Roles of Specialized Pro-Resolving Lipid Mediators in Autophagy and Inflammation




Abstract
1. Introduction
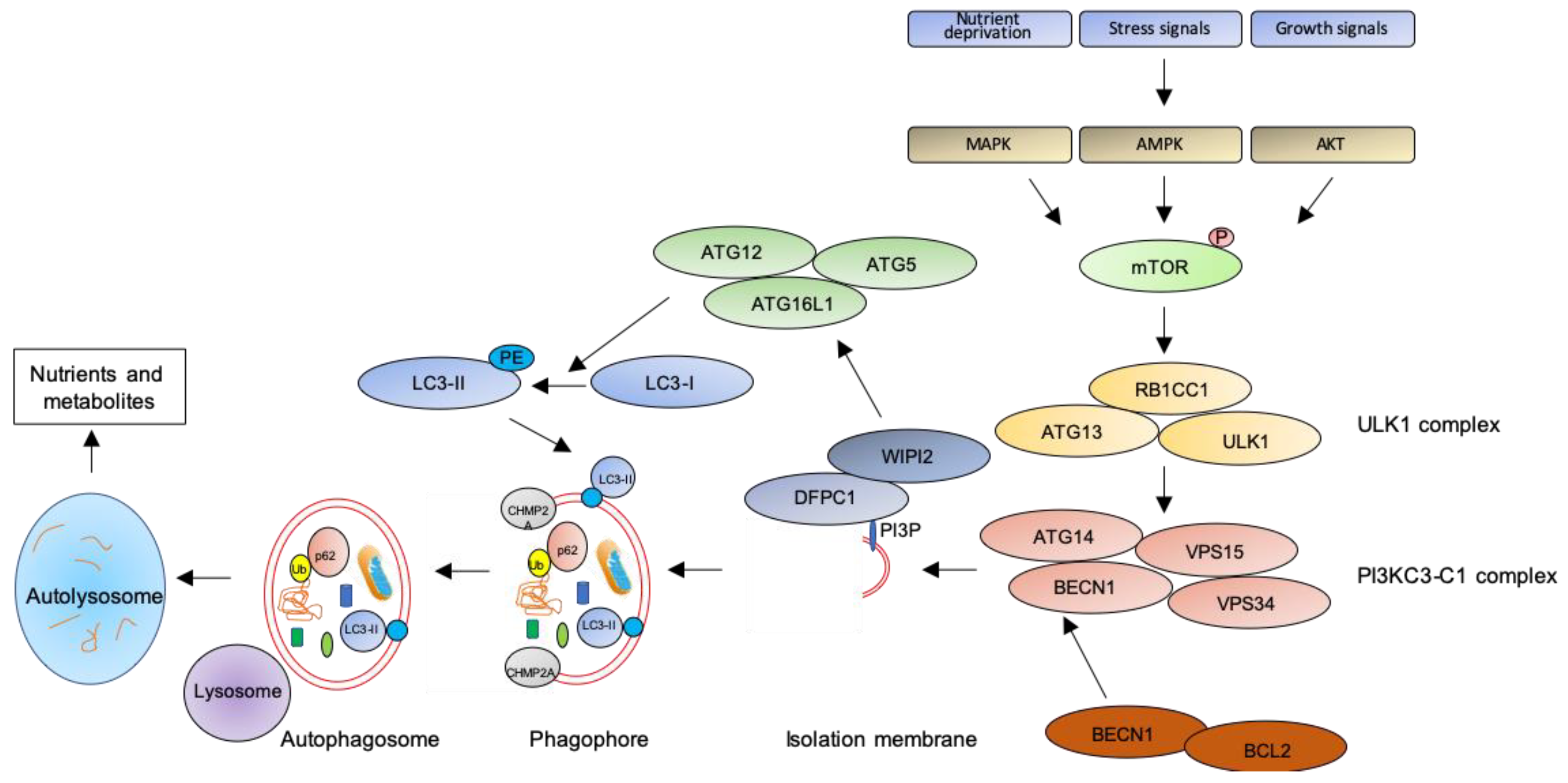
2. The Autophagic Pathway
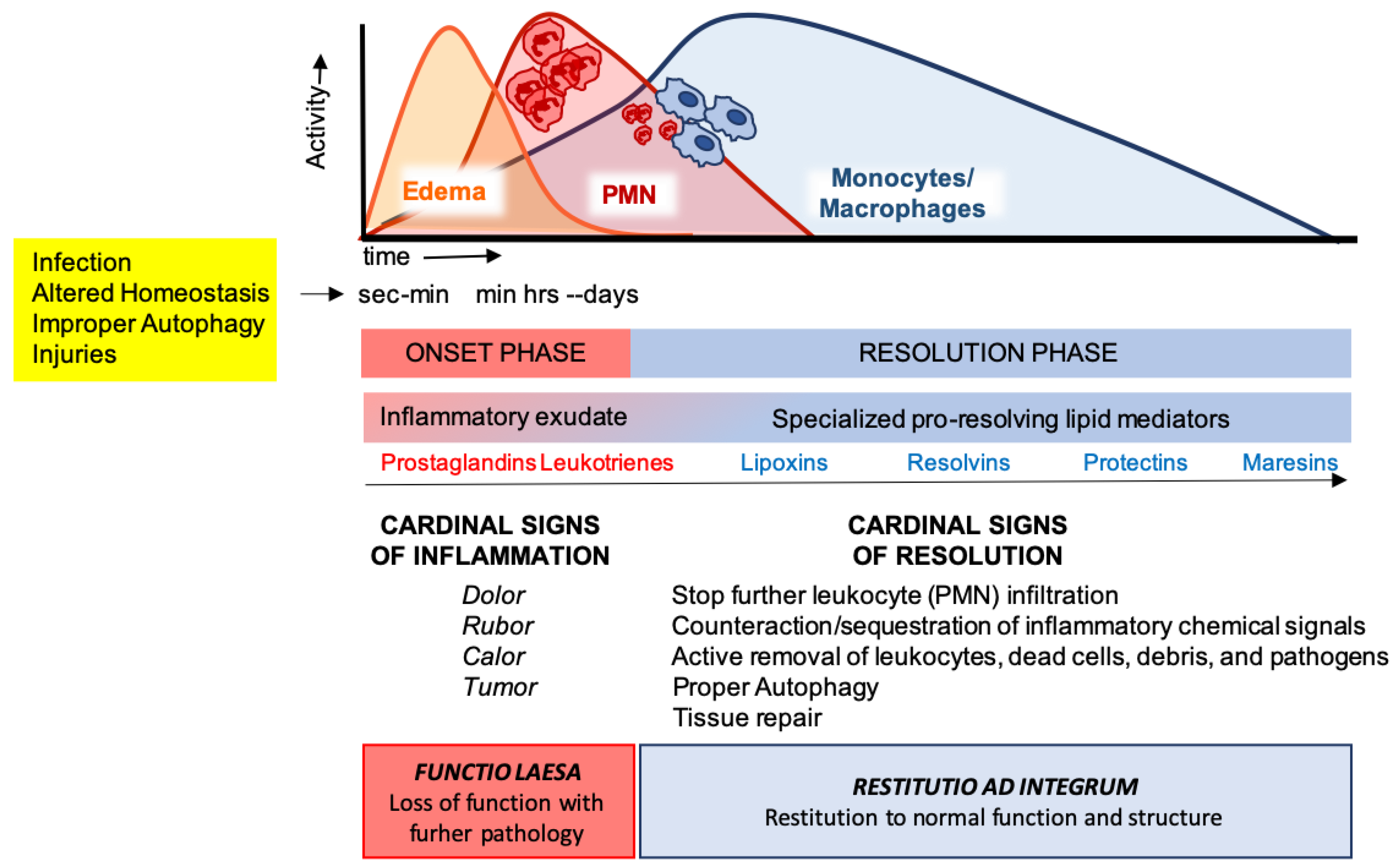
3. The Ideal Outcome of Inflammation: Resolution
3.1. Key Aspects of Acute Inflammation and Resolution
3.2. SPM Biosynthesis
3.3. SPM Receptors and Functions in Resolution of Inflammation
4. Autophagy in Chronic Inflammatory Diseases
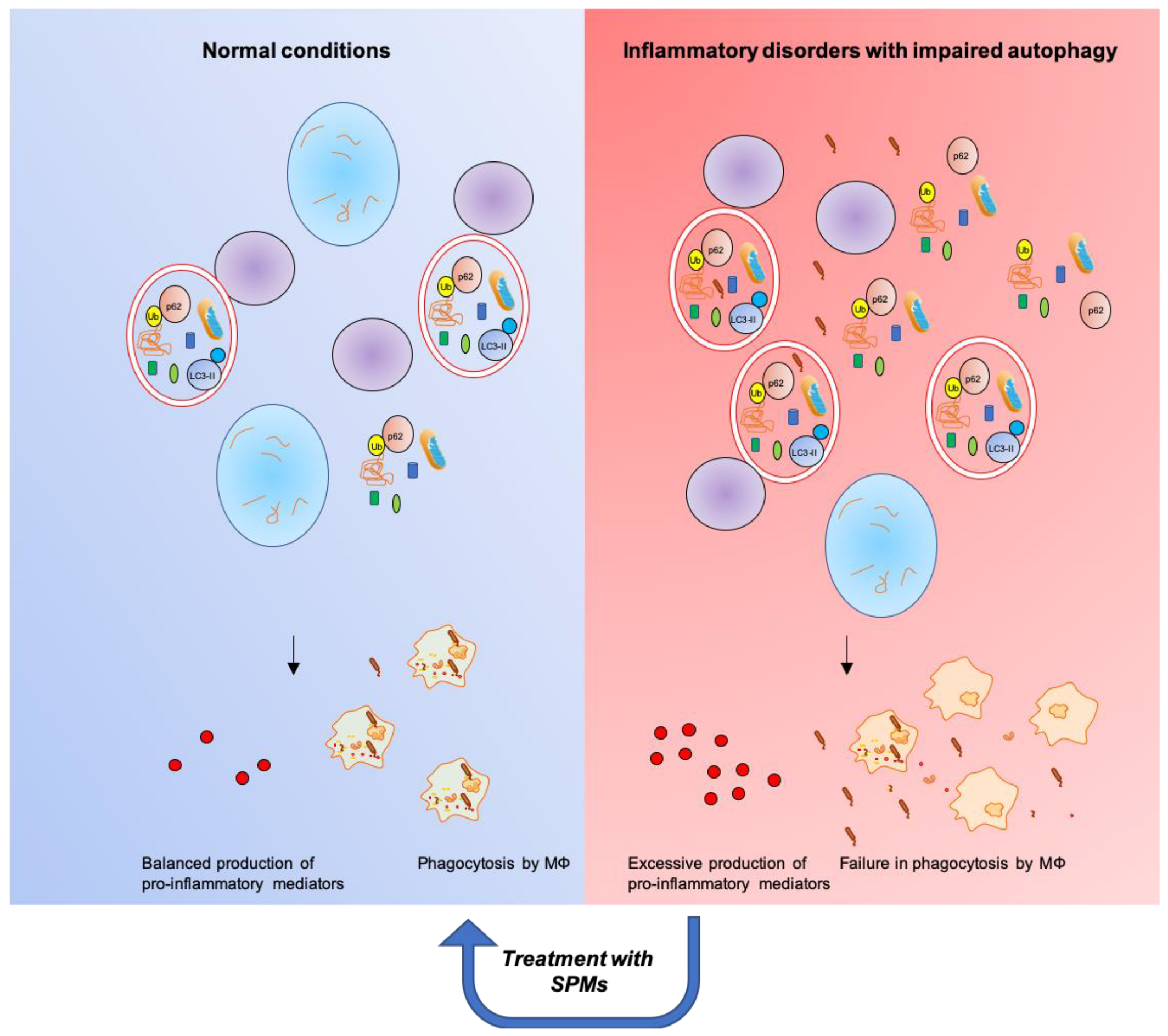
4.1. Autophagy and Inflammation
4.2. Chronic Inflammatory Diseases Characterized by Autophagy Dysfunction: Crohn’s Disease
4.3. Chronic Inflammatory Diseases Characterized by Autophagy Dysfunction: Cystic Fibrosis
4.4. Chronic Inflammatory Diseases Characterized by Autophagy Dysfunction: Cancer
4.5. Chronic Inflammatory Diseases Characterized by Autophagy Dysfunction: Alzheimer’s Disease
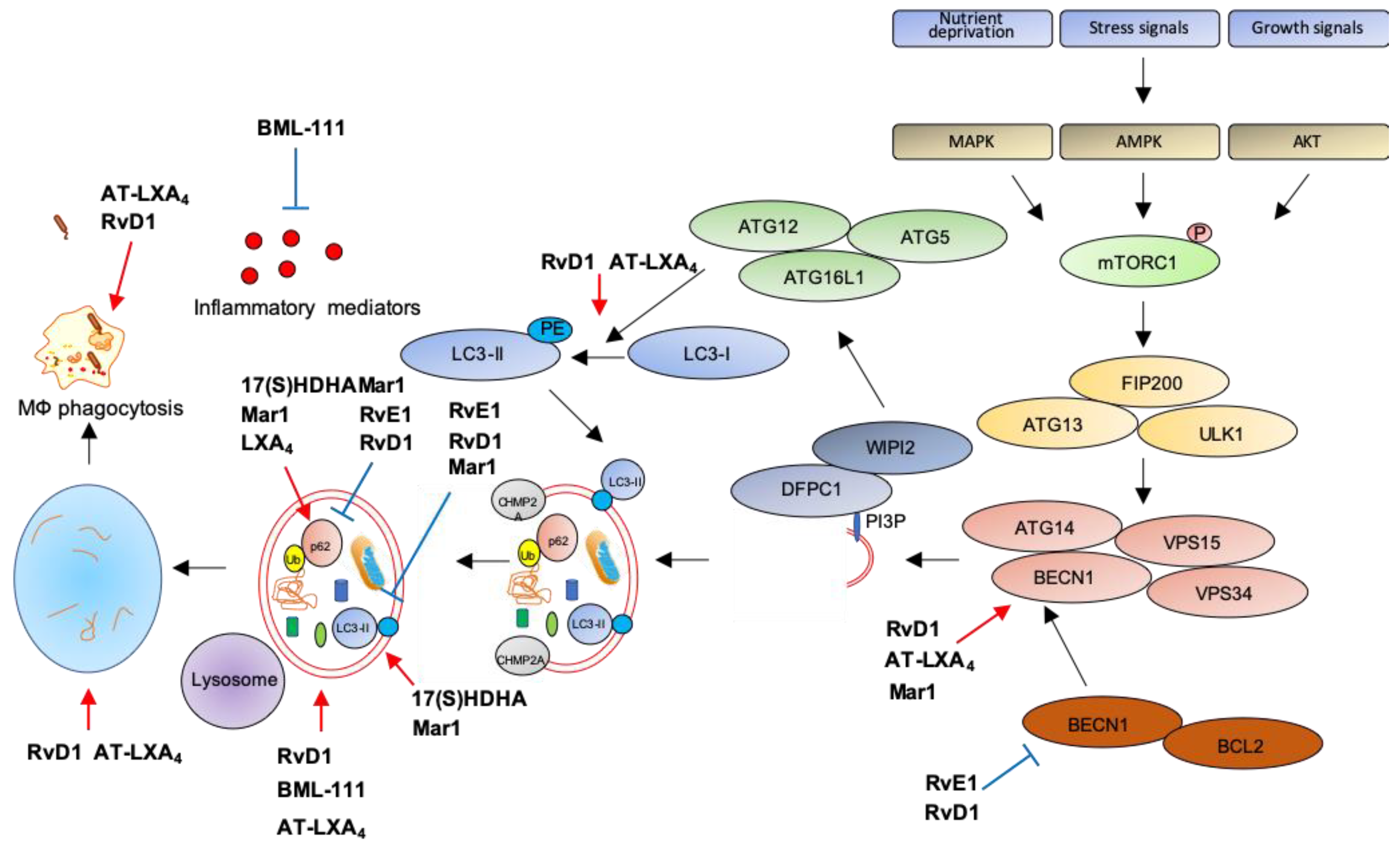
5. Stimulation of Resolution Restores Autophagy and Dampens Chronic Inflammation
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- De Duve, C.; Wattiaux, R. Functions of Lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.; Murera, D.; Gros, F. Autophagy in Chronic Inflammation. In Autophagy Networks in Inflammation; Maiuri, M.C., De Stefano, D., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 107–133. ISBN 978-3-319-30077-1. [Google Scholar]
- Matsuzawa-Ishimoto, Y.; Hwang, S.; Cadwell, K. Autophagy and Inflammation. Annu. Rev. Immunol. 2018, 36, 73–101. [Google Scholar] [CrossRef] [PubMed]
- Netea-Maier, R.T.; Plantinga, T.S.; van de Veerdonk, F.L.; Smit, J.W.; Netea, M.G. Modulation of inflammation by autophagy: Consequences for human disease. Autophagy 2016, 12, 245–260. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Fang, X.; Wang, X. Autophagy and inflammation. Clin. Transl. Med. 2017, 6, 24. [Google Scholar] [CrossRef]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef]
- Netea, M.G.; Balkwill, F.; Chonchol, M.; Cominelli, F.; Donath, M.Y.; Giamarellos-Bourboulis, E.J.; Golenbock, D.; Gresnigt, M.S.; Heneka, M.T.; Hoffman, H.M.; et al. A guiding map for inflammation. Nat. Immunol. 2017, 18, 826–831. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef]
- Schett, G.; Neurath, M.F. Resolution of chronic inflammatory disease: Universal and tissue-specific concepts. Nat. Commun. 2018, 9, 3261. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Cianci, E.; Simiele, F.; Recchiuti, A. Lipoxins and aspirin-triggered lipoxins in resolution of inflammation. Eur. J. Pharmacol. 2015, 760, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Basil, M.C.; Levy, B.D. Specialized pro-resolving mediators: Endogenous regulators of infection and inflammation. Nat. Rev. Immunol. 2016, 16, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-T.; Teles, R.; Kantarci, A.; Chen, T.; McCafferty, J.; Starr, J.R.; Brito, L.C.N.; Paster, B.J.; Van Dyke, T.E. Resolvin E1 Reverses Experimental Periodontitis and Dysbiosis. J. Immunol. 2016, 197, 2796–2806. [Google Scholar] [CrossRef]
- Li, C.; Wu, X.; Liu, S.; Shen, D.; Zhu, J.; Liu, K. Role of Resolvins in the Inflammatory Resolution of Neurological Diseases. Front. Pharmacol. 2020, 11, 612. [Google Scholar] [CrossRef]
- Norling, L.V.; Headland, S.E.; Dalli, J.; Arnardottir, H.H.; Haworth, O.; Jones, H.R.; Irimia, D.; Serhan, C.N.; Perretti, M. Proresolving and cartilage-protective actions of resolvin D1 in inflammatory arthritis. JCI Insight 2016, 1. [Google Scholar] [CrossRef]
- Matte, A.; Recchiuti, A.; Federti, E.; Koehl, B.; Mintz, T.; El Nemer, W.; Tharaux, P.-L.; Brousse, V.; Andolfo, I.; Lamolinara, A.; et al. Resolution of sickle cell disease–associated inflammation and tissue damage with 17R-resolvin D1. Blood 2019, 133, 252–265. [Google Scholar] [CrossRef]
- Codagnone, M.; Cianci, E.; Lamolinara, A.; Mari, V.C.; Nespoli, A.; Isopi, E.; Mattoscio, D.; Arita, M.; Bragonzi, A.; Iezzi, M.; et al. Resolvin D1 enhances the resolution of lung inflammation caused by long-term Pseudomonas aeruginosa infection. Mucosal Immunol. 2018, 11, 35–49. [Google Scholar] [CrossRef]
- Recchiuti, A.; Mattoscio, D.; Isopi, E. Roles, Actions, and Therapeutic Potential of Specialized Pro-resolving Lipid Mediators for the Treatment of Inflammation in Cystic Fibrosis. Front. Pharmacol. 2019, 10, 252. [Google Scholar] [CrossRef]
- Isopi, E.; Mattoscio, D.; Codagnone, M.; Mari, V.C.; Lamolinara, A.; Patruno, S.; D’Aurora, M.; Cianci, E.; Nespoli, A.; Franchi, S.; et al. Resolvin D1 Reduces Lung Infection and Inflammation Activating Resolution in Cystic Fibrosis. Front. Immunol. 2020, 11, 581. [Google Scholar] [CrossRef]
- Fredman, G.; Tabas, I. Boosting Inflammation Resolution in Atherosclerosis. Am. J. Pathol. 2017, 187, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Wu, S.-H.; Zhang, L.; Chen, X.-Q. Pilot application of lipoxin A4 analog and lipoxin A4 receptor agonist in asthmatic children with acute episodes. Exp. Ther. Med. 2017, 14, 2284–2290. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.H.; Chen, X.Q.; Liu, B.; Wu, H.J.; Dong, L. Efficacy and safety of 15(R/S)-methyl-lipoxin A(4) in topical treatment of infantile eczema. Br. J. Dermatol. 2013, 168, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Motwani, M.P.; Colas, R.A.; George, M.J.; Flint, J.D.; Dalli, J.; Richard-Loendt, A.; De Maeyer, R.P.H.; Serhan, C.N.; Gilroy, D.W. Pro-resolving mediators promote resolution in a human skin model of UV-killed Escherichia coli–driven acute inflammation. JCI Insight 2018, 3, e94463. [Google Scholar] [CrossRef]
- Oku, M.; Sakai, Y. Three Distinct Types of Microautophagy Based on Membrane Dynamics and Molecular Machineries. BioEssays 2018, 40, 1800008. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The Role of Atg Proteins in Autophagosome Formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Hurley, J.H.; Young, L.N. Mechanisms of Autophagy Initiation. Annu. Rev. Biochem. 2017, 86, 225–244. [Google Scholar] [CrossRef] [PubMed]
- Burman, C.; Ktistakis, N.T. Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett. 2010, 584, 1302–1312. [Google Scholar] [CrossRef] [PubMed]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Dooley, H.C.; Razi, M.; Polson, H.E.J.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 Links LC3 Conjugation with PI3P, Autophagosome Formation, and Pathogen Clearance by Recruiting Atg12–5-16L1. Mol. Cell 2014, 55, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Weidberg, H.; Shvets, E.; Shpilka, T.; Shimron, F.; Shinder, V.; Elazar, Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010, 29, 1792–1802. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; He, H.; Tang, Z.; Hattori, T.; Liu, Y.; Young, M.M.; Serfass, J.M.; Chen, L.; Gebru, M.; Chen, C.; et al. An autophagy assay reveals the ESCRT-III component CHMP2A as a regulator of phagophore closure. Nat. Commun. 2018, 9, 2855. [Google Scholar] [CrossRef]
- Nakamura, S.; Yoshimori, T. New insights into autophagosome–lysosome fusion. J. Cell Sci. 2017, 130, 1209–1216. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef]
- Pankiv, S.; Alemu, E.A.; Brech, A.; Bruun, J.-A.; Lamark, T.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end–directed vesicle transport. J. Cell Biol. 2010, 188, 253–269. [Google Scholar] [CrossRef]
- Kimura, S.; Noda, T.; Yoshimori, T. Dynein-dependent Movement of Autophagosomes Mediates Efficient Encounters with Lysosomes. Cell Struct. Funct. 2008, 33, 109–122. [Google Scholar] [CrossRef]
- Jiang, P.; Nishimura, T.; Sakamaki, Y.; Itakura, E.; Hatta, T.; Natsume, T.; Mizushima, N. The HOPS complex mediates autophagosome–lysosome fusion through interaction with syntaxin 17. Mol. Biol. Cell 2014, 25, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Inflammation 2010: New Adventures of an Old Flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- deCathelineau, A.M.; Henson, P.M. The final step in programmed cell death: Phagocytes carry apoptotic cells to the grave. Essays Biochem. 2003, 39, 105–117. [Google Scholar] [PubMed]
- Serhan, C.N.; Brain, S.D.; Buckley, C.D.; Gilroy, D.W.; Haslett, C.; O’Neill, L.A.; Perretti, M.; Rossi, A.G.; Wallace, J.L. Resolution of inflammation: State of the art, definitions and terms. FASEB J. 2007, 21, 325–332. [Google Scholar] [CrossRef]
- Watanabe, S.; Alexander, M.; Misharin, A.V.; Budinger, G.R.S. The role of macrophages in the resolution of inflammation. J. Clin. Investig. 2019, 129, 2619–2628. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, M.A.; Vago, J.P.; Perretti, M.; Teixeira, M.M. Mediators of the Resolution of the Inflammatory Response. Trends Immunol. 2019, 40, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Gomez, A.; Perretti, M.; Soehnlein, O. Resolution of inflammation: An integrated view. EMBO Mol. Med. 2013, 5, 661–674. [Google Scholar] [CrossRef]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669. [Google Scholar] [CrossRef]
- Levy, B.D.; Clish, C.B.; Schmidt, B.; Gronert, K.; Serhan, C.N. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat. Immunol. 2001, 2, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Recchiuti, A.; Serhan, C.N. Pro-Resolving Lipid Mediators (SPMs) and Their Actions in Regulating miRNA in Novel Resolution Circuits in Inflammation. Front. Immunol. 2012, 3, 298. [Google Scholar] [CrossRef]
- Serhan, C.N.; Hamberg, M.; Samuelsson, B. Lipoxins: Novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc. Natl. Acad. Sci. USA 1984, 81, 5335–5339. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Sheppard, K.A. Lipoxin formation during human neutrophil-platelet interactions. Evidence for the transformation of leukotriene A4 by platelet 12-lipoxygenase in vitro. J. Clin. Investig. 1990, 85, 772–780. [Google Scholar] [CrossRef]
- Romano, M.; Chen, X.S.; Takahashi, Y.; Yamamoto, S.; Funk, C.D.; Serhan, C.N. Lipoxin synthase activity of human platelet 12-lipoxygenase. Biochem. J. 1993, 296, 127–133. [Google Scholar] [CrossRef]
- Edenius, C.; Kumlin, M.; Björk, T.; Änggård, A.; Lindgren, J.Å. Lipoxin formation in human nasal polyps and bronchial tissue. FEBS Lett. 1990, 272, 25–28. [Google Scholar] [CrossRef]
- Levy, B.D.; Romano, M.; Chapman, H.A.; Reilly, J.J.; Drazen, J.; Serhan, C.N. Human alveolar macrophages have 15-lipoxygenase and generate 15(S)-hydroxy-5,8,11-cis-13-trans-eicosatetraenoic acid and lipoxins. J. Clin. Investig. 1993, 92, 1572–1579. [Google Scholar] [CrossRef]
- Claria, J.; Serhan, C.N. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc. Natl. Acad. Sci. USA 1995, 92, 9475–9479. [Google Scholar] [CrossRef]
- Serhan, C.N. Treating inflammation and infection in the 21st century: New hints from decoding resolution mediators and mechanisms. FASEB J. 2017, 31, 1273–1288. [Google Scholar] [CrossRef]
- Serhan, C.N.; Clish, C.B.; Brannon, J.; Colgan, S.P.; Chiang, N.; Gronert, K. Novel Functional Sets of Lipid-Derived Mediators with Antiinflammatory Actions Generated from Omega-3 Fatty Acids via Cyclooxygenase 2–Nonsteroidal Antiinflammatory Drugs and Transcellular Processing. J. Exp. Med. 2000, 192, 1197–1204. [Google Scholar] [CrossRef]
- Dalli, J.; Chiang, N.; Serhan, C.N. Elucidation of novel 13-series resolvins that increase with atorvastatin and clear infections. Nat. Med. 2015, 21, 1071–1075. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Dalli, J.; Colas, R.A.; Winkler, J.W.; Chiang, N. Protectins and maresins: New pro-resolving families of mediators in acute inflammation and resolution bioactive metabolome. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2015, 1851, 397–413. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Yang, R.; Martinod, K.; Kasuga, K.; Pillai, P.S.; Porter, T.F.; Oh, S.F.; Spite, M. Maresins: Novel macrophage mediators with potent antiinflammatory and proresolving actions. J. Exp. Med. 2009, 206, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Dalli, J.; Ramon, S.; Norris, P.C.; Colas, R.A.; Serhan, C.N. Novel proresolving and tissue-regenerative resolvin and protectin sulfido-conjugated pathways. FASEB J. 2015, 29, 2120–2136. [Google Scholar] [CrossRef] [PubMed]
- Dalli, J.; Vlasakov, I.; Riley, I.R.; Rodriguez, A.R.; Spur, B.W.; Petasis, N.A.; Chiang, N.; Serhan, C.N. Maresin conjugates in tissue regeneration biosynthesis enzymes in human macrophages. Proc. Natl. Acad. Sci. USA 2016, 113, 12232–12237. [Google Scholar] [CrossRef] [PubMed]
- Dalli, J.; Colas, R.A.; Serhan, C.N. Novel n-3 Immunoresolvents: Structures and Actions. Sci. Rep. 2013, 3, 1940. [Google Scholar] [CrossRef]
- Chiang, N.; Serhan, C.N. Structural elucidation and physiologic functions of specialized pro-resolving mediators and their receptors. Mol. Asp. Med. 2017, 58, 114–129. [Google Scholar] [CrossRef]
- Krishnamoorthy, S.; Recchiuti, A.; Chiang, N.; Yacoubian, S.; Lee, C.H.; Yang, R.; Petasis, N.A.; Serhan, C.N. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc. Natl. Acad. Sci. USA 2010, 107, 1660–1665. [Google Scholar] [CrossRef]
- Arita, M.; Bianchini, F.; Aliberti, J.; Sher, A.; Chiang, N.; Hong, S.; Yang, R.; Petasis, N.A.; Serhan, C.N. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J. Exp. Med. 2005, 201, 713–722. [Google Scholar] [CrossRef]
- Ohira, T.; Arita, M.; Omori, K.; Recchiuti, A.; Van Dyke, T.E.; Serhan, C.N. Resolvin E1 receptor activation signals phosphorylation and phagocytosis. J. Biol. Chem. 2010, 285, 3451–3461. [Google Scholar] [CrossRef]
- Fiore, S.; Ryeom, S.W.; Weller, P.F.; Serhan, C.N. Lipoxin recognition sites. Specific binding of labeled lipoxin A4 with human neutrophils. J. Biol. Chem. 1992, 267, 16168–16176. [Google Scholar]
- Romano, M.; Recchia, I.; Recchiuti, A. Lipoxin receptors. Sci. World J. 2007, 7, 1393–1412. [Google Scholar] [CrossRef]
- Romano, M.; Patruno, S.; Pomilio, A.; Recchiuti, A. Proresolving Lipid Mediators and Receptors in Stem Cell Biology: Concise Review. STEM CELLS Transl. Med. 2019, 8, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, S.; Recchiuti, A.; Chiang, N.; Fredman, G.; Serhan, C.N. Resolvin D1 receptor stereoselectivity and regulation of inflammation and proresolving microRNAs. Am. J. Pathol. 2012, 180, 2018–2027. [Google Scholar] [CrossRef]
- Chiang, N.; Fredman, G.; Bäckhed, F.; Oh, S.F.; Vickery, T.; Schmidt, B.A.; Serhan, C.N. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 2012, 484, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Dalli, J.; Winkler, J.W.; Colas, R.A.; Arnardottir, H.; Cheng, C.-Y.C.; Chiang, N.; Petasis, N.A.; Serhan, C.N. Resolvin D3 and Aspirin-Triggered Resolvin D3 Are Potent Immunoresolvents. Chem. Biol. 2013, 20, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Arnardottir, H.H.; Dalli, J.; Norling, L.V.; Colas, R.A.; Perretti, M.; Serhan, C.N. Resolvin D3 Is Dysregulated in Arthritis and Reduces Arthritic Inflammation. J. Immunol. 2016, 197, 2362–2368. [Google Scholar] [CrossRef] [PubMed]
- Corminboeuf, O.; Leroy, X. FPR2/ALXR Agonists and the Resolution of Inflammation. J. Med. Chem. 2015, 58, 537–559. [Google Scholar] [CrossRef]
- Dufton, N.; Hannon, R.; Brancaleone, V.; Dalli, J.; Patel, H.B.; Gray, M.; D’Acquisto, F.; Buckingham, J.C.; Perretti, M.; Flower, R.J. Anti-Inflammatory Role of the Murine Formyl-Peptide Receptor 2: Ligand-Specific Effects on Leukocyte Responses and Experimental Inflammation. J. Immunol. 2010, 184, 2611–2619. [Google Scholar] [CrossRef]
- Devchand, P.R.; Arita, M.; Hong, S.; Bannenberg, G.; Moussignac, R.; Gronert, K.; Serhan, C.N. Human ALX receptor regulates neutrophil recruitment in transgenic mice: Roles in inflammation and host defense. FASEB J. 2003, 17, 652–659. [Google Scholar] [CrossRef]
- Simiele, F.; Recchiuti, A.; Mattoscio, D.; De Luca, A.; Cianci, E.; Franchi, S.; Gatta, V.; Parolari, A.; Werba, J.P.; Camera, M.; et al. Transcriptional regulation of the human FPR2/ALX gene: Evidence of a heritable genetic variant that impairs promoter activity. FASEB J. 2012, 26, 1323–1333. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lu, Y.; Sun, G.; Teng, F.; Luo, N.; Jiang, J.; Wen, A. The common promoter polymorphism rs11666254 downregulates FPR2/ALX expression and increases risk of sepsis in patients with severe trauma. Crit. Care 2017, 21, 171. [Google Scholar] [CrossRef]
- Chiang, N.; Dalli, J.; Colas, R.A.; Serhan, C.N. Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J. Exp. Med. 2015, 212, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.; de la Rosa, X.; Libreros, S.; Serhan, C.N. Novel Resolvin D2 Receptor Axis in Infectious Inflammation. J. Immunol. 2017, 198, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Arita, M.; Ohira, T.; Sun, Y.-P.; Elangovan, S.; Chiang, N.; Serhan, C.N. Resolvin E1 Selectively Interacts with Leukotriene B4 Receptor BLT1 and ChemR23 to Regulate Inflammation. J. Immunol. 2007, 178, 3912–3917. [Google Scholar] [CrossRef]
- Chiang, N.; Libreros, S.; Norris, P.C.; de la Rosa, X.; Serhan, C.N. Maresin 1 activates LGR6 receptor promoting phagocyte immunoresolvent functions. J. Clin. Investig. 2019, 129, 5294–5311. [Google Scholar] [CrossRef]
- Bang, S.; Xie, Y.-K.; Zhang, Z.-J.; Wang, Z.; Xu, Z.-Z.; Ji, R.-R. GPR37 regulates macrophage phagocytosis and resolution of inflammatory pain. J. Clin. Investig. 2018, 128, 3568–3582. [Google Scholar] [CrossRef]
- Flak, M.B.; Koenis, D.S.; Sobrino, A.; Smith, J.; Pistorius, K.; Palmas, F.; Dalli, J. GPR101 mediates the pro-resolving actions of RvD5n-3 DPA in arthritis and infections. J. Clin. Investig. 2019, 130, 359–373. [Google Scholar] [CrossRef]
- Liao, Z.; Dong, J.; Wu, W.; Yang, T.; Wang, T.; Guo, L.; Chen, L.; Xu, D.; Wen, F. Resolvin D1 attenuates inflammation in lipopolysaccharide-induced acute lung injury through a process involving the PPARγ/NF-κB pathway. Respir. Res. 2012, 13, 110. [Google Scholar] [CrossRef]
- Keinan, D.; Leigh, N.; Nelson, J.; De Oleo, L.; Baker, O. Understanding Resolvin Signaling Pathways to Improve Oral Health. Int. J. Mol. Sci. 2013, 14, 5501–5518. [Google Scholar] [CrossRef]
- Recchiuti, A.; Krishnamoorthy, S.; Fredman, G.; Chiang, N.; Serhan, C.N. MicroRNAs in resolution of acute inflammation: Identification of novel resolvin D1-miRNA circuits. FASEB J. 2011, 25, 544–560. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Chiang, N.; Dalli, J. The resolution code of acute inflammation: Novel pro-resolving lipid mediators in resolution. Semin. Immunol. 2015, 27, 200–215. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.D.; Torres, V.J.; Cadwell, K. Autophagy and microbial pathogenesis. Cell Death Differ. 2020, 27, 872–886. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.H.; Song, H.K. A Structural View of Xenophagy, a Battle between Host and Microbes. Mol. Cells 2018, 41, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Heckmann, B.L.; Boada-Romero, E.; Cunha, L.D.; Magne, J.; Green, D.R. LC3-Associated Phagocytosis and Inflammation. J. Mol. Biol. 2017, 429, 3561–3576. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef]
- Hamacher-Brady, A.; Brady, N.R. Mitophagy programs: Mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell Mol. Life Sci. 2016, 73, 775–795. [Google Scholar] [CrossRef]
- Sun, Q.; Fan, J.; Billiar, T.R.; Scott, M.J. Inflammasome and Autophagy Regulation: A Two-way Street. Mol. Med. 2017, 23, 188–195. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.-G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, M.-L.; Zhao, J. Crosstalk between Autophagy and Type I Interferon Responses in Innate Antiviral Immunity. Viruses 2019, 11, 132. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Seo, G.J.; Choi, Y.J.; Kwak, M.-J.; Ge, J.; Rodgers, M.A.; Shi, M.; Leslie, B.J.; Hopfner, K.-P.; Ha, T.; et al. Crosstalk between the cGAS DNA Sensor and Beclin-1 Autophagy Protein Shapes Innate Antimicrobial Immune Responses. Cell Host Microbe 2014, 15, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Fujita, N.; Hayashi, T.; Takahara, K.; Satoh, T.; Lee, H.; Matsunaga, K.; Kageyama, S.; Omori, H.; Noda, T.; et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl. Acad. Sci. USA 2009, 106, 20842–20846. [Google Scholar] [CrossRef]
- Konno, H.; Konno, K.; Barber, G.N. Cyclic Dinucleotides Trigger ULK1 (ATG1) Phosphorylation of STING to Prevent Sustained Innate Immune Signaling. Cell 2013, 155, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Tal, M.C.; Sasai, M.; Lee, H.K.; Yordy, B.; Shadel, G.S.; Iwasaki, A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 2770–2775. [Google Scholar] [CrossRef] [PubMed]
- Jounai, N.; Takeshita, F.; Kobiyama, K.; Sawano, A.; Miyawaki, A.; Xin, K.-Q.; Ishii, K.J.; Kawai, T.; Akira, S.; Suzuki, K.; et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. USA 2007, 104, 14050–14055. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Israel, A. The IKK Complex, a Central Regulator of NF- B Activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a000158. [Google Scholar] [CrossRef]
- Shibata, Y.; Oyama, M.; Kozuka-Hata, H.; Han, X.; Tanaka, Y.; Gohda, J.; Inoue, J. p47 negatively regulates IKK activation by inducing the lysosomal degradation of polyubiquitinated NEMO. Nat. Commun. 2012, 3, 1061. [Google Scholar] [CrossRef]
- Liu, K.; Zhang, L.; Zhao, Q.; Zhao, Z.; Zhi, F.; Qin, Y.; Cui, J. SKP2 attenuates NF-κB signaling by mediating IKKβ degradation through autophagy. J. Mol. Cell Biol. 2018, 10, 205–215. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, X.; Yan, W.; Chen, Y.; Ke, M.; Cheng, C.; Zhu, X.; Xue, W.; Zhou, Q.; Zheng, L.; et al. ANGPTL8 negatively regulates NF-κB activation by facilitating selective autophagic degradation of IKKγ. Nat. Commun. 2017, 8, 2164. [Google Scholar] [CrossRef]
- Germic, N.; Frangez, Z.; Yousefi, S.; Simon, H.-U. Regulation of the innate immune system by autophagy: Neutrophils, eosinophils, mast cells, NK cells. Cell Death Differ. 2019, 26, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Rožman, S.; Yousefi, S.; Oberson, K.; Kaufmann, T.; Benarafa, C.; Simon, H.U. The generation of neutrophils in the bone marrow is controlled by autophagy. Cell Death Differ. 2015, 22, 445–456. [Google Scholar] [CrossRef]
- Itoh, H.; Matsuo, H.; Kitamura, N.; Yamamoto, S.; Higuchi, T.; Takematsu, H.; Kamikubo, Y.; Kondo, T.; Yamashita, K.; Sasada, M.; et al. Enhancement of neutrophil autophagy by an IVIG preparation against multidrug-resistant bacteria as well as drug-sensitive strains. J. Leukoc. Biol. 2015, 98, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Remijsen, Q.; Vanden Berghe, T.; Wirawan, E.; Asselbergh, B.; Parthoens, E.; De Rycke, R.; Noppen, S.; Delforge, M.; Willems, J.; Vandenabeele, P. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011, 21, 290–304. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Wei, Q.; Shin, J.N.; Abdel Fattah, E.; Bonilla, D.L.; Xiang, Q.; Eissa, N.T. Autophagy Is Required for Neutrophil-Mediated Inflammation. Cell Rep. 2015, 12, 1731–1739. [Google Scholar] [CrossRef] [PubMed]
- Germic, N.; Frangez, Z.; Yousefi, S.; Simon, H.-U. Regulation of the innate immune system by autophagy: Monocytes, macrophages, dendritic cells and antigen presentation. Cell Death Differ. 2019, 26, 715–727. [Google Scholar] [CrossRef]
- Zhang, Y.; Morgan, M.J.; Chen, K.; Choksi, S.; Liu, Z. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood 2012, 119, 2895–2905. [Google Scholar] [CrossRef]
- Jacquel, A.; Obba, S.; Boyer, L.; Dufies, M.; Robert, G.; Gounon, P.; Lemichez, E.; Luciano, F.; Solary, E.; Auberger, P. Autophagy is required for CSF-1–induced macrophagic differentiation and acquisition of phagocytic functions. Blood 2012, 119, 4527–4531. [Google Scholar] [CrossRef]
- Wu, M.-Y.; Lu, J.-H. Autophagy and Macrophage Functions: Inflammatory Response and Phagocytosis. Cells 2019, 9, 70. [Google Scholar] [CrossRef]
- Crotzer, V.L.; Blum, J.S. Autophagy and adaptive immunity: Autophagy and immunity. Immunology 2010, 131, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Münz, C. Autophagy in immunity. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2020; Volume 172, pp. 67–85. ISBN 978-0-12-822021-4. [Google Scholar]
- Guan, Q. A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease. J. Immunol. Res. 2019, 2019, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; De La Vega, F.M.; Briggs, J.; et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2007, 39, 207–211. [Google Scholar] [CrossRef]
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W.; et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007, 39, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Lassen, K.G.; Kuballa, P.; Conway, K.L.; Patel, K.K.; Becker, C.E.; Peloquin, J.M.; Villablanca, E.J.; Norman, J.M.; Liu, T.-C.; Heath, R.J.; et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc. Natl. Acad. Sci. USA 2014, 111, 7741–7746. [Google Scholar] [CrossRef]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef]
- Zhang, H.; Zheng, L.; McGovern, D.P.B.; Hamill, A.M.; Ichikawa, R.; Kanazawa, Y.; Luu, J.; Kumagai, K.; Cilluffo, M.; Fukata, M.; et al. Myeloid ATG16L1 Facilitates Host-Bacteria Interactions in Maintaining Intestinal Homeostasis. J. Immunol. Baltim. Md 1950 2017, 198, 2133–2146. [Google Scholar] [CrossRef]
- Racanelli, A.C.; Kikkers, S.A.; Choi, A.M.K.; Cloonan, S.M. Autophagy and inflammation in chronic respiratory disease. Autophagy 2018, 14, 221–232. [Google Scholar] [CrossRef]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Balázs, A.; Mall, M.A. Mucus obstruction and inflammation in early cystic fibrosis lung disease: Emerging role of the IL-1 signaling pathway. Pediatr. Pulmonol. 2019, 54. [Google Scholar] [CrossRef]
- Cantin, A.M.; Hartl, D.; Konstan, M.W.; Chmiel, J.F. Inflammation in cystic fibrosis lung disease: Pathogenesis and therapy. J. Cyst. Fibros. 2015, 14, 419–430. [Google Scholar] [CrossRef]
- Karp, C.L.; Flick, L.M.; Park, K.W.; Softic, S.; Greer, T.M.; Keledjian, R.; Yang, R.; Uddin, J.; Guggino, W.B.; Atabani, S.F.; et al. Defective lipoxin-mediated anti-inflammatory activity in the cystic fibrosis airway. Nat. Immunol. 2004, 5, 388–392. [Google Scholar] [CrossRef]
- Mattoscio, D.; Evangelista, V.; De Cristofaro, R.; Recchiuti, A.; Pandolfi, A.; Di Silvestre, S.; Manarini, S.; Martelli, N.; Rocca, B.; Petrucci, G.; et al. Cystic fibrosis transmembrane conductance regulator (CFTR) expression in human platelets: Impact on mediators and mechanisms of the inflammatory response. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 3970–3980. [Google Scholar] [CrossRef] [PubMed]
- Luciani, A.; Villella, V.R.; Esposito, S.; Brunetti-Pierri, N.; Medina, D.; Settembre, C.; Gavina, M.; Pulze, L.; Giardino, I.; Pettoello-Mantovani, M.; et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat. Cell Biol. 2010, 12, 863–875. [Google Scholar] [CrossRef]
- Iannitti, R.G.; Napolioni, V.; Oikonomou, V.; De Luca, A.; Galosi, C.; Pariano, M.; Massi-Benedetti, C.; Borghi, M.; Puccetti, M.; Lucidi, V.; et al. IL-1 receptor antagonist ameliorates inflammasome-dependent inflammation in murine and human cystic fibrosis. Nat. Commun. 2016, 7, 10791. [Google Scholar] [CrossRef]
- Mayer, M.L.; Blohmke, C.J.; Falsafi, R.; Fjell, C.D.; Madera, L.; Turvey, S.E.; Hancock, R.E.W. Rescue of Dysfunctional Autophagy Attenuates Hyperinflammatory Responses from Cystic Fibrosis Cells. J. Immunol. 2013, 190, 1227–1238. [Google Scholar] [CrossRef]
- Villella, V.R.; Esposito, S.; Ferrari, E.; Monzani, R.; Tosco, A.; Rossin, F.; Castaldo, A.; Silano, M.; Marseglia, G.L.; Romani, L.; et al. Autophagy suppresses the pathogenic immune response to dietary antigens in cystic fibrosis. Cell Death Dis. 2019, 10, 258. [Google Scholar] [CrossRef] [PubMed]
- Caution, K.; Pan, A.; Krause, K.; Badr, A.; Hamilton, K.; Vaidya, A.; Gosu, H.; Daily, K.; Estfanous, S.; Gavrilin, M.A.; et al. Methylomic correlates of autophagy activity in cystic fibrosis. J. Cyst. Fibros. 2019, 18, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Tazi, M.F.; Dakhlallah, D.A.; Caution, K.; Gerber, M.M.; Chang, S.-W.; Khalil, H.; Kopp, B.T.; Ahmed, A.E.; Krause, K.; Davis, I.; et al. Elevated Mirc1/Mir17-92 cluster expression negatively regulates autophagy and CFTR (cystic fibrosis transmembrane conductance regulator) function in CF macrophages. Autophagy 2016, 12, 2026–2037. [Google Scholar] [CrossRef]
- Abdulrahman, B.A.; Khweek, A.A.; Akhter, A.; Caution, K.; Kotrange, S.; Abdelaziz, D.H.A.; Newland, C.; Rosales-Reyes, R.; Kopp, B.; McCoy, K.; et al. Autophagy stimulation by rapamycin suppresses lung inflammation and infection by Burkholderia cenocepacia in a model of cystic fibrosis. Autophagy 2011, 7, 1359–1370. [Google Scholar] [CrossRef]
- Abdulrahman, B.A.; Khweek, A.A.; Akhter, A.; Caution, K.; Tazi, M.; Hassan, H.; Zhang, Y.; Rowland, P.D.; Malhotra, S.; Aeffner, F.; et al. Depletion of the ubiquitin-binding adaptor molecule SQSTM1/p62 from macrophages harboring cftr ΔF508 mutation improves the delivery of Burkholderia cenocepacia to the autophagic machinery. J. Biol. Chem. 2013, 288, 2049–2058. [Google Scholar] [CrossRef] [PubMed]
- Junkins, R.D.; Shen, A.; Rosen, K.; McCormick, C.; Lin, T.-J. Autophagy Enhances Bacterial Clearance during P. aeruginosa Lung Infection. PLoS ONE 2013, 8, e72263. [Google Scholar] [CrossRef] [PubMed]
- Renna, M.; Schaffner, C.; Brown, K.; Shang, S.; Tamayo, M.H.; Hegyi, K.; Grimsey, N.J.; Cusens, D.; Coulter, S.; Cooper, J.; et al. Azithromycin blocks autophagy and may predispose cystic fibrosis patients to mycobacterial infection. J. Clin. Investig. 2011, 121, 3554–3563. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, D.; Villella, V.R.; Esposito, S.; Tosco, A.; Sepe, A.; De Gregorio, F.; Salvadori, L.; Grassia, R.; Leone, C.A.; De Rosa, G.; et al. Restoration of CFTR function in patients with cystic fibrosis carrying the F508del-CFTR mutation. Autophagy 2014, 10, 2053–2074. [Google Scholar] [CrossRef]
- Tosco, A.; De Gregorio, F.; Esposito, S.; De Stefano, D.; Sana, I.; Ferrari, E.; Sepe, A.; Salvadori, L.; Buonpensiero, P.; Di Pasqua, A.; et al. A novel treatment of cystic fibrosis acting on-target: Cysteamine plus epigallocatechin gallate for the autophagy-dependent rescue of class II-mutated CFTR. Cell Death Differ. 2016, 23, 1380–1393. [Google Scholar] [CrossRef]
- Zhang, S.; Stoll, G.; Pedro, J.M.B.S.; Sica, V.; Sauvat, A.; Obrist, F.; Kepp, O.; Li, Y.; Maiuri, L.; Zamzami, N.; et al. Evaluation of autophagy inducers in epithelial cells carrying the ΔF508 mutation of the cystic fibrosis transmembrane conductance regulator CFTR. Cell Death Dis. 2018, 9, 191. [Google Scholar] [CrossRef]
- Bodas, M.; Vij, N. Adapting Proteostasis and Autophagy for Controlling the Pathogenesis of Cystic Fibrosis Lung Disease. Front. Pharmacol. 2019, 10, 20. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Singh, S.S.; Vats, S.; Chia, A.Y.-Q.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef]
- Mattoscio, D.; Casadio, C.; Miccolo, C.; Maffini, F.; Raimondi, A.; Tacchetti, C.; Gheit, T.; Tagliabue, M.; Galimberti, V.E.; De Lorenzi, F.; et al. Autophagy regulates UBC9 levels during viral-mediated tumorigenesis. PLoS Pathog. 2017, 13, e1006262. [Google Scholar] [CrossRef] [PubMed]
- Mattoscio, D.; Medda, A.; Chiocca, S. Human Papilloma Virus and Autophagy. Int. J. Mol. Sci. 2018, 19, 1775. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.S.; Sansanaphongpricha, K.; Xie, Y.; Donnelly, C.R.; Luo, X.; Heath, B.R.; Zhao, X.; Bellile, E.; Hu, H.; Chen, H.; et al. Mitigating SOX2-potentiated Immune Escape of Head and Neck Squamous Cell Carcinoma with a STING-inducing Nanosatellite Vaccine. Clin. Cancer Res. 2018, 24, 4242–4255. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Donnelly, C.R.; Gong, W.; Heath, B.R.; Hao, Y.; Donnelly, L.A.; Moghbeli, T.; Tan, Y.S.; Lin, X.; Bellile, E.; et al. HPV16 drives cancer immune escape via NLRX1-mediated degradation of STING. J. Clin. Investig. 2020, 130, 1635–1652. [Google Scholar] [CrossRef]
- Yang, S.; Imamura, Y.; Jenkins, R.W.; Canadas, I.; Kitajima, S.; Aref, A.; Brannon, A.; Oki, E.; Castoreno, A.; Zhu, Z.; et al. Autophagy Inhibition Dysregulates TBK1 Signaling and Promotes Pancreatic Inflammation. Cancer Immunol. Res. 2016, 4, 520–530. [Google Scholar] [CrossRef]
- Mathew, R.; Khor, S.; Hackett, S.R.; Rabinowitz, J.D.; Perlman, D.H.; White, E. Functional Role of Autophagy-Mediated Proteome Remodeling in Cell Survival Signaling and Innate Immunity. Mol. Cell 2014, 55, 916–930. [Google Scholar] [CrossRef]
- Rao, S.; Tortola, L.; Perlot, T.; Wirnsberger, G.; Novatchkova, M.; Nitsch, R.; Sykacek, P.; Frank, L.; Schramek, D.; Komnenovic, V.; et al. A dual role for autophagy in a murine model of lung cancer. Nat. Commun. 2014, 5, 3056. [Google Scholar] [CrossRef]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-Dependent Anticancer Immune Responses Induced by Chemotherapeutic Agents in Mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef]
- Jin, S.-M.; Jang, H.W.; Sohn, S.Y.; Kim, N.K.; Joung, J.Y.; Cho, Y.Y.; Kim, S.W.; Chung, J.H. Role of autophagy in the resistance to tumour necrosis factor-related apoptosis-inducing ligand-induced apoptosis in papillary and anaplastic thyroid cancer cells. Endocrine 2014, 45, 256–262. [Google Scholar] [CrossRef]
- Isopi, E.; Granzotto, A.; Corona, C.; Bomba, M.; Ciavardelli, D.; Curcio, M.; Canzoniero, L.M.T.; Navarra, R.; Lattanzio, R.; Piantelli, M.; et al. Pyruvate prevents the development of age-dependent cognitive deficits in a mouse model of Alzheimer’s disease without reducing amyloid and tau pathology. Neurobiol. Dis. 2015, 81, 214–224. [Google Scholar] [CrossRef]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the Autophagy-Inflammation-Cell Death Axis in Organismal Aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Bostancıklıoğlu, M. An update on the interactions between Alzheimer’s disease, autophagy and inflammation. Gene 2019, 705, 157–166. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Prieto, P.; Rosales-Mendoza, C.E.; Terrón, V.; Toledano, V.; Cuadrado, A.; López-Collazo, E.; Bannenberg, G.; Martín-Sanz, P.; Fernández-Velasco, M.; Boscá, L. Activation of autophagy in macrophages by pro-resolving lipid mediators. Autophagy 2015, 11, 1729–1744. [Google Scholar] [CrossRef]
- Liu, H.; Zhou, K.; Liao, L.; Zhang, T.; Yang, M.; Sun, C. Lipoxin A4 receptor agonist BML-111 induces autophagy in alveolar macrophages and protects from acute lung injury by activating MAPK signaling. Respir. Res. 2018, 19, 243. [Google Scholar] [CrossRef]
- Xiao, J.; Feng, X.; Huang, X.-Y.; Huang, Z.; Huang, Y.; Li, C.; Li, G.; Nong, S.; Wu, R.; Huang, Y.; et al. Spautin-1 Ameliorates Acute Pancreatitis via inhibiting impaired Autophagy and Alleviating Calcium Overload. Mol. Med. 2016, 22, 643–652. [Google Scholar] [CrossRef]
- Wang, B.; Hu, C.; Mei, Y.; Bao, J.; Ding, S.; Liu, X.; Mei, Q.; Xu, J. Resolvin D1 Resolve Inflammation in Experimental Acute Pancreatitis by Restoring Autophagic Flux. Dig. Dis. Sci. 2018, 63, 3359–3366. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, M.; Ding, W.; Zhao, M.; Ye, J.; Xu, Y.; Wang, Z.; Ye, D.; Li, D.; Liu, J.; et al. Resolvin E1 protects against doxorubicin-induced cardiotoxicity by inhibiting oxidative stress, autophagy and apoptosis by targeting AKT/mTOR signaling. Biochem. Pharmacol. 2020, 180, 114188. [Google Scholar] [CrossRef]
- Börgeson, E.; Johnson, A.M.F.; Lee, Y.S.; Till, A.; Syed, G.H.; Ali-Shah, S.T.; Guiry, P.J.; Dalli, J.; Colas, R.A.; Serhan, C.N.; et al. Lipoxin A4 Attenuates Obesity-Induced Adipose Inflammation and Associated Liver and Kidney Disease. Cell Metab. 2015, 22, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Stienstra, R.; Haim, Y.; Riahi, Y.; Netea, M.; Rudich, A.; Leibowitz, G. Autophagy in adipose tissue and the beta cell: Implications for obesity and diabetes. Diabetologia 2014, 57, 1505–1516. [Google Scholar] [CrossRef] [PubMed]
- Laiglesia, L.M.; Lorente-Cebrián, S.; López-Yoldi, M.; Lanas, R.; Sáinz, N.; Martínez, J.A.; Moreno-Aliaga, M.J. Maresin 1 inhibits TNF-alpha-induced lipolysis and autophagy in 3T3-L1 adipocytes. J. Cell. Physiol. 2018, 233, 2238–2246. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Li, Y.; Liu, W. Maresin 1 regulates autophagy and inflammation in human periodontal ligament cells through glycogen synthase kinase–3β/β-catenin pathway under inflammatory conditions. Arch. Oral Biol. 2018, 87, 242–247. [Google Scholar] [CrossRef]
- Yin, P.; Wang, X.; Wang, S.; Wei, Y.; Feng, J.; Zhu, M. Maresin 1 Improves Cognitive Decline and Ameliorates Inflammation in a Mouse Model of Alzheimer’s Disease. Front. Cell. Neurosci. 2019, 13, 466. [Google Scholar] [CrossRef]
- Yazıcı, D.; Sezer, H. Insulin Resistance, Obesity and Lipotoxicity. In Obesity and Lipotoxicity; Engin, A.B., Engin, A., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2017; Volume 960, pp. 277–304. ISBN 978-3-319-48380-1. [Google Scholar]
- López-Vicario, C.; Alcaraz-Quiles, J.; García-Alonso, V.; Rius, B.; Hwang, S.H.; Titos, E.; Lopategi, A.; Hammock, B.D.; Arroyo, V.; Clària, J. Inhibition of soluble epoxide hydrolase modulates inflammation and autophagy in obese adipose tissue and liver: Role for omega-3 epoxides. Proc. Natl. Acad. Sci. USA 2015, 112, 536–541. [Google Scholar] [CrossRef]
- Pereira, S.S.; Alvarez-Leite, J.I. Low-Grade Inflammation, Obesity, and Diabetes. Curr. Obes. Rep. 2014, 3, 422–431. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SPMs | Disease Model | Molecular Effect | Effect on Autophagy | Refs |
|---|---|---|---|---|
| RvD1 and AT-LXA4 | MΦs inflammation | BECN1 dissociation and activation MAPK dependent, autolysosome formation | Activation | [168] |
| BML-111 | Acute lung injury, impaired autophagy | LC3-II accumulation, p62 degradation MAPK dependent | Activation | [169] |
| RvD1 | Acute pancreatitis, impaired autophagy | Reduction in BECN1, p62, LC3-II | Reactivation of impaired autophagic flux | [171] |
| RvE1 | Cardiotoxicity, impaired autophagy | Reduction in BECN1, p62, LC3-II | Reactivation of impaired autophagic flux | [172] |
| LXA4 | Obesity, increased autophagy | LC3-II decrease, p62 increase | Inhibition | [173] |
| Mar1 | Obesity, increased autophagy | LC3-II decrease, p62 increase | Inhibition | [175] |
| 17(S)-HDHA | Obesity, increased autophagy | LC3-II and p62 accumulation in autophagic flux experiments | Inhibition | [179] |
| Mar1 | Periodontitis | Increase in LC3-II and BECN1, decrease in p62 | Activation | [176] |
| Mar1 | Alzheimer, impaired autophagy | Increase in LC3-II and BECN1, decrease in p62 | Activation | [177] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Recchiuti, A.; Isopi, E.; Romano, M.; Mattoscio, D. Roles of Specialized Pro-Resolving Lipid Mediators in Autophagy and Inflammation. Int. J. Mol. Sci. 2020, 21, 6637. https://doi.org/10.3390/ijms21186637
Recchiuti A, Isopi E, Romano M, Mattoscio D. Roles of Specialized Pro-Resolving Lipid Mediators in Autophagy and Inflammation. International Journal of Molecular Sciences. 2020; 21(18):6637. https://doi.org/10.3390/ijms21186637
Chicago/Turabian StyleRecchiuti, Antonio, Elisa Isopi, Mario Romano, and Domenico Mattoscio. 2020. "Roles of Specialized Pro-Resolving Lipid Mediators in Autophagy and Inflammation" International Journal of Molecular Sciences 21, no. 18: 6637. https://doi.org/10.3390/ijms21186637
APA StyleRecchiuti, A., Isopi, E., Romano, M., & Mattoscio, D. (2020). Roles of Specialized Pro-Resolving Lipid Mediators in Autophagy and Inflammation. International Journal of Molecular Sciences, 21(18), 6637. https://doi.org/10.3390/ijms21186637