mRNA Vaccine Era—Mechanisms, Drug Platform and Clinical Prospection

Abstract
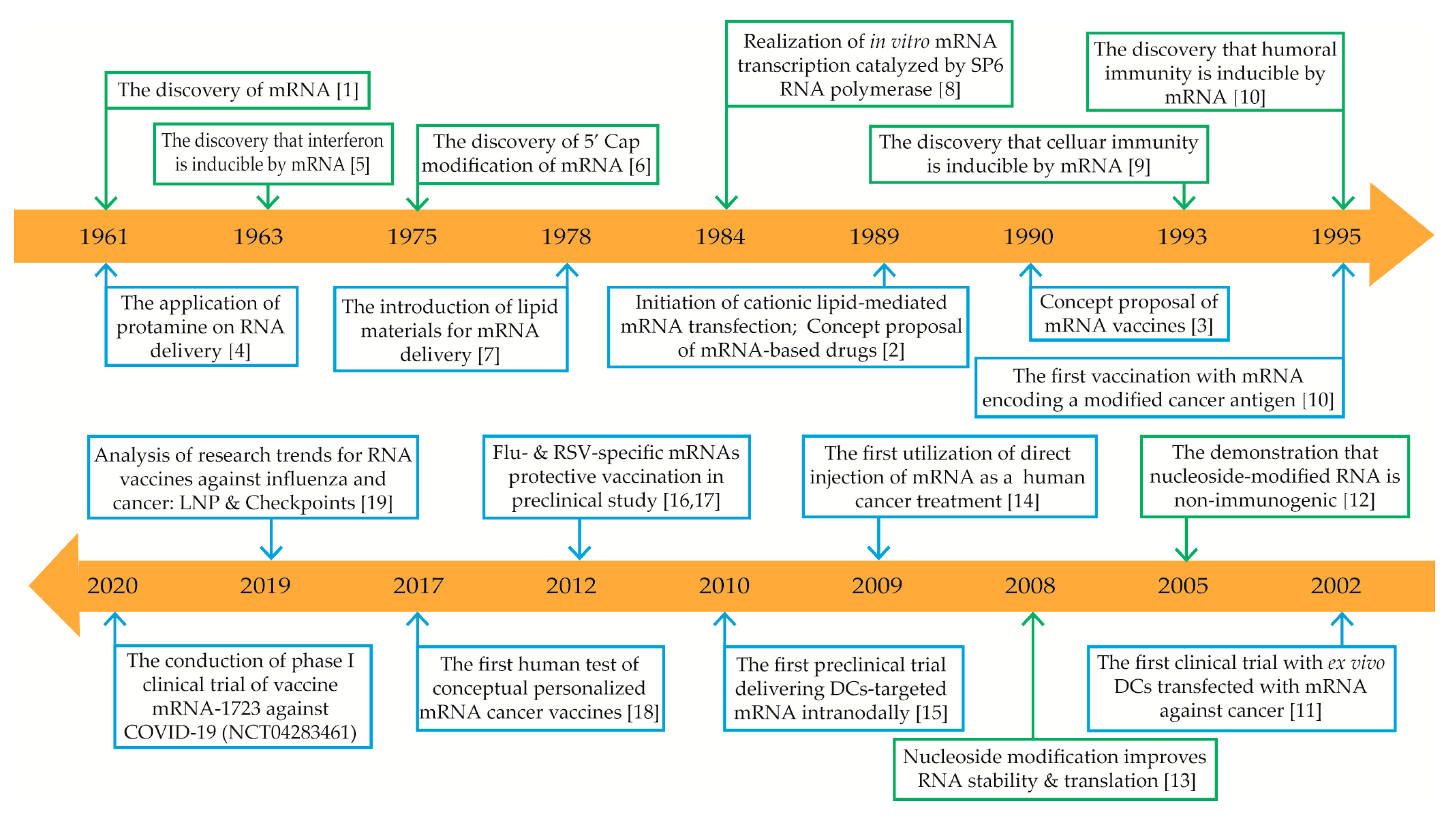
1. Introduction
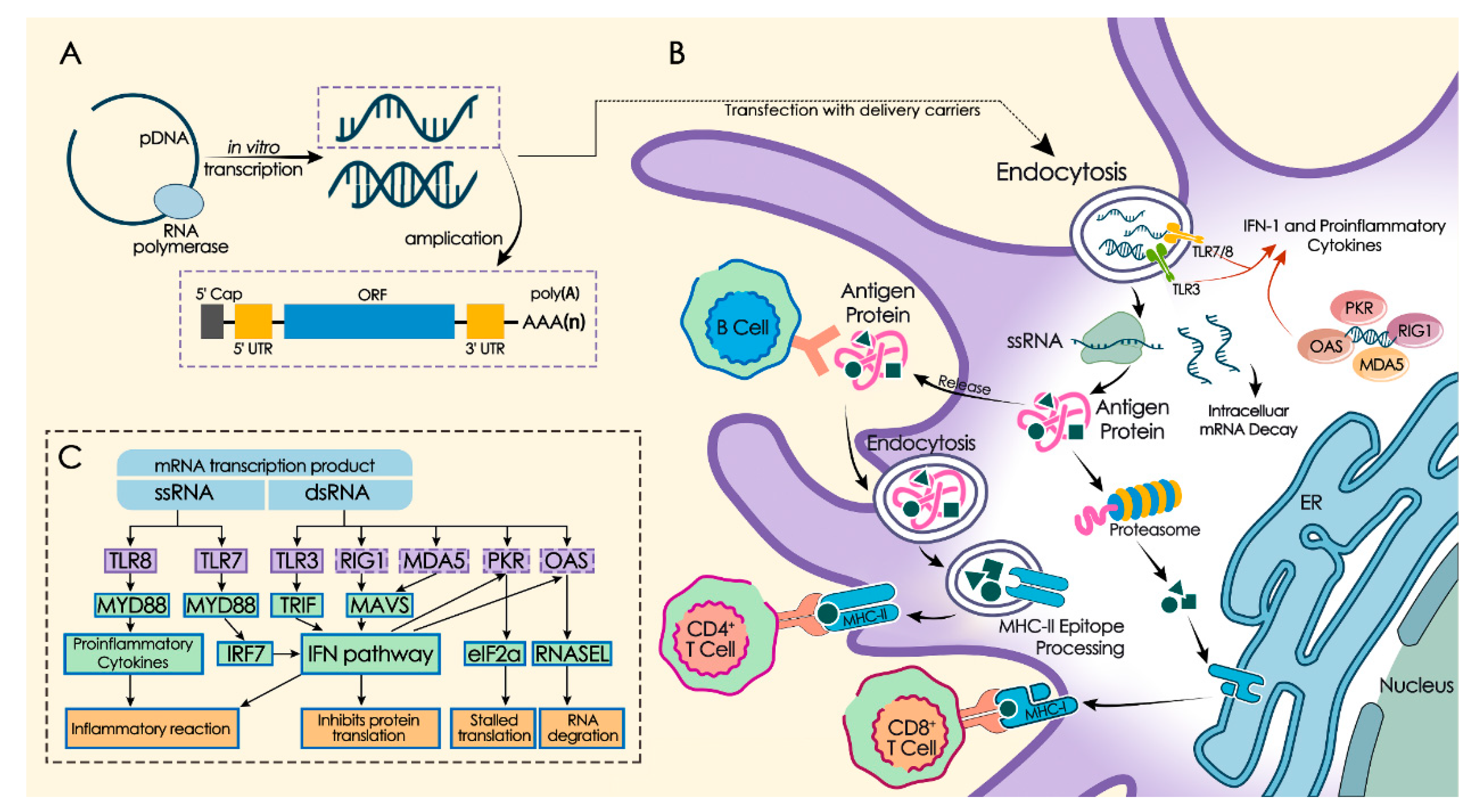
2. mRNA In Vitro Synthesis and Modification
2.1. Five-Prime Cap (5′ cap) and Modification
2.2. Optimization of Untranslated Regions (UTRs)
2.3. Codon Optimization of Open Reading Frame (ORF)
2.4. Poly (A) Tail and mRNA Stability
3. Immunogenic Regulation of mRNA
3.1. mRNA Purification Modulates Self-Adjuvanting Property
3.2. Optimization of mRNA Sequence to Regulate Self-Adjuvanting Property
3.3. Adding Adjuvants to Optimize mRNA Immunogenicity
4. mRNA Delivery System
4.1. Naked mRNA Delivery System
4.1.1. Direct Injection of Naked mRNA
4.1.2. Physical Delivery of Naked mRNA
4.2. Ex Vivo Loading of DCs Delivery System
4.3. Protamine-Formulated Delivery System
4.4. Cationic Lipid-Based Delivery System
4.5. Polymer-Based Delivery System
5. Applications of mRNA as a Drug Platform
5.1. mRNA Vaccines Against Infectious Diseases
5.1.1. Influenza Virus
5.1.2. HIV
5.1.3. Coronavirus
5.1.4. Other Viral Pathogens
5.1.5. Bacterial Pathogens
5.2. mRNA Cancer Vaccines
6. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| mRNA | messenger ribonucleic acid |
| DOTMA | N-[1-(2,3-dioleyloxy) propyl]-N,N,N-trimethylammonium chloride |
| TNF-α | tumor necrosis factor-α |
| IFN-α | interferon-α |
| COVID-19 | coronavirus disease 2019 |
| 5′ cap | five-prime cap |
| 5′ UTR | five-prime untranslated region |
| ORF | open reading frame |
| 3′ UTR | three-prime untranslated region |
| eIF | eukaryotic translation initiation factor |
| ARCA | anti-reverse cap analogs |
| DCs | dendritic cells |
| PABP | Poly (A) binding protein |
| APCs | antigen-presenting cells |
| PRRs | pattern recognition receptors |
| TLR | Toll-like receptor |
| dsRNA | double-stranded RNA |
| RIG-I | Retinoic-acid-inducible gene I |
| RLRs | RIG-I-like receptors |
| MDA5 | melanoma differentiation-associated 5 |
| IFN | type I interferon |
| CD | cluster of differentiation |
| ssRNA | single-stranded RNA |
| PAMP | pathogen-associated molecular pattern |
| ISGs | IFN-stimulated genes |
| IFIT | IFN-inducible protein with tetratricoid repeats |
| OAS | 2′-5′-oligoadenylate synthetase |
| CNE | cationic nanoemulsion |
| AIDS | acquired immune deficiency syndrome |
| HIV | human immunodeficiency virus |
| DOTAP | 1,2-dioleoyl-3trimethylammonium-propane |
| DOPE | 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine |
| Th1 | T-help 1 cell |
| NSCLC | Non-small cell lung cancer |
| ZIKV | Zika virus |
| LNP | lipid nanoparticles |
| PEG | polyethylene glycol |
| DC-Cholesterol | 3β-[N-(N’,N’-dimethylaminoethane) carbamoyl] |
| PBAE | poly(β-amino ester) |
| PSA | polyethyleneimine-stearic acid |
| PEI | polyethylenimine |
| DEAE | diethylaminoethyl |
| hPBAEs | hyperbranched poly(beta amino esters) |
| PEG[Glu(DET)]2 | N-substituted polyethylene glycol-diblock-polyglutamide |
| PLGA | poly(lactic-co-glycolic acid) |
| CLAN | cationic lipid-assisted nanoparticles |
| BHEM-cholesterol | N-bis(2-hydroxyethyl)-N-methyl-N-(2-cholesteryloxycarbonyl aminoethyl) ammonium bromide |
| MHC | major histocompatibility complex |
| CTLs | cytotoxic T-lymphocytes |
| siRNA | short interfering RNA |
| SAM | self-amplifying mRNA |
| gp100 | glycoprotein 100 |
| HVJ | hemagglutinating virus of Japan |
| TB | tuberculosis |
| CMV | cytomegalovirus |
| gB | herpesvirus glycoprotein |
| HA | hemagglutinin |
| hMPV | human metapneumovirus |
| PIV3 | parainfluenza virus 3 |
| RABV-G | rabies virus glycoprotein |
| SARS-CoV | severe acute respiratory syndrome coronavirus |
| MERS-CoV | Middle East respiratory syndrome coronavirus |
| S protein | spike protein |
| prM | pre-membrane |
| E | envelope |
| VEEV | Venezuelan equine encephalitis virus |
| GAS | Group A Streptococci |
| BCG | Mycobacterium bovis Bacillus Calmette–Guérin |
| WT1 | Wilms’ Tumor-1 |
| hTERT | human telomerase reverse transcriptase |
| LAMP | lysosome-associated membrane protein |
| CEA | carcinoembryonic antigens |
| MUC1 | tumor marker expressed by MUC1 gene |
| Survivin | one of the apoptosis inhibitory protein family |
| pp65 | 65K phosphoprotein |
| MiHA | minor histocompatibility antigens |
| Melan-A | melanoma antigen recognized by T cells |
| TRP2 | tyrosinase-related protein 2 |
| SOCS 1 | cytokine signaling 1 |
| NY-ESO-1 | New York esophageal squamous cell carcinoma 1 |
| PAP | prostatic acid phosphatase |
| AML | acute myelocytic leukemia |
| GBM | glioblastoma |
| CAR | chimeric antigen receptor |
| CLDN6 | claudin 6 |
| PIPAC | pressurized intraperitoneal aerosol chemotherapy |
| FCS | fluorescence correlation spectroscopy |
References
- Brenner, S.; Jacob, F.; Meselson, M. An Unstable Intermediate Carrying Information from Genes to Ribosomes for Protein Synthesis. Nature 1961, 190, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Malone, R.W.; Felgner, P.L.; Verma, I.M. Cationic liposome-mediated RNA transfection. Proc. Natl. Acad. Sci. USA 1989, 86, 6077–6081. [Google Scholar] [CrossRef] [PubMed]
- Wolff, J.; Malone, R.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- Smull, C.E.; Mallette, M.; Ludwig, E. The use of basic proteins to increase the infectivity of enterovirus ribonucleic acid. Biochem. Biophys. Res. Commun. 1961, 5, 247–249. [Google Scholar] [CrossRef]
- Isaacs, A.; Cox, R.; Rotem, Z. Foreign Nucleic Acids as the Stimulus to Make Interferon. Lancet 1963, 282, 113–116. [Google Scholar] [CrossRef]
- Furuichi, Y.; Miura, K.-I. A blocked structure at the 5′ terminus of mRNA from cytoplasmic polyhedrosis virus. Nature 1975, 253, 374–375. [Google Scholar] [CrossRef]
- Dimitriadis, G.J. Translation of rabbit globin mRNA introduced by liposomes into mouse lymphocytes. Nature 1978, 274, 923–924. [Google Scholar] [CrossRef] [PubMed]
- Krieg, P.; Melton, D. Functional messenger RNAs are produced by SP6in vitrotranscription of cloned cDNAs. Nucleic Acids Res. 1984, 12, 7057–7070. [Google Scholar] [CrossRef]
- Martinon, F.; Krishnan, S.; Lenzen, G.; Magné, R.; Gomard, E.; Guillet, J.-G.; Levy, J.-P.; Meulien, P. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur. J. Immunol. 1993, 23, 1719–1722. [Google Scholar] [CrossRef]
- Conry, R.M.; LoBuglio, A.F.; Wright, M.; Sumerel, L.; Pike, M.J.; Johanning, F.; Benjamin, R.; Lu, D.; Curiel, D.T. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res. 1995, 55, 1397–1400. [Google Scholar]
- Heiser, A.; Coleman, D.; Dannull, J.; Yancey, D.; Maurice, M.A.; Lallas, C.D.; Dahm, P.; Niedzwiecki, D.; Gilboa, E.; Vieweg, J. Autologous dendritic cells transfected with prostate-specific antigen RNA stimulate CTL responses against metastatic prostate tumors. J. Clin. Investig. 2002, 109, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, E. Suppression of RNA Recognition by Toll-like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of Pseudouridine Into mRNA Yields Superior Nonimmunogenic Vector With Increased Translational Capacity and Biological Stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef] [PubMed]
- Weide, B.; Pascolo, S.; Scheel, B.; Derhovanessian, E.; Pflugfelder, A.; Eigentler, T.K.; Pawelec, G.; Hoerr, I.; Rammensee, H.-G.; Garbe, C. Direct Injection of Protamine-protected mRNA: Results of a Phase 1/2 Vaccination Trial in Metastatic Melanoma Patients. J. Immunother. 2009, 32, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, S.; Selmi, A.; Diken, M.; Koslowski, M.; Britten, C.M.; Huber, C.; Türeci, Ö; Şahin, U. Intranodal Vaccination with Naked Antigen-Encoding RNA Elicits Potent Prophylactic and Therapeutic Antitumoral Immunity. Cancer Res. 2010, 70, 9031–9040. [Google Scholar] [CrossRef]
- Petsch, B.; Schnee, M.; Vogel, A.B.; Lange, E.; Hoffmann, B.; Voss, D.; Schlake, T.; Thess, A.; Kallen, K.-J.; Stitz, L.; et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat. Biotechnol. 2012, 30, 1210–1216. [Google Scholar] [CrossRef]
- Geall, A.J.; Verma, A.; Otten, G.R.; Shaw, C.A.; Hekele, A.; Banerjee, K.; Cu, Y.; Beard, C.W.; Brito, L.A.; Krucker, T.; et al. Nonviral delivery of self-amplifying RNA vaccines. Proc. Natl. Acad. Sci. USA 2012, 109, 14604–14609. [Google Scholar] [CrossRef]
- Şahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Verbeke, R.; Lentacker, I.; De Smedt, S.C.; Dewitte, H. Three decades of messenger RNA vaccine development. Nano Today 2019, 28, 100766. [Google Scholar] [CrossRef]
- Schlake, T.; Thess, A.; Fotin-Mleczek, M.; Kallen, K.-J. Developing mRNA-vaccine technologies. RNA Biol. 2012, 9, 1319–1330. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Linares-Fernández, S.; Lacroix, C.; Exposito, J.-Y.; Verrier, B. Tailoring mRNA Vaccine to Balance Innate/Adaptive Immune Response. Trends Mol. Med. 2020, 26, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, S.; Renmans, D.; Broos, K.; Dewitte, H.; Lentacker, I.; Heirman, C.; Breckpot, K.; Thielemans, K. The ReNAissanCe of mRNA-based cancer therapy. Expert Rev. Vaccines 2014, 14, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Iavarone, C.; O’Hagan, D.T.; Yu, N.; Delahaye, N.F.; Ulmer, J.B. Mechanism of action of mRNA-based vaccines. Expert Rev. Vaccines 2017, 16, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Şahin, U.; Kariko, K.; Türeci, Ö. mRNA-based therapeutics—Developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S.; et al. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Central Sci. 2020, 6, 315–331. [Google Scholar] [CrossRef]
- Xia, J.; Lu, G.; Lu, J.; Zhang, J.; Feng, L.; Wang, B.; Yu, H.; Xu, Y.; Lin, J. Towards an effective mRNA vaccine against 2019-nCoV: Demonstration of virus-like particles expressed from a modified mRNA cocktail. Chinaxiv 2020. [Google Scholar] [CrossRef]
- Pardi, N.; Muramatsu, H.; Weissman, D.; Karikó, K. In Vitro Transcription of Long RNA Containing Modified Nucleosides. In Synthetic Messenger RNA and Cell Metabolism Modulation: Methods and Protocols; Rabinovich, P.M., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 29–42. [Google Scholar]
- Karikó, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef]
- Weissman, E. mRNA transcript therapy. Expert Rev. Vaccines 2014, 14, 265–281. [Google Scholar] [CrossRef]
- A Martin, S.; Paoletti, E.; Moss, B. Purification of mRNA guanylyltransferase and mRNA (guanine-7-) methyltransferase from vaccinia virions. J. Biol. Chem. 1975, 250, 9322–9329. [Google Scholar]
- Stepinski, J.; Waddell, C.; Stolarski, R.; Darzynkiewicz, E.; E Rhoads, R. Synthesis and properties of mRNAs containing the novel “anti-reverse” cap analogs 7-methyl(3′-O-methyl)GpppG and 7-methyl (3′-deoxy)GpppG. RNA 2001, 7, 1486–1495. [Google Scholar] [PubMed]
- Jemielity, J.; Fowler, T.; Zuberek, J.; Stepinski, J.; Lewdorowicz, M.; Niedźwiecka-Kornaś, A.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R.E. Novel “anti-reverse” cap analogs with superior translational properties. RNA 2003, 9, 1108–1122. [Google Scholar] [CrossRef] [PubMed]
- Grudzien-Nogalska, E.; Stepinski, J.; Jemielity, J.; Zuberek, J.; Stolarski, R.; Rhoads, R.E.; Darzynkiewicz, E. Synthesis of Anti-Reverse Cap Analogs (ARCAs) and their Applications in mRNA Translation and Stability. Methods Enzymol. 2007, 431, 203–227. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, A.N.; Diken, M.; Kreiter, S.; Selmi, A.; Kowalska, J.; Jemielity, J.; Darzynkiewicz, E.; Huber, C.; Türeci, Ö.; Şahin, U. Phosphorothioate cap analogs increase stability and translational efficiency of RNA vaccines in immature dendritic cells and induce superior immune responses in vivo. Gene Ther. 2010, 17, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Grudzien-Nogalska, E.; Jemielity, J.; Kowalska, J.; Darzynkiewicz, E.; Rhoads, R.E. Phosphorothioate cap analogs stabilize mRNA and increase translational efficiency in mammalian cells. RNA 2007, 13, 1745–1755. [Google Scholar] [CrossRef]
- Rydzik, A.; Kulis, M.; Lukaszewicz, M.; Kowalska, J.; Zuberek, J.; Darzynkiewicz, Z.; Darzynkiewicz, E.; Jemielity, J. Synthesis and properties of mRNA cap analogs containing imidodiphosphate moiety—Fairly mimicking natural cap structure, yet resistant to enzymatic hydrolysis. Bioorganic Med. Chem. 2012, 20, 1699–1710. [Google Scholar] [CrossRef]
- Strenkowska, M.; Grzela, R.; Majewski, M.; Wnek, K.; Kowalska, J.; Lukaszewicz, M.; Zuberek, J.; Darzynkiewicz, E.; Kuhn, A.N.; Şahin, U.; et al. Cap analogs modified with 1,2-dithiodiphosphate moiety protect mRNA from decapping and enhance its translational potential. Nucleic Acids Res. 2016, 44, 9578–9590. [Google Scholar] [CrossRef]
- Vaidyanathan, S.; Azizian, K.T.; Haque, A.A.; Henderson, J.M.; Hendel, A.; Shore, S.; Antony, J.S.; Hogrefe, R.I.; Kormann, M.S.; Porteus, M.H.; et al. Uridine Depletion and Chemical Modification Increase Cas9 mRNA Activity and Reduce Immunogenicity without HPLC Purification. Mol. Ther. Nucleic Acids 2018, 12, 530–542. [Google Scholar] [CrossRef]
- Murray, E.L.; Schoenberg, D.R. A+U-Rich Instability Elements Differentially Activate 5′-3′ and 3′-5′ mRNA Decay. Mol. Cell. Biol. 2007, 27, 2791–2799. [Google Scholar] [CrossRef]
- Louis, I.V.-S.; Bohjanen, P.R. Coordinate regulation of mRNA decay networks by GU-rich elements and CELF1. Curr. Opin. Genet. Dev. 2011, 21, 444–451. [Google Scholar] [CrossRef]
- Ferizi, M.; Leonhardt, C.; Meggle, C.; Aneja, M.K.; Rudolph, C.; Plank, C.; Rädler, J.O. Stability analysis of chemically modified mRNA using micropattern-based single-cell arrays. Lab Chip 2015, 15, 3561–3571. [Google Scholar] [CrossRef]
- Von Niessen, A.G.O.; Poleganov, M.A.; Rechner, C.; Plaschke, A.; Kranz, L.M.; Fesser, S.; Diken, M.; Löwer, M.; Vallazza, B.; Beissert, T.; et al. Improving mRNA-Based Therapeutic Gene Delivery by Expression-Augmenting 3′ UTRs Identified by Cellular Library Screening. Mol. Ther. 2019, 27, 824–836. [Google Scholar] [CrossRef] [PubMed]
- Gray, N.K.; Wickens, M. Control of Translation Initiation in Animals. Annu. Rev. Cell Dev. Biol. 1998, 14, 399–458. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. At least six nucleotides preceding the AUG initiator codon enhance translation in mammalian cells. J. Mol. Biol. 1987, 196, 947–950. [Google Scholar] [CrossRef]
- Pelletier, J.; Sonenberg, N. Insertion mutagenesis to increase secondary structure within the 5′ noncoding region of a eukaryotic mRNA reduces translational efficiency. Cell 1985, 40, 515–526. [Google Scholar] [CrossRef]
- Gustafsson, C.; Govindarajan, S.; Minshull, J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004, 22, 346–353. [Google Scholar] [CrossRef]
- Gallie, D.R. The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency. Genes Dev. 1991, 5, 2108–2116. [Google Scholar] [CrossRef]
- Lima, S.A.; Chipman, L.; Nicholson, A.; Chen, Y.-H.; Yee, B.A.; Yeo, G.W.; Coller, J.; Pasquinelli, A.E. Short poly(A) tails are a conserved feature of highly expressed genes. Nat. Struct. Mol. Biol. 2017, 24, 1057–1063. [Google Scholar] [CrossRef]
- Holtkamp, S.; Kreiter, S.; Selmi, A.; Simon, P.; Koslowski, M.; Huber, C.; Türeci, Ö.; Şahin, U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 2006, 108, 4009–4017. [Google Scholar] [CrossRef]
- Grier, A.; Burleigh, S.; Sahni, J.; Clough, C.; Cardot, V.; Choe, D.C.; Krutein, M.C.; Rawlings, D.J.; Jensen, M.C.; Scharenberg, A.M.; et al. pEVL: A Linear Plasmid for Generating mRNA IVT Templates With Extended Encoded Poly(A) Sequences. Mol. Ther. Nucleic Acids 2016, 5, e306. [Google Scholar] [CrossRef]
- Park, J.-E.; Yi, H.; Kim, Y.; Chang, H.; Kim, V.N. Regulation of Poly(A) Tail and Translation during the Somatic Cell Cycle. Mol. Cell 2016, 62, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef] [PubMed]
- Kallen, K.-J.; Heidenreich, R.; Schnee, M.; Petsch, B.; Schlake, T.; Thess, A.; Baumhof, P.; Scheel, B.; Koch, S.D.; Fotin-Mleczek, M. A novel, disruptive vaccination technology. Hum. Vaccines Immunother. 2013, 9, 2263–2276. [Google Scholar] [CrossRef] [PubMed]
- De Beuckelaer, A.; Grooten, J.; De Koker, S. Type I Interferons Modulate CD8 + T Cell Immunity to mRNA Vaccines. Trends Mol. Med. 2017, 23, 216–226. [Google Scholar] [CrossRef]
- Bourquin, C.; Schmidt, L.; Hornung, V.; Wurzenberger, C.; Anz, D.; Sandholzer, N.; Schreiber, S.; Voelkl, A.; Hartmann, G.; Endres, S. Immunostimulatory RNA oligonucleotides trigger an antigen-specific cytotoxic T-cell and IgG2a response. Blood 2006, 109, 2953–2960. [Google Scholar] [CrossRef]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-Specific Recognition of Single-Stranded RNA via Toll-like Receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef]
- De Beuckelaer, A.; Pollard, C.; Van Lint, S.; Roose, K.; Van Hoecke, L.; Naessens, T.; Udhayakumar, V.K.; Smet, M.; Sanders, N.; Lienenklaus, S.; et al. Type I Interferons Interfere with the Capacity of mRNA Lipoplex Vaccines to Elicit Cytolytic T Cell Responses. Mol. Ther. 2016, 24, 2012–2020. [Google Scholar] [CrossRef]
- Baiersdörfer, M.; Boros, G.; Muramatsu, H.; Mahiny, A.; Vlatkovic, I.; Sahin, U.; Karikó, K. A Facile Method for the Removal of dsRNA Contaminant from In vitro-Transcribed mRNA. Mol. Ther. Nucleic Acids 2019, 15, 26–35. [Google Scholar] [CrossRef]
- Kumar, P.; Sweeney, T.R.; Skabkin, M.A.; Skabkina, O.V.; Hellen, C.U.T.; Pestova, T.V. Inhibition of translation by IFIT family members is determined by their ability to interact selectively with the 5′-terminal regions of cap0-, cap1- and 5′ppp- mRNAs. Nucleic Acids Res. 2013, 42, 3228–3245. [Google Scholar] [CrossRef]
- Pollard, C.; Rejman, J.; De Haes, W.; Verrier, B.; Van Gulck, E.; Naessens, T.; De Smedt, S.C.; Bogaert, P.; Grooten, J.; Vanham, G.; et al. Type I IFN Counteracts the Induction of Antigen-Specific Immune Responses by Lipid-Based Delivery of mRNA Vaccines. Mol. Ther. 2012, 21, 251–259. [Google Scholar] [CrossRef]
- Hornung, V.; Ellegast, J.M.; Kim, S.; Brzózka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.-K.; Schlee, M.; et al. 5′-Triphosphate RNA Is the Ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.R.; Muramatsu, H.; Jha, B.K.; Silverman, R.H.; Weissman, D.; Karikó, K. Nucleoside modifications in RNA limit activation of 2′-5′-oligoadenylate synthetase and increase resistance to cleavage by RNase L. Nucleic Acids Res. 2011, 39, 9329–9338. [Google Scholar] [CrossRef] [PubMed]
- Kormann, M.S.; Hasenpusch, G.; Aneja, M.K.; Nica, G.; Flemmer, A.W.; Herber-Jonat, S.; Huppmann, M.; E Mays, L.; Illenyi, M.; Schams, A.; et al. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat. Biotechnol. 2011, 29, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Mays, L.E.; Ammon-Treiber, S.; Mothes, B.; Alkhaled, M.; Rottenberger, J.; Müller-Hermelink, E.S.; Grimm, M.; Mezger, M.; Beer-Hammer, S.; Von Stebut, E.; et al. Modified Foxp3 mRNA protects against asthma through an IL-10-dependent mechanism. J. Clin. Investig. 2013, 123, 1216–1228. [Google Scholar] [CrossRef]
- A Brito, L.; Chan, M.; A Shaw, C.; Hekele, A.; Carsillo, T.; Schaefer, M.; Archer, J.; Seubert, A.; Otten, G.R.; Beard, C.W.; et al. A Cationic Nanoemulsion for the Delivery of Next-generation RNA Vaccines. Mol. Ther. 2014, 22, 2118–2129. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, S.; Goyvaerts, C.; Maenhout, S.; Goethals, L.; Disy, A.; Benteyn, D.; Pen, J.; Bonehill, A.; Heirman, C.; Breckpot, K.; et al. Preclinical Evaluation of TriMix and Antigen mRNA-Based Antitumor Therapy. Cancer Res. 2012, 72, 1661–1671. [Google Scholar] [CrossRef]
- Bonehill, A.; Tuyaerts, S.; Van Nuffel, A.M.; Heirman, C.; Bos, T.J.; Fostier, K.; Neyns, B.; Thielemans, K. Enhancing the T-cell Stimulatory Capacity of Human Dendritic Cells by Co-electroporation With CD40L, CD70 and Constitutively Active TLR4 Encoding mRNA. Mol. Ther. 2008, 16, 1170–1180. [Google Scholar] [CrossRef]
- Leal, L.; Guardo, A.C.; Morón-López, S.; Salgado, M.; Mothe, B.; Heirman, C.; Pannus, P.; Vanham, G.; Ham, H.J.V.D.; Gruters, R.; et al. Phase I clinical trial of an intranodally administered mRNA-based therapeutic vaccine against HIV-1 infection. AIDS 2018, 32, 2533–2545. [Google Scholar] [CrossRef]
- Lonez, C.; Vandenbranden, M.; Ruysschaert, J.-M. Cationic lipids activate intracellular signaling pathways. Adv. Drug Deliv. Rev. 2012, 64, 1749–1758. [Google Scholar] [CrossRef]
- Scheel, B.; Teufel, R.; Probst, J.; Carralot, J.-P.; Geginat, J.; Radsak, M.; Jarrossay, D.; Wagner, H.; Rammensee, H.-G.; Hoerr, I.; et al. Toll-like receptor-dependent activation of several human blood cell types by protamine-condensed mRNA. Eur. J. Immunol. 2005, 35, 1557–1566. [Google Scholar] [CrossRef]
- Kowalczyk, A.; Doener, F.; Zanzinger, K.; Noth, J.; Baumhof, P.; Fotin-Mleczek, M.; Heidenreich, R. Self-adjuvanted mRNA vaccines induce local innate immune responses that lead to a potent and boostable adaptive immunity. Vaccine 2016, 34, 3882–3893. [Google Scholar] [CrossRef]
- Weng, Y.; Li, C.; Yang, T.; Hu, B.; Zhang, M.; Guo, S.; Xiao, H.; Liang, X.-J.; Huang, Y. The challenge and prospect of mRNA therapeutics landscape. Biotechnol. Adv. 2020, 40, 107534. [Google Scholar] [CrossRef]
- Wadhwa, A.; Aljabbari, A.; Lokras, A.; Foged, C.; Thakur, A. Opportunities and Challenges in the Delivery of mRNA-Based Vaccines. Pharmaceutics 2020, 12, 102. [Google Scholar] [CrossRef]
- Probst, J.; Weide, B.; Scheel, B.; Pichler, B.J.; Hoerr, I.; Rammensee, H.-G.; Pascolo, S. Spontaneous cellular uptake of exogenous messenger RNA in vivo is nucleic acid-specific, saturable and ion dependent. Gene Ther. 2007, 14, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Selmi, A.; Vascotto, F.; Kautz-Neu, K.; Türeci, Ö.; Şahin, U.; Von Stebut, E.; Diken, M.; Kreiter, S. Uptake of synthetic naked RNA by skin-resident dendritic cells via macropinocytosis allows antigen expression and induction of T-cell responses in mice. Cancer Immunol. Immunother. 2016, 65, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Weide, B.; Carralot, J.-P.; Reese, A.; Scheel, B.; Eigentler, T.K.; Hoerr, I.; Rammensee, H.-G.; Garbe, C.; Pascolo, S. Results of the First Phase I/II Clinical Vaccination Trial With Direct Injection of mRNA. J. Immunother. 2008, 31, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Phua, K.K.L.; Leong, K.W.; Nair, S.K. Transfection efficiency and transgene expression kinetics of mRNA delivered in naked and nanoparticle format. J. Control. Release 2013, 166, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Bialkowski, L.; Van Weijnen, A.; Van Der Jeught, K.; Renmans, D.; Daszkiewicz, L.; Heirman, C.; Stangé, G.; Breckpot, K.; Aerts, J.L.; Thielemans, K. Intralymphatic mRNA vaccine induces CD8 T-cell responses that inhibit the growth of mucosally located tumours. Sci. Rep. 2016, 6, 22509. [Google Scholar] [CrossRef] [PubMed]
- Johansson, D.X.; Ljungberg, K.; Kakoulidou, M.; Liljeström, P. Intradermal Electroporation of Naked Replicon RNA Elicits Strong Immune Responses. PLoS ONE 2012, 7, e29732. [Google Scholar] [CrossRef]
- McLenachan, S.; Zhang, D.; Palomo, A.B.A.; Edel, M.J.; Chen, F.K. mRNA Transfection of Mouse and Human Neural Stem Cell Cultures. PLoS ONE 2013, 8, e83596. [Google Scholar] [CrossRef]
- Steitz, J.; Britten, C.M.; Wolfel, T.; Tüting, T. Effective induction of anti-melanoma immunity following genetic vaccination with synthetic mRNA coding for the fusion protein EGFP.TRP2. Cancer Immunol. Immunother. 2005, 55, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Peking, P.; Koller, U.; Hainzl, S.; Kitzmueller, S.; Kocher, T.; Mayr, E.; Nyström, A.; Lener, T.; Reichelt, J.; Bauer, J.W.; et al. A Gene Gun-mediated Nonviral RNA trans-splicing Strategy for Col7a1 Repair. Mol. Ther. Nucleic Acids 2016, 5, e287. [Google Scholar] [CrossRef]
- Dewitte, H.; Van Lint, S.; Heirman, C.; Thielemans, K.; De Smedt, S.C.; Breckpot, K.; Lentacker, I. The potential of antigen and TriMix sonoporation using mRNA-loaded microbubbles for ultrasound-triggered cancer immunotherapy. J. Control. Release 2014, 194, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Koh, K.J.; Liu, Y.; Lim, S.H.; Loh, X.J.; Kang, L.; Lim, C.Y.; Phua, K.K.L. Formulation, characterization and evaluation of mRNA-loaded dissolvable polymeric microneedles (RNApatch). Sci. Rep. 2018, 8, 11842. [Google Scholar] [CrossRef] [PubMed]
- Golombek, S.; Pilz, M.; Steinle, H.; Kochba, E.; Levin, Y.; Lunter, D.; Schlensak, C.; Wendel, H.P.; Avci-Adali, M. Intradermal Delivery of Synthetic mRNA Using Hollow Microneedles for Efficient and Rapid Production of Exogenous Proteins in Skin. Mol. Ther. Nucleic Acids 2018, 11, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Khoury, H.J.; Collins, R.H.; Blum, W.; Stiff, P.J.; Elias, L.; Lebkowski, J.S.; Reddy, A.; Nishimoto, K.P.; Sen, D.; Wirth, E.D.; et al. Immune responses and long-term disease recurrence status after telomerase-based dendritic cell immunotherapy in patients with acute myeloid leukemia. Cancer 2017, 123, 3061–3072. [Google Scholar] [CrossRef]
- Batich, K.A.; Reap, E.A.; Archer, G.E.; Sanchez-Perez, L.; Nair, S.K.; Schmittling, R.J.; Norberg, P.; Xie, W.; Herndon, J.E.; Healy, P.; et al. Long-term Survival in Glioblastoma with Cytomegalovirus pp65-Targeted Vaccination. Clin. Cancer Res. 2017, 23, 1898–1909. [Google Scholar] [CrossRef]
- Kübler, H.; Scheel, B.; Gnad-Vogt, U.; Miller, K.; Schultze-Seemann, W.; Dorp, F.V.; Parmiani, G.; Hampel, C.; Wedel, S.; Trojan, L.; et al. Self-adjuvanted mRNA vaccination in advanced prostate cancer patients: A first-in-man phase I/IIa study. J. Immunother. Cancer 2015, 3, 26. [Google Scholar] [CrossRef]
- Alberer, M.; Gnad-Vogt, U.; Hong, H.S.; Mehr, K.T.; Backert, L.; Finak, G.; Gottardo, R.; Bica, M.A.; Garofano, A.; Koch, S.D.; et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: An open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet 2017, 390, 1511–1520. [Google Scholar] [CrossRef]
- Papachristofilou, A.; Hipp, M.M.; Klinkhardt, U.; Früh, M.; Sebastian, M.; Weiss, C.; Pless, M.; Cathomas, R.; Hilbe, W.; Pall, G.; et al. Phase Ib evaluation of a self-adjuvanted protamine formulated mRNA-based active cancer immunotherapy, BI1361849 (CV9202), combined with local radiation treatment in patients with stage IV non-small cell lung cancer. J. Immunother. Cancer 2019, 7, 38. [Google Scholar] [CrossRef]
- Sebastian, M.; Schröder, A.; Scheel, B.; Hong, H.S.; Muth, A.; Von Boehmer, L.; Zippelius, A.; Mayer, F.; Reck, M.; Atanackovic, D.; et al. A phase I/IIa study of the mRNA-based cancer immunotherapy CV9201 in patients with stage IIIB/IV non-small cell lung cancer. Cancer Immunol. Immunother. 2019, 68, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Tuyishime, S.; Muramatsu, H.; Kariko, K.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Weissman, D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control. Release 2015, 217, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Bahl, K.; Senn, J.J.; Yuzhakov, O.; Bulychev, A.; Brito, L.A.; Hassett, K.J.; Laska, M.E.; Smith, M.; Almarsson, Ö.; Thompson, J.; et al. Preclinical and Clinical Demonstration of Immunogenicity by mRNA Vaccines against H10N8 and H7N9 Influenza Viruses. Mol. Ther. 2017, 25, 1316–1327. [Google Scholar] [CrossRef]
- Feldman, R.A.; Fuhr, R.; Smolenov, I.V.; Ribeiro, A.M.; Panther, L.; Watson, M.; Senn, J.J.; Smith, M.; Almarsson, Ö.; Pujar, H.S.; et al. mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine 2019, 37, 3326–3334. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Pelc, R.S.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251. [Google Scholar] [CrossRef]
- Richner, J.M.; Himansu, S.; Dowd, K.A.; Butler, S.L.; Salazar, V.; Fox, J.M.; Julander, J.G.; Tang, W.W.; Shresta, S.; Pierson, T.C.; et al. Modified mRNA Vaccines Protect against Zika Virus Infection. Cell 2017, 168, 1114–1125. [Google Scholar] [CrossRef]
- Arteta, M.Y.; Kjellman, T.; Bartesaghi, S.; Wallin, S.; Wu, X.; Kvist, A.J.; Dabkowska, A.; Szekely, N.; Radulescu, A.; Bergenholtz, J.; et al. Successful reprogramming of cellular protein production through mRNA delivered by functionalized lipid nanoparticles. Proc. Natl. Acad. Sci. USA 2018, 115, E3351–E3360. [Google Scholar] [CrossRef]
- Robinson, E.; Macdonald, K.D.; Slaughter, K.; McKinney, M.; Patel, S.; Sun, C.; Sahay, G. Lipid Nanoparticle-Delivered Chemically Modified mRNA Restores Chloride Secretion in Cystic Fibrosis. Mol. Ther. 2018, 26, 2034–2046. [Google Scholar] [CrossRef]
- Sedic, M.; Senn, J.J.; Lynn, A.; Laska, M.; Smith, M.; Platz, S.J.; Bolen, J.; Hoge, S.; Bulychev, A.; Jacquinet, E.; et al. Safety Evaluation of Lipid Nanoparticle–Formulated Modified mRNA in the Sprague-Dawley Rat and Cynomolgus Monkey. Veter. Pathol. 2017, 55, 341–354. [Google Scholar] [CrossRef]
- Rybakova, Y.; Kowalski, P.S.; Huang, Y.; Gonzalez, J.; Heartlein, M.W.; DeRosa, F.; Delcassian, D.; Anderson, D.G. mRNA Delivery for Therapeutic Anti-HER2 Antibody Expression In vivo. Mol. Ther. 2019, 27, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Michel, T.; Luft, D.; Abraham, M.-K.; Reinhardt, S.; Medina, M.L.S.; Kurz, J.; Schaller, M.; Avci-Adali, M.; Schlensak, C.; Peter, K.; et al. Cationic Nanoliposomes Meet mRNA: Efficient Delivery of Modified mRNA Using Hemocompatible and Stable Vectors for Therapeutic Applications. Mol. Ther. Nucleic Acids 2017, 8, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Rosigkeit, S.; Meng, M.; Grunwitz, C.; Gomes, P.; Kreft, A.; Hayduk, N.; Heck, R.; Pickert, G.; Ziegler, K.; Abassi, Y.; et al. Monitoring Translation Activity of mRNA-Loaded Nanoparticles in Mice. Mol. Pharm. 2018, 15, 3909–3919. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Li, L.; Huang, Y.; Delcassian, D.; Chahal, J.; Han, J.; Shi, Y.; Sadtler, K.; Gao, W.; Lin, J.; et al. Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat. Biotechnol. 2019, 37, 1174–1185. [Google Scholar] [CrossRef] [PubMed]
- Sunshine, J.; Sunshine, S.B.; Bhutto, I.; Handa, J.T.; Green, J.J. Poly(β-Amino Ester)-Nanoparticle Mediated Transfection of Retinal Pigment Epithelial Cells In vitro and In vivo. PLoS ONE 2012, 7, e37543. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, J.C.; Patel, A.; Kauffman, K.J.; Fenton, O.S.; Webber, M.J.; Heartlein, M.W.; DeRosa, F.; Anderson, D.G. Polymer-Lipid Nanoparticles for Systemic Delivery of mRNA to the Lungs. Angew. Chem. Int. Ed. 2016, 55, 13808–13812. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Dorkin, J.R.; Wang, W.; Chang, P.H.; Webber, M.J.; Tang, B.C.; Yang, J.; Abutbul-Ionita, I.; Danino, D.; DeRosa, F.; et al. Poly(glycoamidoamine) Brushes Formulated Nanomaterials for Systemic siRNA and mRNA Delivery in Vivo. Nano Lett. 2016, 16, 842–848. [Google Scholar] [CrossRef]
- Zhao, M.; Li, M.; Zhang, Z.-R.; Gong, T.; Sun, X. Induction of HIV-1 gag specific immune responses by cationic micelles mediated delivery of gag mRNA. Drug Deliv. 2015, 23, 2596–2607. [Google Scholar] [CrossRef]
- Dunn, A.W.; Kalinichenko, V.V.; Shi, D. Highly Efficient In Vivo Targeting of the Pulmonary Endothelium Using Novel Modifications of Polyethylenimine: An Importance of Charge. Adv. Heal. Mater. 2018, 7, 1800876. [Google Scholar] [CrossRef]
- Schumann, C.; Nguyen, D.X.; Norgard, M.; Bortnyak, Y.; Korzun, T.; Chan, S.; Lorenz, A.S.; Moses, A.S.; Albarqi, H.A.; Wong, L.; et al. Increasing lean muscle mass in mice via nanoparticle-mediated hepatic delivery of follistatin mRNA. Theranostics 2018, 8, 5276–5288. [Google Scholar] [CrossRef]
- Patel, A.; Kaczmarek, J.C.; Bose, S.; Kauffman, K.J.; Mir, F.; Heartlein, M.W.; DeRosa, F.; Langer, R.; Anderson, D.G. Inhaled Nanoformulated mRNA Polyplexes for Protein Production in Lung Epithelium. Adv. Mater. 2019, 31. [Google Scholar] [CrossRef] [PubMed]
- Siewert, C.; Haas, H.; Nawroth, T.; Ziller, A.; Nogueira, S.; Schroer, M.A.; Blanchet, C.; Svergun, D.; Radulescu, A.; Bates, F.; et al. Investigation of charge ratio variation in mRNA—DEAE-dextran polyplex delivery systems. Biomaterials 2019, 192, 612–620. [Google Scholar] [CrossRef]
- Yasar, H.; Biehl, A.; De Rossi, C.; Koch, M.; Murgia, X.; Loretz, B.; Lehr, C. Kinetics of mRNA delivery and protein translation in dendritic cells using lipid-coated PLGA nanoparticles. J. Nanobiotechnology 2018, 16, 72. [Google Scholar] [CrossRef] [PubMed]
- Prieve, M.G.; Harvie, P.; Monahan, S.D.; Roy, D.; Li, A.G.; Blevins, T.L.; Paschal, A.E.; Waldheim, M.; Bell, E.C.; Galperin, A.; et al. Targeted mRNA Therapy for Ornithine Transcarbamylase Deficiency. Mol. Ther. 2018, 26, 801–813. [Google Scholar] [CrossRef]
- Fan, Y.; Li, M.; Luo, Y.-L.; Chen, Q.; Wang, L.; Zhang, H.; Shen, S.; Gu, Z.; Wang, J. Cationic lipid-assisted nanoparticles for delivery of mRNA cancer vaccine. Biomater. Sci. 2018, 6, 3009–3018. [Google Scholar] [CrossRef] [PubMed]
- Granot-Matok, Y.; Kon, E.; Dammes, N.; Mechtinger, G.; Peer, D. Therapeutic mRNA delivery to leukocytes. J. Control. Release 2019, 305, 165–175. [Google Scholar] [CrossRef]
- Hu, Z.; Ott, P.A.; Wu, C.J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 2017, 18, 168–182. [Google Scholar] [CrossRef]
- Sun, X.; Zeng, L.; Huang, Y. Transcutaneous delivery of DNA/mRNA for cancer therapeutic vaccination. J. Gene Med. 2019, 21, e3089. [Google Scholar] [CrossRef]
- E Broderick, K.; Humeau, L.M. Electroporation-enhanced delivery of nucleic acid vaccines. Expert Rev. Vaccines 2014, 14, 195–204. [Google Scholar] [CrossRef]
- Callis, J.; Fromm, M.; Walbot, V.; Cellis, J. Expression of mRNA electroporated into plant and animal cells. Nucleic Acids Res. 1987, 15, 5823–5831. [Google Scholar] [CrossRef][Green Version]
- Qiu, P.; Ziegelhoffer, P.; Sun, J.; Yang, N.S. Gene gun delivery of mRNA in situ results in efficient transgene expression and genetic immunization. Gene Ther. 1996, 3, 262–268. [Google Scholar]
- Vacchelli, E.; Vitale, I.; Eggermont, A.; Fridman, W.H.; Fucikova, J.; Cremer, I.; Galon, J.; Tartour, E.; Zitvogel, L.; Kroemer, G.; et al. Trial watch: Dendritic cell-based interventions for cancer therapy. OncoImmunology 2013, 2, e25771. [Google Scholar] [CrossRef] [PubMed]
- Wykes, M.; Pombo, A.; Jenkins, C.; MacPherson, G. Dendritic cells interact directly with naive B lymphocytes to transfer antigen and initiate class switching in a primary T-dependent response. J. Immunol. 1998, 161, 1313–1319. [Google Scholar] [PubMed]
- Wilgenhof, S.; Corthals, J.; Heirman, C.; Van Baren, N.; Lucas, S.; Kvistborg, P.; Thielemans, K.; Neyns, B. Phase II Study of Autologous Monocyte-Derived mRNA Electroporated Dendritic Cells (TriMixDC-MEL) Plus Ipilimumab in Patients With Pretreated Advanced Melanoma. J. Clin. Oncol. 2016, 34, 1330–1338. [Google Scholar] [CrossRef]
- Lesterhuis, W.J.; De Vries, I.J.M.; Schuurhuis, D.H.; Boullart, A.C.I.; Jacobs, J.F.; De Boer, A.J.; Scharenborg, N.M.; Brouwer, H.M.H.; Van De Rakt, M.W.M.M.; Figdor, C.G.; et al. Vaccination of colorectal cancer patients with CEA-loaded dendritic cells: Antigen-specific T cell responses in DTH skin tests. Ann. Oncol. 2006, 17, 974–980. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.F.; Palucka, A.K. Dendritic cells as therapeutic vaccines against cancer. Nat. Rev. Immunol. 2005, 5, 296–306. [Google Scholar] [CrossRef]
- Benteyn, D.; Heirman, C.; Bonehill, A.; Thielemans, K.; Breckpot, K. mRNA-based dendritic cell vaccines. Expert Rev. Vaccines 2014, 14, 161–176. [Google Scholar] [CrossRef]
- Gay, C.L.; Debenedette, M.A.; Tcherepanova, I.Y.; Gamble, A.; Lewis, W.E.; Cope, A.B.; Kuruc, J.D.; McGee, K.S.; Kearney, M.; Coffin, J.M.; et al. Immunogenicity of AGS-004 Dendritic Cell Therapy in Patients Treated During Acute HIV Infection. AIDS Res. Hum. Retroviruses 2018, 34, 111–122. [Google Scholar] [CrossRef]
- Amos, H. Protamine enhancement of RNA uptake by cultured chick cells. Biochem. Biophys. Res. Commun. 1961, 5, 1–4. [Google Scholar] [CrossRef]
- Fotin-Mleczek, M.; Duchardt, K.M.; Lorenz, C.; Pfeiffer, R.; Ojkić-Zrna, S.; Probst, J.; Kallen, K.-J. Messenger RNA-based Vaccines With Dual Activity Induce Balanced TLR-7 Dependent Adaptive Immune Responses and Provide Antitumor Activity. J. Immunother. 2011, 34, 1–15. [Google Scholar] [CrossRef]
- Christensen, D.; Agger, E.M.; Andreasen, L.V.; Kirby, D.J.; Andersen, P.; Perrie, Y. Liposome-based cationic adjuvant formulations (CAF): Past, present, and future. J. Liposome Res. 2009, 19, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, K.J.; Webber, M.J.; Anderson, D.G. Materials for non-viral intracellular delivery of messenger RNA therapeutics. J. Control. Release 2016, 240, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Frank-Kamenetsky, M.; Grefhorst, A.; Anderson, N.N.; Racie, T.S.; Bramlage, B.; Akinc, A.; Butler, D.; Charisse, K.; Dorkin, R.; Fan, Y.; et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc. Natl. Acad. Sci. USA 2008, 105, 11915–11920. [Google Scholar] [CrossRef] [PubMed]
- Hajj, K.A.; A Whitehead, K. Tools for translation: Non-viral materials for therapeutic mRNA delivery. Nat. Rev. Mater. 2017, 2, 17056. [Google Scholar] [CrossRef]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef]
- Lungwitz, U.; Breunig, M.; Blunk, T.; Göpferich, A. Polyethylenimine-based non-viral gene delivery systems. Eur. J. Pharm. Biopharm. 2005, 60, 247–266. [Google Scholar] [CrossRef]
- Rejman, J.; Tavernier, G.; Bavarsad, N.; Demeester, J.; De Smedt, S.C. mRNA transfection of cervical carcinoma and mesenchymal stem cells mediated by cationic carriers. J. Control. Release 2010, 147, 385–391. [Google Scholar] [CrossRef]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 2006, 114, 100–109. [Google Scholar] [CrossRef]
- Wilson, D.R.; Mosenia, A.; Suprenant, M.P.; Upadhya, R.; Routkevitch, D.; Meyer, R.A.; Quinones-Hinojosa, A.; Green, J.J. Continuous microfluidic assembly of biodegradable poly(beta-amino ester)/DNA nanoparticles for enhanced gene delivery. J. Biomed. Mater. Res. Part A 2017, 105, 1813–1825. [Google Scholar] [CrossRef]
- Zugates, G.T.; Peng, W.; Zumbuehl, A.; Jhunjhunwala, S.; Huang, Y.-H.; Langer, R.; A Sawicki, J.; Anderson, D.G. Rapid Optimization of Gene Delivery by Parallel End-modification of Poly(β-amino ester)s. Mol. Ther. 2007, 15, 1306–1312. [Google Scholar] [CrossRef]
- Kaczmarek, J.C.; Kauffman, K.J.; Fenton, O.S.; Sadtler, K.; Patel, A.; Heartlein, M.W.; DeRosa, F.; Anderson, D.G. Optimization of a Degradable Polymer–Lipid Nanoparticle for Potent Systemic Delivery of mRNA to the Lung Endothelium and Immune Cells. Nano Lett. 2018, 18, 6449–6454. [Google Scholar] [CrossRef] [PubMed]
- Palmiero, U.C.; Kaczmarek, J.C.; Fenton, O.S.; Anderson, D.G. Poly(β-amino ester)-co -poly(caprolactone) Terpolymers as Nonviral Vectors for mRNA Delivery In Vitro and In Vivo. Adv. Heal. Mater. 2018, 7, 1800249. [Google Scholar] [CrossRef] [PubMed]
- Perri, S.; Greer, C.E.; Thudium, K.; Doe, B.; Legg, H.; Liu, H.; Romero, R.E.; Tang, Z.; Bin, Q.; Dubensky, T.W.; et al. An Alphavirus Replicon Particle Chimera Derived from Venezuelan Equine Encephalitis and Sindbis Viruses Is a Potent Gene-Based Vaccine Delivery Vector. J. Virol. 2003, 77, 10394–10403. [Google Scholar] [CrossRef] [PubMed]
- Ulmer, J.B.; Mason, P.W.; Geall, A.; Mandl, C.W. RNA-based vaccines. Vaccine 2012, 30, 4414–4418. [Google Scholar] [CrossRef]
- Mandl, C.W.; Aberle, J.; Aberle, S.W.; Holzmann, H.; Allison, S.L.; Heinz, F.X. In vitro-synthesized infectious RNA as an attenuated live vaccine in a flavivirus model. Nat. Med. 1998, 4, 1438–1440. [Google Scholar] [CrossRef]
- Boczkowski, D.; Nair, S.K.; Snyder, D.; Gilboa, E. Dendritic cells pulsed with RNA are potent antigen-presenting cells in vitro and in vivo. J. Exp. Med. 1996, 184, 465–472. [Google Scholar] [CrossRef]
- Zhou, W.-Z.; Hoon, D.S.B.; Huang, S.; Fujii, S.; Hashimoto, K.; Morishita, R.; Kaneda, Y. RNA Melanoma Vaccine: Induction of Antitumor Immunity by Human Glycoprotein 100 mRNA Immunization. Hum. Gene Ther. 1999, 10, 2719–2724. [Google Scholar] [CrossRef]
- Ekiert, D.C.; Kashyap, A.K.; Steel, J.; Rubrum, A.; Bhabha, G.; Khayat, R.; Lee, J.H.; Dillon, M.A.; O’Neil, R.E.; Faynboym, A.M.; et al. Neutralization of influenza A viruses by insertion of a single antibody loop into the receptor binding site. Nature 2012, 489, 526–532. [Google Scholar] [CrossRef]
- Brazzoli, M.; Magini, D.; Bonci, A.; Buccato, S.; Giovani, C.; Kratzer, R.; Zurli, V.; Mangiavacchi, S.; Casini, D.; Brito, L.M.; et al. Induction of Broad-Based Immunity and Protective Efficacy by Self-amplifying mRNA Vaccines Encoding Influenza Virus Hemagglutinin. J. Virol. 2015, 90, 332–344. [Google Scholar] [CrossRef]
- Pardi, N.; Parkhouse, K.; Kirkpatrick, E.; McMahon, M.; Zost, S.J.; Mui, B.L.; Tam, Y.K.; Karikó, K.; Barbosa, C.J.; Madden, T.D.; et al. Nucleoside-modified mRNA immunization elicits influenza virus hemagglutinin stalk-specific antibodies. Nat. Commun. 2018, 9, 3361. [Google Scholar] [CrossRef]
- Willis, E.; Pardi, N.; Parkhouse, K.; Mui, B.L.; Tam, Y.K.; Weissman, D.; Hensley, S.E. Nucleoside-modified mRNA vaccination partially overcomes maternal antibody inhibition of de novo immune responses in mice. Sci. Transl. Med. 2020, 12, eaav5701. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, R.T.; Kwon, D.S.; Macklin, E.A.; Shopis, J.R.; McLean, A.P.; McBrine, N.; Flynn, T.; Peter, L.; Sbrolla, A.; Kaufmann, D.E.; et al. Immunization of HIV-1-Infected Persons With Autologous Dendritic Cells Transfected With mRNA Encoding HIV-1 Gag and Nef: Results of a Randomized, Placebo-Controlled Clinical Trial. J. Acquir. Immune Defic. Syndr. 2016, 71, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Bogers, W.M.; Oostermeijer, H.; Mooij, P.; Koopman, G.; Verschoor, E.J.; Davis, D.; Ulmer, J.B.; Brito, L.A.; Cu, Y.; Banerjee, K.; et al. Potent immune responses in rhesus macaques induced by nonviral delivery of a self-amplifying RNA vaccine expressing HIV type 1 envelope with a cationic nanoemulsion. J. Infect. Dis. 2014, 211, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Guardo, A.C.; Joe, P.T.; Miralles, L.; Bargalló, M.E.; Mothe, B.; Krasniqi, A.; Heirman, C.; García, F.; Thielemans, K.; Brander, C.; et al. Preclinical evaluation of an mRNA HIV vaccine combining rationally selected antigenic sequences and adjuvant signals (HTI-TriMix). AIDS 2017, 31, 321–332. [Google Scholar] [CrossRef]
- De Jong, W.; Leal, L.; Buyze, J.; Pannus, P.; Guardo, A.C.; Salgado, M.; Mothe, B.; Molto, J.; Moron-Lopez, S.; Gálvez, C.; et al. Therapeutic Vaccine in Chronically HIV-1-Infected Patients: A Randomized, Double-Blind, Placebo-Controlled Phase IIa Trial with HTI-TriMix. Vaccines 2019, 7, 209. [Google Scholar] [CrossRef]
- Mulligan, M.J.; Lyke, K.E.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.P.; Neuzil, K.; Raabe, V.; Bailey, R.; Swanson, K.A.; et al. Phase 1/2 Study to Describe the Safety and Immunogenicity of a COVID-19 RNA Vaccine Candidate (BNT162b1) in Adults 18 to 55 Years of Age: Interim Report. medRxiv 2020. [Google Scholar] [CrossRef]
- Schnee, M.; Vogel, A.B.; Voss, D.; Petsch, B.; Baumhof, P.; Kramps, T.; Stitz, L. An mRNA Vaccine Encoding Rabies Virus Glycoprotein Induces Protection against Lethal Infection in Mice and Correlates of Protection in Adult and Newborn Pigs. PLoS Neglected Trop. Dis. 2016, 10, e0004746. [Google Scholar] [CrossRef]
- Armbruster, N.; Jasny, E.; Petsch, B. Advances in RNA Vaccines for Preventive Indications: A Case Study of A Vaccine Against Rabies. Vaccines 2019, 7, 132. [Google Scholar] [CrossRef]
- Samsa, M.M.; Dupuy, L.C.; Beard, C.W.; Six, C.M.; Schmaljohn, C.S.; Mason, P.W.; Geall, A.J.; Ulmer, J.B.; Yu, D. Self-Amplifying RNA Vaccines for Venezuelan Equine Encephalitis Virus Induce Robust Protective Immunogenicity in Mice. Mol. Ther. 2019, 27, 850–865. [Google Scholar] [CrossRef]
- Awasthi, S.; Hook, L.M.; Pardi, N.; Wang, F.; Myles, A.; Cancro, M.P.; Cohen, G.H.; Weissman, D.; Friedman, H.M. Nucleoside-modified mRNA encoding HSV-2 glycoproteins C, D, and E prevents clinical and subclinical genital herpes. Sci. Immunol. 2019, 4, eaaw7083. [Google Scholar] [CrossRef]
- Shaw, C.; Panther, L.; August, A.; Zaks, T.; Smolenov, I.; Bart, S.; Watson, M. Safety and immunogenicity of a mRNA-based chikungunya vaccine in a phase 1 dose-ranging trial. Int. J. Infect. Dis. 2019, 79, 17. [Google Scholar] [CrossRef]
- Shaw, C.; Lee, H.; Knightly, C.; Kalidindi, S.; Zaks, T.; Smolenov, I.; Panther, L. 2754. Phase 1 Trial of an mRNA-Based Combination Vaccine Against hMPV and PIV3. Open Forum Infect. Dis. 2019, 6, S970. [Google Scholar] [CrossRef]
- Pepini, T.; Pulichino, A.-M.; Carsillo, T.; Carlson, A.L.; Sari-Sarraf, F.; Ramsauer, K.; Debasitis, J.C.; Maruggi, G.; Otten, G.R.; Geall, A.J.; et al. Induction of an IFN-Mediated Antiviral Response by a Self-Amplifying RNA Vaccine: Implications for Vaccine Design. J. Immunol. 2017, 198, 4012–4024. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Catani, J.P.P.; Mc Cafferty, S.; Couck, L.; Broeck, W.V.D.; Gorlé, N.; Vandenbroucke, R.E.; Devriendt, B.; Ulbert, S.; Cnops, L.; et al. Immunogenicity and Protection Efficacy of a Naked Self-Replicating mRNA-Based Zika Virus Vaccine. Vaccines 2019, 7, 96. [Google Scholar] [CrossRef] [PubMed]
- Chahal, J.S.; Khan, O.F.; Cooper, C.L.; McPartlan, J.S.; Tsosie, J.K.; Tilley, L.D.; Sidik, S.M.; Lourido, S.; Langer, R.; Bavari, S.; et al. Dendrimer-RNA nanoparticles generate protective immunity against lethal Ebola, H1N1 influenza, and Toxoplasma gondii challenges with a single dose. Proc. Natl. Acad. Sci. USA 2016, 113, E4133–E4142. [Google Scholar] [CrossRef] [PubMed]
- Maruggi, G.; Chiarot, E.; Giovani, C.; Buccato, S.; Bonacci, S.; Frigimelica, E.; Margarit, I.; Geall, A.; Bensi, G.; Maione, D. Immunogenicity and protective efficacy induced by self-amplifying mRNA vaccines encoding bacterial antigens. Vaccine 2017, 35, 361–368. [Google Scholar] [CrossRef]
- Garcia, A.B.; Siu, E.; Sun, T.; Exler, V.; Brito, L.; Hekele, A.; Otten, G.; Augustijn, K.; Janse, C.J.; Ulmer, J.B.; et al. Neutralization of the Plasmodium-encoded MIF ortholog confers protective immunity against malaria infection. Nat. Commun. 2018, 9, 2714. [Google Scholar] [CrossRef]
- Maruggi, G.; Zhang, C.; Li, J.; Ulmer, J.B.; Yu, D. mRNA as a Transformative Technology for Vaccine Development to Control Infectious Diseases. Mol. Ther. 2019, 27, 757–772. [Google Scholar] [CrossRef]
- Zhang, L.; Ru, H.-W.; Chen, F.-Z.; Jin, C.-Y.; Sun, R.-F.; Fan, X.-Y.; Guo, M.; Mai, J.-T.; Xu, W.-X.; Lin, Q.-X.; et al. Variable Virulence and Efficacy of BCG Vaccine Strains in Mice and Correlation With Genome Polymorphisms. Mol. Ther. 2016, 24, 398–405. [Google Scholar] [CrossRef]
- WHO. Global Tuberculosis Report 2019; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Tameris, M.D.; Hatherill, M.; Landry, B.S.; Scriba, T.J.; Snowden, M.A.; Lockhart, S.P.; E Shea, J.; McClain, J.B.; Hussey, G.D.; A Hanekom, W.; et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: A randomised, placebo-controlled phase 2b trial. Lancet 2013, 381, 1021–1028. [Google Scholar] [CrossRef]
- Kose, N.; Fox, J.M.; Sapparapu, G.; Bombardi, R.; Tennekoon, R.N.; De Silva, A.M.; Elbashir, S.M.; Theisen, M.A.; Humphris, E.L.; Ciaramella, G.; et al. A lipid-encapsulated mRNA encoding a potently neutralizing human monoclonal antibody protects against chikungunya infection. Sci. Immunol. 2019, 4, eaaw6647. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, S.; Vormehr, M.; Van De Roemer, N.; Diken, M.; Löwer, M.; Diekmann, J.; Boegel, S.; Schrörs, B.; Vascotto, F.; Castle, J.C.; et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015, 520, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Türeci, Ö.; Vormehr, M.; Diken, M.; Kreiter, S.; Huber, C.; Şahin, U. Targeting the Heterogeneity of Cancer with Individualized Neoepitope Vaccines. Clin. Cancer Res. 2016, 22, 1885–1896. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Coulie, P.G.; Eynde, B.V.D.; Van Der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef]
- Castle, J.C.; Kreiter, S.; Diekmann, J.; Löwer, M.; Van De Roemer, N.; De Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the Mutanome for Tumor Vaccination. Cancer Res. 2012, 72, 1081–1091. [Google Scholar] [CrossRef]
- Kreiter, S.; Castle, J.C.; Türeci, Ö.; Şahin, U. Targeting the tumor mutanome for personalized vaccination therapy. Oncoimmunology 2012, 1, 768–769. [Google Scholar] [CrossRef]
- Heesen, L.; Frenzel, K.; Bolte, S.; Bukur, V.; Diken, M.; Derhovanessian, E.; Kreiter, S.; Kuhn, A.N.; Kühlcke, K.; Löwer, M.; et al. Abstract CT221: Mutanome engineered RNA immuno-therapy (MERIT) for patients with triple negative breast cancer (TNBC). Clin. Trials 2019. [Google Scholar] [CrossRef]
- Van Lint, S.; Renmans, D.; Broos, K.; Goethals, L.; Maenhout, S.; Benteyn, D.; Goyvaerts, C.; Du Four, S.; Van Der Jeught, K.; Bialkowski, L.; et al. Intratumoral Delivery of TriMix mRNA Results in T-cell Activation by Cross-Presenting Dendritic Cells. Cancer Immunol. Res. 2015, 4, 146–156. [Google Scholar] [CrossRef]
- De Keersmaecker, B.; Claerhout, S.; Carrasco, J.; Bar, I.; Corthals, J.; Wilgenhof, S.; Neyns, B.; Thielemans, K. TriMix and tumor antigen mRNA electroporated dendritic cell vaccination plus ipilimumab: Link between T-cell activation and clinical responses in advanced melanoma. J. Immunother. Cancer 2020, 8, e000329. [Google Scholar] [CrossRef]
- Reinhard, K.; Rengstl, B.; Oehm, P.; Michel, K.; Billmeier, A.; Hayduk, N.; Klein, O.; Kuna, K.; Ouchan, Y.; Wöll, S.; et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science 2020, 367, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Mukalel, A.J.; Riley, R.S.; Zhang, R.; Mitchell, M.J. Nanoparticles for nucleic acid delivery: Applications in cancer immunotherapy. Cancer Lett. 2019, 458, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Jabulowsky, R.A.; Loquai, C.; Mitzel-Rink, H.; Utikal, J.; Gebhardt, C.; Hassel, J.C.; Kaufmann, R.; Pinter, A.; Derhovanessian, E.; Anft, C.; et al. Abstract CT156: A first-in-human phase I/II clinical trial assessing novel mRNA-lipoplex nanoparticles encoding shared tumor antigens for immunotherapy of malignant melanoma. Clin. Trials 2018, 78, CT156. [Google Scholar] [CrossRef]
- Wagenaar, T.R.; Hotz, C.; Gieseke, F.; Cao, H.; Diekmann, J.; Diken, M.; Grunwitz, C.; Hebert, A.; Hsu, K.; Jordan, M.; et al. Abstract LB-130: Combinatorial treatment with intratumoral cytokine mRNAs results in high frequency of tumor rejection and development of anti-tumor immunity across a range of preclinical cancer models. Immunology 2018, 78. [Google Scholar] [CrossRef]
- Shariati, M.; Zhang, H.; Van De Sande, L.; Descamps, B.; Vanhove, C.; Willaert, W.; Ceelen, W.; De Smedt, S.C.; Remaut, K. High Pressure Nebulization (PIPAC) Versus Injection for the Intraperitoneal Administration of mRNA Complexes. Pharm. Res. 2019, 36, 126. [Google Scholar] [CrossRef]
- Jones, K.L.; Drane, D.; Gowans, E.J. Long-term storage of DNA-free RNA for use in vaccine studies. Biotechniques 2007, 43, 675–681. [Google Scholar] [CrossRef]
- Coolen, A.-L.; Lacroix, C.; Mercier-Gouy, P.; Delaune, E.; Monge, C.; Exposito, J.-Y.; Verrier, B. Poly(lactic acid) nanoparticles and cell-penetrating peptide potentiate mRNA-based vaccine expression in dendritic cells triggering their activation. Biomaterials 2019, 195, 23–37. [Google Scholar] [CrossRef]
- Zhang, H.; De Smedt, S.C.; Remaut, K. Fluorescence Correlation Spectroscopy to find the critical balance between extracellular association and intracellular dissociation of mRNA complexes. Acta Biomater. 2018, 75, 358–370. [Google Scholar] [CrossRef]
- Zhang, H.; Rombouts, K.; Raes, L.; Xiong, R.; De Smedt, S.C.; Braeckmans, K.; Remaut, K. Fluorescence-Based Quantification of Messenger RNA and Plasmid DNA Decay Kinetics in Extracellular Biological Fluids and Cell Extracts. Adv. Biosyst. 2020, 4, e2000057. [Google Scholar] [CrossRef]
- AlQuraishi, M. AlphaFold at CASP13. Bioinformatics 2019, 35, 4862–4865. [Google Scholar] [CrossRef]
- Wei, G.-W. Protein structure prediction beyond AlphaFold. Nat. Mach. Intell. 2019, 1, 336–337. [Google Scholar] [CrossRef]
- Şahin, U.; Türeci, Ö. Personalized vaccines for cancer immunotherapy. Science 2018, 359, 1355–1360. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Delivery System (Delivery Methods/Materials) | Administration | Target Host | Disease(s) | Reference(s) |
|---|---|---|---|---|
| Direct Injection | ||||
| Intradermal | Mice | -- | [75,76] | |
| Intradermal | Human | Melanoma | [77] | |
| Intranodal | Mice | Cancer | [15] | |
| Intranodal | Mice | -- | [67] | |
| Subcutaneous, intranasal, intravenous | Mice | -- | [78] | |
| Subcutaneous tumors, intranodal | Mice | Cervical cancer | [79] | |
| Intranodal | Human | Melanoma | [18] | |
| Physical Delivery Methods | ||||
| Electroporation | ||||
| Intradermal | Mice | -- | [80] | |
| -- | Neurosphere | -- | [81] | |
| -- | DCs | Melanoma | [18] | |
| Gene gun | ||||
| -- | Mice | Melanoma | [82] | |
| -- | Mice | Epidermolysis bullosa | [83] | |
| Sonophoresis | ||||
| -- | DCs | -- | [84] | |
| Microneedles | ||||
| -- | Mice | -- | [85] | |
| Intradermal | Pig | -- | [86] | |
| Ex Vivo Loading of DCs Delivery | ||||
| Subcutaneous | Mice | Different tumors | [84] | |
| Intradermal | Human | Acute myeloid leukemia | [87] | |
| -- | Mice | Glioblastoma | [88] | |
| Protamine-Formulated Delivery | ||||
| Intradermal | Human | Melanoma | [14] | |
| Intradermal, Intranodal | Mice, ferret, pig | Cancer, infectious diseases | [54] | |
| Intradermal | Human | Prostate cancer | [89] | |
| Intradermal, intramuscular | Human | Rabies | [90] | |
| Intradermal | Human | NSCLC | [91] | |
| Intradermal | Human | NSCLC | [92] | |
| Lipid-Based Delivery | ||||
| LNP | Intramuscular | Mice, rat | Respiratory syncytial virus infection | [17] |
| LNP | Intravenous, intraperitoneal, subcutaneous, intramuscular, intradermal, intratracheal | Mice | -- | [93] |
| LNP | Intravenous | Human | Melanoma | [94] |
| LNP | Intramuscular | Human | H10N8 and H7N9 | [95,96] |
| LNP | Intradermal, intravenous, subcutaneous | Mice, rhesus macaque | ZIKV | [97] |
| LNP | Intramuscular | Mice | ZIKV | [98] |
| Lipid-Based Delivery | ||||
| LNP | Intravenous | Human adipocyte, hepatocyte | Anemia | [99] |
| LNP | Nasal pumping | Mice | Cystic fibrosis | [100] |
| LNP | Intravenous | Rat, monkey | Anemia | [101] |
| LNP | Intravenous | Mice | Cancer | [102] |
| DOTAP/DOPE | Subcutaneous | Mice | AIDS | [61] |
| DOPE/DC-Cholesterol (2:1) | -- | A549 Cells | -- | [103] |
| DOTMA/DOPE or DOTMA/ cholesterol | Intravenous | Mice | -- | [104] |
| Lipid library | -- | DCs, HeLa cells | Melanoma | [105] |
| Polymer-Based Delivery | ||||
| PBAE | Subretinal injections | Mice | Retina diseases | [106] |
| PBAE, lipid-PEG | Intravenous | Mice | -- | [107] |
| Poly(glycoamidoamine) | Intravenous | Mice | Anemia, myelodysplasia | [108] |
| PSA, PEI | Subcutaneous | Mice | AIDS | [109] |
| PEI-PEG | Intravenous | Mice | Pulmonary vascular disease | [110] |
| PEG[Glu(DET)]2 | Subcutaneous | Mice | Muscle atrophy | [111] |
| hPBAEs | Inhalation | Mice | -- | [112] |
| DEAE-Dextran | -- | DCs | -- | [113] |
| Lipid and Polymer Hybrid | ||||
| DOTMA, PLGA | -- | DCs | -- | [114] |
| LNP and polymer micelle | Intravenous | Mice | Ornithine transcarbamylase deficiency | [115] |
| CLAN (PEG-PLGA, PLGA, BHEM-cholesterol) | Intravenous | Mice, DCs | Lymphoma | [116] |
| Study Product | Antigen | Delivery Carrier | Administration | Phase | NCT Identifier | Status | Target |
|---|---|---|---|---|---|---|---|
| -- | -- | DCs | intradermal | Ⅰ/Ⅱ | NCT00833781 | Completed | AIDS |
| iHIVARNA-01 | HTI | DCs | inguinal intranodal | Ⅰ | NCT02413645 | Completed | AIDS |
| iHIVARNA-01 | HTI | DCs | intranasal | Ⅱ | NCT02888756 | Terminated | AIDS |
| mRNA-1647/mRNA-1443 | CMV associated antigens | -- | -- | Ⅰ | NCT03382405 | Active, not recruiting | CMV infection |
| mRNA-1647 | gB, pentamer complex | -- | -- | Ⅱ | NCT04232280 | Recruiting | CMV infection |
| mRNA-1273 | Spike protein | lipsome | intramuscular | Ⅰ | NCT04283461 | Active, not recruiting | COVID-19 |
| mRNA-1273 | Spike protein | lipsome | -- | Ⅱ | NCT04405076 | Active, not recruiting | COVID-19 |
| BNT162a1/BNT162b1/BNT162b2/BNT162c2 | Spike protein | LNP | intramuscular | Ⅰ/Ⅱ | NCT04380701 | Recruiting | COVID-19 |
| BNT162a1/BNT162b1/BNT162b2/BNT162c2 | Spike protein | LNP | intramuscular | Ⅰ/Ⅱ | NCT04368728 | Recruiting | COVID-19 |
| CVnCoV Vaccine | Spike protein | -- | intramuscular | Ⅰ | NCT04449276 | Recruiting | COVID-19 |
| VAL-506440 | H10N8 HA | LNP | intramuscular/intradermal | Ⅰ | NCT03076385 | Completed | Influenza |
| VAL-339851 | H7N9 HA | LNP | intramuscular | Ⅰ | NCT03345043 | Active, not recruiting | Influenza |
| mRNA- 1653 | hMPV, PIV3 | -- | -- | Ⅰ | NCT03392389 | Completed | hMPV infection |
| mRNA- 1653 | hMPV, PIV3 | -- | -- | Ⅰ | NCT04144348 | Recruiting | hMPV infection |
| CV7201 | Rabies virus glycoprotein | RNActive® | -- | Ⅰ | NCT02241135 | Completed | Rabies |
| CV7202 | RABV-G protein antigens | -- | intramuscular | Ⅰ | NCT03713086 | Active, not recruiting | Rabies |
| mRNA- 1325 | -- | -- | -- | Ⅰ | NCT03014089 | Completed | Zika virus |
| mRNA- 1893 | Zika virus associated antigen | -- | -- | Ⅰ | NCT04064905 | Recruiting | Zika virus |
| Study Product | Antigen | Delivery Carrier | Administration | Phase | NCT Identifier | Status | Target |
|---|---|---|---|---|---|---|---|
| -- | WT1 | DCs | intradermal | Ⅰ | NCT00834002 | Completed | AML |
| -- | WT1 | DCs | intradermal | Ⅱ | NCT01686334 | Recruiting | AML |
| -- | Leukemia associated antigens, CMV antigen | DCs | intradermal | Ⅰ/Ⅱ | NCT01734304 | Completed | AML |
| -- | WT1 | DCs | -- | Ⅰ/Ⅱ | NCT03083054 | Active, not recruiting | AML |
| GRNVAC1 | hTERT, LAMP-1 | DCs | -- | Ⅱ | NCT00510133 | Completed | AML |
| -- | Leukemia associated antigens | DCs | -- | Ⅰ | NCT00514189 | Terminated | AML |
| -- | -- | DCs | -- | Ⅰ | NCT02808416 | Active, not recruiting | Brain metastases |
| -- | CEA | DCs | intravenous/intradermal | Ⅰ/Ⅱ | NCT00228189 | Completed | Colorectal cancer, liver metastases |
| -- | MUC1, survivin | DCs | -- | Ⅰ/Ⅱ | NCT02693236 | Unknown * | Esophagus cancer |
| -- | -- | DCs | intradermal | Ⅰ/Ⅱ | NCT00846456 | Completed | GBM |
| -- | Human CMV pp65-LAMP, HIV-Gag | DCs | intradermal | Ⅱ | NCT03688178 | Suspended | GBM |
| -- | Human CMV pp65-LAMP | DCs | intradermal | Ⅱ | NCT02366728 | Active, not recruiting | GBM |
| -- | WT1 | DCs | intradermal | Ⅰ/Ⅱ | NCT02649582 | Recruiting | GBM |
| -- | -- | DCs | Intravenous/intradermal | Ⅰ | NCT02709616 | Active, not recruiting | GBM |
| PerCellVac2 | Glioma associated antigens | DCs | -- | Ⅰ | NCT02808364 | Active, not recruiting | GBM |
| DEN-STEM | hTERT, survivin, autologous tumor antigens | DCs | intradermal | Ⅱ/Ⅲ | NCT03548571 | Recruiting | GBM |
| pp65 DC | pp65 | DCs | subcutaneous | Ⅱ | NCT02465268 | Recruiting | GBM |
| I-ATTAC | Human CMV pp65-LAMP | DCs | intradermal | Ⅱ | NCT03927222 | Recruiting | GBM |
| -- | CMV pp65-LAMP | DCs | intradermal | Ⅰ | NCT00639639 | Active, not recruiting | GBM |
| -- | WT1 | DCs | intradermal | Ⅰ/Ⅱ | NCT01291420 | Unknown * | GBM |
| -- | Brain tumor stem cell specific antigens | DCs | intradermal | Ⅰ | NCT00890032 | Completed | GBM |
| -- | MiHA | DCs | intravenous | Ⅰ/Ⅱ | NCT02528682 | Recruiting | Hematological malignancies |
| -- | CMV pp65-LAMP | DCs | intradermal | Ⅰ | NCT00626483 | Completed | Malignant neoplasms Brain |
| -- | WT1 | DCs | intradermal | Ⅰ/Ⅱ | NCT02649829 | Recruiting | Malignant pleural mesothelioma |
| -- | -- | DCs | intradermal/intranasal | Ⅰ/Ⅱ | NCT01278940 | Completed | Melanoma |
| -- | gp100, tyrosinase | DCs | -- | Ⅰ/Ⅱ | NCT00243529 | Completed | Melanoma |
| -- | Melan-A, Mage-A1, Mage-A3, survivin, gp100, tyrosinase | -- | intradermal | Ⅰ/Ⅱ | NCT00204607 | Completed | Melanoma |
| -- | Melan-A, Mage-A1, Mage-A3, Survivin, gp100, tyrosinase | -- | intradermal | Ⅰ/Ⅱ | NCT00204516 | Completed | Melanoma |
| -- | gp100, tyrosinase | DCs | intradermal/intravenous | Ⅰ/Ⅱ | NCT00940004 | Completed | Melanoma |
| -- | hTERT, survivin | DCs | -- | Ⅰ/Ⅱ | NCT00961844 | Terminated | Melanoma |
| -- | hTERT, survivin, p53 | DCs | intradermal | Ⅰ | NCT00978913 | Completed | Melanoma |
| -- | -- | DCs | intravenous/intranasal | Ⅰ | NCT01066390 | Completed | Melanoma |
| -- | gp 100, tyrosinase | DCs | intranasal | Ⅰ/Ⅱ | NCT01530698 | Completed | Melanoma |
| -- | gp 100, tyrosinase | DCs | Intradermal/intravenous | Ⅱ | NCT02285413 | Completed | Melanoma |
| -- | TRP2 | DCs | subcutaneous | Ⅰ | NCT01456104 | Active, not recruiting | Melanoma |
| mRNA- 4157 | multiple neoantigens | -- | -- | Ⅱ | NCT03897881 | Recruiting | Melanoma |
| NCI-4650 | -- | -- | intramuscular | Ⅰ/Ⅱ | NCT03480152 | Terminated | Melanoma |
| -- | CT7, Mage-A3, WT1 | DCs | subcutaneous | Ⅰ | NCT01995708 | Active, not recruiting | Multiple myeloma |
| CV9201 | -- | RNActive® | -- | Ⅰ/Ⅱ | NCT00923312 | Completed | NSCLC |
| DC-CIK | SOCS 1, MUC1, survivin | DCs | -- | Ⅰ/Ⅱ | NCT02688686 | Unknown * | NSCLC |
| BI 1361849 | -- | -- | -- | Ⅰ/Ⅱ | NCT03164772 | Recruiting | NSCLC |
| DC-006 vaccine | hTERT, survivin | DCs | intradermal | Ⅰ/Ⅱ | NCT01334047 | Terminated | Ovarian cancer |
| W_ova1 Vaccine | -- | Liposome | intravenous | Ⅰ | NCT04163094 | Recruiting | Ovarian cancer |
| -- | TERT | DCs | -- | Ⅰ | NCT01456065 | Unknown * | Ovarian cancer |
| -- | hTERT, survivin | DCs | -- | Ⅰ/Ⅱ | NCT01197625 | Active, not recruiting | Prostate cancer |
| -- | NY-ESO-1, MUC1 PepTivator® | protamine and DCs | intranasal | Ⅱ | NCT02692976 | Completed | Prostate cancer |
| -- | hTERT, survivin, PSA, PAP | DCs | intradermal | Ⅱ | NCT01446731 | Completed | Prostate cancer |
| CV9104 | Prostate associated antigens | RNActive® | intradermal | Ⅱ | NCT02140138 | Terminated | Prostate cancer |
| mRNA- 4157 | multiple neoantigens | -- | intramuscular | Ⅰ | NCT03313778 | Recruiting | Solid tumors |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, S.; Yang, K.; Li, R.; Zhang, L. mRNA Vaccine Era—Mechanisms, Drug Platform and Clinical Prospection. Int. J. Mol. Sci. 2020, 21, 6582. https://doi.org/10.3390/ijms21186582
Xu S, Yang K, Li R, Zhang L. mRNA Vaccine Era—Mechanisms, Drug Platform and Clinical Prospection. International Journal of Molecular Sciences. 2020; 21(18):6582. https://doi.org/10.3390/ijms21186582
Chicago/Turabian StyleXu, Shuqin, Kunpeng Yang, Rose Li, and Lu Zhang. 2020. "mRNA Vaccine Era—Mechanisms, Drug Platform and Clinical Prospection" International Journal of Molecular Sciences 21, no. 18: 6582. https://doi.org/10.3390/ijms21186582
APA StyleXu, S., Yang, K., Li, R., & Zhang, L. (2020). mRNA Vaccine Era—Mechanisms, Drug Platform and Clinical Prospection. International Journal of Molecular Sciences, 21(18), 6582. https://doi.org/10.3390/ijms21186582