Google Scholar] [CrossRef] [PubMed]
- Botos, I.; Segal, D.M.; Davies, D.R. The Structural Biology of Toll-Like Receptors. Structure 2011, 19, 447–459. [Google Scholar] [CrossRef] [PubMed]
- UniProt. Toll-Like Receptor 1. Available online: https://www.uniprot.org/uniprot/Q15399 (accessed on 10 August 2020).
- Protein Data Bank in Europe. Crystal Structure of the TLR1-TLR2 Heterodimer Induced by Binding of a tri-Acylatedlipopeptide. Available online: https://www.ebi.ac.uk/pdbe/entry/pdb/2z7x (accessed on 10 August 2020).
- UniProt. Toll-Like Receptor 2. Available online: https://www.uniprot.org/uniprot/O60603 (accessed on 10 August 2020).
- Protein Data Bank in Europe. Crystal Structure of TLR2-TLR6-Pam2CSK4 Complex. Available online: https://www.ebi.ac.uk/pdbe/entry/pdb/3a79 (accessed on 10 August 2020).
- UniProt. Toll-Like Receptor 3. Available online: https://www.uniprot.org/uniprot/O15455 (accessed on 10 August 2020).
- Protein Data Bank in Europe. Crystal Structure of the Complex of TLR3 and bi-Specific Diabody. Available online: https://www.ebi.ac.uk/pdbe/entry/pdb/5gs0 (accessed on 10 August 2020).
- UniProt. Toll-Like Receptor 4. Available online: https://www.uniprot.org/uniprot/O00206 (accessed on 10 August 2020).
- Protein Data Bank in Europe. The Crystal Structure of Mouse TLR4/MD-2/neoseptin-3. Available online: https://www.ebi.ac.uk/pdbe/entry/pdb/5ijc (accessed on 10 August 2020).
- UniProt. Toll-Like Receptor 5. Available online: https://www.uniprot.org/uniprot/O60602 (accessed on 10 August 2020).
- Protein Data Bank in Europe. Homology Model of Human Toll-Like Receptor 5 Fitted into an Electron Microscopy Single Particle Reconstruction. Available online: https://www.ebi.ac.uk/pdbe/entry/pdb/3j0a (accessed on 10 August 2020).
- UniProt. Toll-Like Receptor 6. Available online: https://www.uniprot.org/uniprot/Q9Y2C9 (accessed on 10 August 2020).
- Protein Data Bank in Europe. Crystal Structure of TIR Domain TLR6. Available online: https://www.ebi.ac.uk/pdbe/entry/pdb/4om7 (accessed on 10 August 2020).
- UniProt. Toll-Like Receptor 7. Available online: https://www.uniprot.org/uniprot/Q9NYK1 (accessed on 10 August 2020).
- UniProt. Toll-Like Receptor 8. Available online: https://www.uniprot.org/uniprot/Q9NR97 (accessed on 10 August 2020).
- Protein Data Bank in Europe. Crystal Structure of Human TLR8 in Complex with XG-1-236. Available online: https://www.ebi.ac.uk/pdbe/entry/pdb/4qc0 (accessed on 10 August 2020).
- UniProt. Toll-Like Receptor 9. Available online: https://www.uniprot.org/uniprot/Q9NR96 (accessed on 10 August 2020).
- UniProt. Toll-Like Receptor 10. Available online: https://www.uniprot.org/uniprot/Q9BXR5 (accessed on 10 August 2020).
- Protein Data Bank in Europe. The TIR Domain of Human Toll-Like Receptor 10 (TLR10). Available online: https://www.ebi.ac.uk/pdbe/entry/pdb/2j67 (accessed on 10 August 2020).
- UniProt. Toll-Like Receptor 11. Available online: https://www.uniprot.org/uniprot/Q6R5P0 (accessed on 10 August 2020).
- UniProt. Toll-Like Receptor 12. Available online: https://www.uniprot.org/uniprot/Q6QNU9 (accessed on 10 August 2020).
- UniProt. Toll-Like Receptor 13. Available online: https://www.uniprot.org/uniprot/Q6R5N8 (accessed on 10 August 2020).
- Farhat, K.; Riekenberg, S.; Heine, H.; Debarry, J.; Lang, R.; Mages, J.; Buwitt-Beckmann, U.; Röschmann, K.; Jung, G.; Wiesmüller, K.-H.; et al. Heterodimerization of TLR2 with TLR1 or TLR6 expands the ligand spectrum but does not lead to differential signaling. J. Leukoc. Biol. 2008, 83, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, Y.; Zhang, Q.; Wang, F.; Zhang, D. Toll-like receptors and prostate cancer. Front. Immunol. 2014, 5, 352. [Google Scholar] [CrossRef]
- Goulopoulou, S.; McCarthy, C.G.; Clinton Webb, R. Toll-like receptors in the vascular system: Sensing the dangers within. Pharmacol. Rev. 2016, 68, 142–167. [Google Scholar] [CrossRef]
- Schaefer, L. Complexity of danger: The diverse nature of damage-associated molecular patterns. J. Biol. Chem. 2014, 289, 35237–35245. [Google Scholar] [CrossRef]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate immune pattern recognition: A cell biological perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef]
- Miao, E.A.; Andersen-Nissen, E.; Warren, S.E.; Aderem, A. TLR5 and Ipaf: Dual sensors of bacterial flagellin in the innate immune system. Semin. Immunopathol. 2007, 29, 275–288. [Google Scholar] [CrossRef]
- Takeuchi, O.; Kawai, T.; Sanjo, H.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Takeda, K.; Akira, S. TLR6: A novel member of an expanding Toll-like receptor family. Gene 1999, 231, 59–65. [Google Scholar] [CrossRef]
- Oosting, M.; Cheng, S.C.; Bolscher, J.M.; Vestering-Stenger, R.; Plantinga, T.S.; Verschueren, I.C.; Arts, P.; Garritsen, A.; van Eenennaam, H.; Sturm, P.; et al. Human TLR10 is an anti-inflammatory pattern-recognition receptor. Proc. Natl. Acad. Sci. USA 2014, 111, E4478–E4484. [Google Scholar] [CrossRef] [PubMed]
- Raetz, M.; Kibardin, A.; Sturge, C.R.; Pifer, R.; Li, H.; Burstein, E.; Ozato, K.; Larin, S.; Yarovinsky, F. Cooperation of TLR12 and TLR11 in the IRF8-Dependent IL-12 Response to Toxoplasma gondii Profilin. J. Immunol. 2013, 191, 4818–4827. [Google Scholar] [CrossRef] [PubMed]
- Yarovinsky, F.; Zhang, D.; Andersen, J.F.; Bannenberg, G.L.; Serhan, C.N.; Hayden, M.S.; Hieny, S.; Sutterwala, F.S.; Flavell, R.A.; Ghosh, S.; et al. Immunology: TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 2005, 308, 1626–1629. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Zhao, P.; Rodriguez-Pinto, D.; Qi, D.; Henegariu, O.; Alexopoulou, L.; Flavell, R.A.; Wong, F.S.; Wen, L. Inflammatory Regulation by TLR3 in Acute Hepatitis. J. Immunol. 2009, 183, 3712–3719. [Google Scholar] [CrossRef] [PubMed]
- Negishi, H.; Osawa, T.; Ogami, K.; Ouyang, X.; Sakaguchi, S.; Koshiba, R.; Yanai, H.; Seko, Y.; Shitara, H.; Bishop, K.; et al. A critical link between Toll-like receptor 3 and type II interferon signaling pathways in antiviral innate immunity. Proc. Natl. Acad. Sci. USA 2008, 105, 20446–20451. [Google Scholar] [CrossRef]
- Hemmi, H.; Kaisho, T.; Takeuchi, O.; Sato, S.; Sanjo, H.; Hoshino, K.; Horiuchi, T.; Tomizawa, H.; Takeda, K.; Akira, S. Small-antiviral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat. Immunol. 2002, 3, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Guo, Z.; Kiniwa, Y.; Voo, K.S.; Peng, W.; Fu, T.; Wang, D.Y.; Li, Y.; Wang, H.Y.; Wang, R.F. Immunology: Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science 2005, 309, 1380–1384. [Google Scholar] [CrossRef]
- Notley, C.A.; Jordan, C.K.; McGovern, J.L.; Brown, M.A.; Ehrenstein, M.R. DNA methylation governs the dynamic regulation of inflammation by apoptotic cells during efferocytosis. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Kanherkar, R.R.; Bhatia-Dey, N.; Csoka, A.B. Epigenetics across the human lifespan. Front. Cell Dev. Biol. 2014, 2, 49. [Google Scholar] [CrossRef]
- Anders, H.-J.; Banas, B.; Schlöndorff, D. Signaling Danger: Toll-Like Receptors and their Potential Roles in Kidney Disease. J. Am. Soc. Nephrol. 2004, 15, 854–867. [Google Scholar] [CrossRef]
- Verstak, B.; Stack, J.; Ve, T.; Mangan, M.; Hjerrild, K.; Jeon, J.; Stahl, R.; Latz, E.; Gay, N.; Kobe, B.; et al. The TLR signaling adaptor TRAM interacts with TRAF6 to mediate activation of the inflammatory response by TLR4. J. Leukoc. Biol. 2014, 96, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Phung, Q.; Chan, S.; Chaudhari, R.; Quan, C.; O’Rourke, K.M.; Eby, M.; Pietras, E.; Cheng, G.; Bazan, J.F.; et al. DUBA: A deubiquitinase that regulates type I interferon production. Science 2007, 318, 1628–1632. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Jin, J.; Xu, S.; Liu, H.; Li, N.; Cao, X. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat. Immunol. 2010, 11, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Skaug, B.; Chen, J.; Du, F.; He, J.; Ma, A.; Chen, Z.J. Direct, noncatalytic mechanism of IKK inhibition by A20. Mol. Cell 2011, 44, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Yuk, J.M.; Shin, D.M.; Lee, H.M.; Kim, J.J.; Kim, S.W.; Jin, H.S.; Yang, C.S.; Park, K.A.; Chanda, D.; Kim, D.K.; et al. The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by Toll-like receptors. Nat. Immunol. 2011, 12, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Kawai, T.; Akira, S. Dissecting negative regulation of Toll-like receptor signaling. Trends Immunol. 2012, 33, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Tun-Kyi, A.; Ryo, A.; Yamamoto, M.; Finn, G.; Fujita, T.; Akira, S.; Yamamoto, N.; Lu, K.P.; Yamaoka, S. Negative regulation of interferon-regulatory factor 3-dependent innate antiviral response by the prolyl isomerase Pin1. Nat. Immunol. 2006, 7, 598–605. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 1–9. [Google Scholar] [CrossRef]
- Couser, W.G.; Johnson, R.J. The etiology of glomerulonephritis: Roles of infection and autoimmunity. Kidney Int. 2014, 86, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Latz, E.; Ontiveros, F.; Kono, H. The sterile inflammatory response. Annu. Rev. Immunol. 2010, 28, 321–342. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, Y.; Kitaura, J.; Izawa, K.; Kaitani, A.; Komeno, Y.; Nakamura, M.; Yamazaki, S.; Enomoto, Y.; Oki, T.; Akiba, H.; et al. TIM1 is an endogenous ligand for LMIR5/CD300b: LMIR5 deficiency ameliorates mouse kidney ischemia/reperfusion injury. J. Exp. Med. 2010, 207, 1501–1511. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.J. Toll-Like Receptors and Danger Signaling in Kidney Injury. J. Am. Soc. Nephrol. 2010, 21, 1270–1274. [Google Scholar] [CrossRef]
- Rosin, D.L.; Okusa, M.D. Dangers Within: DAMP Responses to Damage and Cell Death in Kidney Disease. J. Am. Soc. Nephrol. 2011, 22, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Mertowski, S.; Grywalska, E.; Gosik, K.; Smarz-Widelska, I.; Hymos, A.; Dworacki, G.; Niedźwiedzka-Rystwej, P.; Drop, B.; Roliński, J.; Załuska, W. TLR2 Expression on Select Lymphocyte Subsets as a New Marker in Glomerulonephritis. J. Clin. Med. 2020, 9, 541. [Google Scholar] [CrossRef]
- Rosenberg, A.Z.; Kopp, J.B. Focal Segmental Glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2017, 12, 502–517. [Google Scholar] [CrossRef]
- Reggiani, F.; Ponticelli, C. Focal segmental glomerular sclerosis: Do not overlook the role of immune response. J. Nephrol. 2016, 29, 525–534. [Google Scholar] [CrossRef]
- Eardley, K.S.; Kubal, C.; Zehnder, D.; Quinkler, M.; Lepenies, J.; Savage, C.O.; Howie, A.J.; Kaur, K.; Cooper, M.S.; Adu, D.; et al. The role of capillary density, macrophage infiltration and interstitial scarring in the pathogenesis of human chronic kidney disease. Kidney Int. 2008, 74, 495–504. [Google Scholar] [CrossRef]
- Wu, X.; Dolecki, G.J.; Sherry, B.; Zagorski, J.; Lefkowith, J.B. Chemokines are expressed in a myeloid cell-dependent fashion and mediate distinct functions in immune complex glomerulonephritis in rat. J. Immunol. 1997, 158, 3917–3924. [Google Scholar]
- Wang, H.; Zheng, C.; Xu, X.; Zhao, Y.; Lu, Y.; Liu, Z. Fibrinogen links podocyte injury with Toll-like receptor 4 and is associated with disease activity in FSGS patients. Nephrology 2018, 23, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Gaman, A.M.; Moisa, C.; Diaconu, C.C.; Gaman, M.A. Crosstalk between Oxidative Stress, Chronic Inflammation and Disease Progression in Essential Thrombocythemia. Rev. Chim. 2019, 70, 3486–3489. [Google Scholar] [CrossRef]
- Găman, M.A.; Epîngeac, M.E.; Diaconu, C.C.; Găman, A.M. Evaluation of oxidative stress levels in obesity and diabetes by the free oxygen radical test and free oxygen radical defence assays and correlations with anthropometric and laboratory parameters. World J. Diabetes 2020, 11, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Yacov, N.; Feldman, B.; Volkov, A.; Ishai, E.; Breitbart, E.; Mendel, I. Treatment with lecinoxoids attenuates focal and segmental glomerulosclerosis development in nephrectomized rats. Basic Clin. Pharmacol. Toxicol. 2019, 124, 131–143. [Google Scholar] [CrossRef]
- Segarra-Medrano, A.; Carnicer-Cáceres, C.; Arbós-Via, M.A.; Quiles-Pérez, M.T.; Agraz-Pamplona, I.; Ostos-Roldán, E. Biological markers of nephrotic syndrome: A few steps forward in the long way. Nefrología 2012, 32, 558–572. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.R.; Choi, M. Minimal Change Disease in Adults. In Glomerulonephritis; Trachtman, H., Herlitz, L.C., Lerma, E.V., Hogan, J.J., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 97–114. ISBN 978-3-319-49379-4. [Google Scholar]
- Uwaezuoke, S.N. Biomarkers of Common Childhood Renal Diseases. In Biomarker—Indicator of Abnormal Physiological Process; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef]
- Srivastava, T.; Sharma, M.; Yew, K.H.; Sharma, R.; Duncan, R.S.; Saleem, M.A.; McCarthy, E.T.; Kats, A.; Cudmore, P.A.; Alon, U.S.; et al. LPS and PAN-induced podocyte injury in an in vitro model of minimal change disease: Changes in TLR profile. J. Cell Commun. Signal. 2013, 7, 49–60. [Google Scholar] [CrossRef]
- Mishra, O.P.; Kumar, R.; Narayan, G.; Srivastava, P.; Abhinay, A.; Prasad, R.; Singh, A.; Batra, V.V. Toll-like receptor 3 (TLR-3), TLR-4 and CD80 expression in peripheral blood mononuclear cells and urinary CD80 levels in children with idiopathic nephrotic syndrome. Pediatr. Nephrol. 2017, 32, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, T.; Shimada, M.; Araya, C.E.; Huskey, J.; Garin, E.H.; Johnson, R.J. Minimal Change Disease: A CD80 podocytopathy? Semin. Nephrol. 2011, 31, 320–325. [Google Scholar] [CrossRef]
- Lai, W.L.; Yeh, T.H.; Chen, P.M.; Chan, C.K.; Chiang, W.C.; Chen, Y.M.; Wu, K.D.; Tsai, T.J. Membranous nephropathy: A review on the pathogenesis, diagnosis, and treatment. J. Formos. Med. Assoc. 2015, 114, 102–111. [Google Scholar] [CrossRef]
- Couser, W.G. Primary Membranous Nephropathy. Clin. J. Am. Soc. Nephrol. 2017, 12, 983–997. [Google Scholar] [CrossRef]
- Liu, W.; Gao, C.; Dai, H.; Zheng, Y.; Dong, Z.; Gao, Y.; Liu, F.; Zhang, Z.; Liu, Z.; Liu, W.; et al. Immunological Pathogenesis of Membranous Nephropathy: Focus on PLA2R1 and Its Role. Front. Immunol. 2019, 10, 1809. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Chen, C.H.; Huang, Y.C.; Chan, C.J.; Chen, D.C.; Tsai, F.J. Genetic susceptibility to idiopathic membranous nephropathy in high-prevalence Area, Taiwan. Biomedicine 2014, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Nafar, M.; Samavat, S. Biomarkers in IgA Nephropathy. In Biomarkers in Kidney Disease; Patel, V.B., Ed.; Springer: Dordrecht, The Netherlands, 2015; pp. 1–29. ISBN 978-94-007-7743-9. [Google Scholar]
- Liu, Y.; Ma, X.; Lv, J.; Shi, S.; Liu, L.; Chen, Y.; Zhang, H. Risk factors for pregnancy outcomes in patients with IgA nephropathy: A matched cohort study. Am. J. Kidney Dis. 2014, 64, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Coppo, R.; Camilla, R.; Amore, A.; Peruzzi, L. Oxidative Stress in IgA Nephropathy. Nephron Clin. Pract. 2010, 116, c196–c199. [Google Scholar] [CrossRef]
- Suzuki, H.; Suzuki, Y.; Narita, I.; Aizawa, M.; Kihara, M.; Yamanaka, T.; Kanou, T.; Tsukaguchi, H.; Novak, J.; Horikoshi, S.; et al. Toll-Like Receptor 9 Affects Severity of IgA Nephropathy. J. Am. Soc. Nephrol. 2008, 19, 2384–2395. [Google Scholar] [CrossRef]
- Rollino, C.; Vischini, G.; Coppo, R. IgA nephropathy and infections. J. Nephrol. 2016, 29, 463–468. [Google Scholar] [CrossRef]
- Merkle, M.; Ribeiro, A.; Köppel, S.; Pircher, J.; Mannell, H.; Roeder, M.; Wörnle, M. TLR3-dependent immune regulatory functions of human mesangial cells. Cell. Mol. Immunol. 2012, 9, 334–340. [Google Scholar] [CrossRef]
- Coppo, R.; Amore, A.; Peruzzi, L.; Vergano, L.; Camilla, R. Innate immunity and IgA nephropathy. J. Nephrol. 2010, 23, 626–632. [Google Scholar]
- Sheng, X.; Zuo, X.; Liu, X.; Zhou, Y.; Sun, X. Crosstalk between TLR4 and Notch1 signaling in the IgA nephropathy during inflammatory response. Int. Urol. Nephrol. 2018, 50, 779–785. [Google Scholar] [CrossRef]
- Lim, B.J.; Lee, D.; Hong, S.W.; Jeong, H.J. Toll-Like Receptor 4 Signaling is Involved in IgA-Stimulated Mesangial Cell Activation. Yonsei Med. J. 2011, 52, 610–615. [Google Scholar] [CrossRef]
- Chebotareva, N.; Bobkova, I.; Shilov, E. Heat shock proteins and kidney disease: Perspectives of HSP therapy. Cell Stress Chaperones 2017, 22, 319–343. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, T.; Mii, A.; Fukui, M.; Nagahama, K.; Shimizu, A.; Tsuruoka, S. IgA Nephropathy and Psoriatic Arthritis that Improved with Steroid Pulse Therapy and Mizoribine in Combination with Treatment for Chronic Tonsillitis and Epipharyngitis. Intern. Med. 2015, 54, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.J.; Lock, H.R.; Wolfs, T.G.A.M.; Buurman, W.A.; Sacks, S.H.; Robson, M.G. Toll-like receptor 4 ligation on intrinsic renal cells contributes to the induction of antibody-mediated glomerulonephritis via CXCL1 and CXCL2. J. Am. Soc. Nephrol. 2007, 18, 1732–1739. [Google Scholar] [CrossRef] [PubMed]
- Myllymäki, J.; Syrjänen, J.; Helin, H.; Pasternack, A.; Kattainen, A.; Mustonen, J. Vascular diseases and their risk factors in IgA nephropathy. Nephrol. Dial. Transplant. 2006, 21, 1876–1882. [Google Scholar] [CrossRef] [PubMed]
- Feehally, J.; Barratt, J. The Genetics of IgA Nephropathy: An Overview from Western Countries. Kidney Dis. 2015, 1, 33–41. [Google Scholar] [CrossRef]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-Like Receptors. Annu. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef] [PubMed]
- Sano, N.; Kitazawa, K.; Sugisaki, T. Localization and roles of CD44, hyaluronic acid and osteopontin in IgA nephropathy. Nephron 2001, 89, 416–421. [Google Scholar] [CrossRef]
- Zhang, Y.-M.; Zhou, X.-J.; Zhang, H. What Genetics Tells Us About the Pathogenesis of IgA Nephropathy: The Role of Immune Factors and Infection. Kidney Int. Rep. 2017, 2, 318–331. [Google Scholar] [CrossRef]
- Cui, Y.; Liu, S.; Cui, W.; Gao, D.; Zhou, W.; Luo, P. Identification of potential biomarkers and therapeutic targets for human IgA nephropathy and hypertensive nephropathy by bioinformatics analysis. Mol. Med. Rep. 2017, 16, 3087–3094. [Google Scholar] [CrossRef]
- Tycová, I.; Hrubá, P.; Maixnerová, D.; Girmanová, E.; Mrázová, P.; Straňavová, L.; Zachoval, R.; Merta, M.; Slatinská, J.; Kollár, M.; et al. Molecular profiling in IgA nephropathy and focal and segmental glomerulosclerosis. Physiol. Res. 2018, 67, 93–105. [Google Scholar] [CrossRef]
- Wardle, E.N. B Lymphocyte Stimulator and Autoimmune Disease. Saudi J. Kidney Dis. Transplant. 2004, 15, 155. [Google Scholar]
- Li, W.; Peng, X.; Liu, Y.; Liu, H.; Liu, F.; He, L.; Liu, Y.; Zhang, F.; Guo, C.; Chen, G.; et al. TLR9 and BAFF: Their expression in patients with IgA nephropathy. Mol. Med. Rep. 2014, 10, 1469–1474. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Wu, W.; Wen, Y.; Li, X. Hydroxychloroquine alleviates persistent proteinuria in IgA nephropathy. Int. Urol. Nephrol. 2017, 49, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Chen, C.S.; Yiang, G.T.; Cheng, P.W.; Chen, Y.L.; Chiu, H.C.; Liu, K.H.; Lee, W.C.; Li, C.J. The Emerging Role of Pathogenesis of IgA Nephropathy. J. Clin. Med. 2018, 7, 225. [Google Scholar] [CrossRef] [PubMed]
- Coppo, R. Treatment of IgA nephropathy: Recent advances and prospects. Nephrol. Ther. 2018, 14, S13–S21. [Google Scholar] [CrossRef]
- Yuling, H.; Ruijing, X.; Xiang, J.; Yanping, J.; Lang, C.; Li, L.; Dingping, Y.; Xinti, T.; Jingyi, L.; Zhiqing, T.; et al. CD19+CD5+ B Cells in Primary IgA Nephropathy. J. Am. Soc. Nephrol. 2008, 19, 2130–2139. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Zhang, L.; Zhao, P.W.; Ma, L.; Li, C.; Zou, H.B.; Jiang, Y.F. Functional implications of regulatory B cells in human IgA nephropathy. Scand. J. Immunol. 2014, 79, 51–60. [Google Scholar] [CrossRef]
- Coppo, R.; Camilla, R.; Amore, A.; Peruzzi, L.; Daprà, V.; Loiacono, E.; Vatrano, S.; Rollino, C.; Sepe, V.; Rampino, T.; et al. Toll-like receptor 4 expression is increased in circulating mononuclear cells of patients with immunoglobulin A nephropathy. Clin. Exp. Immunol. 2010, 159, 73–81. [Google Scholar] [CrossRef]
- Almaani, S.; Meara, A.; Rovin, B.H. Update on Lupus Nephritis. Clin. J. Am. Soc. Nephrol. 2017, 12, 825–835. [Google Scholar] [CrossRef]
- Ruiz Irastorza, G.; Espinosa, G.; Frutos, M.A.; Jiménez Alonso, J.; Praga, M.; Pallarés, L.; Rivera, F.; Robles Marhuenda, A.; Segarra, A.; Quereda, C.; et al. Diagnosis and treatment of lupus nephritis. Consensus document from the systemic auto-immune disease group (GEAS) of the Spanish Society of Internal Medicine (SEMI) and Spanish Society of Nephrology (S.E.N.). Nefrologia 2012, 32 (Suppl. 1), 1–35. [Google Scholar] [CrossRef]
- Klonowska-Szymczyk, A.; Kulczycka-Siennicka, L.; Robak, T.; Smolewski, P.; Cebula-Obrzut, B.; Robak, E. The impact of agonists and antagonists of TLR3 and TLR9 on concentrations of IL-6, IL10 and sIL-2R in culture supernatants of peripheral blood mononuclear cells derived from patients with systemic lupus erythematosus. Adv. Hyg. Exp. Med. 2017, 71, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Aragón, C.C.; Tafúr, R.A.; Suárez-Avellaneda, A.; Martínez, M.D.T.; de las Salas, A.; Tobón, G.J. Urinary biomarkers in lupus nephritis. J. Transl. Autoimmun. 2020, 3, 100042. [Google Scholar] [CrossRef] [PubMed]
- Patole, P.S.; Pawar, R.D.; Lech, M.; Zecher, D.; Schmidt, H.; Segerer, S.; Ellwart, A.; Henger, A.; Kretzler, M.; Anders, H.-J. Expression and regulation of Toll-like receptors in lupus-like immune complex glomerulonephritis of MRL-Fas(lpr) mice. Nephrol. Dial. Transplant. 2006, 21, 3062–3073. [Google Scholar] [CrossRef] [PubMed]
- Pawar, R.D.; Patole, P.S.; Zecher, D.; Segerer, S.; Kretzler, M.; Schlöndorff, D.; Anders, H.J. Toll-Like Receptor-7 Modulates Immune Complex Glomerulonephritis. J. Am. Soc. Nephrol. 2006, 17, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Yu, W.; Wen, Y.; Li, H. Beta2-glycoprotein I Expression in Lupus Nephritis Patients with Antiphospholipid-associated Nephropathy. J. Rheumatol. 2016, 43, 2026–2032. [Google Scholar] [CrossRef]
- Kwok, S.K.; Tsokos, G.C. New insights into the role of renal resident cells in the pathogenesis of lupus nephritis. Korean J. Intern. Med. 2018, 33, 284–289. [Google Scholar] [CrossRef]
- Urbonaviciute, V.; Starke, C.; Pirschel, W.; Pohle, S.; Frey, S.; Daniel, C.; Amann, K.; Schett, G.; Herrmann, M.; Voll, R.E. Toll-like Receptor 2 Is Required for Autoantibody Production and Development of Renal Disease in Pristane-Induced Lupus. Arthritis Rheum. 2013, 65, 1612–1623. [Google Scholar] [CrossRef]
- Conti, F.; Spinelli, F.R.; Truglia, S.; Miranda, F.; Alessandri, C.; Ceccarelli, F.; Bombardieri, M.; Giannakakis, K.; Valesini, G. Kidney Expression of Toll Like Receptors in Lupus Nephritis: Quantification and Clinicopathological Correlations. Mediat. Inflamm. 2016, 2016, 7697592. [Google Scholar] [CrossRef]
- Lorenz, G.; Lech, M.; Anders, H.J. Toll-like receptor activation in the pathogenesis of lupus nephritis. Clin. Immunol. 2017, 185, 86–94. [Google Scholar] [CrossRef]
- Pawar, R.D.; Patole, P.S.; Ellwart, A.; Lech, M.; Segerer, S.; Schlondorff, D.; Anders, H.J. Ligands to Nucleic Acid–Specific Toll-Like Receptors and the Onset of Lupus Nephritis. J. Am. Soc. Nephrol. 2006, 17, 3365–3373. [Google Scholar] [CrossRef]
- Lorenz, G.; Anders, H.J. Neutrophils, Dendritic Cells, Toll-Like Receptors, and Interferon-α in Lupus Nephritis. Semin. Nephrol. 2015, 35, 410–426. [Google Scholar] [CrossRef]
- Chen, D.N.; Fan, L.; Wu, Y.X.; Zhou, Q.; Chen, W.; Yu, X.Q. A Predictive Model for Estimation Risk of Proliferative Lupus Nephritis. Chin. Med. J. 2018, 131, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Pisetsky, D.S. HMGB1: A smoking gun in lupus nephritis? Arthritis Res. Ther. 2012, 14, 112. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sun, F.; Teng, J.; Yu, P.; Li, W.; Chang, J.; Xu, H. Involvement of TWEAK and the NF-κB signaling pathway in lupus nephritis. Exp. Ther. Med. 2018, 15, 2611–2619. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Watanabe, K.S.; Liu, K.; Hiramatsu, S.; Zeggar, S.; Katsuyama, E.; Tatebe, N.; Akahoshi, A.; Takenaka, F.; Hanada, T.; et al. Anti-high Mobility Group Box 1 Antibody Ameliorates Albuminuria in MRL/lpr Lupus-Prone Mice. Mol. Ther. Meth. Clin. Dev. 2017, 6, 31–39. [Google Scholar] [CrossRef]
- Ma, K.; Li, J.; Fang, Y.; Lu, L. Roles of B Cell-Intrinsic TLR Signals in Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2015, 16, 13084–13105. [Google Scholar] [CrossRef] [PubMed]
- Wirestam, L.; Schierbeck, H.; Skogh, T.; Gunnarsson, I.; Ottosson, L.; Erlandsson-Harris, H.; Wetterö, J.; Sjöwall, C. Antibodies against High Mobility Group Box protein-1 (HMGB1) versus other anti-nuclear antibody fine-specificities and disease activity in systemic lupus erythematosus. Arthritis Res. Ther. 2015, 17, 338. [Google Scholar] [CrossRef]
- Qing, X.; Pitashny, M.; Thomas, D.B.; Barrat, F.J.; Hogarth, M.P.; Putterman, C. Pathogenic anti-DNA antibodies modulate gene expression in mesangial cells: Involvement of HMGB1 in anti-DNA antibody-induced renal injury. Immunol. Lett. 2008, 121, 61–73. [Google Scholar] [CrossRef]
- Banas, M.C.; Banas, B.; Hudkins, K.L.; Wietecha, T.A.; Iyoda, M.; Bock, E.; Hauser, P.; Pippin, J.W.; Shankland, S.J.; Smith, K.D.; et al. TLR4 Links Podocytes with the Innate Immune System to Mediate Glomerular Injury. J. Am. Soc. Nephrol. 2008, 19, 704–713. [Google Scholar] [CrossRef]
- Jończyk, M.; Kuliczkowska-Płaksej, J.; Mierzwicka, A.; Bolanowski, M. The polycystic ovarian syndrome and chronic inflammation: The role of Toll-like receptors. Postepy Hig. Med. Dosw. 2018, 72, 1199–1207. [Google Scholar] [CrossRef]
- Lartigue, A.; Colliou, N.; Calbo, S.; François, A.; Jacquot, S.; Arnoult, C.; Tron, F.; Gilbert, D.; Musette, P. Critical role of TLR2 and TLR4 in autoantibody production and glomerulonephritis in lpr mutation-induced mouse lupus. J. Immunol. 2009, 183, 6207–6216. [Google Scholar] [CrossRef] [PubMed]
- Horton, C.G.; Pan, Z.; Farris, A.D. Targeting toll-like receptors for treatment of SLE. Mediat. Inflamm. 2010, 2010, 498980. [Google Scholar] [CrossRef] [PubMed]
- Wardle, E.N. Toll-Like Receptors and Glomerulonephritis. Saudi J. Kidney Dis. Transpl. 2007, 18, 159–172. [Google Scholar]
- Lichtnekert, J.; Vielhauer, V.; Zecher, D.; Kulkarni, O.; Clauss, S.; Hornung, V.; Mayadas, T.; Beutler, B.; Akira, S.; Anders, H.-J. Trif is not required for immune complex glomerulonephritis: Dying cells activate mesangial cells via Tlr2/Myd88 rather than Tlr3/Trif. Am. J. Physiol. Renal. Physiol. 2009, 296, F867–F874. [Google Scholar] [CrossRef]
- Pradhan, V.; Patwardhan, M.; Ghosh, K. Fc gamma receptor polymorphisms in systemic lupus erythematosus and their correlation with the clinical severity of the disease. Indian J. Hum. Genet. 2008, 14, 77–81. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mackay, M.; Stanevsky, A.; Wang, T.; Aranow, C.; Li, M.; Koenig, S.; Ravetch, J.V.; Diamond, B. Selective dysregulation of the FcγIIB receptor on memory B cells in SLE. J. Exp. Med. 2006, 203, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.K. Diabetic nephropathy—Complications and treatment. Int. J. Nephrol. Renovasc. Dis. 2014, 7, 361–381. [Google Scholar] [CrossRef] [PubMed]
- Mudaliar, H.; Pollock, C.; Panchapakesan, U. Role of Toll-like receptors in diabetic nephropathy. Clin. Sci. 2014, 126, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Conserva, F.; Gesualdo, L.; Papale, M. A Systems Biology Overview on Human Diabetic Nephropathy: From Genetic Susceptibility to Post-Transcriptional and Post-Translational Modifications. J. Diabetes Res. 2016, 2016, 7934504. [Google Scholar] [CrossRef] [PubMed]
- Umanath, K.; Lewis, J.B. Update on Diabetic Nephropathy: Core Curriculum 2018. Am. J. Kidney Dis. 2018, 71, 884–895. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, J.; Qin, X.; Zhang, J.; Liu, J.; Qin, X. Advances in early biomarkers of diabetic nephropathy. Rev. Assoc. Med. Bras. 2018, 64, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Mansour, I.; Thajudeen, B. Overview of Diabetic Nephropathy. J. R. Soc. Med. 2017, 1–21. [Google Scholar] [CrossRef]
- Wifi, M.-N.A.; Assem, M.; Elsherif, R.H.; El-Azab, H.A.-F.; Saif, A. Toll-like receptors-2 and -9 (TLR2 and TLR9) gene polymorphism in patients with type 2 diabetes and diabetic foot. Medicine 2017, 96, e6760. [Google Scholar] [CrossRef] [PubMed]
- Eknoyan, G. A Historical Overview of Diabetic Nephropathy. In Diabetic Nephropathy; Springer: Cham, Switzerland, 2019; pp. 3–19. [Google Scholar] [CrossRef]
- Chen, X.; Ma, J.; Kwan, T.; Stribos, E.G.D.; Messchendorp, A.L.; Loh, Y.W.; Wang, X.; Paul, M.; Cunningham, E.C.; Habib, M.; et al. Blockade of HMGB1 Attenuates Diabetic Nephropathy in Mice. Sci. Rep. 2018, 8, 8319. [Google Scholar] [CrossRef]
- Shi, H.; Che, Y.; Bai, L.; Zhang, J.; Fan, J.; Mao, H. High mobility group box 1 in diabetic nephropathy (Review). Exp. Ther. Med. 2017, 14, 2431–2433. [Google Scholar] [CrossRef][Green Version]
- Bellini, S.; Barutta, F.; Mastrocola, R.; Imperatore, L.; Bruno, G.; Gruden, G. Heat Shock Proteins in Vascular Diabetic Complications: Review and Future Perspective. Int. J. Mol. Sci. 2017, 18, 2709. [Google Scholar] [CrossRef]
- Guo, C.; Zhang, L.; Nie, L.; Zhang, N.; Xiao, D.; Ye, X.; Ou, M.; Liu, Y.; Zhang, B.; Wang, M.; et al. Association of polymorphisms in the MyD88, IRAK4 and TRAF6 genes and susceptibility to type 2 diabetes mellitus and diabetic nephropathy in a southern Han Chinese population. Mol. Cell. Endocrinol. 2016, 429, 114–119. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Zhang, T.; Chi, Y.; Liu, M.; Liu, Y. Genistein and Myd88 Activate Autophagy in High Glucose-Induced Renal Podocytes In Vitro. Med. Sci. Monit. 2018, 24, 4823–4831. [Google Scholar] [CrossRef]
- Murea, M.; Register, T.C.; Divers, J.; Bowden, D.W.; Carr, J.J.; Hightower, C.R.; Xu, J.; Smith, S.C.; Hruska, K.A.; Langefeld, C.D.; et al. Relationships between serum MCP-1 and subclinical kidney disease: African American-Diabetes Heart Study. BMC Nephrol. 2012, 13, 148. [Google Scholar] [CrossRef]
- Suryavanshi, S.V.; Kulkarni, Y.A. NF-κβ: A Potential Target in the Management of Vascular Complications of Diabetes. Front. Pharmacol. 2017, 8, 798. [Google Scholar] [CrossRef]
- Lu, Q.; Wang, W.-W.; Zhang, M.-Z.; Ma, Z.-X.; Qiu, X.-R.; Shen, M.; Yin, X.-X. ROS induces epithelial-mesenchymal transition via the TGF-β1/PI3K/Akt/mTOR pathway in diabetic nephropathy. Exp. Ther. Med. 2019, 17, 835–846. [Google Scholar] [CrossRef]
- Tong, Y.; Chuan, J.; Bai, L.; Shi, J.; Zhong, L.; Duan, X.; Zhu, Y. The protective effect of shikonin on renal tubular epithelial cell injury induced by high glucose. Biomed. Pharmacother. 2018, 98, 701–708. [Google Scholar] [CrossRef]
- Li, F.; Yang, N.; Zhang, L.; Tan, H.; Huang, B.; Liang, Y.; Chen, M.; Yu, X. Increased Expression of Toll-Like Receptor 2 in Rat Diabetic Nephropathy. Am. J. Nephrol. 2010, 32, 179–186. [Google Scholar] [CrossRef]
- Cha, J.J.; Hyun, Y.Y.; Lee, M.H.; Kim, J.E.; Nam, D.H.; Song, H.K.; Kang, Y.S.; Lee, J.E.; Kim, H.W.; Han, J.Y.; et al. Renal protective effects of toll-like receptor 4 signaling blockade in type 2 diabetic mice. Endocrinology 2013, 154, 2144–2155. [Google Scholar] [CrossRef]
- Stadler, K.; Goldberg, I.J.; Susztak, K. The Evolving Understanding of the Contribution of Lipid Metabolism to Diabetic Kidney Disease. Curr. Diabetes Rep. 2015, 15, 40. [Google Scholar] [CrossRef]
- Cao, A.; Wang, L.; Chen, X.; Guo, H.; Chu, S.; Zhang, X.; Peng, W. Ursodeoxycholic Acid Ameliorated Diabetic Nephropathy by Attenuating Hyperglycemia-Mediated Oxidative Stress. Biol. Pharm. Bull. 2016, 39, 1300–1308. [Google Scholar] [CrossRef]
- Kaur, H.; Chien, A.; Jialal, I. Hyperglycemia induces Toll like receptor 4 expression and activity in mouse mesangial cells: Relevance to diabetic nephropathy. Am. J. Physiol. Renal Physiol. 2012, 303, F1145–F1150. [Google Scholar] [CrossRef][Green Version]
- Takata, S.; Sawa, Y.; Uchiyama, T.; Ishikawa, H. Expression of Toll-Like Receptor 4 in Glomerular Endothelial Cells under Diabetic Conditions. Acta Histochem. Cytochem. 2013, 46, 35–42. [Google Scholar] [CrossRef]
- Panchapakesan, U.; Pollock, C. The role of toll-like receptors in diabetic kidney disease. Curr. Opin. Nephrol. Hypertens. 2018, 27, 30–34. [Google Scholar] [CrossRef]
- Aluksanasuwan, S.; Sueksakit, K.; Fong-Ngern, K.; Thongboonkerd, V. Role of HSP60 (HSPD1) in diabetes-induced renal tubular dysfunction: Regulation of intracellular protein aggregation, ATP production, and oxidative stress. FASEB J. 2017, 31, 2157–2167. [Google Scholar] [CrossRef]
- Zyzak, J.; Matuszyk, J.; Siednienko, J. Multilevel maturation of Toll-like receptor 9. Postepy Hig. Med. Dosw. 2013, 67, 1034–1046. [Google Scholar] [CrossRef] [PubMed]
- Zipris, D. Toll-like receptors and type 1 diabetes. Adv. Exp. Med. Biol. 2010, 654, 585–610. [Google Scholar] [CrossRef]
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute kidney injury. Lancet 2019, 394, 1949–1964. [Google Scholar] [CrossRef]
- Baer, P.C.; Koch, B.; Geiger, H. Kidney Inflammation, Injury and Regeneration. Int. J. Mol. Sci. 2020, 21, 1164. [Google Scholar] [CrossRef] [PubMed]
- Coca, S.G.; Yusuf, B.; Shlipak, M.G.; Garg, A.X.; Parikh, C.R. Long-term risk of mortality and other adverse outcomes after acute kidney injury: A systematic review and meta-analysis. Am. J. Kidney Dis. 2009, 53, 961–973. [Google Scholar] [CrossRef]
- Hsu, C.; Chertow, G.M.; McCulloch, C.E.; Fan, D.; Ordoñez, J.D.; Go, A.S. Nonrecovery of kidney function and death after acute on chronic renal failure. Clin. J. Am. Soc. Nephrol. 2009, 4, 891–898. [Google Scholar] [CrossRef]
- Wald, R.; Quinn, R.R.; Luo, J.; Li, P.; Scales, D.C.; Mamdani, M.M.; Ray, J.G.; University of Toronto Acute Kidney Injury Research Group. Chronic dialysis and death among survivors of acute kidney injury requiring dialysis. JAMA 2009, 302, 1179–1185. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, Q.; Wen, J.; Chen, T.; He, L.; Wang, Y.; Yin, J.; Wu, R.; Xue, R.; Li, S.; et al. Ischemic Duration and Frequency Determines AKI-to-CKD Progression Monitored by Dynamic Changes of Tubular Biomarkers in IRI Mice. Front. Physiol. 2019, 10, 153. [Google Scholar] [CrossRef]
- Leemans, J.C.; Stokman, G.; Claessen, N.; Rouschop, K.M.; Teske, G.J.D.; Kirschning, C.J.; Akira, S.; van der Poll, T.; Weening, J.J.; Florquin, S. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J. Clin. Investig. 2005, 115, 2894–2903. [Google Scholar] [CrossRef]
- Kim, B.S.; Lim, S.W.; Li, C.; Kim, J.S.; Sun, B.K.; Ahn, K.O.; Han, S.W.; Kim, J.; Yang, C.W. Ischemia-reperfusion injury activates innate immunity in rat kidneys. Transplantation 2005, 79, 1370–1377. [Google Scholar] [CrossRef]
- Wolfs, T.G.A.M.; Buurman, W.A.; van Schadewijk, A.; de Vries, B.; Daemen, M.A.R.C.; Hiemstra, P.S.; van’t Veer, C. In vivo expression of Toll-like receptor 2 and 4 by renal epithelial cells: IFN-gamma and TNF-alpha mediated up-regulation during inflammation. J. Immunol. 2002, 168, 1286–1293. [Google Scholar] [CrossRef]
- Paulus, P.; Rupprecht, K.; Baer, P.; Obermüller, N.; Penzkofer, D.; Reissig, C.; Scheller, B.; Holfeld, J.; Zacharowski, K.; Dimmeler, S.; et al. The early activation of toll-like receptor (TLR)-3 initiates kidney injury after ischemia and reperfusion. PLoS ONE 2014, 15, e94366. [Google Scholar] [CrossRef] [PubMed]
- Good, D.W.; George, T.; Watts, B.A. Toll-like Receptor 2 Is Required for LPS-induced Toll-like Receptor 4 Signaling and Inhibition of Ion Transport in Renal Thick Ascending Limb. J. Biol. Chem. 2012, 287, 20208–20220. [Google Scholar] [CrossRef] [PubMed]
- Good, D.W.; George, T.; Watts, B.A. Toll-like receptor 2 mediates inhibition of HCO3− absorption by bacterial lipoprotein in medullary thick ascending limb. Am. J. Physiol. Renal Physiol. 2010, 299, F536–F544. [Google Scholar] [CrossRef] [PubMed]
- Vallés, P.G.; Lorenzo, A.G.; Bocanegra, V.; Vallés, R. Acute kidney injury: What part do toll-like receptors play? Int. J. Nephrol. Renovasc. Dis. 2014, 7, 241–251. [Google Scholar] [CrossRef]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef]
- Devarajan, P. Update on Mechanisms of Ischemic Acute Kidney Injury. J. Am. Soc. Nephrol. 2006, 17, 1503–1520. [Google Scholar] [CrossRef]
- Kinsey, G.R.; Li, L.; Okusa, M.D. Inflammation in acute kidney injury. Nephron Exp. Nephrol. 2008, 109, e102–e107. [Google Scholar] [CrossRef]
- Venkatachalam, M.A.; Griffin, K.A.; Lan, R.; Geng, H.; Saikumar, P.; Bidani, A.K. Acute kidney injury: A springboard for progression in chronic kidney disease. Am. J. Physiol. Renal Physiol. 2010, 298, F1078–F1094. [Google Scholar] [CrossRef]
- Lee, S.; Huen, S.; Nishio, H.; Nishio, S.; Lee, H.K.; Choi, B.-S.; Ruhrberg, C.; Cantley, L.G. Distinct macrophage phenotypes contribute to kidney injury and repair. J. Am. Soc. Nephrol. 2011, 22, 317–326. [Google Scholar] [CrossRef]
- Wang, Y.; Harris, D.C.H. Macrophages in Renal Disease. J. Am. Soc. Nephrol. 2011, 22, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.Y.; Paterson, D.J.; Atkins, R.C. Initiation and evolution of interstitial leukocytic infiltration in experimental glomerulonephritis. Kidney Int. 1991, 40, 425–433. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Location of Coding Genes | Location in the Cell | The Number of Amino Acids | Molecular Weight (kDa) | Number of LLR | Reference |
|---|---|---|---|---|---|---|
| TLR1 | Chromosome 4 | Golgi apparatus, Phagosome, Cell membrane | 786aa | 90.31 | 19 | [25,26,27] |
| TLR2 | Chromosome 4 | Phagosome | 784aa | 89.83 | 19 | [25,28,29] |
| TLR3 | Chromosome 4 | Early endosome, ER | 904aa | 103.82 | 23 | [25,30,31] |
| TLR4 | Chromosome 9 | Cell membrane, Early endosome | 839aa | 95.68 | 21 | [25,32,33] |
| TLR5 | Chromosome 1 | No data | 858aa | 97.83 | 20 | [25,34,35] |
| TLR6 | Chromosome 4 | Golgi apparatus, Cell membrane, Phagosome | 796aa | 91.88 | 19 | [25,36,37] |
| TLR7 | Chromosome X | Endosomes, Lysosomes, ER, Phagosome | 1049aa | 120.92 | 25 | [25,38] |
| TLR8 | Chromosome X | No data | 1041aa | 119.82 | 25 | [25,39,40] |
| TLR9 | Chromosome 3 | Endosomes, Lysosomes, ER, Phagosome | 1032aa | 115.86 | 25 | [25,41] |
| TLR10 | Chromosome 4 | No data | 811aa | 94.56 | 19 | [25,42,43] |
| TLR11 | Expression in mice | No data | 926aa | 105.83 | 10 | [44] |
| TLR12 | Expression in mice | No data | 906aa | 99.94 | 17 | [45] |
| TLR13 | Expression in mice | Endosomes | 991aa | 114.44 | 25 | [46] |
| Name | Occurrence | Ligand PAMP | The Origin of PAMP | Ligand DAMP | Reference |
|---|---|---|---|---|---|
| Extracellular | |||||
| TLR1 | Macrophages Neutrophils B lymphocytes Dendritic cells | Lipopeptides Soluble factors (lipoproteins) | Bacteria | No data | [47,48] |
| TLR2 | Macrophages Neutrophils B lymphocytes Dendritic cells NK cells | Bacterial lipopeptides Teichoic acids, LAM Moduline, Glycolipids of bacteria, Porins, LPS, | Bacteria | Apolipoprotein CIII, Heparin sulphate, Hyaluronic acid, Hsp60, Hsp70, Peroxiredoxin | [47,48,49,50] |
| Glycosinositolphospholipids | Protozoa, e.g., Trypanosoma cruzi | ||||
| Zymosan | Fungi | ||||
| Hemagglutinin | Measles virus | ||||
| Protein | Herpesvirus | ||||
| Hsp70 proteins | Host organism | ||||
| TLR4 | Macrophages Neutrophils B lymphocytes Dendritic cells NK cells Treg cells | LPS | Bacteria | C-reactive protein, Fibronectin, Fibrinogen, Heparin sulphate, Neutrophil, Elastase, Angiotensin II, Hsp60 | [48,49,50,51] |
| Fusion proteins, Proteins present in the coating | Viruses, e.g., RSV virus | ||||
| Taxol | Plants | ||||
| Hsp60 protein Hsp70 protein A fragment of the A domain of fibronectin Hyaluronic acid oligosaccharide Fibrinogen Heparan sulphate | Host organism | ||||
| TLR5 | Macrophages B lymphocytes Dendritic cells Treg cells | Flagellin | Bacteria (Gram-negative) | No data | [48,52] |
| TLR6 | Macrophages Neutrophils Dendritic cells | Diacyl lipopeptides Lipoteichoic acids Zymosan | Bacteria Fungi | Versican | [47,48,49,50,53] |
| TLR10 | Dendritic cells | No data | No data | No data | [54] |
| TLR11 | Macrophages Dendritic cells | Flagellin Profilin | Bacteria Protozoa, e.g., Toxoplasma gondii | No data | [55,56] |
| TLR12 | Dendritic cells | Profilin | Protozoa, e.g., Toxoplasma gondii | No data | [55] |
| Intracellular | |||||
| TLR3 | Macrophages Neutrophils B lymphocytes Dendritic cells | Double-stranded RNA | Viruses | Own double-stranded RNA | [57,58] |
| TLR7 | Macrophages Neutrophils Dendritic cells Treg cells | Single-stranded RNA Antiviral and anticancer compounds | Viruses Synthetic | Own single-stranded RNA | [48,59] |
| TLR8 | Dendritic cells Treg cells | Single-stranded RNA Antiviral compounds | Viruses Synthetic | Own single-stranded RNA | [48,60] |
| TLR9 | Macrophages Neutrophils Dendritic cells | Double-stranded DNA (containing unmethylated CpG sequences) | Bacteria, viruses and synthetic | HMGB1 Mitochondrial DNA | [48,49,50,61] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mertowski, S.; Lipa, P.; Morawska, I.; Niedźwiedzka-Rystwej, P.; Bębnowska, D.; Hrynkiewicz, R.; Grywalska, E.; Roliński, J.; Załuska, W. Toll-Like Receptor as a Potential Biomarker in Renal Diseases. Int. J. Mol. Sci. 2020, 21, 6712. https://doi.org/10.3390/ijms21186712
Mertowski S, Lipa P, Morawska I, Niedźwiedzka-Rystwej P, Bębnowska D, Hrynkiewicz R, Grywalska E, Roliński J, Załuska W. Toll-Like Receptor as a Potential Biomarker in Renal Diseases. International Journal of Molecular Sciences. 2020; 21(18):6712. https://doi.org/10.3390/ijms21186712
Chicago/Turabian StyleMertowski, Sebastian, Paulina Lipa, Izabela Morawska, Paulina Niedźwiedzka-Rystwej, Dominika Bębnowska, Rafał Hrynkiewicz, Ewelina Grywalska, Jacek Roliński, and Wojciech Załuska. 2020. "Toll-Like Receptor as a Potential Biomarker in Renal Diseases" International Journal of Molecular Sciences 21, no. 18: 6712. https://doi.org/10.3390/ijms21186712
APA StyleMertowski, S., Lipa, P., Morawska, I., Niedźwiedzka-Rystwej, P., Bębnowska, D., Hrynkiewicz, R., Grywalska, E., Roliński, J., & Załuska, W. (2020). Toll-Like Receptor as a Potential Biomarker in Renal Diseases. International Journal of Molecular Sciences, 21(18), 6712. https://doi.org/10.3390/ijms21186712