Neuroinflammation-Associated Alterations of the Brain as Potential Neural Biomarkers in Anxiety Disorders



Abstract
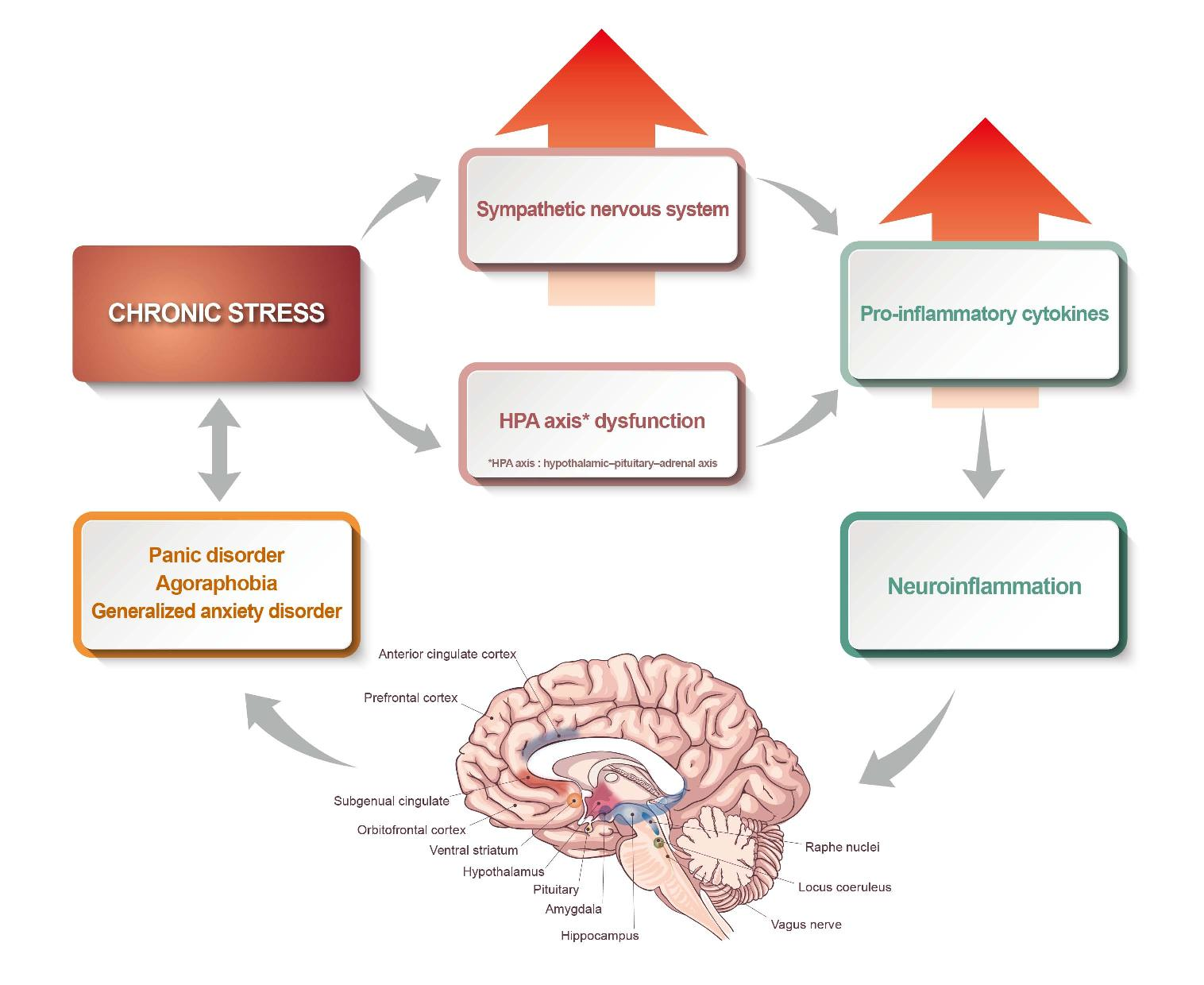
:
{kind=link}
{kind=link}
1. Introduction
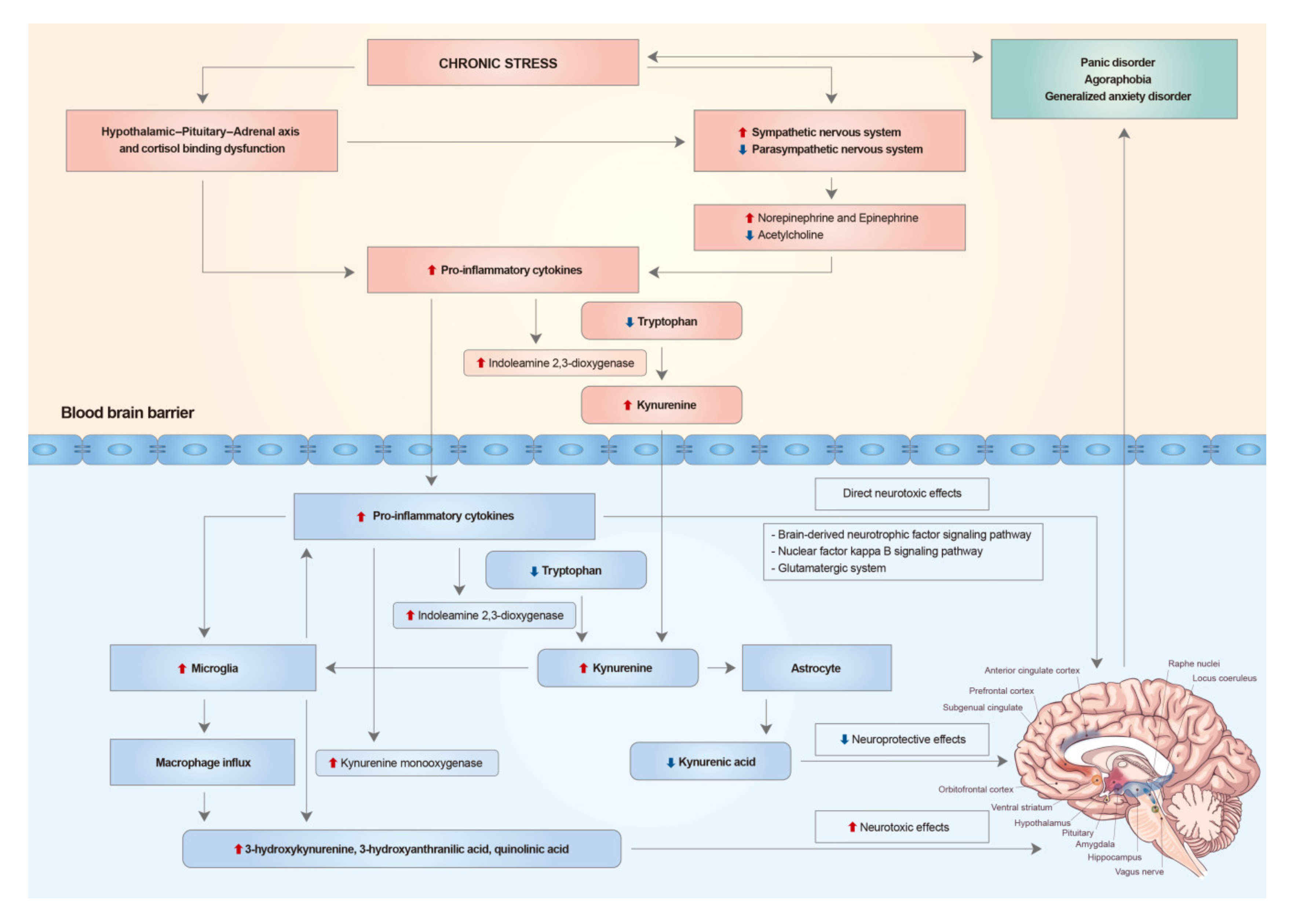
2. Chronic Stress, the Hypothalamic-Pituitary-Adrenal Axis, and the Autonomic Nervous System
3. The Immune System in Anxiety Disorders
4. Systemic Inflammation and Neuroinflammation
5. Neurotoxic Cytokine Effects on the Brain
6. Alterations of Limbic and Pre-Frontal Structures of the Brain in Anxiety Disorders
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Giacobbe, P.; Flint, A. Diagnosis and Management of Anxiety Disorders. Continuum. Minneap. Minn. 2018, 24, 893–919. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5), 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- Singewald, N.; Schmuckermair, C.; Whittle, N.; Holmes, A.; Ressler, K.J. Pharmacology of cognitive enhancers for exposure-based therapy of fear, anxiety and trauma-related disorders. Pharmacol. Ther. 2015, 149, 150–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, J.; Charney, D. Comorbidity of mood and anxiety disorders. Depress. Anxiety. 2000, 12, 69–76. [Google Scholar] [CrossRef]
- Michopoulos, V.; Powers, A.; Gillespie, C.F.; Ressler, K.J.; Jovanovic, T. Inflammation in Fear- and Anxiety-Based Disorders: PTSD, GAD, and Beyond. Neuropsychopharmacology 2017, 42, 254–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patriquin, M.A.; Mathew, S.J. The Neurobiological Mechanisms of Generalized Anxiety Disorder and Chronic Stress. Chronic. Stress (Thousand Oaks) 2017, 1. [Google Scholar] [CrossRef] [PubMed]
- Yehuda, R. Status of glucocorticoid alterations in post-traumatic stress disorder. Ann. N. Y. Acad. Sci. 2009, 1179, 56–69. [Google Scholar] [CrossRef]
- Hendrickson, R.C.; Raskind, M.A. Noradrenergic dysregulation in the pathophysiology of PTSD. Exp. Neurol. 2016, 284, 181–195. [Google Scholar] [CrossRef] [Green Version]
- Nance, D.M.; Sanders, V.M. Autonomic innervation and regulation of the immune system (1987–2007). Brain. Behav. Immun. 2007, 21, 736–745. [Google Scholar] [CrossRef] [Green Version]
- Felger, J.C. Imaging the Role of Inflammation in Mood and Anxiety-related Disorders. Curr. Neuropharmacol. 2018, 16, 533–558. [Google Scholar] [CrossRef]
- Mochcovitch, M.D.; da Rocha Freire, R.C.; Garcia, R.F.; Nardi, A.E. A systematic review of fMRI studies in generalized anxiety disorder: Evaluating its neural and cognitive basis. J. Affect. Disord. 2014, 167, 336–342. [Google Scholar] [CrossRef]
- Lin, E.; Tsai, S.J. Gene-Environment Interactions and Role of Epigenetics in Anxiety Disorders. Adv. Exp. Med. Biol. 2020, 1191, 93–102. [Google Scholar] [PubMed]
- Conway, C.C.; Rutter, L.A.; Brown, T.A. Chronic environmental stress and the temporal course of depression and panic disorder: A trait-state-occasion modeling approach. J. Abnorm. Psychol. 2016, 125, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Wade, S.L.; Monroe, S.M.; Michelson, L.K. Chronic life stress and treatment outcome in agoraphobia with panic attacks. Am. J. Psychiatry 1993, 150, 1491–1495. [Google Scholar] [PubMed]
- Stephens, M.A.; Wand, G. Stress and the HPA axis: Role of glucocorticoids in alcohol dependence. Alcohol Res. 2012, 34, 468–483. [Google Scholar] [PubMed]
- Hannibal, K.E.; Bishop, M.D. Chronic stress, cortisol dysfunction, and pain: A psychoneuroendocrine rationale for stress management in pain rehabilitation. Phys. Ther. 2014, 94, 1816–1825. [Google Scholar] [CrossRef]
- Boumpas, D.T.; Chrousos, G.P.; Wilder, R.L.; Cupps, T.R.; Balow, J.E. Glucocorticoid therapy for immune-mediated diseases: Basic and clinical correlates. Ann. Intern. Med. 1993, 119, 1198–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory action of glucocorticoids--new mechanisms for old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrousos, G.P. The role of stress and the hypothalamic-pituitary-adrenal axis in the pathogenesis of the metabolic syndrome: Neuro-endocrine and target tissue-related causes. Int. J. Obes. Relat. Metab. Disord. 2000, 24, S50–S55. [Google Scholar] [CrossRef] [Green Version]
- Norman, M.; Hearing, S.D. Glucocorticoid resistance—What is known? Curr. Opin. Pharmacol. 2002, 2, 723–729. [Google Scholar] [CrossRef]
- Sorrells, S.F.; Caso, J.R.; Munhoz, C.D.; Sapolsky, R.M. The stressed CNS: When glucocorticoids aggravate inflammation. Neuron 2009, 64, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Ray, D.W.; Matthews, L.C. Current concepts in glucocorticoid resistance. Steroids 2012, 77, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Fries, E.; Hesse, J.; Hellhammer, J.; Hellhammer, D.H. A new view on hypocortisolism. Psychoneuroendocrinology 2005, 30, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, A.; Truitt, W.; Rainnie, D.; Sajdyk, T. Role of stress, corticotrophin releasing factor (CRF) and amygdala plasticity in chronic anxiety. Stress 2005, 8, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Gustavson, S.M.; Sandoval, D.A.; Ertl, A.C.; Bao, S.; Raj, S.R.; Davis, S.N. Stimulation of both type I and type II corticosteroid receptors blunts counterregulatory responses to subsequent hypoglycemia in healthy man. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E506–E512. [Google Scholar] [CrossRef]
- Condren, R.M.; O’Neill, A.; Ryan, M.C.; Barrett, P.; Thakore, J.H. HPA axis response to a psychological stressor in generalised social phobia. Psychoneuroendocrinology 2002, 27, 693–703. [Google Scholar] [CrossRef]
- Schreiber, W.; Lauer, C.J.; Krumrey, K.; Holsboer, F.; Krieg, J.C. Dysregulation of the hypothalamic-pituitary-adrenocortical system in panic disorder. Neuropsychopharmacology 1996, 15, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Erhardt, A.; Ising, M.; Unschuld, P.G.; Kern, N.; Lucae, S.; Putz, B.; Uhr, M.; Binder, E.B.; Holsboer, F.; Keck, M.E. Regulation of the hypothalamic-pituitary-adrenocortical system in patients with panic disorder. Neuropsychopharmacology 2006, 31, 2515–2522. [Google Scholar] [CrossRef] [Green Version]
- Griffin, M.G.; Resick, P.A.; Yehuda, R. Enhanced cortisol suppression following dexamethasone administration in domestic violence survivors. Am. J. Psychiatry 2005, 162, 1192–1199. [Google Scholar] [CrossRef]
- Risbrough, V.B.; Stein, M.B. Role of corticotropin releasing factor in anxiety disorders: A translational research perspective. Horm. Behav. 2006, 50, 550–561. [Google Scholar] [CrossRef] [Green Version]
- Zorn, J.V.; Schur, R.R.; Boks, M.P.; Kahn, R.S.; Joels, M.; Vinkers, C.H. Cortisol stress reactivity across psychiatric disorders: A systematic review and meta-analysis. Psychoneuroendocrinology 2017, 77, 25–36. [Google Scholar] [CrossRef]
- Mantella, R.C.; Butters, M.A.; Amico, J.A.; Mazumdar, S.; Rollman, B.L.; Begley, A.E.; Reynolds, C.F.; Lenze, E.J. Salivary cortisol is associated with diagnosis and severity of late-life generalized anxiety disorder. Psychoneuroendocrinology 2008, 33, 773–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staufenbiel, S.M.; Penninx, B.W.; Spijker, A.T.; Elzinga, B.M.; van Rossum, E.F. Hair cortisol, stress exposure, and mental health in humans: A systematic review. Psychoneuroendocrinology 2013, 38, 1220–1235. [Google Scholar] [CrossRef] [PubMed]
- Vreeburg, S.A.; Hartman, C.A.; Hoogendijk, W.J.; van Dyck, R.; Zitman, F.G.; Ormel, J.; Penninx, B.W. Parental history of depression or anxiety and the cortisol awakening response. Br. J. Psychiatry 2010, 197, 180–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coryell, W.; Noyes, R., Jr.; Schlechte, J. The significance of HPA axis disturbance in panic disorder. Biol. Psychiatry 1989, 25, 989–1002. [Google Scholar] [CrossRef]
- Jansen, A.S.; Nguyen, X.V.; Karpitskiy, V.; Mettenleiter, T.C.; Loewy, A.D. Central command neurons of the sympathetic nervous system: Basis of the fight-or-flight response. Science 1995, 270, 644–646. [Google Scholar] [CrossRef]
- Jones, B.E.; Yang, T.Z. The efferent projections from the reticular formation and the locus coeruleus studied by anterograde and retrograde axonal transport in the rat. J. Comp. Neurol. 1985, 242, 56–92. [Google Scholar] [CrossRef]
- Kalk, N.J.; Nutt, D.J.; Lingford-Hughes, A.R. The role of central noradrenergic dysregulation in anxiety disorders: Evidence from clinical studies. J. Psychopharmacol. 2011, 25, 3–16. [Google Scholar] [CrossRef]
- Sand, P.G.; Mori, T.; Godau, C.; Stober, G.; Flachenecker, P.; Franke, P.; Nothen, M.M.; Fritze, J.; Maier, W.; Lesch, K.P.; et al. Norepinephrine transporter gene (NET) variants in patients with panic disorder. Neurosci. Lett. 2002, 333, 41–44. [Google Scholar] [CrossRef]
- Aunis, D. Exocytosis in chromaffin cells of the adrenal medulla. Int. Rev. Cytol. 1998, 181, 213–320. [Google Scholar]
- Charney, D.S.; Woods, S.W.; Goodman, W.K.; Heninger, G.R. Neurobiological mechanisms of panic anxiety: Biochemical and behavioral correlates of yohimbine-induced panic attacks. Am. J. Psychiatry 1987, 144, 1030–1036. [Google Scholar]
- Charney, D.S.; Woods, S.W.; Heninger, G.R. Noradrenergic function in generalized anxiety disorder: Effects of yohimbine in healthy subjects and patients with generalized anxiety disorder. Psychiatry Res. 1989, 27, 173–182. [Google Scholar] [CrossRef]
- Baldwin, D.S.; Anderson, I.M.; Nutt, D.J.; Bandelow, B.; Bond, A.; Davidson, J.R.; den Boer, J.A.; Fineberg, N.A.; Knapp, M.; Scott, J.; et al. Evidence-based guidelines for the pharmacological treatment of anxiety disorders: Recommendations from the British Association for Psychopharmacology. J. Psychopharmacol. 2005, 19, 567–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blechert, J.; Michael, T.; Grossman, P.; Lajtman, M.; Wilhelm, F.H. Autonomic and respiratory characteristics of posttraumatic stress disorder and panic disorder. Psychosom. Med. 2007, 69, 935–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thayer, J.F.; Lane, R.D. Claude Bernard and the heart-brain connection: Further elaboration of a model of neurovisceral integration. Neurosci. Biobehav. Rev. 2009, 33, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Laborde, S.; Mosley, E.; Thayer, J.F. Heart Rate Variability and Cardiac Vagal Tone in Psychophysiological Research—Recommendations for Experiment Planning, Data Analysis, and Data Reporting. Front. Psychol. 2017, 8, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thayer, J.F.; Friedman, B.H.; Borkovec, T.D. Autonomic characteristics of generalized anxiety disorder and worry. Biol. Psychiatry 1996, 39, 255–266. [Google Scholar] [CrossRef]
- Chalmers, J.A.; Quintana, D.S.; Abbott, M.J.; Kemp, A.H. Anxiety Disorders are Associated with Reduced Heart Rate Variability: A Meta-Analysis. Front. Psychiatry 2014, 5, 80. [Google Scholar] [CrossRef] [Green Version]
- Pepys, M.B.; Hirschfield, G.M. C-reactive protein: A critical update. J. Clin. Invest. 2003, 111, 1805–1812. [Google Scholar] [CrossRef]
- Vogelzangs, N.; Beekman, A.T.; de Jonge, P.; Penninx, B.W. Anxiety disorders and inflammation in a large adult cohort. Transl. Psychiatry 2013, 3, e249. [Google Scholar] [CrossRef] [Green Version]
- Copeland, W.E.; Shanahan, L.; Worthman, C.; Angold, A.; Costello, E.J. Generalized anxiety and C-reactive protein levels: A prospective, longitudinal analysis. Psychol. Med. 2012, 42, 2641–2650. [Google Scholar] [CrossRef] [Green Version]
- Bankier, B.; Barajas, J.; Martinez-Rumayor, A.; Januzzi, J.L. Association between C-reactive protein and generalized anxiety disorder in stable coronary heart disease patients. Eur. Heart. J. 2008, 29, 2212–2217. [Google Scholar] [CrossRef] [PubMed]
- Vieira, M.M.; Ferreira, T.B.; Pacheco, P.A.; Barros, P.O.; Almeida, C.R.; Araujo-Lima, C.F.; Silva-Filho, R.G.; Hygino, J.; Andrade, R.M.; Linhares, U.C.; et al. Enhanced Th17 phenotype in individuals with generalized anxiety disorder. J. Neuroimmunol. 2010, 229, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Boyman, O.; Sprent, J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 2012, 12, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Woodward, E.A.; Prele, C.M.; Nicholson, S.E.; Kolesnik, T.B.; Hart, P.H. The anti-inflammatory effects of interleukin-4 are not mediated by suppressor of cytokine signalling-1 (SOCS1). Immunology 2010, 131, 118–127. [Google Scholar] [CrossRef]
- Wingo, A.P.; Gibson, G. Blood gene expression profiles suggest altered immune function associated with symptoms of generalized anxiety disorder. Brain. Behav. Immun. 2015, 43, 184–191. [Google Scholar] [CrossRef] [Green Version]
- De Berardis, D.; Serroni, N.; Campanella, D.; Marini, S.; Rapini, G.; Valchera, A.; Iasevoli, F.; Mazza, M.; Fornaro, M.; Perna, G.; et al. Alexithymia, Suicide Ideation, C-Reactive Protein, and Serum Lipid Levels Among Outpatients with Generalized Anxiety Disorder. Arch. Suicide Res. 2017, 21, 100–112. [Google Scholar] [CrossRef]
- Wagner, E.Y.; Wagner, J.T.; Glaus, J.; Vandeleur, C.L.; Castelao, E.; Strippoli, M.P.; Vollenweider, P.; Preisig, M.; von Kanel, R. Evidence for chronic low-grade systemic inflammation in individuals with agoraphobia from a population-based prospective study. PLoS ONE 2015, 10, e0123757. [Google Scholar] [CrossRef] [Green Version]
- Hoge, E.A.; Brandstetter, K.; Moshier, S.; Pollack, M.H.; Wong, K.K.; Simon, N.M. Broad spectrum of cytokine abnormalities in panic disorder and posttraumatic stress disorder. Depress. Anxiety 2009, 26, 447–455. [Google Scholar] [CrossRef]
- Manfro, G.G.; Pollack, M.H.; Otto, M.W.; Worthington, J.J.; Rosenbaum, J.F.; Scott, E.L.; Kradin, R.L. Cell-surface expression of L-selectin (CD62L) by blood lymphocytes: Correlates with affective parameters and severity of panic disorder. Depress. Anxiety 2000, 11, 31–37. [Google Scholar] [CrossRef]
- Rapaport, M.H. Circulating lymphocyte phenotypic surface markers in anxiety disorder patients and normal volunteers. Biol. Psychiatry 1998, 43, 458–463. [Google Scholar] [CrossRef]
- Quan, N.; Banks, W.A. Brain-immune communication pathways. Brain Behav. Immun. 2007, 21, 727–735. [Google Scholar] [CrossRef]
- Ericsson, A.; Kovacs, K.J.; Sawchenko, P.E. A functional anatomical analysis of central pathways subserving the effects of interleukin-1 on stress-related neuroendocrine neurons. J. Neurosci. 1994, 14, 897–913. [Google Scholar] [CrossRef] [PubMed]
- Bluthe, R.M.; Walter, V.; Parnet, P.; Laye, S.; Lestage, J.; Verrier, D.; Poole, S.; Stenning, B.E.; Kelley, K.W.; Dantzer, R. Lipopolysaccharide induces sickness behaviour in rats by a vagal mediated mechanism. C. R. Acad. Sci. III 1994, 317, 499–503. [Google Scholar] [PubMed]
- Luheshi, G.N.; Bluthe, R.M.; Rushforth, D.; Mulcahy, N.; Konsman, J.P.; Goldbach, M.; Dantzer, R. Vagotomy attenuates the behavioural but not the pyrogenic effects of interleukin-1 in rats. Auton. Neurosci. 2000, 85, 127–132. [Google Scholar] [CrossRef]
- Roth, J.; de Souza, G.E. Fever induction pathways: Evidence from responses to systemic or local cytokine formation. Braz. J. Med. Biol. Res. 2001, 34, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Borovikova, L.V.; Ivanova, S.; Zhang, M.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.V. Circumventricular organs: Integrators of circulating signals controlling hydration, energy balance, and immune function. In Neurobiology of Body Fluid Homeostasis: Transduction and Integration; de Luca, L.A., Jr., Menani, J.V., Johnson, A.K., Eds.; CRC Press: Boca Raton, FL, USA, 2014. [Google Scholar]
- Guerrero, J.M.; Reiter, R.J. A brief survey of pineal gland-immune system interrelationships. Endocr. Res. 1992, 18, 91–113. [Google Scholar] [CrossRef]
- Breder, C.D.; Dinarello, C.A.; Saper, C.B. Interleukin-1 immunoreactive innervation of the human hypothalamus. Science 1988, 240, 321–324. [Google Scholar] [CrossRef]
- Komaki, G.; Arimura, A.; Koves, K. Effect of intravenous injection of IL-1 beta on PGE2 levels in several brain areas as determined by microdialysis. Am. J. Physiol. 1992, 262, E246–E251. [Google Scholar] [CrossRef]
- Peruzzo, B.; Pastor, F.E.; Blazquez, J.L.; Schobitz, K.; Pelaez, B.; Amat, P.; Rodriguez, E.M. A second look at the barriers of the medial basal hypothalamus. Exp. Brain Res. 2000, 132, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Kastin, A.J.; Durham, D.A. Bidirectional transport of interleukin-1 alpha across the blood-brain barrier. Brain Res. Bull. 1989, 23, 433–437. [Google Scholar] [CrossRef]
- Xiang, S.; Pan, W.; Kastin, A.J. Strategies to create a regenerating environment for the injured spinal cord. Curr. Pharm. Des. 2005, 11, 1267–1277. [Google Scholar] [CrossRef] [PubMed]
- Fabry, Z.; Fitzsimmons, K.M.; Herlein, J.A.; Moninger, T.O.; Dobbs, M.B.; Hart, M.N. Production of the cytokines interleukin 1 and 6 by murine brain microvessel endothelium and smooth muscle pericytes. J. Neuroimmunol. 1993, 47, 23–34. [Google Scholar] [CrossRef]
- Gyoneva, S.; Davalos, D.; Biswas, D.; Swanger, S.A.; Garnier-Amblard, E.; Loth, F.; Akassoglou, K.; Traynelis, S.F. Systemic inflammation regulates microglial responses to tissue damage in vivo. Glia 2014, 62, 1345–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biber, K.; Neumann, H.; Inoue, K.; Boddeke, H.W. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007, 30, 596–602. [Google Scholar] [CrossRef]
- D’Mello, C.; Le, T.; Swain, M.G. Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J. Neurosci. 2009, 29, 2089–2102. [Google Scholar] [CrossRef] [Green Version]
- Peracoli, M.T.; Kurokawa, C.S.; Calvi, S.A.; Mendes, R.P.; Pereira, P.C.; Marques, S.A.; Soares, A.M. Production of pro- and anti-inflammatory cytokines by monocytes from patients with paracoccidioidomycosis. Microbes Infect. 2003, 5, 413–418. [Google Scholar] [CrossRef]
- Barrientos, R.M.; Sprunger, D.B.; Campeau, S.; Higgins, E.A.; Watkins, L.R.; Rudy, J.W.; Maier, S.F. Brain-derived neurotrophic factor mRNA downregulation produced by social isolation is blocked by intrahippocampal interleukin-1 receptor antagonist. Neuroscience 2003, 121, 847–853. [Google Scholar] [CrossRef]
- Ben Menachem-Zidon, O.; Goshen, I.; Kreisel, T.; Ben Menahem, Y.; Reinhartz, E.; Ben Hur, T.; Yirmiya, R. Intrahippocampal transplantation of transgenic neural precursor cells overexpressing interleukin-1 receptor antagonist blocks chronic isolation-induced impairment in memory and neurogenesis. Neuropsychopharmacology 2008, 33, 2251–2262. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.W.; Chen, Y.C.; Yu, L.; Chen, H.I.; Jen, C.J.; Huang, A.M.; Tsai, H.J.; Chang, Y.T.; Kuo, Y.M. Treadmill exercise counteracts the suppressive effects of peripheral lipopolysaccharide on hippocampal neurogenesis and learning and memory. J. Neurochem. 2007, 103, 2471–2481. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.R.; Wang, M.; Ribeiro, D.; Cho, H.J.; Olmstead, R.; Breen, E.C.; Martinez-Maza, O.; Cole, S. Sleep loss activates cellular inflammatory signaling. Biol. Psychiatry 2008, 64, 538–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, J.W.; Duman, R.S. IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc. Natl. Acad. Sci. USA 2008, 105, 751–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ida, T.; Hara, M.; Nakamura, Y.; Kozaki, S.; Tsunoda, S.; Ihara, H. Cytokine-induced enhancement of calcium-dependent glutamate release from astrocytes mediated by nitric oxide. Neurosci. Lett. 2008, 432, 232–236. [Google Scholar] [CrossRef]
- Gavillet, M.; Allaman, I.; Magistretti, P.J. Modulation of astrocytic metabolic phenotype by proinflammatory cytokines. Glia 2008, 56, 975–989. [Google Scholar] [CrossRef]
- Thornton, P.; Pinteaux, E.; Gibson, R.M.; Allan, S.M.; Rothwell, N.J. Interleukin-1-induced neurotoxicity is mediated by glia and requires caspase activation and free radical release. J. Neurochem. 2006, 98, 258–266. [Google Scholar] [CrossRef]
- Maddison, D.C.; Giorgini, F. The kynurenine pathway and neurodegenerative disease. Semin. Cell. Dev. Biol. 2015, 40, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Murakami, Y.; Hoshi, M.; Imamura, Y.; Arioka, Y.; Yamamoto, Y.; Saito, K. Remarkable role of indoleamine 2,3-dioxygenase and tryptophan metabolites in infectious diseases: Potential role in macrophage-mediated inflammatory diseases. Mediators Inflamm. 2013, 2013, 391984. [Google Scholar] [CrossRef] [Green Version]
- Myint, A.M.; Kim, Y.K. Network beyond IDO in psychiatric disorders: Revisiting neurodegeneration hypothesis. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 48, 304–313. [Google Scholar] [CrossRef]
- Dang, Y.; Dale, W.E.; Brown, O.R. Comparative effects of oxygen on indoleamine 2,3-dioxygenase and tryptophan 2,3-dioxygenase of the kynurenine pathway. Free Radic. Biol. Med. 2000, 28, 615–624. [Google Scholar] [CrossRef]
- Gal, E.M.; Sherman, A.D. L-kynurenine: Its synthesis and possible regulatory function in brain. Neurochem. Res. 1980, 5, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; O’Connor, J.C.; Lawson, M.A.; Kelley, K.W. Inflammation-associated depression: From serotonin to kynurenine. Psychoneuroendocrinology 2011, 36, 426–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, C.M.; Hale, P.T.; Carlin, J.M. The role of IFN-gamma and TNF-alpha-responsive regulatory elements in the synergistic induction of indoleamine dioxygenase. J. Interferon Cytokine Res. 2005, 25, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Mellor, A.L.; Munn, D.H. Tryptophan catabolism and T-cell tolerance: Immunosuppression by starvation? Immunol. Today 1999, 20, 469–473. [Google Scholar] [CrossRef]
- Heyes, M.P.; Achim, C.L.; Wiley, C.A.; Major, E.O.; Saito, K.; Markey, S.P. Human microglia convert l-tryptophan into the neurotoxin quinolinic acid. Biochem. J. 1996, 320, 595–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zunszain, P.A.; Anacker, C.; Cattaneo, A.; Choudhury, S.; Musaelyan, K.; Myint, A.M.; Thuret, S.; Price, J.; Pariante, C.M. Interleukin-1beta: A new regulator of the kynurenine pathway affecting human hippocampal neurogenesis. Neuropsychopharmacology 2012, 37, 939–949. [Google Scholar] [CrossRef] [Green Version]
- Eren, I.; Tukel, R.; Polat, A.; Karaman, R.; Unal, S. Evaluation of regional cerebral blood flow changes in panic disorder with Tc99m-HMPAO SPECT. Turk. Psikiyatri. Derg. 2002, 13, 89–97. [Google Scholar] [CrossRef]
- Shang, J.; Fu, Y.; Ren, Z.; Zhang, T.; Du, M.; Gong, Q.; Lui, S.; Zhang, W. The common traits of the ACC and PFC in anxiety disorders in the DSM-5: Meta-analysis of voxel-based morphometry studies. PLoS ONE 2014, 9, e93432. [Google Scholar] [CrossRef] [Green Version]
- Slavich, G.M.; Way, B.M.; Eisenberger, N.I.; Taylor, S.E. Neural sensitivity to social rejection is associated with inflammatory responses to social stress. Proc. Natl. Acad. Sci. USA 2010, 107, 14817–14822. [Google Scholar] [CrossRef] [Green Version]
- Kullmann, J.S.; Grigoleit, J.S.; Lichte, P.; Kobbe, P.; Rosenberger, C.; Banner, C.; Wolf, O.T.; Engler, H.; Oberbeck, R.; Elsenbruch, S.; et al. Neural response to emotional stimuli during experimental human endotoxemia. Hum. Brain. Mapp. 2013, 34, 2217–2227. [Google Scholar] [CrossRef]
- Eisenberger, N.I.; Berkman, E.T.; Inagaki, T.K.; Rameson, L.T.; Mashal, N.M.; Irwin, M.R. Inflammation-induced anhedonia: Endotoxin reduces ventral striatum responses to reward. Biol. Psychiatry 2010, 68, 748–754. [Google Scholar] [CrossRef] [Green Version]
- Harrison, N.A.; Brydon, L.; Walker, C.; Gray, M.A.; Steptoe, A.; Critchley, H.D. Inflammation causes mood changes through alterations in subgenual cingulate activity and mesolimbic connectivity. Biol. Psychiatry 2009, 66, 407–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muscatell, K.A.; Dedovic, K.; Slavich, G.M.; Jarcho, M.R.; Breen, E.C.; Bower, J.E.; Irwin, M.R.; Eisenberger, N.I. Greater amygdala activity and dorsomedial prefrontal-amygdala coupling are associated with enhanced inflammatory responses to stress. Brain Behav. Immun. 2015, 43, 46–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, N.A.; Brydon, L.; Walker, C.; Gray, M.A.; Steptoe, A.; Dolan, R.J.; Critchley, H.D. Neural origins of human sickness in interoceptive responses to inflammation. Biol. Psychiatry 2009, 66, 415–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inagaki, T.K.; Muscatell, K.A.; Irwin, M.R.; Cole, S.W.; Eisenberger, N.I. Inflammation selectively enhances amygdala activity to socially threatening images. Neuroimage 2012, 59, 3222–3226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dresler, T.; Guhn, A.; Tupak, S.V.; Ehlis, A.C.; Herrmann, M.J.; Fallgatter, A.J.; Deckert, J.; Domschke, K. Revise the revised? New dimensions of the neuroanatomical hypothesis of panic disorder. J. Neural Transm. (Vienna) 2013, 120, 3–29. [Google Scholar] [CrossRef] [PubMed]
- Asami, T.; Yamasue, H.; Hayano, F.; Nakamura, M.; Uehara, K.; Otsuka, T.; Roppongi, T.; Nihashi, N.; Inoue, T.; Hirayasu, Y. Sexually dimorphic gray matter volume reduction in patients with panic disorder. Psychiatry Res. 2009, 173, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Sobanski, T.; Wagner, G.; Peikert, G.; Gruhn, U.; Schluttig, K.; Sauer, H.; Schlosser, R. Temporal and right frontal lobe alterations in panic disorder: A quantitative volumetric and voxel-based morphometric MRI study. Psychol. Med. 2010, 40, 1879–1886. [Google Scholar] [CrossRef]
- Yoo, H.K.; Kim, M.J.; Kim, S.J.; Sung, Y.H.; Sim, M.E.; Lee, Y.S.; Song, S.Y.; Kee, B.S.; Lyoo, I.K. Putaminal gray matter volume decrease in panic disorder: An optimized voxel-based morphometry study. Eur. J. Neurosci. 2005, 22, 2089–2094. [Google Scholar] [CrossRef]
- Protopopescu, X.; Pan, H.; Tuescher, O.; Cloitre, M.; Goldstein, M.; Engelien, A.; Yang, Y.; Gorman, J.; LeDoux, J.; Stern, E.; et al. Increased brainstem volume in panic disorder: A voxel-based morphometric study. Neuroreport 2006, 17, 361–363. [Google Scholar] [CrossRef]
- Roppongi, T.; Nakamura, M.; Asami, T.; Hayano, F.; Otsuka, T.; Uehara, K.; Fujiwara, A.; Saeki, T.; Hayasaka, S.; Yoshida, T.; et al. Posterior orbitofrontal sulcogyral pattern associated with orbitofrontal cortex volume reduction and anxiety trait in panic disorder. Psychiatry Clin. Neurosci. 2010, 64, 318–326. [Google Scholar] [CrossRef]
- Lai, C.H.; Hsu, Y.Y. A subtle grey-matter increase in first-episode, drug-naive major depressive disorder with panic disorder after 6 weeks’ duloxetine therapy. Int. J. Neuropsychopharmacol. 2011, 14, 225–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asami, T.; Hayano, F.; Nakamura, M.; Yamasue, H.; Uehara, K.; Otsuka, T.; Roppongi, T.; Nihashi, N.; Inoue, T.; Hirayasu, Y. Anterior cingulate cortex volume reduction in patients with panic disorder. Psychiatry Clin. Neurosci. 2008, 62, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Uchida, R.R.; del-Ben, C.M.; Busatto, G.F.; Duran, F.L.; Guimaraes, F.S.; Crippa, J.A.; Araujo, D.; Santos, A.C.; Graeff, F.G. Regional gray matter abnormalities in panic disorder: A voxel-based morphometry study. Psychiatry Res. 2008, 163, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Hayano, F.; Nakamura, M.; Asami, T.; Uehara, K.; Yoshida, T.; Roppongi, T.; Otsuka, T.; Inoue, T.; Hirayasu, Y. Smaller amygdala is associated with anxiety in patients with panic disorder. Psychiatry Clin. Neurosci. 2009, 63, 266–276. [Google Scholar] [CrossRef]
- Lai, C.H.; Hsu, Y.Y.; Wu, Y.T. First episode drug-naive major depressive disorder with panic disorder: Gray matter deficits in limbic and default network structures. Eur. Neuropsychopharmacol. 2010, 20, 676–682. [Google Scholar] [CrossRef]
- Massana, G.; Serra-Grabulosa, J.M.; Salgado-Pineda, P.; Gasto, C.; Junque, C.; Massana, J.; Mercader, J.M.; Gomez, B.; Tobena, A.; Salamero, M. Amygdalar atrophy in panic disorder patients detected by volumetric magnetic resonance imaging. Neuroimage 2003, 19, 80–90. [Google Scholar] [CrossRef]
- Uchida, R.R.; del-Ben, C.M.; Santos, A.C.; Araujo, D.; Crippa, J.A.; Guimaraes, F.S.; Graeff, F.G. Decreased left temporal lobe volume of panic patients measured by magnetic resonance imaging. Braz. J. Med. Biol. Res. 2003, 36, 925–929. [Google Scholar] [CrossRef] [Green Version]
- Vythilingam, M.; Anderson, E.R.; Goddard, A.; Woods, S.W.; Staib, L.H.; Charney, D.S.; Bremner, J.D. Temporal lobe volume in panic disorder-a quantitative magnetic resonance imaging study. Psychiatry Res. 2000, 99, 75–82. [Google Scholar] [CrossRef]
- Massana, G.; Serra-Grabulosa, J.M.; Salgado-Pineda, P.; Gasto, C.; Junque, C.; Massana, J.; Mercader, J.M. Parahippocampal gray matter density in panic disorder: A voxel-based morphometric study. Am. J. Psychiatry 2003, 160, 566–568. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.H. Gray matter deficits in panic disorder: A pilot study of meta-analysis. J. Clin. Psychopharmacol. 2011, 31, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Radua, J.; van den Heuvel, O.A.; Surguladze, S.; Mataix-Cols, D. Meta-analytical comparison of voxel-based morphometry studies in obsessive-compulsive disorder vs other anxiety disorders. Arch. Gen. Psychiatry 2010, 67, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, A.; Yoshida, T.; Otsuka, T.; Hayano, F.; Asami, T.; Narita, H.; Nakamura, M.; Inoue, T.; Hirayasu, Y. Midbrain volume increase in patients with panic disorder. Psychiatry Clin. Neurosci. 2011, 65, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Han, D.H.; Renshaw, P.F.; Dager, S.R.; Chung, A.; Hwang, J.; Daniels, M.A.; Lee, Y.S.; Lyoo, I.K. Altered cingulate white matter connectivity in panic disorder patients. J. Psychiatr. Res. 2008, 42, 399–407. [Google Scholar] [CrossRef]
- Sakai, Y.; Kumano, H.; Nishikawa, M.; Sakano, Y.; Kaiya, H.; Imabayashi, E.; Ohnishi, T.; Matsuda, H.; Yasuda, A.; Sato, A.; et al. Cerebral glucose metabolism associated with a fear network in panic disorder. Neuroreport 2005, 16, 927–931. [Google Scholar] [CrossRef]
- Bisaga, A.; Katz, J.L.; Antonini, A.; Wright, C.E.; Margouleff, C.; Gorman, J.M.; Eidelberg, D. Cerebral glucose metabolism in women with panic disorder. Am. J. Psychiatry 1998, 155, 1178–1183. [Google Scholar] [CrossRef]
- Lee, Y.S.; Hwang, J.; Kim, S.J.; Sung, Y.H.; Kim, J.; Sim, M.E.; Bae, S.C.; Kim, M.J.; Lyoo, I.K. Decreased blood flow of temporal regions of the brain in subjects with panic disorder. J. Psychiatr. Res. 2006, 40, 528–534. [Google Scholar] [CrossRef]
- De Cristofaro, M.T.; Sessarego, A.; Pupi, A.; Biondi, F.; Faravelli, C. Brain perfusion abnormalities in drug-naive, lactate-sensitive panic patients: A SPECT study. Biol. Psychiatry 1993, 33, 505–512. [Google Scholar] [CrossRef]
- Nordahl, T.E.; Semple, W.E.; Gross, M.; Mellman, T.A.; Stein, M.B.; Goyer, P.; King, A.C.; Uhde, T.W.; Cohen, R.M. Cerebral glucose metabolic differences in patients with panic disorder. Neuropsychopharmacology 1990, 3, 261–272. [Google Scholar]
- Na, K.S.; Ham, B.J.; Lee, M.S.; Kim, L.; Kim, Y.K.; Lee, H.J.; Yoon, H.K. Decreased gray matter volume of the medial orbitofrontal cortex in panic disorder with agoraphobia: A preliminary study. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 45, 195–200. [Google Scholar] [CrossRef]
- Lueken, U.; Straube, B.; Konrad, C.; Wittchen, H.U.; Strohle, A.; Wittmann, A.; Pfleiderer, B.; Uhlmann, C.; Arolt, V.; Jansen, A.; et al. Neural substrates of treatment response to cognitive-behavioral therapy in panic disorder with agoraphobia. Am. J. Psychiatry 2013, 170, 1345–1355. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, A.; Schlagenhauf, F.; John, T.; Guhn, A.; Rehbein, H.; Siegmund, A.; Stoy, M.; Held, D.; Schulz, I.; Fehm, L.; et al. A new paradigm (Westphal-Paradigm) to study the neural correlates of panic disorder with agoraphobia. Eur. Arch. Psychiatry Clin. Neurosci. 2011, 261, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Lueken, U.; Straube, B.; Reinhardt, I.; Maslowski, N.I.; Wittchen, H.U.; Strohle, A.; Wittmann, A.; Pfleiderer, B.; Konrad, C.; Ewert, A.; et al. Altered top-down and bottom-up processing of fear conditioning in panic disorder with agoraphobia. Psychol. Med. 2014, 44, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, A.; Schlagenhauf, F.; Guhn, A.; Lueken, U.; Gaehlsdorf, C.; Stoy, M.; Bermpohl, F.; Fydrich, T.; Pfleiderer, B.; Bruhn, H.; et al. Anticipating agoraphobic situations: The neural correlates of panic disorder with agoraphobia. Psychol. Med. 2014, 44, 2385–2396. [Google Scholar] [CrossRef]
- De Bellis, M.D.; Casey, B.J.; Dahl, R.E.; Birmaher, B.; Williamson, D.E.; Thomas, K.M.; Axelson, D.A.; Frustaci, K.; Boring, A.M.; Hall, J.; et al. A pilot study of amygdala volumes in pediatric generalized anxiety disorder. Biol. Psychiatry 2000, 48, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Cha, J.; Greenberg, T.; Song, I.; Blair Simpson, H.; Posner, J.; Mujica-Parodi, L.R. Abnormal hippocampal structure and function in clinical anxiety and comorbid depression. Hippocampus 2016, 26, 545–553. [Google Scholar] [CrossRef] [Green Version]
- De Bellis, M.D.; Keshavan, M.S.; Shifflett, H.; Iyengar, S.; Dahl, R.E.; Axelson, D.A.; Birmaher, B.; Hall, J.; Moritz, G.; Ryan, N.D. Superior temporal gyrus volumes in pediatric generalized anxiety disorder. Biol. Psychiatry 2002, 51, 553–562. [Google Scholar] [CrossRef]
- Schienle, A.; Ebner, F.; Schafer, A. Localized gray matter volume abnormalities in generalized anxiety disorder. Eur. Arch. Psychiatry Clin. Neurosci. 2011, 261, 303–307. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, L.; Yu, R.; Liu, J.; Tang, J.; Tan, L.; Liao, M.; Yang, F.; Shan, B. White matter integrity alterations in first episode, treatment-naive generalized anxiety disorder. J. Affect. Disord. 2013, 148, 196–201. [Google Scholar] [CrossRef]
- Terlevic, R.; Isola, M.; Ragogna, M.; Meduri, M.; Canalaz, F.; Perini, L.; Rambaldelli, G.; Travan, L.; Crivellato, E.; Tognin, S.; et al. Decreased hypothalamus volumes in generalized anxiety disorder but not in panic disorder. J. Affect. Disord. 2013, 146, 390–394. [Google Scholar] [CrossRef]
- Moon, C.M.; Kim, G.W.; Jeong, G.W. Whole-brain gray matter volume abnormalities in patients with generalized anxiety disorder: Voxel-based morphometry. Neuroreport 2014, 25, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Hilbert, K.; Pine, D.S.; Muehlhan, M.; Lueken, U.; Steudte-Schmiedgen, S.; Beesdo-Baum, K. Gray and white matter volume abnormalities in generalized anxiety disorder by categorical and dimensional characterization. Psychiatry Res. 2015, 234, 314–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, K.M.; Drevets, W.C.; Dahl, R.E.; Ryan, N.D.; Birmaher, B.; Eccard, C.H.; Axelson, D.; Whalen, P.J.; Casey, B.J. Amygdala response to fearful faces in anxious and depressed children. Arch. Gen. Psychiatry 2001, 58, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Monk, C.S.; Telzer, E.H.; Mogg, K.; Bradley, B.P.; Mai, X.; Louro, H.M.; Chen, G.; McClure-Tone, E.B.; Ernst, M.; Pine, D.S. Amygdala and ventrolateral prefrontal cortex activation to masked angry faces in children and adolescents with generalized anxiety disorder. Arch. Gen. Psychiatry 2008, 65, 568–576. [Google Scholar] [CrossRef]
- Chen, A.C.; Etkin, A. Hippocampal network connectivity and activation differentiates post-traumatic stress disorder from generalized anxiety disorder. Neuropsychopharmacology 2013, 38, 1889–1898. [Google Scholar] [CrossRef] [Green Version]
- McClure, E.B.; Monk, C.S.; Nelson, E.E.; Parrish, J.M.; Adler, A.; Blair, R.J.; Fromm, S.; Charney, D.S.; Leibenluft, E.; Ernst, M.; et al. Abnormal attention modulation of fear circuit function in pediatric generalized anxiety disorder. Arch. Gen. Psychiatry 2007, 64, 97–106. [Google Scholar] [CrossRef]
- Paulesu, E.; Sambugaro, E.; Torti, T.; Danelli, L.; Ferri, F.; Scialfa, G.; Sberna, M.; Ruggiero, G.M.; Bottini, G.; Sassaroli, S. Neural correlates of worry in generalized anxiety disorder and in normal controls: A functional MRI study. Psychol. Med. 2010, 40, 117–124. [Google Scholar] [CrossRef]
- Blair, K.; Shaywitz, J.; Smith, B.W.; Rhodes, R.; Geraci, M.; Jones, M.; McCaffrey, D.; Vythilingam, M.; Finger, E.; Mondillo, K.; et al. Response to emotional expressions in generalized social phobia and generalized anxiety disorder: Evidence for separate disorders. Am. J. Psychiatry 2008, 165, 1193–1202. [Google Scholar] [CrossRef] [Green Version]
- Krain, A.L.; Gotimer, K.; Hefton, S.; Ernst, M.; Castellanos, F.X.; Pine, D.S.; Milham, M.P. A functional magnetic resonance imaging investigation of uncertainty in adolescents with anxiety disorders. Biol. Psychiatry 2008, 63, 563–568. [Google Scholar] [CrossRef]
- Cha, J.; Carlson, J.M.; Dedora, D.J.; Greenberg, T.; Proudfit, G.H.; Mujica-Parodi, L.R. Hyper-reactive human ventral tegmental area and aberrant mesocorticolimbic connectivity in overgeneralization of fear in generalized anxiety disorder. J. Neurosci. 2014, 34, 5855–5860. [Google Scholar] [CrossRef] [Green Version]
- Buff, C.; Schmidt, C.; Brinkmann, L.; Gathmann, B.; Tupak, S.; Straube, T. Directed threat imagery in generalized anxiety disorder. Psychol. Med. 2018, 48, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Hoehn-Saric, R.; Schlund, M.W.; Wong, S.H. Effects of citalopram on worry and brain activation in patients with generalized anxiety disorder. Psychiatry Res. 2004, 131, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Hilbert, K.; Lueken, U.; Beesdo-Baum, K. Neural structures, functioning and connectivity in Generalized Anxiety Disorder and interaction with neuroendocrine systems: A systematic review. J. Affect. Disord. 2014, 158, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Etkin, A.; Prater, K.E.; Schatzberg, A.F.; Menon, V.; Greicius, M.D. Disrupted amygdalar subregion functional connectivity and evidence of a compensatory network in generalized anxiety disorder. Arch. Gen. Psychiatry 2009, 66, 1361–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, A.K.; Fudge, J.L.; Kelly, C.; Perry, J.S.; Daniele, T.; Carlisi, C.; Benson, B.; Castellanos, F.X.; Milham, M.P.; Pine, D.S.; et al. Intrinsic functional connectivity of amygdala-based networks in adolescent generalized anxiety disorder. J. Am. Acad. Child Adolesc. Psychiatry 2013, 52, 290–299.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohler, O.; Krogh, J.; Mors, O.; Benros, M.E. Inflammation in Depression and the Potential for Anti-Inflammatory Treatment. Curr. Neuropharmacol. 2016, 14, 732–742. [Google Scholar] [CrossRef] [Green Version]
- Goyal, M.; Singh, S.; Sibinga, E.M.; Gould, N.F.; Rowland-Seymour, A.; Sharma, R.; Berger, Z.; Sleicher, D.; Maron, D.D.; Shihab, H.M.; et al. Meditation programs for psychological stress and well-being: A systematic review and meta-analysis. JAMA Intern. Med. 2014, 174, 357–368. [Google Scholar] [CrossRef] [Green Version]
- Bower, J.E.; Irwin, M.R. Mind-body therapies and control of inflammatory biology: A descriptive review. Brain Behav. Immun. 2016, 51, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Morgan, N.; Irwin, M.R.; Chung, M.; Wang, C. The effects of mind-body therapies on the immune system: Meta-analysis. PLoS ONE 2014, 9, e100903. [Google Scholar] [CrossRef] [Green Version]
- Bower, J.E.; Crosswell, A.D.; Stanton, A.L.; Crespi, C.M.; Winston, D.; Arevalo, J.; Ma, J.; Cole, S.W.; Ganz, P.A. Mindfulness meditation for younger breast cancer survivors: A randomized controlled trial. Cancer 2015, 121, 1231–1240. [Google Scholar] [CrossRef]
- Bower, J.E.; Greendale, G.; Crosswell, A.D.; Garet, D.; Sternlieb, B.; Ganz, P.A.; Irwin, M.R.; Olmstead, R.; Arevalo, J.; Cole, S.W. Yoga reduces inflammatory signaling in fatigued breast cancer survivors: A randomized controlled trial. Psychoneuroendocrinology 2014, 43, 20–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cryan, J.F.; Dinan, T.G. Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 2012, 13, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Dinan, T.G.; Stanton, C.; Cryan, J.F. Psychobiotics: A novel class of psychotropic. Biol. Psychiatry 2013, 74, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, O.; Keshteli, A.H.; Afshar, H.; Esmaillzadeh, A.; Adibi, P. Adherence to Mediterranean dietary pattern is inversely associated with depression, anxiety and psychological distress. Nutr. Neurosci. 2019, 1–12. [Google Scholar] [CrossRef]
- Dai, J.; Jones, D.P.; Goldberg, J.; Ziegler, T.R.; Bostick, R.M.; Wilson, P.W.; Manatunga, A.K.; Shallenberger, L.; Jones, L.; Vaccarino, V. Association between adherence to the Mediterranean diet and oxidative stress. Am. J. Clin. Nutr. 2008, 88, 1364–1370. [Google Scholar]
- Del Colle, S.; Morello, F.; Rabbia, F.; Milan, A.; Naso, D.; Puglisi, E.; Mulatero, P.; Veglio, F. Antihypertensive drugs and the sympathetic nervous system. J. Cardiovasc. Pharmacol. 2007, 50, 487–496. [Google Scholar] [CrossRef]
- Black, J.W.; Crowther, A.F.; Shanks, R.G.; Smith, L.H.; Dornhorst, A.C. A New Adrenergic Betareceptor Antagonist. Lancet 1964, 1, 1080–1081. [Google Scholar] [CrossRef]
- Steenen, S.A.; van Wijk, A.J.; van der Heijden, G.J.; van Westrhenen, R.; de Lange, J.; de Jongh, A. Propranolol for the treatment of anxiety disorders: Systematic review and meta-analysis. J. Psychopharmacol. 2016, 30, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Meibach, R.C.; Dunner, D.; Wilson, L.G.; Ishiki, D.; Dager, S.R. Comparative efficacy of propranolol, chlordiazepoxide, and placebo in the treatment of anxiety: A double-blind trial. J. Clin. Psychiatry 1987, 48, 355–358. [Google Scholar]
- Grosz, H.J. Narcotic withdrawal symptoms in heroin users treated with propranolol. Lancet 1972, 2, 564–566. [Google Scholar] [CrossRef]
- Fleminger, S.; Greenwood, R.J.; Oliver, D.L. Pharmacological management for agitation and aggression in people with acquired brain injury. Cochrane Database Syst. Rev. 2006, CD003299. [Google Scholar] [CrossRef] [PubMed]
- Drew, P.J.; Barnes, J.N.; Evans, S.J. The effect of acute beta-adrenoceptor blockade on examination performance. Br. J. Clin. Pharmacol. 1985, 19, 783–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brantigan, C.O.; Brantigan, T.A.; Joseph, N. Effect of beta blockade and beta stimulation on stage fright. Am. J. Med. 1982, 72, 88–94. [Google Scholar] [CrossRef]
- Clark, D.B.; Agras, W.S. The assessment and treatment of performance anxiety in musicians. Am. J. Psychiatry 1991, 148, 598–605. [Google Scholar]
- Elman, M.J.; Sugar, J.; Fiscella, R.; Deutsch, T.A.; Noth, J.; Nyberg, M.; Packo, K.; Anderson, R.J. The effect of propranolol versus placebo on resident surgical performance. Trans. Am. Ophthalmol. Soc. 1998, 96, 283–291. [Google Scholar] [PubMed]
- Mealy, K.; Ngeh, N.; Gillen, P.; Fitzpatrick, G.; Keane, F.B.; Tanner, A. Propranolol reduces the anxiety associated with day case surgery. Eur. J. Surg. 1996, 162, 11–14. [Google Scholar]
- Wohleb, E.S.; Hanke, M.L.; Corona, A.W.; Powell, N.D.; Stiner, L.M.; Bailey, M.T.; Nelson, R.J.; Godbout, J.P.; Sheridan, J.F. Beta-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J. Neurosci. 2011, 31, 6277–6288. [Google Scholar] [CrossRef] [Green Version]
- Sugama, S.; Takenouchi, T.; Hashimoto, M.; Ohata, H.; Takenaka, Y.; Kakinuma, Y. Stress-induced microglial activation occurs through beta-adrenergic receptor: Noradrenaline as a key neurotransmitter in microglial activation. J. Neuroinflamm. 2019, 16, 266. [Google Scholar] [CrossRef] [Green Version]
- Armstead, W.M.; Vavilala, M.S. Propranolol protects cerebral autoregulation and reduces hippocampal neuronal cell death through inhibition of interleukin-6 upregulation after traumatic brain injury in pigs. Br. J. Anaesth. 2019, 123, 610–617. [Google Scholar] [CrossRef]
- Lin, S.Y.; Wang, Y.Y.; Chang, C.Y.; Wu, C.C.; Chen, W.Y.; Kuan, Y.H.; Liao, S.L.; Chen, C.J. Effects of beta-Adrenergic Blockade on Metabolic and Inflammatory Responses in a Rat Model of Ischemic Stroke. Cells 2020, 9, 1373. [Google Scholar] [CrossRef]
- Hoover, D.B. Cholinergic modulation of the immune system presents new approaches for treating inflammation. Pharmacol. Ther. 2017, 179, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Ballina, M.; Valdes-Ferrer, S.I.; Dancho, M.E.; Ochani, M.; Katz, D.; Cheng, K.F.; Olofsson, P.S.; Chavan, S.S.; Al-Abed, Y.; Tracey, K.J.; et al. Xanomeline suppresses excessive pro-inflammatory cytokine responses through neural signal-mediated pathways and improves survival in lethal inflammation. Brain Behav. Immun. 2015, 44, 19–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niederbichler, A.D.; Papst, S.; Claassen, L.; Jokuszies, A.; Ipaktchi, K.; Reimers, K.; Hirsch, T.; Steinstraesser, L.; Kraft, T.; Vogt, P.M. Burn-induced organ dysfunction: Vagus nerve stimulation improves cardiac function. Eplasty 2010, 10, e45. [Google Scholar] [PubMed]
- Koopman, F.A.; Schuurman, P.R.; Vervoordeldonk, M.J.; Tak, P.P. Vagus nerve stimulation: A new bioelectronics approach to treat rheumatoid arthritis? Best Pract. Res. Clin. Rheumatol. 2014, 28, 625–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meregnani, J.; Clarencon, D.; Vivier, M.; Peinnequin, A.; Mouret, C.; Sinniger, V.; Picq, C.; Job, A.; Canini, F.; Jacquier-Sarlin, M.; et al. Anti-inflammatory effect of vagus nerve stimulation in a rat model of inflammatory bowel disease. Auton. Neurosci. 2011, 160, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Jin, K.; Uteshev, V.V. A type-II positive allosteric modulator of alpha7 nAChRs reduces brain injury and improves neurological function after focal cerebral ischemia in rats. PLoS ONE 2013, 8, e73581. [Google Scholar]
- Dash, P.K.; Zhao, J.; Kobori, N.; Redell, J.B.; Hylin, M.J.; Hood, K.N.; Moore, A.N. Activation of Alpha 7 Cholinergic Nicotinic Receptors Reduce Blood-Brain Barrier Permeability following Experimental Traumatic Brain Injury. J. Neurosci. 2016, 36, 2809–2818. [Google Scholar] [CrossRef] [Green Version]
- O’Reardon, J.P.; Cristancho, P.; Peshek, A.D. Vagus Nerve Stimulation (VNS) and Treatment of Depression: To the Brainstem and Beyond. Psychiatry (Edgmont) 2006, 3, 54–63. [Google Scholar]
- George, M.S.; Ward, H.E., Jr.; Ninan, P.T.; Pollack, M.; Nahas, Z.; Anderson, B.; Kose, S.; Howland, R.H.; Goodman, W.K.; Ballenger, J.C. A pilot study of vagus nerve stimulation (VNS) for treatment-resistant anxiety disorders. Brain Stimul. 2008, 1, 112–121. [Google Scholar] [CrossRef]
- Kox, M.; van Eijk, L.T.; Verhaak, T.; Frenzel, T.; Kiers, H.D.; Gerretsen, J.; van der Hoeven, J.G.; Kornet, L.; Scheiner, A.; Pickkers, P. Transvenous vagus nerve stimulation does not modulate the innate immune response during experimental human endotoxemia: A randomized controlled study. Arthritis Res. Ther. 2015, 17, 150. [Google Scholar] [CrossRef] [Green Version]
- Clancy, J.A.; Mary, D.A.; Witte, K.K.; Greenwood, J.P.; Deuchars, S.A.; Deuchars, J. Non-invasive vagus nerve stimulation in healthy humans reduces sympathetic nerve activity. Brain Stimul. 2014, 7, 871–877. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Won, E.; Kim, Y.-K. Neuroinflammation-Associated Alterations of the Brain as Potential Neural Biomarkers in Anxiety Disorders. Int. J. Mol. Sci. 2020, 21, 6546. https://doi.org/10.3390/ijms21186546
Won E, Kim Y-K. Neuroinflammation-Associated Alterations of the Brain as Potential Neural Biomarkers in Anxiety Disorders. International Journal of Molecular Sciences. 2020; 21(18):6546. https://doi.org/10.3390/ijms21186546
Chicago/Turabian StyleWon, Eunsoo, and Yong-Ku Kim. 2020. "Neuroinflammation-Associated Alterations of the Brain as Potential Neural Biomarkers in Anxiety Disorders" International Journal of Molecular Sciences 21, no. 18: 6546. https://doi.org/10.3390/ijms21186546
APA StyleWon, E., & Kim, Y.-K. (2020). Neuroinflammation-Associated Alterations of the Brain as Potential Neural Biomarkers in Anxiety Disorders. International Journal of Molecular Sciences, 21(18), 6546. https://doi.org/10.3390/ijms21186546