Abstract
There are still major challenges regarding the early diagnosis and treatment of chronic kidney disease (CKD), which is in part due to the fact that its pathophysiology is very complex and not clarified in detail. The diagnosis of CKD commonly is made after kidney damage has occurred. This highlights the need for better mechanistic insight into CKD as well as improved clinical tools for both diagnosis and treatment. In the last decade, many studies have focused on microRNAs (miRs) as novel diagnostic tools or clinical targets. MiRs are small non-coding RNA molecules that are involved in post-transcriptional gene regulation and many have been studied in CKD. A wide array of pre-clinical and clinical studies have highlighted the potential role for miRs in the pathogenesis of hypertensive nephropathy, diabetic nephropathy, glomerulonephritis, kidney tubulointerstitial fibrosis, and some of the associated cardiovascular complications. In this review, we will provide an overview of the miRs studied in CKD, especially highlighting miR-103a-3p, miR-192-5p, the miR-29 family and miR-21-5p as these have the greatest potential to result in novel therapeutic and diagnostic strategies.
1. Introduction
Chronic kidney disease (CKD) is a major contributor to morbidity and mortality. It is assumed to become the fifth most common cause of death worldwide in the year 2040 [1]. CKD is classified into five stages based on estimated glomerular filtration rate (eGFR) (G1-5) and albumin-to-creatinine ratio (ACR) (A1-3) as shown in Table 1 and Table 2 [2]. The underlying causes of CKD genesis and progression vary and besides genetic causes include for example diabetes and hypertension resulting in diabetic kidney disease and hypertensive kidney disease, respectively. Sustained or repeated kidney injury over time leads to maladaptive responses, such as extracellular matrix (ECM) deposition in the glomeruli and tubulointerstitium. This results in the common features of CKD: tubulointerstitial fibrosis and glomerulosclerosis. These pathological changes provoke an abnormal kidney structure, hypoxia, microvascular capillary rarefaction, and tubular atrophy, which all together leads to a decrease in renal filtration capacity and eventually to end-stage kidney disease (ESKD) [3,4].

Table 1.
Estimated glomerular filtration rate (eGFR) categories in CKD.
Table 2.
Albuminuria categories in CKD.
Although CKD has a great impact on health worldwide, CKD itself is generally not the direct cause of death in industrialized countries in contrast to many other parts of the world, due to effective treatment options. However, CKD impacts global health indirectly by considerably increasing the risk of cardiovascular diseases (CVDs) [5,6]. Furthermore, many studies have already clearly shown that heart failure additionally leads to kidney failure and vice versa, as reviewed elsewhere [7,8,9]. Some predisposing factors for developing CVDs and CKD are shared, such as obesity, diabetes, and age, and some risk factors for CVDs are CKD-related, such as increased uremic toxin concentrations, proteinuria, and altered mineral metabolism [10]. The most common CKD-related CVD death is ischemic heart disease, which includes myocardial infarction, angina pectoris and sudden cardiac death [11]. Ischemic heart disease accounts for approximately 55% of all CVD deaths in CKD stage 2–4 patients and for about 75% of deaths in stage 5 (ESKD) patients [12]. Another commonly found cardiac pathology in CKD patients is uremic cardiomyopathy, a term not very well defined, which is reviewed elsewhere in detail [13].
Patients with mild to moderate CKD are often asymptomatic, leaving them untreated and consequently many of these patients progress to advanced stages of CKD [14]. Currently, albuminuria and eGFR constitute the central tools for the diagnosis of CKD and its stages. However, these tools mostly enable diagnosis of CKD after kidney damage has already occurred, they do not provide etiologic clues and only limited information on disease activity in the kidneys [15]. Hence, earlier and more personalized diagnosis and therapy of CKD may be more suitable to limit the CKD-associated mortality. For this, more detailed mechanistic insights are needed into CKD development and progression. Over the last years, multiple microRNA (miR) species have been identified to play a crucial role in this pathology. This review will highlight four miRs that have been widely studied and shown potential to be used in the clinic in the future as diagnostic or even therapeutic tools.
1.1. MicroRNA Synthesis and Function
MiRs are small non-coding RNA fragments of approximately 22 nucleotides that are involved in post-transcriptional gene regulation by targeting the 3′ UTR of mRNAs [16]. The stepwise process of synthesis of miRs starts with primary miR (pri-miR) that is transcribed from the DNA by RNA polymerase II (Pol II) (Figure 1). Subsequently, the Drosha/DiGeorge syndrome critical region gene 8 (DGCR8) microprocessor complex crops the pri-miR to produce precursor miR (pre-miR). Exportin-5 and GTP-binding nuclear protein Ran (Ran-GTP) then mediate the export of the pre-miR from the nucleus into the cytoplasm where the pre-miR is processed by Dicer and Tar RNA binding protein (TRBP) eventually producing a miR duplex containing the mature 5p and 3p miR strands. In the nomenclature of miRs, the additions of ‘-5p’ or ‘-3p’ behind the specific miR number (e.g., miR-21-5p) indicate which strand is loaded into a RNA-induced silencing complex (RISC). The RISC consists of Dicer, TRBP, and Argonaute 1 (AGO1) amongst other things and is subsequently guided towards the target mRNA [17,18]. The miR binds to the 3′ UTR of mRNA if the seed sequence-a highly conserved sequence at positions 2–7 from the miR 5′ end-is a match and has either a partial or full sequential complementarity to its target, resulting in translational repression or mRNA degradation respectively [16,17]. In this manner, miRs can modulate the expression of a wide range of proteins and are implicated in various processes like cell proliferation, apoptosis, and differentiation.
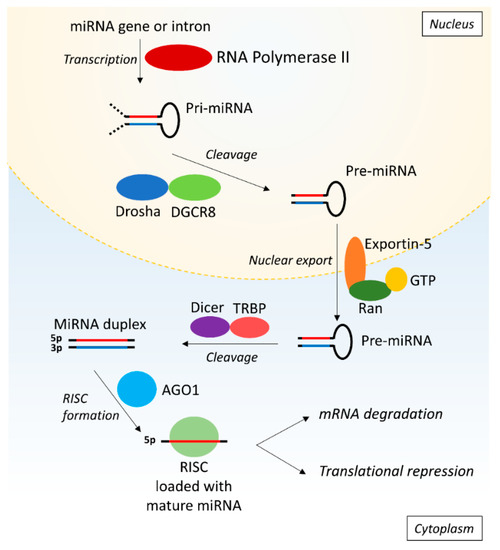
Figure 1.
MicroRNA biosynthesis. First, the pri-miR is synthesized from the DNA by RNA polymerase II and is subsequently cleaved by Drosha and DGCR8 into pre-miR. The pre-miR is exported into the cytoplasm by Exportin-5 and Ran-GTP, followed by cleavage into a miR duplex by Dicer and TRBP. One strand is loaded into RISC consisting of AGO1, Dicer and TRBP. The RISC is guided to the target mRNA where the miR induces target degradation or translational repression of the mRNA. AGO1: argonaute 1; DGCR8: Drosha/DiGeorge syndrome critical region gene 8; pre-miR: primary microRNA; pre-mRNA: precursor messenger ribonucleic acid; Ran-GTP: GTP-binding nuclear protein Ran; RISC: RNA-induced silencing complex; RNA: ribonucleic acid; TRBP: tar RNA binding protein.
1.2. miRs as Diagnostic Tools
Different sources of miRs can be used for biomarker research. For example, miRs can be extracted from plasma, serum, urine or exosomes, i.e., small membrane vesicles containing varies proteins, lipids, and nucleic acids [19]. MiRs are relatively stable in both serum and urine under a variety of storage conditions [20,21]. However, the stability of exosomal miRs isolated from plasma depends on the method of exosome isolation and storage, indicating the importance of optimized extraction methods [22]. If handled correctly, exosomal miRs seem to be more stable than non-exosomal miRs and therefore are preferred as in biomarker research [23]. Besides their diagnostic use, miRs could also serve as prognostic biomarkers to support physicians decision-making in therapy as reviewed in [24]. Conventional detection methods include polymerase chain reaction (PCR), microarrays, RNA sequencing (RNA-seq), and in situ hybridization (ISH), which will be briefly highlighted in this section [25].
PCR is the classical miR detection method and basically amplifies small segments of DNA. Besides regular PCR more specialized PCR methods are available like droplet digital PCR, enabling more sensitive and reproducible analyses [26,27]. However, these more specialized PCR techniques are labor intensive and costly, and thereby mostly used for non-clinical, small scale research. A major limitation of PCR is that it is hypothesis-driven as one can only check for a specific or small set of miRs.
Microarrays, i.e., biochips enabling high-throughput screening, overcome the limitations of PCR as this method can map miR expression profiles in a unbiased manner [28]. The method is expensive, but can simultaneously provide high throughput analyses of various microRNAs between various organs or tissues or between health and disease [28]. Because of their time-, cost-, and labor-intensive features, microarrays are unlikely to be routinely used in the clinic.
RNA-Seq is a method to isolate and sequence miRs in a high-throughput manner and is for example used to determine miR expression patterns. Unlike microarrays, which contain a specific set of known targets, RNA-Seq is not dependent on pre-chosen targets and has therefore an even wider coverage than microarrays. Although the method has disadvantages such as high costs and run length [29], the current, existing, and publicly available RNA-seq data sets could be used to overcome these hurdles. The existing datasets are often based on kidney biopsies from CKD patients, but have largely been analyzed to only evaluate protein coding genes. Therefore, it would be worthwhile to take another look at these datasets especially focusing on miRs, which could yield valuable information and at the same time reduce the cost of RNA-seq, assuming that the samples are prepared without poly(A) polymerase which is unfortunately often not the case.
ISH is a powerful method to directly detect and localize miR expression levels within tissue or individual cells. Of all miR detection techniques, ISH is the only method providing spatial information. To increase specificity and stability, locked nucleic acids probes are used for this method [30], which are RNA derivative nucleotides in which the ribose ring is locked in a specific conformation.
1.3. miRs as Therapeutic Tools
Therapeutically, miR levels have to be either raised or their binding capacity to mRNAs has to be decreased, depending on the desired effects [31,32]. Oligonucleotide manipulation techniques, including antagomiRs and locked nucleic acids, are already widely employed to inhibit miR binding to mRNA in research settings or clinical trials [31,33]. Besides miR targeting, small interfering RNAs (siRNAs) are also promising therapeutic agents [34]. SiRNAs and miRs share many similarities as both induce gene silencing at the post-transcriptional level, though their mechanisms of action are distinct. The major difference between these two short duplex RNA molecules is that siRNAs target one mRNA, whereas miRs can have multiple targets [34].
AntagomiRs are modified oligonucleotides and have characteristic complementary sequences, which extend beyond the seed sequence. They can thereby scavenge the miR away from its target mRNA [35]. Furthermore, miR expression can be inhibited by locked nucleic acids, which have an exceptional binding affinity that allows to use short sequences complementary to just the seed region of the miR [36]. Finally, miR mimics can be used to elevate miR levels [37]. These double-stranded RNA fragments act similarly to the targeted miR and bind to the same mRNA target, thereby increasing the potential to modulate the translation process [37].
Increasing miR levels with mimics or inhibiting miRs by antagomiRs is already being evaluated in clinical trials [38,39] in, for example, indications such as cancer or hepatitis C [40,41,42]. All of these trials showed some improvements in the clinical outcome of treated patients, but are still ongoing to assess their long-term efficacy and the safety of miR interference in particular [39].
2. miRs in Kidney Disease
A large number of miRs has been identified that may play a role in CKD, although the exact relevance in the majority of cases currently remains elusive. Table 3 summarizes current insights into the involvement of miRs in CKD, with a special emphasis on hypertensive nephropathy, Table 4 on diabetic kidney disease/diabetic nephropathy and Table 5 on kidney tubulointerstitial fibrosis. Additionally, Table 6 highlights the effects that these miRs have in CKD-associated CVD. The role of miRs in other kidney diseases—such as acute kidney injury, glomerulonephritides, or systemic autoimmune diseases—have been reviewed elsewhere [43,44,45,46,47], and will therefore not be discussed in the current review.
Table 3.
Overview of miRs that are involved in hypertensive nephropathy (only ex-vivo/in-vivo evidence).
Table 4.
Overview of miRs that are involved in diabetic kidney disease/diabetic nephropathy (only ex-vivo/in-vivo evidence).
Table 5.
Overview of miRs that are involved in kidney fibrosis and CKD (only ex-vivo/in-vivo evidence).
Table 6.
Overview of miRs that are involved in CKD-related CVD (only ex-vivo/in-vivo evidence).
Several studies have focused on changes in miR expression patterns during the various stages of CKD, including ESKD, kidney transplantation, and hemodialysis patients [48,49,50]. Since this topic has been covered in other reviews [51,52], it will not be further discussed here.
This review will highlight the four miRs, which, based on data of therapeutically relevant animal models and clinical studies, have been identified as the most promising ones to be used in the clinic in the future as diagnostic or even therapeutic tools in the context of CKD and associated CVD.
2.1. miR-103a-3p
Recently, Lu et al. investigated the role of miR-103a-3p in angiotensin-II-induced kidney inflammation and fibrosis [55]. Angiotensin-II exerts vasoconstrictive effects and pro-inflammatory actions on post-glomerular arteries, resulting in glomerular injury and kidney fibrosis [133,134]. Although the pathological role of angiotensin-II in hypertensive nephropathy is well established [135], the underlying molecular mechanisms remain poorly understood. Over the last years, some miRs have been associated with hypertensive nephropathy (Table 3), although most of these studies only demonstrated changes in miR expression upon disease development and progression and did not elucidate whether miRs themselves have a causal role in disease development. Comparing patients with hypertensive nephropathy to normotensive healthy controls, Lu et al. demonstrated that both serum and urine miR-103a-3p levels were significantly higher in hypertensive nephropathy patients [55]. Additionally, when mice were injected with different doses of angiotensin-II, this increased urine and serum miR-103a-3p levels independently of effects on arterial blood pressure.
To further study the role of miR-103a-3p in angiotensin-II-induced kidney injury, mice that either overexpressed miR-103a-3p by using a recombinant adeno-associated virus or mice with a knockdown of miR-103a-3p by the means of a locked nucleic acid-anti-miR-103a-3p were investigated [55]. Overexpression of miR-103a-3p in angiotensin-II-infused mice resulted in albuminuria, kidney inflammation, and kidney fibrosis, whereas knockdown of miR-103a-3p resulted in decreased angiotensin-II-induced effects on the kidney. These effects were mediated by SNRK, which was identified as a novel target of miR-103a-3p. In line with the fact that miR-103a-3p downregulates Snrk expression, kidneys of patients with hypertensive nephropathy expressed lower levels of SNRK compared to control kidneys. Further supporting this mechanism, it was shown that angiotensin-II infusion in mice reduced SNRK levels in a miR-103a-3p dependent manner.
All in all, this study identified a novel miR-103a-3p/SNRK dependent mechanism by which angiotensin-II induces kidney injury. Although the role of miR-103a-3p in the development of hypertensive nephropathy has been clearly demonstrated, it would be interesting to perform further studies to elucidate whether miR-103a-3p knockdown in already established hypertensive nephropathy can prevent further progression of the disease. Such studies would be required to validate miR-103a-3p as new therapeutic target in hypertensive and potentially other nephropathies.
2.2. mir-192-5p
One essential miR, involved in the development of diabetic kidney disease (Table 4), is miR-192-5p. Overexpression of miR-192-5p in proximal tubular epithelial cells suppressed the expression of ZEB1 (also known as δEF1) and ZEB2 (also known as SIP1) in-vitro, thereby opposing the TGF-β-mediated downregulation of E-cadherin [89]. Further supporting the role of miR-192-5p in fibrosis is the finding that increased abundance of miR-192-5p reduced the expression of ZEB1/2 and thereby increased collagen 1 alpha 2 (Col1a2) expression in mesangial cells [85].
The in-vivo relevance of the above findings was confirmed in mouse models of type 1 (streptozotocin (STZ)-injection) and type 2 (db/db) diabetes. Compared to non-diabetic control mice, the expression of miR-192-5p was significantly increased in isolated glomeruli from diabetic mice, and this coincided with increased levels of TGF-β1 and Col1a2 [85]. TGF-β1-induced miR-192-5p expression is also relevant in the context of glomerular mesangial cell survival and hypertrophy [87]. These effects are mediated by a complex mechanism in which reduced ZEB2 resulted in increased expression of miR-216a-5p/miR-217-5p, thereby inhibiting phosphatase and tensin homolog (PTEN) and increasing Akt expression and causing mesangial cell hypertrophy [87,136]. Interestingly, it could also be observed that a positive feedback loop exists in this system as miR-192-5p upregulated TGF-β1 [91]. This feedback loop in turn was at least partly driven by miR-200b/c-3p, which was identified as a downstream target of miR-192-5p.
The in-vivo relevance of miR-192-5p in diabetic nephropathy was assessed further in mice with a genetic deletion of miR-192-5p [90]. STZ-injected miR-192-5p deficient mice displayed attenuated kidney TGF-β1 expression and had reduced kidney fibrosis, glomerular hypertrophy, and albuminuria compared to diabetic wild-type mice [90]. To evaluate the therapeutic potential of targeting miR-192-5p, locked nucleic acid-anti-miR-192-5p was tested in a mouse model of STZ-induced diabetic nephropathy [86]. In the kidneys of diabetic mice, inhibition of miR-192-5p significantly increased ZEB1/2 expression, while decreasing the expression of TGF-β1, collagen, and fibronectin, resulting in less kidney fibrosis and attenuated albuminuria. This further supports the notion that locked nucleic acid-anti-miR-192-5p is an interesting approach for the prevention or perhaps even treatment of diabetic kidney disease [86].
Interestingly, in contrast to the animal studies described above, in a cohort of patients with established diabetic kidney disease, miR-192-5p expression was lowest in kidney biopsies of patients with advanced diabetic kidney disease [89]. In addition, low levels of kidney miR-192-5p correlated with tubulointerstitial fibrosis and impaired eGFR [89]. Analyses of extracellular vesicles isolated from urine of patients with type 2 diabetes demonstrated that the expression of miR-192-5p was significantly increased in microalbuminuric patients compared to normoalbuminuric diabetics and healthy controls. Notably, miR-192-5p expression levels dropped significantly again in macroalbuminuric patients. These findings suggest that miR-192-5p could also play a disease stage-dependent role in diabetic kidney disease, which needs further confirmation [88]. This could explain the apparently controversial findings between the in vivo animal studies and the human study, as the animal models present a less advanced disease stage than the patients. Furthermore, receiver operating characteristic (ROC) curve analyses revealed that miR-192-5p had an area under the curve (AUC) of 0.802 in discriminating the normoalbuminuric from microalbuminuric patients [88].
Besides its role in diabetic kidney disease, miR-192-5p has also been implicated in non-diabetic kidney fibrosis (Table 5). Kidney miR-192-5p was enriched in mice with unilateral ureteral obstruction (UUO) and rats with 5/6 nephrectomy, both models of progressive kidney fibrosis [137]. In vitro, in rat tubular epithelial cells, miR-192-5p stimulated TGF-β1-induced collagen expression [119]. Additionally, TGF-β1 also induced miR-192-5p expression, which was shown to be mediated via Smad3. Finally, in-vitro studies revealed that miR-192-5p targeting may have therapeutic potential, as treatment of rat tubular epithelial cells with a miR-192-5p inhibitor blocked TGF-β1-induced collagen production [119]. Taken together, these data suggest that miR-192-5p might play a causal role in non-diabetic kidney fibrosis and CKD as well.
2.3. miR-29 Family
The miR-29 family consists of three members, miR-29a-3p, miR-29b-3p and miR-29c-3p. While all three family members share a common seed sequence, they also have unique sequence features causing functional differences [138]. In a mouse model of early STZ-induced diabetic nephropathy, the kidney expression of all three miR-29 family members was decreased compared to non-diabetic controls [79]. In advanced diabetic nephropathy, induced by uninephrectomy plus STZ-injection in rats, both kidney miR-29a-3p and miR-29c-3p expression was significantly decreased [79]. MiR-29 family members repress the expression of TGF-β1 and the collagen genes col1a2 and col4a1 in proximal tubular cells, podocytes, as well as mesangial cells [79]. As for miR-192-5p, the actions of miR-29 family members are mediated via Smad3, and knockdown of miR-29b-3p in mesangial cells enhanced TGF-β1/Smad3-mediated fibrosis in-vitro [77]. The decreased kidney miR-29b-3p expression diabetic nephropathy mice (db/db) also coincided with increased microalbuminuria, kidney fibrosis, and kidney inflammation [77]. In line with this and strongly supporting a role of miR-29b-3p in DN, overexpression of miR-29b-3p by ultrasound-microbubble-mediated gene transfer normalized microalbuminuria and attenuated kidney injury [77]. In contrast to the animal studies, a human study in two independent cohorts of diabetic patients demonstrated that the urinary expression of miR-29b-3p was increased in diabetic kidney disease patients compared to control subjects [76]. This further highlights the importance of the location at which expression is measured, as in all the animal studies miR-29 expression was measured in kidney tissue while in the human cohort its expression was measured in urine samples.
MiR-29 family members also play a role in the development of non-diabetic CKD. In different CKD models (adenine nephropathy, UUO) miR-29 expression decreased upon disease development [79,109,110]. As in experimental diabetic nephropathy, overexpression of miR-29b-3p using ultrasound-microbubble-mediated gene transfer again prevented intrakidney collagen III upregulation and kidney fibrosis in a mouse model of UUO [109]. These beneficial effects were confirmed by intramuscular injection of exosomes containing miR-29a/b/c-3p into mice with UUO, resulting in an attenuation of kidney fibrosis and decreased blood urea levels, which again seemed to be mediated by reduced TGF-β3 and collagen production [111].
2.4. miR-21-5p
The role of miR-21-5p in diabetic kidney disease was supported by studies that showed increased expression of miR-21-5p in serum or kidney of diabetic kidney disease patients or rodents with diabetic nephropathy [62,64,65,66,68,69]. This increased expression in diabetic kidney disease and diabetic nephropathy was correlated with tubulointerstitial fibrosis, kidney injury, and lower eGFR [62,64,65,68,69].
In-vivo targeting of miR-21-5p using locked nucleic acid, ultrasound-microbubble-mediated gene transfer or lentiviral vectors has been used to validate its role in the development of DN. Knockdown of miR-21-5p by using locked nucleic acid-anti-miR-21-5p in STZ-induced murine diabetes, resulted in decreased interstitial fibrosis due to reduced expression of the collagen genes Col1a2 and Col III [69]. MiR-21-5p knockdown also ameliorated kidney inflammation, namely infiltration by F4/80 positive macrophages, possibly via reducing the expression of monocyte chemotactic protein-1 (MCP-1), a well-known chemokine that mainly attracts monocytes and macrophages to sites of injury [69]. Additionally, miR-21-5p silencing prevented podocyte loss and reduced albuminuria compared to control mice [69]. Comparable results were obtained using an approach, where miR-21-5p was knocked down using ultrasound-microbubble-mediated gene transfer [65]. Transfer of miR-21-5p knockdown plasmids into kidneys of diabetic db/db mice ameliorated kidney fibrosis as well as overexpression of TGF-β1, collagen I and IV. Furthermore, expression of the pro-inflammatory cytokines tumor necrosis factor-α (TNF-α) and MCP-1 was reduced upon miR-21-5p knockdown, which coincided with a reduced infiltration of F4/80 positive macrophages [65].
Using various in-vitro studies, Smad7 was identified as a key underlying regulator of both miR-21-5p induced fibrosis and inflammation [65]. Overexpression of miR-21-5p in rat tubular epithelial cells and mesenchymal cells decreased Smad7 expression, while knockdown of miR-21-5p restored Smad7 levels [65]. Furthermore, knockdown of Smad7 in-vitro increased the expression of inflammatory cytokines, collagen, and fibronectin, confirming that Smad7 plays a protective role in kidney fibrosis and inflammation [65]. Several other studies confirmed these effects of miR-21-5p on kidney fibrosis and inflammation in diabetic nephropathy models, for example, inhibition of miR-21-5p in a mouse model (KK-Ay mice) by lentiviral vectors [67] and a mouse (KK-Ay mice) and rat (STZ injection) model in which suppression of miR-21-5p via antagomiR-21-5p reduced kidney fibrosis and inflammation [66,68]. These studies targeting miR-21-5p clearly establish the potential to halt experimental diabetic nephropathy but it is unknown so far whether targeting of miR-21-5p also has therapeutic potential for prevention or reversal of DN.
The impact of miR-21-5p on non-diabetic kidney fibrosis has been tested in the UUO and the unilateral ischemia reperfusion injury (IRI) model using mice genetically lacking miR-21-5p or treated with anti-miR-21-5p oligonucleotides [107]. The lack or silencing of miR-21-5p in both models ameliorated albuminuria, kidney fibrosis and injury [107]. Similarly, ultrasound-microbubble-mediated gene transfer of miR-21-5p-knockdown plasmids in mice with UUO reduced the development of kidney fibrosis [106]. Finally, the therapeutic potential of this targeting approach was assessed by knocking down miR-21-5p in established kidney fibrosis by injecting the miR-21-5p knockdown plasmid 4 days after UUO. Under these circumstances, miR-21-5p-knockdown also halted the progression of fibrosis [106], suggesting that miR-21-5p targeting may become a new therapeutic option to combat kidney fibrosis.
Alport syndrome is a genetic disorder caused by inherited mutations in COL4A3, COL4A4 and COL4A5 genes. Its key features include hearing loss, glomerulonephritis and ESKD [139]. Mice deficient in Col4a3 have been used as a murine model for Alport nephropathy [140]. The expression level of miR-21-5p was elevated in the kidney, glomeruli, and tubules of these mice compared to controls. Additionally, miR-21-5p expression was increased from 3 weeks of age in Col4a3−/− kidneys and increased further along with disease progression. In the same study, Col4a3−/− mice were injected subcutaneously with anti-miR-21 oligonucleotides [140]. Inhibition of miR-21-5p resulted in significantly lower albuminuria and blood urea nitrogen (BUN), which reflect improved kidney function, as compared to vehicle control. Notably, anti-miR-21 treatment prolonged median survival of mice by 32 days and did not show any adverse side-effects. Moreover, miR-21-5p silencing enhanced PPARα/retinoid X receptor (PPARα/RXR) activity, which was likely the cause of the observed protection against TGF-β-induced fibrogenesis and inflammation in glomerular and interstitial cells [140]. Tubular function was also preserved by miR-21-5p silencing by enhanced mitochondrial function resulting in reduced ROS production [140]. Another pre-clinical study showed the association of miR-21-5p with Alport syndrome in dogs. Dogs suffering from X-linked hereditary nephropathy (XLHN) were used in this study as it is the equivalent to human Alport syndrome. Kidney samples were harvested at several time points during disease progression, including ESKD. In XLHN dogs, kidney miR-21-5p was first significantly upregulated at the time point at which serum creatinine was increased [141]. Additionally, kidney miR-21-5p expression levels were correlated with clinical markers of kidney function. More specifically, miR-21-5p expression was positively correlated with the serum creatinine concentration, whilst it was negatively correlated with GFR [141]. The expression pattern of miR-21-5p has also been examined in the kidneys of Alport syndrome patients [142]. The miR expression was significantly increased in kidney samples from Alport syndrome patients compared to controls and miR-21-5p levels positively correlated with serum BUN, serum creatinine, proteinuria, and overall severity of kidney pathology. MiR-21-5p was also shown to be localized to damaged tubular epithelial cells and glomeruli. Furthermore, human kidney samples of patients with Alport syndrome showed abnormal expression of genes involved in fibrosis, mitochondrial function, inflammation, and kidney injury [142]. Worth mentioning are the clinical trials focusing on miR-21-5p in Alport syndrome. A phase I and phase II clinical trial included the usage of the drugs RG012 and SAR339375, respectively to target miR-21-5p. Despite their potential and interesting set-up, the trials were delayed due to re-organization within companies and have yet to publish results (NCT03373786 and NCT02855268). Overall, both animal and human studies suggest that miR-21-5p plays a pivotal role in the progression of CKD caused by Alport syndrome. It also suggests that therapeutic interference of this miR may become a new way of battling the consequences of Alport syndrome.
Besides these kidney effects, miR-21-5p also impacts CKD-related cardiovascular effects (Table 6). For example, in 5/6 nephrectomized rats concentric left-ventricular hypertrophy, not driven by volume expansion, ensued within 4 to 5 weeks after CKD induction [130]. Additionally, the lipid content in left ventricular tissue increased at week 7 after surgery. Although there was no overall left ventricular fibrosis, perivascular fibrosis increased at 7 weeks post CKD induction. At this time point, small RNA deep sequencing in left ventricular tissue revealed an upregulation of miR-21-5p. To study the significance of increased left ventricular miR-21-5p expression, anti-miR-21-5p oligonucleotides were injected intravenously into 5/6-nephrectomized and sham operated rats at week 1 and 4 post-surgery. Suppression of miR-21-5p reduced left ventricular wall thickening and improved cardiac function, without significant effects on blood pressure or the kidney pathology, indicating that the changes in cardiac phenotype were not driven by a reduction in kidney injury [130]. This lack of kidney effects of miR-21-5p suppression is in sharp contrast with the kidney studies discussed above and is likely caused by substantial differences in disease models and treatment protocols.
Further analysis revealed altered peroxisome proliferator-activated receptor alpha (PPARα) expression in the left ventricle upon miR-21-5p suppression. Involvement of PPARα was further demonstrated by delivering a low dose of the PPARα agonist clofibrate via daily injections. Clofibrate treatment in nephrectomized rats prevented left ventricular hypertrophy and improved cardiac function, similar to rats treated with anti-miR-21-5p. This study therefore unveiled a new pathway involved in mediating CKD-related cardiovascular effects.
3. Limitations and Future Perspectives
In this review, we discussed the role of miRNA in CKD and highlighted four miRs that have the potential to serve as novel biomarkers and therapeutic targets in CKD and associated CVDs. Besides these specific miRs, many more miRs have been demonstrated to play a role in CKD. Although miRs are currently not yet being used in such a manner in the clinic, this review highlights their enormous potential and thereby the need for further pre-clinical and especially clinical research in this field. However, before miRs can be routinely used as biomarkers or therapeutics in the clinic, first some crucial aspects need to be addressed in future research.
One important aspect which should be taken into consideration is the fact that the number of miRs varies between species. The human genome contains more than 2500 miRs, while rats have less than 1000 and mice a little less than 2000 miRs [143]. In order to enable proper translation into the clinic, one should already in a pre-clinical stage select the miRs, which are highly conserved between species.
Furthermore, it is necessary to determine the optimal miR detection method and in which type of sample the miRs should be measured. As shown before, different methods are being used—e.g., RT-PCR, RNA-seq, or ISH—which all have specific advantages and disadvantages. Additionally, studies have either used serum, plasma, or urine samples or even only isolated exosomes from these liquids. In order to optimize miR use as a biomarker, pre-clinical studies should focus on all of these potential sources to select the most optimal one to be validated in the clinic. On another note, in kidney diseases urine seems to have a great advantage over blood as a source for biomarkers. Urine has direct access to the damaged tissues of the kidney and urinary tract, making it a closer, non-invasive source of biomarkers than peripheral blood.
Moreover, a common note is the wide range of targets of miRs. A single miR is known to be able to regulate several mRNA targets and a single mRNA target can be regulated by several miRs. These targets are regulated and expressed in a tissue- and even cell-dependent manner. Thus, if one would target these miRs with mimics or antagomiRs, the chances of inducing off-target effects would be greatly increased as compared to standard drugs. To reduce these side-effects researchers should focus on methods to deliver miR therapeutics to specific tissues or even cells. Currently, carefully engineered nanoparticles are being studied for this purpose. These particles are coated with antigens that can be recognized by receptors specifically expressed on the targeted cells. Although promising, nanoparticle treatment needs to be optimized as some obstacles, such as low efficiencies in cell internalization and in the release of antagomiRs into the cytoplasm, remain [144].
Another note of caution for researchers is the variability between patients. Variables such as age, gender, and ethnicity can already have an impact on the biomarker profile and response to therapy. Other variables such as the use of medication or having comorbidities could also affect biomarker results or treatment efficiency. Furthermore, related to therapies, disease conditions have been shown to increase pharmacological effects of antagomiRs [145]. The disease severity and type could therefore influence therapeutic actions of miR-related therapies. However, these variables might actually make the use of miRs as biomarkers and therapeutics highly attractive, as it adds to a common goal in medicine: personalized medicine.
Besides the limitations mentioned above, our review unveiled another area in CKD-related miR research that needs to be addressed. Figure 2 provides an overview of the miRs already studied in kidney fibrosis, CKD (including diabetic and hypertensive) and CKD-related CVD. As clearly visible in the figure, miR research has primarily focussed on DKD, kidney fibrosis and CKD, while CKD-related CVD and HKD have received comparably little attention up until now. Future studies should therefore focus on these neglected areas, especially since CVD and CKD are interlinked with one another and represent a high burden on global health. Another interesting aspect to highlight in Figure 2 is the overlap between the subtypes of CKD and CKD-related CVD, like for example miR-21-5p which is involved in all four described pathologies. It is worthwhile to research miRs that could connect two or more CKD subtypes, as it might unveil important common targets, underlying pathways or biomarkers.
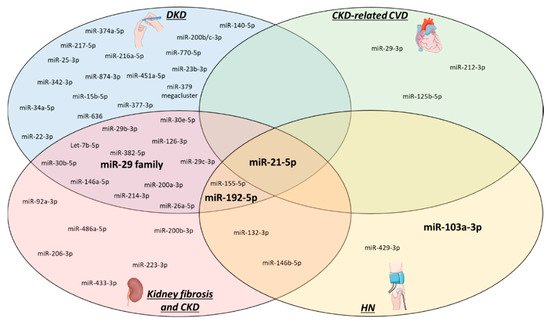
Figure 2.
CKD-related microRNAs. All miRs that have until now been studied in CKD (ex-vivo or in-vivo models only) are depicted based on their relation to the specific pathologies. From all of these, the four relevant miRs that are highlighted in this review are visualized in bold. HN: hypertensive nephropathy, DKD: diabetic kidney disease, CVD: cardiovascular disease, CKD: chronic kidney disease.
4. Conclusions
Various therapies are being employed to slow down the progression of CKD [146]. However, none of the current therapies completely halt progression and none are capable of reversing the disease [147]. Although, miRs are currently not yet being used in the clinic, this review highlights four miRs, which have the potential for clinical application in the near future like the antagonism of miR-21-5p in diabetic nephropathy or the locked nucleic acid-anti-miR-103a-3p in hypertensive nephropathy. However, these studies investigate the effect of miR modulation on the initial development of CKD, rather than the real therapeutic effects of miR modulation on already established diseases, but clearly demonstrate the potential of miR modulation using agomiRs and antagomiRs. Currently, better methods for synthesis and delivery of miR are being developed to provide safer and more accurate delivery, such as lipid-based nanoparticles, FDA-approved poly-lactic-co-glycolic acid (PLGA)-based nanoparticles and ultrasound microbubble-mediated gene transfer [148,149]. Such methods are required to enable human clinical trials based on miR targeting strategies to fully evaluate its therapeutic potential.
All in all, it has been suggested that miRs can play an important role in a clinical diagnostic setting, although validation studies are still required [150]. Therefore, MiRs might have a significant beneficial impact on present-day diagnostic and therapeutic strategies and they bear great potential in CKD and associated CVD in the near future.
Funding
The authors’ research is supported by a grant from the Interdisciplinary Center for Clinical Research within the faculty of Medicine at the RWTH Aachen University, the DZHK (German Centre for Cardiovascular Research) and the BMBF (German Ministry of Education and Research), and NWO-ZonMw Veni (91619053) to E.P.C.v.d.V and by the DFG (SFB/TRR219 TP C-04, S-03) to J.J. and by the DFG (SFB/TRR219 TP C-01, M-01) to J.F.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Foreman, K.J.; Marquez, N.; Dolgert, A.; Fukutaki, K.; Fullman, N.; McGaughey, M.A.; Pletcher, M.E.; Smith, A.; Tang, K.; Yuan, C.-W.; et al. Forecasting life expectancy, years of life lost, and all-Cause and cause-Specific mortality for 250 causes of death: Reference and alternative scenarios for 2016-40 for 195 countries and territories. Lancet 2018, 392, 2052–2090. [Google Scholar] [CrossRef]
- Inker, L.A.; Astor, B.C.; Fox, C.H.; Isakova, T.; Lash, J.P.; Peralta, C.A.; Kurella Tamura, M.; Feldman, H.I. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for the evaluation and management of CKD. American journal of kidney diseases. Off. J. Natl. Kidney Found 2014, 63, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Mullins, L.J.; Conway, B.R.; Menzies, R.I.; Denby, L.; Mullins, J.J. Renal disease pathophysiology and treatment: Contributions from the rat. Dis. Models Mech. 2016, 9, 1419–1433. [Google Scholar] [CrossRef] [PubMed]
- Couser, W.G.; Remuzzi, G.; Mendis, S.; Tonelli, M. The contribution of chronic kidney disease to the global burden of major noncommunicable diseases. Kidney Int. 2011, 80, 1258–1270. [Google Scholar] [CrossRef]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef]
- Cruz, D.N.; Bagshaw, S.M. Heart-kidney interaction: Epidemiology of cardiorenal syndromes. Int. J. Nephrol. 2010, 2011, 351291. [Google Scholar] [CrossRef]
- Ronco, C.; Di Lullo, L. Cardiorenal Syndrome in Western Countries: Epidemiology, Diagnosis and Management Approaches. Kidney Dis. 2017, 2, 151–163. [Google Scholar] [CrossRef]
- Uduman, J. Epidemiology of Cardiorenal Syndrome. Adv. Chronic Kidney Dis. 2018, 25, 391–399. [Google Scholar] [CrossRef]
- Reiss, A.B.; Voloshyna, I.; De Leon, J.; Miyawaki, N.; Mattana, J. Cholesterol Metabolism in CKD. American journal of kidney diseases. Off. J. Natl. Kidney Found. 2015, 66, 1071–1082. [Google Scholar] [CrossRef]
- Burlacu, A.; Genovesi, S.; Basile, C.; Ortiz, A.; Mitra, S.; Kirmizis, D.; Kanbay, M.; Davenport, A.; van der Sande, F.; Covic, A. ERA-EDTA, EWGo: Coronary artery disease in dialysis patients: Evidence synthesis, controversies and proposed management strategies. J. Nephrol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.; James, M.; Wiebe, N.; Hemmelgarn, B.; Manns, B.; Klarenbach, S.; Tonelli, M. Cause of Death in Patients with Reduced Kidney Function. J. Am. Soc. Nephrol. 2015, 26, 2504–2511. [Google Scholar] [CrossRef] [PubMed]
- Lekawanvijit, S. Cardiotoxicity of Uremic Toxins: A Driver of Cardiorenal. Syndr. Toxins 2018, 10, 352. [Google Scholar] [CrossRef] [PubMed]
- Hazzan, A.D.; Halinski, C.; Agoritsas, S.; Fishbane, S.; DeVita, M.V. Epidemiology and Challenges to the Management of Advanced CKD. Adv. Chronic Kidney Dis. 2016, 23, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Al Rubeaan, K.; Siddiqui, K.; Al-Ghonaim, M.A.; Youssef, A.M.; Al-Sharqawi, A.H.; AlNaqeb, D. Assessment of the diagnostic value of different biomarkers in relation to various stages of diabetic nephropathy in type 2 diabetic patients. Sci. Rep. 2017, 7, 2684. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Bartel, B. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Macfarlane, L.-A.; Murphy, P.R. MicroRNA: Biogenesis, Function and Role in Cancer. Curr. Genom. 2010, 11, 537–561. [Google Scholar] [CrossRef]
- Wang, M.; Yu, F.; Ding, H.; Wang, Y.; Li, P.; Wang, K. Emerging Function and Clinical Values of Exosomal MicroRNAs in Cancer. Mol. Ther. Nucleic Acids 2019, 16, 791–804. [Google Scholar] [CrossRef]
- Chen, X.; Ba, Y.; Ma, L.; Cai, X.; Yin, Y.; Wang, K.; Guo, J.; Zhang, Y.; Chen, J.; Guo, X.; et al. Characterization of microRNAs in serum: A novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008, 18, 997–1006. [Google Scholar] [CrossRef]
- Mall, C.; Rocke, D.M.; Durbin-Johnson, B.; Weiss, R.H. Stability of miRNA in human urine supports its biomarker potential. Biomark. Med. 2013, 7, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Rubio, D.; Martin-Burriel, I.; Gil, A.; Cubero, P.; Forner, M.; Khalyfa, A.; Marin, J.M. Stability of Circulating Exosomal miRNAs in Healthy Subjects. Sci. Rep. 2018, 8, 10306. [Google Scholar] [CrossRef] [PubMed]
- Nik, M.K.; Shahidan, N. WNS: Non-Exosomal and Exosomal Circulatory MicroRNAs: Which Are More Valid as Biomarkers? Front. Pharmacol. 2019, 10, 1500. [Google Scholar]
- de Gonzalo-Calvo, D.; Vea, A.; Bar, C.; Fiedler, J.; Couch, L.S.; Brotons, C.; Llorente-Cortes, V.; Thum, T. Circulating non-coding RNAs in biomarker-guided cardiovascular therapy: A novel tool for personalized medicine? Eur. Heartj. 2019, 40, 1643–1650. [Google Scholar] [CrossRef]
- Dave, V.P.; Ngo, T.A.; Pernestig, A.K.; Tilevik, D.; Kant, K.; Nguyen, T.; Wolff, A.; Bang, D.D. MicroRNA amplification and detection technologies: Opportunities and challenges for point of care diagnostics. Lab. Investig. J. Tech. Methods Pathol. 2019, 99, 452–469. [Google Scholar] [CrossRef]
- Robinson, S.; Follo, M.; Haenel, D.; Mauler, M.; Stallmann, D.; Tewari, M.; Duerschmied, D.; Peter, K.; Bode, C.; Ahrens, I.; et al. Droplet digital PCR as a novel detection method for quantifying microRNAs in acute myocardial infarction. Int. J. Cardiol. 2018, 257, 247–254. [Google Scholar] [CrossRef]
- Kramer, M.F. Stem-loop RT-qPCR for miRNAs. Curr. Protoc. Mol. Biol. 2011, 95, 15.10.1–15.10.15. [Google Scholar] [CrossRef]
- Kaliyappan, K.; Palanisamy, M.; Govindarajan, R.; Duraiyan, J. Microarray and its applications. J. Pharm. Bioallied Sci. 2012, 4, S310–S312. [Google Scholar] [CrossRef]
- Motameny, S.; Wolters, S.; Nurnberg, P.; Schumacher, B. Next Generation Sequencing of miRNAs—Strategies, Resources and Methods. Genes 2010, 1, 70–84. [Google Scholar] [CrossRef]
- Andachi, Y.; Kohara, Y.; Okamura, K.; Nakanishi, K. MicroRNA Detection by Whole-Mount In Situ Hybridization in C. elegans. In Advanced Structural Safety Studies; Springer Science and Business Media LLC: Berlin, Germany, 2017; Volume 1680, pp. 75–86. [Google Scholar]
- Broderick, J.A.; Zamore, P.D. MicroRNA therapeutics. Gene Ther. 2011, 18, 1104–1110. [Google Scholar] [CrossRef]
- Huang, C.K.; Kafert-Kasting, S.; Thum, T. Preclinical and Clinical Development of Noncoding RNA Therapeutics for Cardiovascular Disease. Circ. Res. 2020, 126, 663–678. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.; Hossain, G.S.; Kocerha, J. The Potential for microRNA Therapeutics and Clinical Research. Front. Genet. 2019, 10, 478. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.K.; Chow, M.Y.; Zhang, Y.; Leung, S.W. siRNA Versus miRNA as Therapeutics for Gene Silencing. Mol. Ther. Nucleic Acids 2015, 4, e252. [Google Scholar] [CrossRef]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Obad, S.; dos Santos, C.O.; Petri, A.; Heidenblad, M.; Broom, O.; Ruse, C.; Fu, C.; Lindow, M.; Stenvang, J.; Straarup, E.M.; et al. Silencing of microRNA families by seed-targeting tiny LNAs. Nat. Genet. 2011, 43, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Nogimori, T.; Furutachi, K.; Ogami, K.; Hosoda, N.; Hoshino, S. A novel method for stabilizing microRNA mimics. Biochem. Biophys. Res. Commun. 2019, 511, 422–426. [Google Scholar] [CrossRef]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Doss, C.G.P.; Lee, S.-S. Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol. Ther. Nucleic Acids 2017, 8, 132–143. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203. [Google Scholar] [CrossRef]
- Janssen, H.L.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Rodriguez-Torres, K.; van der Meer, A.J.; van der Meer, A.K.; Chen, A.; Zhou, Y.; Zhou, R.; et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef]
- Beg, M.S.; Brenner, A.; Sachdev, J.; Ejadi, S.; Borad, M.; Kang, Y.-K.; Lim, H.; Kim, T.-Y.; Bader, A.; Stoudemire, J.; et al. Abstract C43: Safety, tolerability, and clinical activity of MRX34, the first-in-class liposomal miR-34 mimic, in patients with advanced solid tumors. Clin. Trials 2015, 14. [Google Scholar] [CrossRef]
- Hong, D.S.; Kang, Y.; Brenner, A.J.; Sachdev, J.C.; Ejadi, S.; Borad, M.J.; Kim, T.-Y.; Lim, H.Y.; Park, K.; Becerra, C.; et al. MRX34, a liposomal miR-34 mimic, in patients with advanced solid tumors: Final dose-escalation results from a first-in-human phase I trial of microRNA therapy. J. Clin. Oncol. 2016, 34, 2508. [Google Scholar] [CrossRef]
- Brandenburger, T.; Salgado Somoza, A.; Devaux, Y.; Lorenzen, J.M. Noncoding RNAs in acute kidney injury. Kidney Int. 2018, 94, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Ledeganck, K.J.; Gielis, E.M.; Abramowicz, D.; Stenvinkel, P.; Shiels, P.G.; Van Craenenbroeck, A.H. MicroRNAs in AKI and Kidney Transplantation. Clin. J. Am. Soc. Nephrol. 2019, 14, 454–468. [Google Scholar] [CrossRef]
- Szeto, C.C.; Li, P.K. MicroRNAs in IgA nephropathy. Nat. Rev. Nephrol. 2014, 10, 249–256. [Google Scholar] [CrossRef]
- Trionfini, P.; Benigni, A. MicroRNAs as Master Regulators of Glomerular Function in Health and Disease. J. Am. Soc. Nephrol. 2017, 28, 1686–1696. [Google Scholar] [CrossRef] [PubMed]
- Trionfini, P.; Benigni, A.; Remuzzi, G. MicroRNAs in kidney physiology and disease. Nat. Rev. Nephrol. 2015, 11, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Fourdinier, O.; Schepers, E.; Metzinger-Le Meuth, V.; Glorieux, G.; Liabeuf, S.; Verbeke, F.; Vanholder, R.; Brigant, B.; Pletinck, A.; Diouf, M.; et al. European Uremic Toxin Work Group, E: Serum levels of miR-126 and miR-223 and outcomes in chronic kidney disease patients. Sci. Rep. 2019, 9, 4477. [Google Scholar] [CrossRef] [PubMed]
- Brigant, B.; Meuth, V.M.-L.; Massy, Z.A.; McKay, N.; Liabeuf, S.; Pelletier, M.; Sallée, M.; M’Baya-Moutoula, E.; Paul, P.; Drüeke, T.B.; et al. Serum microRNAs are altered in various stages of chronic kidney disease: A preliminary study. Clin. Kidney J. 2017, 10, 578. [Google Scholar] [CrossRef]
- Ulbing, M.; Kirsch, A.H.; Leber, B.; Lemesch, S.; Munzker, J.; Schweighofer, N.; Hofer, D.; Trummer, O.; Rosenkranz, A.R.; Muller, H.; et al. MicroRNAs 223-3p and 93-5p in patients with chronic kidney disease before and after renal transplantation. Bone 2017, 95, 115–123. [Google Scholar] [CrossRef]
- Metzinger-Le Meuth, V.; Metzinger, L. miR-223 and other miRNA’s evaluation in chronic kidney disease: Innovative biomarkers and therapeutic tools. Non-Coding Rna Res. 2019, 4, 30–35. [Google Scholar] [CrossRef]
- Connor, K.L.; Denby, L. MicroRNAs as non-invasive biomarkers of renal disease. Nephrol. Dial. Transplant. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Lu, C.; Qian, Y.; Li, H.; Tan, Y.; Cai, L.; Weng, H. Urinary miR-21 as a potential biomarker of hypertensive kidney injury and fibrosis. Sci. Rep. 2017, 7, 17737. [Google Scholar] [CrossRef] [PubMed]
- Lyu, H.; Li, X.; Wu, Q.; Hao, L. Overexpression of microRNA-21 mediates Ang II-induced renal fibrosis by activating the TGF-beta1/Smad3 pathway via suppressing PPARalpha. J. Pharmacol. Sci. 2019, 141, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Ma, Z.; Ding, Y.; Bedarida, T.; Chen, L.; Xie, Z.; Song, P.; Zou, M.H. Circulating miR-103a-3p contributes to angiotensin II-induced renal inflammation and fibrosis via a SNRK/NF-kappaB/p65 regulatory axis. Nat. Commun. 2019, 10, 2145. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.A.; Wang, F.; Liu, Y.; Kriegel, A.J.; Geurts, A.M.; Usa, K.; Xue, H.; Wang, D.; Kong, Y.; Liang, M. MiR-192-5p in the Kidney Protects Against the Development of Hypertension. Hypertension 2019, 73, 399–406. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, Q.; Wang, W.; Hu, J.; Li, P.-L.; Yi, F.; Li, N. Downregulation of microRNA-429 contributes to angiotensin II-induced profibrotic effect in rat kidney. Am. J. Physiol. Physiol. 2018, 315, F1536–F1541. [Google Scholar] [CrossRef]
- Park, J.T.; Kato, M.; Lanting, L.; Castro, N.; Nam, B.Y.; Wang, M.; Kang, S.W.; Natarajan, R. Repression of let-7 by transforming growth factor-beta1-induced Lin28 upregulates collagen expression in glomerular mesangial cells under diabetic conditions. Am. J. Physiol. Ren. Physiol. 2014, 307, F1390–F1403. [Google Scholar] [CrossRef]
- Wang, B.; Jha, J.C.; Hagiwara, S.; McClelland, A.D.; Jandeleit-Dahm, K.; Thomas, M.C.; Cooper, M.E.; Kantharidis, P. Transforming growth factor-beta1-mediated renal fibrosis is dependent on the regulation of transforming growth factor receptor 1 expression by let-7b. Kidney Int. 2014, 85, 352–361. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Kuo, M.C.; Hung, W.W.; Wu, L.Y.; Wu, P.H.; Chang, W.A.; Kuo, P.L.; Hsu, Y.L. High Glucose Induces Mesangial Cell Apoptosis through miR-15b-5p and Promotes Diabetic Nephropathy by Extracellular Vesicle Delivery. Mol. Ther. J. Am. Soc. Gene Ther. 2020, 28, 963–974. [Google Scholar] [CrossRef]
- Eissa, S.; Matboli, M.; Aboushahba, R.; Bekhet, M.M.; Soliman, Y. Urinary exosomal microRNA panel unravels novel biomarkers for diagnosis of type 2 diabetic kidney disease. J. Diabetes Complicat. 2016, 30, 1585–1592. [Google Scholar] [CrossRef]
- Dey, N.; Das, F.; Mariappan, M.M.; Mandal, C.C.; Ghosh-Choudhury, N.; Kasinath, B.S.; Choudhury, G.G. MicroRNA-21 orchestrates high glucose-induced signals to TOR complex 1, resulting in renal cell pathology in diabetes. J. Biol. Chem. 2011, 286, 25586–25603. [Google Scholar] [CrossRef] [PubMed]
- Zang, J.; Maxwell, A.P.; Simpson, D.A.; McKay, G.J. Differential Expression of Urinary Exosomal MicroRNAs miR-21-5p and miR-30b-5p in Individuals with Diabetic Kidney Disease. Sci. Rep. 2019, 9, 10900. [Google Scholar] [CrossRef] [PubMed]
- McClelland, A.D.; Herman-Edelstein, M.; Komers, R.; Jha, J.C.; Winbanks, C.E.; Hagiwara, S.; Gregorevic, P.; Kantharidis, P.; Cooper, M.E. miR-21 promotes renal fibrosis in diabetic nephropathy by targeting PTEN and SMAD7. Clin. Sci. 2015, 129, 1237–1249. [Google Scholar] [CrossRef]
- Zhong, X.; Chung, A.C.; Chen, H.Y.; Dong, Y.; Meng, X.M.; Li, R.; Yang, W.; Hou, F.F.; Lan, H.Y. miR-21 is a key therapeutic target for renal injury in a mouse model of type 2 diabetes. Diabetologia 2013, 56, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhao, L.; Xing, Y.; Lin, B. Down-regulation of microRNA-21 reduces inflammation and podocyte apoptosis in diabetic nephropathy by relieving the repression of TIMP3 expression. Biomed. Pharmacother. 2018, 108, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Gao, Y.B.; Zhang, N.; Zou, D.; Wang, P.; Zhu, Z.Y.; Li, J.Y.; Zhou, S.N.; Wang, S.C.; Wang, Y.Y.; et al. miR-21 overexpression enhances TGF-beta1-induced epithelial-to-mesenchymal transition by target smad7 and aggravates renal damage in diabetic nephropathy. Mol. Cell. Endocrinol. 2014, 392, 163–172. [Google Scholar] [CrossRef]
- Wang, J.; Duan, L.; Tian, L.; Liu, J.; Wang, S.; Gao, Y.; Yang, J. Serum miR-21 may be a Potential Diagnostic Biomarker for Diabetic Nephropathy. Exp. Clin. Endocrinol. Diabetes 2015, 124, 417–423. [Google Scholar] [CrossRef][Green Version]
- Kolling, M.; Kaucsar, T.; Schauerte, C.; Hubner, A.; Dettling, A.; Park, J.K.; Busch, M.; Wulff, X.; Meier, M.; Scherf, K.; et al. Therapeutic miR-21 Silencing Ameliorates Diabetic Kidney Disease in Mice. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 165–180. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, S.; Wu, D.; Liu, X.; Shi, M.; Wang, Y.; Zhang, F.; Ding, J.; Xiao, Y.; Guo, B. MicroRNA-22 Promotes Renal Tubulointerstitial Fibrosis by Targeting PTEN and Suppressing Autophagy in Diabetic Nephropathy. J. Diabetes Res. 2018, 2018, 1–11. [Google Scholar] [CrossRef]
- Zhao, B.; Li, H.; Liu, J.; Han, P.; Zhang, C.; Bai, H.; Yuan, X.; Wang, X.; Li, L.; Ma, H.; et al. MicroRNA-23b Targets Ras GTPase-Activating Protein SH3 Domain-Binding Protein 2 to Alleviate Fibrosis and Albuminuria in Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 2597–2608. [Google Scholar] [CrossRef]
- Liu, Y.; Li, H.; Liu, J.; Han, P.; Li, X.; Bai, H.; Zhang, C.; Sun, X.; Teng, Y.; Zhang, Y.; et al. Variations in MicroRNA-25 Expression Influence the Severity of Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2017, 28, 3627–3638. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zhang, Y.; Wang, Z.; Wang, L.; Wei, X.; Zhang, B.; Wen, Z.; Fang, H.; Pang, Q.; Yi, F. Regulation of NADPH oxidase activity is associated with miRNA-25-mediated NOX4 expression in experimental diabetic nephropathy. Am. J. Nephrol. 2010, 32, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.J.; Kato, M.; Deshpande, S.; Zhang, E.; Das, S.; Lanting, L.; Wang, M.; Natarajan, R.; Sadhan, D. Inhibition of the processing of miR-25 by HIPK2-Phosphorylated-MeCP2 induces NOX4 in early diabetic nephropathy. Sci. Rep. 2016, 6, 38789. [Google Scholar] [CrossRef] [PubMed]
- Koga, K.; Yokoi, H.; Mori, K.; Kasahara, M.; Kuwabara, T.; Imamaki, H.; Ishii, A.; Mori, K.P.; Kato, Y.; Ohno, S.; et al. MicroRNA-26a inhibits TGF-beta-induced extracellular matrix protein expression in podocytes by targeting CTGF and is downregulated in diabetic nephropathy. Diabetologia 2015, 58, 2169–2180. [Google Scholar] [CrossRef]
- Beltrami, C.; Simpson, K.; Jesky, M.; Wonnacott, A.; Carrington, C.; Holmans, P.; Newbury, L.; Jenkins, R.; Ashdown, T.; Dayan, C.; et al. Association of Elevated Urinary miR-126, miR-155, and miR-29b with Diabetic Kidney Disease. Am. J. Pathol. 2018, 188, 1982–1992. [Google Scholar] [CrossRef]
- Chen, H.Y.; Zhong, X.; Huang, X.R.; Meng, X.M.; You, Y.; Chung, A.C.; Lan, H.Y. MicroRNA-29b inhibits diabetic nephropathy in db/db mice. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 842–853. [Google Scholar] [CrossRef]
- Long, J.; Wang, Y.; Wang, W.; Chang, B.H.; Danesh, F.R. MicroRNA-29c is a signature microRNA under high glucose conditions that targets Sprouty homolog 1, and its in vivo knockdown prevents progression of diabetic nephropathy. J. Biol. Chem. 2011, 286, 11837–11848. [Google Scholar] [CrossRef]
- Wang, B.; Komers, R.; Carew, R.; Winbanks, C.E.; Xu, B.; Herman-Edelstein, M.; Koh, P.; Thomas, M.; Jandeleit-Dahm, K.; Gregorevic, P.; et al. Suppression of microRNA-29 expression by TGF-beta1 promotes collagen expression and renal fibrosis. J. Am. Soc. Nephrol. 2012, 23, 252–265. [Google Scholar] [CrossRef]
- Dieter, C.; Assmann, T.S.; Costa, A.R.; Canani, L.H.; De Souza, B.M.; Bauer, A.C.; Crispim, D. MiR-30e-5p and MiR-15a-5p Expressions in Plasma and Urine of Type 1 Diabetic Patients With Diabetic Kidney Disease. Front. Genet. 2019, 10, 563. [Google Scholar] [CrossRef]
- Zhang, L.; He, S.; Guo, S.; Xie, W.; Xin, R.; Yu, H.; Yang, F.; Qiu, J.; Zhang, D.; Zhou, S.; et al. Down-regulation of miR-34a alleviates mesangial proliferation in vitro and glomerular hypertrophy in early diabetic nephropathy mice by targeting GAS1. J. Diabetes Complicat. 2014, 28, 259–264. [Google Scholar] [CrossRef]
- Su, J.; Ren, J.; Chen, H.; Liu, B. MicroRNA-140-5p ameliorates the high glucose-induced apoptosis and inflammation through suppressing TLR4/NF-kappaB signaling pathway in human renal tubular epithelial cells. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef]
- Bhatt, K.; Lanting, L.L.; Jia, Y.; Yadav, S.; Reddy, M.A.; Magilnick, N.; Boldin, M.; Natarajan, R. Anti-Inflammatory Role of MicroRNA-146a in the Pathogenesis of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 2277–2288. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, Y.; Li, L.; Su, B.; Yang, L.; Fan, W.; Yin, Q.; Chen, L.; Cui, T.; Zhang, J.; et al. Involvement of inflammation-related miR-155 and miR-146a in diabetic nephropathy: Implications for glomerular endothelial injury. Bmc Nephrol. 2014, 15, 142. [Google Scholar] [CrossRef]
- Kato, M.; Zhang, J.; Wang, M.; Lanting, L.; Yuan, H.; Rossi, J.J.; Natarajan, R. MicroRNA-192 in diabetic kidney glomeruli and its function in TGF-beta-induced collagen expression via inhibition of E-box repressors. Proc. Natl. Acad. Sci. USA 2007, 104, 3432–3437. [Google Scholar] [CrossRef]
- Putta, S.; Lanting, L.; Sun, G.; Lawson, G.; Kato, M.; Natarajan, R. Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J. Am. Soc. Nephrol. 2012, 23, 458–469. [Google Scholar] [CrossRef]
- Kato, M.; Putta, S.; Wang, M.; Yuan, H.; Lanting, L.; Nair, I.; Gunn, A.; Nakagawa, Y.; Shimano, H.; Todorov, I.; et al. TGF-beta activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat. Cell Biol. 2009, 11, 881–889. [Google Scholar] [CrossRef]
- Jia, Y.; Guan, M.; Zheng, Z.; Zhang, Q.; Tang, C.; Xu, W.; Xiao, Z.; Wang, L.; Xue, Y. miRNAs in Urine Extracellular Vesicles as Predictors of Early-Stage Diabetic Nephropathy. J. Diabetes Res. 2016, 2016, 7932765. [Google Scholar] [CrossRef]
- Krupa, A.; Jenkins, R.; Luo, D.D.; Lewis, A.; Phillips, A.; Fraser, D. Loss of MicroRNA-192 promotes fibrogenesis in diabetic nephropathy. J. Am. Soc. Nephrol. 2010, 21, 438–447. [Google Scholar] [CrossRef]
- Deshpande, S.D.; Putta, S.; Wang, M.; Lai, J.Y.; Bitzer, M.; Nelson, R.G.; Lanting, L.L.; Kato, M.; Natarajan, R. Transforming growth factor-beta-induced cross talk between p53 and a microRNA in the pathogenesis of diabetic nephropathy. Diabetes 2013, 62, 3151–3162. [Google Scholar] [CrossRef]
- Kato, M.; Arce, L.; Wang, M.; Putta, S.; Lanting, L.; Natarajan, R. A microRNA circuit mediates transforming growth factor-beta1 autoregulation in renal glomerular mesangial cells. Kidney Int. 2011, 80, 358–368. [Google Scholar] [CrossRef]
- Wang, B.; Koh, P.; Winbanks, C.; Coughlan, M.T.; McClelland, A.; Watson, A.; Jandeleit-Dahm, K.; Burns, W.C.; Thomas, M.C.; Cooper, M.E.; et al. miR-200a Prevents renal fibrogenesis through repression of TGF-beta2 expression. Diabetes 2011, 60, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Park, J.T.; Kato, M.; Yuan, H.; Castro, N.; Lanting, L.; Wang, M.; Natarajan, R. FOG2 protein down-regulation by transforming growth factor-beta1-induced microRNA-200b/c leads to Akt kinase activation and glomerular mesangial hypertrophy related to diabetic nephropathy. J. Biol. Chem. 2013, 288, 22469–22480. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shen, E.; Wang, Y.; Li, J.; Cheng, D.; Chen, Y.; Gui, D.; Wang, N. Cross talk between miR-214 and PTEN attenuates glomerular hypertrophy under diabetic conditions. Sci. Rep. 2016, 6, 31506–31511. [Google Scholar] [CrossRef]
- Jiang, Z.H.; Tang, Y.Z.; Song, H.N.; Yang, M.; Li, B.; Ni, C.L. miRNA342 suppresses renal interstitial fibrosis in diabetic nephropathy by targeting SOX6. Int. J. Mol. Med. 2020, 45, 45–52. [Google Scholar]
- Yang, Z.; Guo, Z.; Dong, J.; Sheng, S.; Wang, Y.; Yu, L.; Wang, H.; Tang, L. miR-374a Regulates Inflammatory Response in Diabetic Nephropathy by Targeting MCP-1 Expression. Front. Pharmacol. 2018, 9, 900. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, Y.; Minto, A.W.; Wang, J.; Shi, Q.; Li, X.; Quigg, R.J. MicroRNA-377 is up-regulated and can lead to increased fibronectin production in diabetic nephropathy. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2008, 22, 4126–4135. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, L.J.; Xu, W.L.; Liu, S.; Yu, J.Y. MicroRNA3795p suppresses renal fibrosis by regulating the LIN28/let7 axis in diabetic nephropathy. Int. J. Mol. Med. 2019, 44, 1619–1628. [Google Scholar] [PubMed]
- Kato, M.; Wang, M.; Chen, Z.; Bhatt, K.; Oh, H.J.; Lanting, L.; Deshpande, S.; Jia, Y.; Lai, J.Y.; O’Connor, C.L.; et al. An endoplasmic reticulum stress-regulated lncRNA hosting a microRNA megacluster induces early features of diabetic nephropathy. Nat. Commun. 2016, 7, 12864. [Google Scholar] [CrossRef]
- Wang, S.; Wen, X.; Han, X.R.; Wang, Y.J.; Shen, M.; Fan, S.H.; Zhuang, J.; Zhang, Z.F.; Shan, Q.; Li, M.Q.; et al. Repression of microRNA-382 inhibits glomerular mesangial cell proliferation and extracellular matrix accumulation via FoxO1 in mice with diabetic nephropathy. Cell Prolif. 2018, 51, e12462. [Google Scholar] [CrossRef]
- Sun, Y.; Peng, R.; Peng, H.; Liu, H.; Wen, L.; Wu, T.; Yi, H.; Li, A.; Zhang, Z. miR-451 suppresses the NF-kappaB-mediated proinflammatory molecules expression through inhibiting LMP7 in diabetic nephropathy. Mol. Cell. Endocrinol. 2016, 433, 75–86. [Google Scholar] [CrossRef]
- Mohan, A.; Singh, R.S.; Kumari, M.; Garg, D.; Upadhyay, A.; Ecelbarger, C.M.; Tripathy, S.; Tiwari, S. Urinary Exosomal microRNA-451-5p Is a Potential Early Biomarker of Diabetic Nephropathy in Rats. PLoS ONE 2016, 11, e0154055. [Google Scholar] [CrossRef]
- Wang, L.; Li, H. MiR-770-5p facilitates podocyte apoptosis and inflammation in diabetic nephropathy by targeting TIMP3. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef]
- Yao, T.; Zha, N.; Gao, P.; Shui, H.; Wu, X. MiR-874 alleviates renal injury and inflammatory response in diabetic nephropathy through targeting toll-like receptor-4. J. Cell. Physiol. 2018, 234, 871–879. [Google Scholar] [CrossRef]
- Lange, T.; Artelt, N.; Kindt, F.; Stracke, S.; Rettig, R.; Lendeckel, U.; Chadjichristos, C.E.; Kavvadas, P.; Chatziantoniou, C.; Endlich, K.; et al. MiR-21 is up-regulated in urinary exosomes of chronic kidney disease patients and after glomerular injury. J. Cell. Mol. Med. 2019, 23, 4839–4843. [Google Scholar] [CrossRef]
- Zhong, X.; Chung, A.C.; Chen, H.Y.; Meng, X.M.; Lan, H.Y. Smad3-mediated upregulation of miR-21 promotes renal fibrosis. J. Am. Soc. Nephrol. 2011, 22, 1668–1681. [Google Scholar] [CrossRef]
- Chau, B.N.; Xin, C.; Hartner, J.; Ren, S.; Castano, A.P.; Linn, G.; Li, J.; Tran, P.T.; Kaimal, V.; Huang, X.; et al. MicroRNA-21 Promotes Fibrosis of the Kidney by Silencing Metabolic Pathways. Sci. Transl. Med. 2012, 4, 121ra18. [Google Scholar] [CrossRef]
- Zhang, A.; Wang, H.; Wang, B.; Yuan, Y.; Klein, J.D.; Wang, X.H. Exogenous miR-26a suppresses muscle wasting and renal fibrosis in obstructive kidney disease. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 13590–13601. [Google Scholar] [CrossRef]
- Qin, W.; Chung, A.C.; Huang, X.R.; Meng, X.M.; Hui, D.S.; Yu, C.M.; Sung, J.J.; Lan, H.Y. TGF-beta/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J. Am. Soc. Nephrol. 2011, 22, 1462–1474. [Google Scholar] [CrossRef]
- Jiang, L.; Zhou, Y.; Xiong, M.; Fang, L.; Wen, P.; Cao, H.; Yang, J.; Dai, C.; He, W. Sp1 mediates microRNA-29c-regulated type I collagen production in renal tubular epithelial cells. Exp. Cell Res. 2013, 319, 2254–2265. [Google Scholar] [CrossRef]
- Wang, H.; Wang, B.; Zhang, A.; Hassounah, F.; Seow, Y.; Wood, M.; Ma, F.; Klein, J.D.; Price, S.R.; Wang, X.H. Exosome-Mediated miR-29 Transfer Reduces Muscle Atrophy and Kidney Fibrosis in Mice. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 571–583. [Google Scholar] [CrossRef]
- Jiang, L.; Qiu, W.; Zhou, Y.; Wen, P.; Fang, L.; Cao, H.; Zen, K.; He, W.; Zhang, C.; Dai, C.; et al. microRNA-30e/mitochondrial uncoupling protein 2 axis mediates TGF-beta1-induced tubular epithelial cell extracellular matrix production and kidney fibrosis. Kidney Int. 2013, 84, 285–296. [Google Scholar] [CrossRef]
- Shang, F.; Wang, S.C.; Hsu, C.Y.; Miao, Y.; Martin, M.; Yin, Y.; Wu, C.C.; Wang, Y.T.; Wu, G.; Chien, S.; et al. MicroRNA-92a Mediates Endothelial Dysfunction in CKD. J. Am. Soc. Nephrol. 2017, 28, 3251–3261. [Google Scholar] [CrossRef]
- Bijkerk, R.; de Bruin, R.G.; van Solingen, C.; van Gils, J.M.; Duijs, J.M.; van der Veer, E.P.; Rabelink, T.J.; Humphreys, B.D.; van Zonneveld, A.J. Silencing of microRNA-132 reduces renal fibrosis by selectively inhibiting myofibroblast proliferation. Kidney Int. 2016, 89, 1268–1280. [Google Scholar] [CrossRef]
- Morishita, Y.; Imai, T.; Yoshizawa, H.; Watanabe, M.; Ishibashi, K.; Muto, S.; Nagata, D. Delivery of microRNA-146a with polyethylenimine nanoparticles inhibits renal fibrosis in vivo. Int. J. Nanomed. 2015, 10, 3475–3488. [Google Scholar] [CrossRef]
- Ichii, O.; Otsuka, S.; Sasaki, N.; Namiki, Y.; Hashimoto, Y.; Kon, Y. Altered expression of microRNA miR-146a correlates with the development of chronic renal inflammation. Kidney Int. 2012, 81, 280–292. [Google Scholar] [CrossRef]
- Paterson, M.R.; Geurts, A.M.; Kriegel, A.J. miR-146b-5p has a sex-specific role in renal and cardiac pathology in a rat model of chronic kidney disease. Kidney Int. 2019, 96, 1332–1345. [Google Scholar] [CrossRef]
- Xi, W.; Zhao, X.; Wu, M.; Jia, W.; Li, H. Lack of microRNA-155 ameliorates renal fibrosis by targeting PDE3A/TGF-beta1/Smad signaling in mice with obstructive nephropathy. Cell Biol. Int. 2018, 42, 1523–1532. [Google Scholar] [CrossRef]
- Chung, A.C.; Huang, X.R.; Meng, X.; Lan, H.Y. miR-192 mediates TGF-beta/Smad3-driven renal fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.; Jiang, L.; Zhou, Y.; Qiu, W.; Fang, L.; Tan, R.; Wen, P.; Yang, J. The miR-200 family regulates TGF-beta1-induced renal tubular epithelial to mesenchymal transition through Smad pathway by targeting ZEB1 and ZEB2 expression. Am. J. Physiol. Ren. Physiol. 2012, 302, F369–F379. [Google Scholar] [CrossRef]
- Oba, S.; Kumano, S.; Suzuki, E.; Nishimatsu, H.; Takahashi, M.; Takamori, H.; Kasuya, M.; Ogawa, Y.; Sato, K.; Kimura, K.; et al. miR-200b precursor can ameliorate renal tubulointerstitial fibrosis. PLoS ONE 2010, 5, e13614. [Google Scholar] [CrossRef]
- Zhao, S.Q.; Shen, Z.C.; Gao, B.F.; Han, P. microRNA-206 overexpression inhibits epithelial-mesenchymal transition and glomerulosclerosis in rats with chronic kidney disease by inhibiting JAK/STAT signaling pathway. J. Cell. Biochem. 2019, 120, 14604–14617. [Google Scholar] [CrossRef] [PubMed]
- Bai, M.; Chen, H.; Ding, D.; Song, R.; Lin, J.; Zhang, Y.; Guo, Y.; Chen, S.; Ding, G.; Zhang, Y.; et al. MicroRNA-214 promotes chronic kidney disease by disrupting mitochondrial oxidative phosphorylation. Kidney Int. 2019, 95, 1389–1404. [Google Scholar] [CrossRef] [PubMed]
- Denby, L.; Ramdas, V.; Lu, R.; Conway, B.R.; Grant, J.S.; Dickinson, B.; Aurora, A.B.; McClure, J.D.; Kipgen, D.; Delles, C.; et al. MicroRNA-214 antagonism protects against renal fibrosis. J. Am. Soc. Nephrol. 2014, 25, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Xie, T.; Xue, N.; Kuang, Q.; Wei, Z.; Liang, M.; Ding, X. miR-382 Contributes to Renal Tubulointerstitial Fibrosis by Downregulating HSPD1. Oxidative Med. Cell. Longev. 2017, 2017, 4708516. [Google Scholar] [CrossRef]
- Kriegel, A.J.; Liu, Y.; Cohen, B.; Usa, K.; Liu, Y.; Liang, M. MiR-382 targeting of kallikrein 5 contributes to renal inner medullary interstitial fibrosis. Physiol. Genom. 2012, 44, 259–267. [Google Scholar] [CrossRef]
- Li, R.; Chung, A.C.; Dong, Y.; Yang, W.; Zhong, X.; Lan, H.Y. The microRNA miR-433 promotes renal fibrosis by amplifying the TGF-beta/Smad3-Azin1 pathway. Kidney Int. 2013, 84, 1129–1144. [Google Scholar] [CrossRef]
- Xu, J.; Li, R.; Workeneh, B.; Dong, Y.; Wang, X.; Hu, Z. Transcription factor FoxO1, the dominant mediator of muscle wasting in chronic kidney disease, is inhibited by microRNA-486. Kidney Int. 2012, 82, 401–411. [Google Scholar] [CrossRef]
- Rana, I.; Kompa, A.R.; Skommer, J.; Wang, B.H.; Lekawanvijit, S.; Kelly, D.J.; Krum, H.; Charchar, F.J. Contribution of microRNA to pathological fibrosis in cardio-renal syndrome: Impact of uremic toxins. Physiol. Rep. 2015, 3. [Google Scholar] [CrossRef]
- Chuppa, S.; Liang, M.; Liu, P.; Liu, Y.; Casati, M.C.; Cowley, A.W.; Patullo, L.; Kriegel, A.J. MicroRNA-21 regulates peroxisome proliferator-activated receptor alpha, a molecular mechanism of cardiac pathology in Cardiorenal Syndrome Type 4. Kidney Int. 2018, 93, 375–389. [Google Scholar] [CrossRef]
- Chao, C.T.; Liu, Y.P.; Su, S.F.; Yeh, H.Y.; Chen, H.Y.; Lee, P.J.; Chen, W.J.; Lee, Y.M.; Huang, J.W.; Chiang, C.K.; et al. Circulating MicroRNA-125b Predicts the Presence and Progression of Uremic Vascular Calcification. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1402–1414. [Google Scholar] [CrossRef]
- Sarkozy, M.; Gaspar, R.; Zvara, A.; Siska, A.; Kovari, B.; Szucs, G.; Marvanykovi, F.; Kovacs, M.G.; Dioszegi, P.; Bodai, L.; et al. Chronic kidney disease induces left ventricular overexpression of the pro-hypertrophic microRNA-212. Sci. Rep. 2019, 9, 1302. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.M.; Wong, A.L.; Bezuhly, M. Modulation of angiotensin II signaling in the prevention of fibrosis. Fibrogenesis Tissue Repair 2015, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mezzano, S.; Ruiz-Ortega, M.; Egido, J. Angiotensin II and renal fibrosis. Hypertension 2001, 38, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Taal, M.W.; Brenner, B.M. Renoprotective benefits of RAS inhibition: From ACEI to angiotensin II antagonists. Kidney Int. 2000, 57, 1803–1817. [Google Scholar] [CrossRef]
- Mahimainathan, L.; Das, F.; Venkatesan, B.; Choudhury, G.G. Mesangial cell hypertrophy by high glucose is mediated by downregulation of the tumor suppressor PTEN. Diabetes 2006, 55, 2115–2125. [Google Scholar] [CrossRef]
- Nogueira, A.; Pires, M.J.; Oliveira, P.A. Pathophysiological Mechanisms of Renal Fibrosis: A Review of Animal Models and Therapeutic Strategies. In Vivo 2017, 31, 1–22. [Google Scholar] [CrossRef]
- Kwon, J.J.; Factora, T.D.; Dey, S.; Kota, J. A Systematic Review of miR-29 in Cancer. Mol. Ther. Oncolytics 2019, 12, 173–194. [Google Scholar] [CrossRef]
- Kashtan, C. Alport syndrome: Facts and opinions. F1000Res 2017, 6, 50. [Google Scholar] [CrossRef]
- Gomez, I.G.; MacKenna, D.A.; Johnson, B.G.; Kaimal, V.; Roach, A.; Ren, S.; Nakagawa, N.; Xin, C.; Newitt, R.; Pandya, S.; et al. Anti-microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J. Clin. Investig. 2014, 125, 141–156. [Google Scholar] [CrossRef]
- Clark, S.D.; Song, W.; Cianciolo, R.; Lees, G.; Nabity, M.; Liu, S. Abnormal Expression of miR-21 in Kidney Tissue of Dogs With X-Linked Hereditary Nephropathy: A Canine Model of Chronic Kidney Disease. Veter. Pathol. 2018, 56, 93–105. [Google Scholar] [CrossRef]
- Guo, J.; Song, W.; Boulanger, J.; Xu, E.Y.; Wang, F.; Zhang, Y.; He, Q.; Wang, S.; Yang, L.; Pryce, C.; et al. Dysregulated Expression of microRNA-21 and Disease-Related Genes in Human Patients and in a Mouse Model of Alport Syndrome. Hum. Gene Ther. 2019, 30, 865–881. [Google Scholar] [CrossRef]
- RNA Central. Available online: https://rnacentral.org/ (accessed on 15 August 2020).
- Gilleron, J.; Querbes, W.; Zeigerer, A.; Borodovsky, A.; Marsico, G.; Schubert, U.; Manygoats, K.; Seifert, S.; Andree, C.; Stoter, M.; et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 2013, 31, 638–646. [Google Scholar] [CrossRef]
- Eding, J.E.; Demkes, C.J.; Lynch, J.M.; Seto, A.G.; Montgomery, R.L.; Semus, H.M.; Jackson, A.L.; Isabelle, M.; Chimenti, S.; Van Rooij, E. The Efficacy of Cardiac Anti-miR-208a Therapy Is Stress Dependent. Mol. Ther. 2017, 25, 694–704. [Google Scholar] [CrossRef]
- Sarafidis, P.; Ferro, C.J.; Morales, E.; Ortiz, A.; Malyszko, J.; Hojs, R.; Khazim, K.; Ekart, R.; Valdivielso, J.M.; Fouque, D.; et al. SGLT-2 inhibitors and GLP-1 receptor agonists for nephroprotection and cardioprotection in patients with diabetes mellitus and chronic kidney disease. A consensus statement by the EURECA-m and the DIABESITY working groups of the ERA-EDTA. Nephrol. Dial. Transplant. 2019, 34, 208–230. [Google Scholar] [CrossRef]
- Stanton, R.C. Clinical Challenges in Diagnosis and Management of Diabetic Kidney Disease. Am. J. Kidney Dis. 2014, 63, S3–S21. [Google Scholar] [CrossRef]
- Bai, Z.; Wei, J.; Yu, C.; Han, X.; Qin, X.; Zhang, C.; Zhang, T.-T.; Bai, L.; Huang, W. Non-viral nanocarriers for intracellular delivery of microRNA therapeutics. J. Mater. Chem. B 2019, 7, 1209–1225. [Google Scholar] [CrossRef]
- Sirsi, S.R.; Borden, M.A. Advances in ultrasound mediated gene therapy using microbubble contrast agents. Theranostics 2012, 2, 1208–1222. [Google Scholar] [CrossRef]
- Velu, V.K.; Ramesh, R.; Srinivasan, A.R. Circulating MicroRNAs as Biomarkers in Health and Disease. J. Clin. Diagn. Res. 2012, 6, 1791–1795. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).