Vitamin D and Cardiovascular Disease, with Emphasis on Hypertension, Atherosclerosis, and Heart Failure


{kind=link}
Abstract
1. Introduction
2. Vitamin D Metabolism
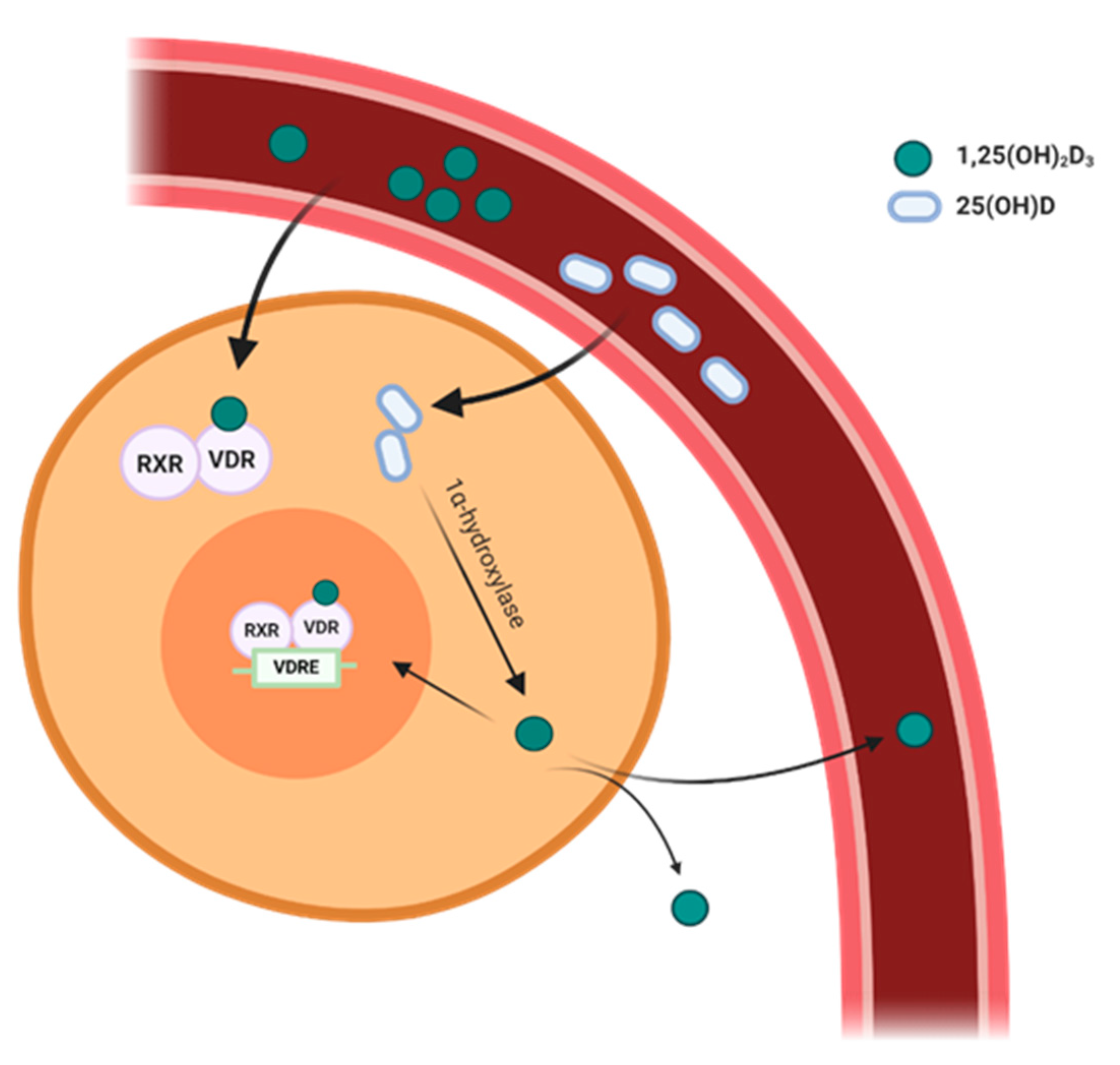
3. Expression of VDR and 1α-Hydroxylase in the Cardiovascular System
4. Hypertension
5. Atherosclerosis
6. Heart Failure
7. Conclusions
Funding
Conflicts of Interest
References
- Holick, M.F.; Biancuzzo, R.M.; Chen, T.C.; Klein, E.K.; Young, A.; Bibuld, D.; Reitz, R.; Salameh, W.; Ameri, A.; Tannenbaum, A.D. Vitamin D2 is as effective as vitamin D3 in maintaining circulating concentrations of 25-hydroxyvitamin D. J. Clin. Endocrinol. Metab. 2008, 93, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Hammami, M.M.; Yusuf, A. Differential effects of vitamin D2 and D3 supplements on 25-hydroxyvitamin D level are dose, sex, and time dependent: A randomized controlled trial. BMC Endocr. Disord. 2017, 17, 12. [Google Scholar] [CrossRef] [PubMed]
- Heaney, R.P.; Recker, R.R.; Grote, J.; Horst, R.L.; Armas, L.A.G. Vitamin D3 Is More Potent Than Vitamin D2 in Humans. J. Clin. Endocrinol. Metab. 2011, 96, E447–E452. [Google Scholar] [CrossRef] [PubMed]
- Houghton, L.A.; Vieth, R. The case against ergocalciferol (vitamin D2) as a vitamin supplement. Am. J. Clin. Nutr. 2006, 84, 694–697. [Google Scholar] [CrossRef] [PubMed]
- Shieh, A.; Chun, R.F.; Ma, C.; Witzel, S.; Meyer, B.; Rafison, B.; Swinkels, L.; Huijs, T.; Pepkowitz, S.; Holmquist, B.; et al. Effects of High-Dose Vitamin D2 Versus D3 on Total and Free 25-Hydroxyvitamin D and Markers of Calcium Balance. J. Clin. Endocrinol. Metab. 2016, 101, 3070–3078. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D. Vitamin D metabolism, mechanism of action, and clinical applications. Chem. Biol. 2014, 21, 319–329. [Google Scholar] [CrossRef]
- O’Riordan, J.L.H.; Bijvoet, O.L.M. Rickets before the discovery of vitamin D. BoneKEy Rep. 2014, 3, 478. [Google Scholar] [CrossRef][Green Version]
- Nair, R.; Maseeh, A. Vitamin D: The “sunshine” vitamin. J. Pharm. Pharm. 2012, 3, 118–126. [Google Scholar]
- Holick, M.F.; Binkley, N.C.; Bischoff-Ferrari, H.A.; Gordon, C.M.; Hanley, D.A.; Heaney, R.P.; Murad, M.H.; Weaver, C.M. Evaluation, Treatment, and Prevention of Vitamin D Deficiency: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2011, 96, 1911–1930. [Google Scholar] [CrossRef]
- Manson, J.E.; Brannon, P.M.; Rosen, C.J.; Taylor, C.L. Vitamin D Deficiency—Is There Really a Pandemic? N. Engl. J. Med. 2016, 375, 1817–1820. [Google Scholar] [CrossRef]
- Scragg, R. Seasonality of Cardiovascular Disease Mortality and the Possible Protective Effect of Ultra-violet Radiation. Int. J. Epidemiol. 1981, 10, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Scragg, R. Seasonal variation of mortality in Queensland. Community Health Stud. 1982, 6, 120–129. [Google Scholar] [CrossRef]
- Scragg, R.; Jackson, R.; Holdaway, I.M.; Lim, T.; Beaglehole, R. Myocardial Infarction Is Inversely Associated With Plasma 25-Hydroxyvitamin D3 Levels: A Community-Based Study. Int. J. Epidemiol. 1990, 19, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Al Mheid, I.; Patel, R.; Murrow, J.; Morris, A.; Rahman, A.; Fike, L.; Kavtaradze, N.; Uphoff, I.; Hooper, C.; Tangpricha, V.; et al. Vitamin D Status Is Associated With Arterial Stiffness and Vascular Dysfunction in Healthy Humans. J. Am. Coll. Cardiol. 2011, 58, 186–192. [Google Scholar] [CrossRef]
- Yiu, Y.F.; Chan, Y.H.; Yiu, K.H.; Siu, C.W.; Li, S.W.; Wong, L.Y.; Lee, S.W.; Tam, S.; Wong, E.W.; Cheung, B.M.; et al. Vitamin D deficiency is associated with depletion of circulating endothelial progenitor cells and endothelial dysfunction in patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 2011, 96, E830–E835. [Google Scholar] [CrossRef]
- Lee, J.H.; Gadi, R.; Spertus, J.A.; Tang, F.; O’Keefe, J.H. Prevalence of vitamin D deficiency in patients with acute myocardial infarction. Am. J. Cardiol. 2011, 107, 1636–1638. [Google Scholar] [CrossRef]
- London, G.M.; Guérin, A.P.; Verbeke, F.H.; Pannier, B.; Boutouyrie, P.; Marchais, S.J.; Mëtivier, F. Mineral Metabolism and Arterial Functions in End-Stage Renal Disease: Potential Role of 25-Hydroxyvitamin D Deficiency. Clin. J. Am. Soc. Nephrol. 2007, 18, 613–620. [Google Scholar] [CrossRef]
- Manousaki, D.; Mokry, L.E.; Ross, S.; Goltzman, D.; Richards, J.B. Mendelian Randomization Studies Do Not Support a Role for Vitamin D in Coronary Artery Disease. Circulation 2016, 9, 349–356. [Google Scholar] [CrossRef]
- Scragg, R.; Stewart, A.W.; Waayer, D.; Lawes, C.M.M.; Toop, L.; Sluyter, J.; Murphy, J.; Khaw, K.-T.; Camargo, C.A. Effect of Monthly High-Dose Vitamin D Supplementation on Cardiovascular Disease in the Vitamin D Assessment Study. JAMA Cardiol. 2017, 2, 608–616. [Google Scholar] [CrossRef]
- Manson, J.E.; Cook, N.R.; Lee, I.M.; Christen, W.; Bassuk, S.S.; Mora, S.; Gibson, H.; Gordon, D.; Copeland, T.; D’Agostino, D.; et al. Vitamin D Supplements and Prevention of Cancer and Cardiovascular Disease. N. Engl. J. Med. 2019, 380, 33–44. [Google Scholar] [CrossRef]
- Djoussé, L.; Cook, N.R.; Kim, E.; Bodar, V.; Walter, J.; Bubes, V.; Luttmann-Gibson, H.; Mora, S.; Joseph, J.; Lee, I.-M.; et al. Supplementation With Vitamin D and Omega-3 Fatty Acids and Incidence of Heart Failure Hospitalization. Circulation 2020, 141, 784–786. [Google Scholar] [CrossRef] [PubMed]
- Christakos, S.; Ajibade, D.V.; Dhawan, P.; Fechner, A.J.; Mady, L.J. Vitamin D: Metabolism. Endocrinol. Metab. Clin. N. Am. 2010, 39, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, S.M.; Schutkowski, A.; Schreier, B.; Rabe, S.; König, B.; Gekle, M.; Stangl, G.I. Vitamin D Receptor Deficiency Does Not Affect Blood Pressure and Heart Function. Front. Physiol. 2019, 10, 1118. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; David, V.; Quarles, L.D. Regulation and function of the FGF23/klotho endocrine pathways. Physiol. Rev. 2012, 92, 131–155. [Google Scholar] [CrossRef]
- Bikle, D.D.; Patzek, S.; Wang, Y. Physiologic and pathophysiologic roles of extra renal CYP27b1: Case report and review. Bone Rep. 2018, 8, 255–267. [Google Scholar] [CrossRef]
- Anderson, P.H.; May, B.K.; Morris, H.A. Vitamin D metabolism: New concepts and clinical implications. Clin. Biochem. Rev. 2003, 24, 13–26. [Google Scholar]
- Kennel, K.A.; Drake, M.T.; Hurley, D.L. Vitamin D deficiency in adults: When to test and how to treat. Mayo Clin. Proc. 2010, 85, 752–758. [Google Scholar] [CrossRef]
- Walters, M.R.; Wicker, D.C.; Riggle, P.C. 1,25-Dihydroxyvitamin D3 receptors identified in the rat heart. J. Mol. Cell Cardiol. 1986, 18, 67–72. [Google Scholar] [CrossRef]
- Tishkoff, D.X.; Nibbelink, K.A.; Holmberg, K.H.; Dandu, L.; Simpson, R.U. Functional Vitamin D Receptor (VDR) in the T-Tubules of Cardiac Myocytes: VDR Knockout Cardiomyocyte Contractility. Endocrinology 2008, 149, 558–564. [Google Scholar] [CrossRef]
- Chen, S.; Glenn, D.J.; Ni, W.; Grigsby, C.L.; Olsen, K.; Nishimoto, M.; Law, C.S.; Gardner, D.G. Expression of the vitamin D receptor is increased in the hypertrophic heart. Hypertension 2008, 52, 1106–1112. [Google Scholar] [CrossRef]
- Merke, J.; Milde, P.; Lewicka, S.; Hügel, U.; Klaus, G.; Mangelsdorf, D.J.; Haussler, M.R.; Rauterberg, E.W.; Ritz, E. Identification and regulation of 1,25-dihydroxyvitamin D3 receptor activity and biosynthesis of 1,25-dihydroxyvitamin D3. Studies in cultured bovine aortic endothelial cells and human dermal capillaries. J. Clin. Investig. 1989, 83, 1903–1915. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.S.K.; Delansorne, R.; Man, R.Y.K.; Vanhoutte, P.M. Vitamin D derivatives acutely reduce endothelium-dependent contractions in the aorta of the spontaneously hypertensive rat. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H289–H296. [Google Scholar] [CrossRef] [PubMed]
- Somjen, D.; Weisman, Y.; Kohen, F.; Gayer, B.; Limor, R.; Sharon, O.; Jaccard, N.; Knoll, E.; Stern, N. 25-Hydroxyvitamin D3 -1α-Hydroxylase Is Expressed in Human Vascular Smooth Muscle Cells and Is Upregulated by Parathyroid Hormone and Estrogenic Compounds. Circulation 2005, 111, 1666–1671. [Google Scholar] [CrossRef] [PubMed]
- Zehnder, D.; Bland, R.; Chana, R.S.; Wheeler, D.C.; Howie, A.J.; Williams, M.C.; Stewart, P.M.; Hewison, M. Synthesis of 1,25-dihydroxyvitamin D(3) by human endothelial cells is regulated by inflammatory cytokines: A novel autocrine determinant of vascular cell adhesion. J. Am. Soc. Nephrol. 2002, 13, 621–629. [Google Scholar]
- Rostand, S.G. Ultraviolet Light May Contribute to Geographic and Racial Blood Pressure Differences. Hypertension 1997, 30, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Fleck, A. Latitude and ischaemic heart disease. Lancet 1989, 1, 613. [Google Scholar] [CrossRef]
- Bunker, J.; Callister, W.; Chang, C.L.; Sever, P.S. How common is true resistant hypertension? J. Hum. Hypertens. 2011, 25, 137–140. [Google Scholar] [CrossRef]
- Li, Y.C.; Kong, J.; Wei, M.; Chen, Z.F.; Liu, S.Q.; Cao, L.P. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J. Clin. Investig. 2002, 110, 229–238. [Google Scholar] [CrossRef]
- Erben, R.G.; Soegiarto, D.W.; Weber, K.; Zeitz, U.; Lieberherr, M.; Gniadecki, R.; Möller, G.; Adamski, J.; Balling, R. Deletion of Deoxyribonucleic Acid Binding Domain of the Vitamin D Receptor Abrogates Genomic and Nongenomic Functions of Vitamin D. Mol. Endocrinol. 2002, 16, 1524–1537. [Google Scholar] [CrossRef]
- Andrukhova, O.; Slavic, S.; Zeitz, U.; Riesen, S.C.; Heppelmann, M.S.; Ambrisko, T.D.; Markovic, M.; Kuebler, W.M.; Erben, R.G. Vitamin D Is a Regulator of Endothelial Nitric Oxide Synthase and Arterial Stiffness in Mice. Mol. Endocrinol. 2014, 28, 53–64. [Google Scholar] [CrossRef]
- Ni, W.; Watts, S.W.; Ng, M.; Chen, S.; Glenn, D.J.; Gardner, D.G. Elimination of Vitamin D Receptor in Vascular Endothelial Cells Alters Vascular Function. Hypertension 2014, 64, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.S.K.; Delansorne, R.; Man, R.Y.K.; Svenningsen, P.; Vanhoutte, P.M. Chronic treatment with vitamin D lowers arterial blood pressure and reduces endothelium-dependent contractions in the aorta of the spontaneously hypertensive rat. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1226–H1234. [Google Scholar] [CrossRef] [PubMed]
- Kassi, E.; Adamopoulos, C.; Basdra, E.K.; Papavassiliou, A.G. Role of Vitamin D in Atherosclerosis. Circulation 2013, 128, 2517–2531. [Google Scholar] [CrossRef] [PubMed]
- Scragg, R.; Sowers, M.; Bell, C. Serum 25-hydroxyvitamin D, Ethnicity, and Blood Pressure in the Third National Health and Nutrition Examination Survey. Am. J. Hypertens. 2007, 20, 713–719. [Google Scholar] [CrossRef] [PubMed]
- He, J.L.; Scragg, R.K. Vitamin D, Parathyroid Hormone, and Blood Pressure in the National Health and Nutrition Examination Surveys. Am. J. Hypertens. 2011, 24, 911–917. [Google Scholar] [CrossRef][Green Version]
- Judd, S.E.; Nanes, M.S.; Ziegler, T.R.; Wilson, P.W.; Tangpricha, V. Optimal vitamin D status attenuates the age-associated increase in systolic blood pressure in white Americans: Results from the third National Health and Nutrition Examination Survey. Am. J. Clin. Nutr. 2008, 87, 136–141. [Google Scholar] [CrossRef]
- Wang, T.J.; Pencina, M.J.; Booth, S.L.; Jacques, P.F.; Ingelsson, E.; Lanier, K.; Benjamin, E.J.; D’Agostino, R.B.; Wolf, M.; Vasan, R.S. Vitamin D Deficiency and Risk of Cardiovascular Disease. Circulation 2008, 117, 503–511. [Google Scholar] [CrossRef]
- Forman, J.P.; Giovannucci, E.; Holmes, M.D.; Bischoff-Ferrari, H.A.; Tworoger, S.S.; Willett, W.C.; Curhan, G.C. Plasma 25-Hydroxyvitamin D Levels and Risk of Incident Hypertension. Hypertension 2007, 49, 1063–1069. [Google Scholar] [CrossRef]
- Ke, L.; Graubard, B.I.; Albanes, D.; Fraser, D.R.; Weinstein, S.J.; Virtamo, J.; Brock, K.E. Hypertension, Pulse, and Other Cardiovascular Risk Factors and Vitamin D Status in Finnish Men. Am. J. Hypertens. 2013, 26, 951–956. [Google Scholar] [CrossRef]
- Kunutsor, S.K.; Apekey, T.A.; Steur, M. Vitamin D and risk of future hypertension: Meta-analysis of 283,537 participants. Eur. J. Epidemiol. 2013, 28, 205–221. [Google Scholar] [CrossRef]
- Snijder, M.B.; Lips, P.; Seidell, J.C.; Visser, M.; Deeg, D.J.H.; Dekker, J.M.; Van Dam, R.M. Vitamin D status and parathyroid hormone levels in relation to blood pressure: A population-based study in older men and women. J. Int. Med. 2007, 261, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Reis, J.P.; Von Muhlen, D.; Kritz-Silverstein, D.; Wingard, D.L.; Barrett-Connor, E. Vitamin D, Parathyroid Hormone Levels, and the Prevalence of Metabolic Syndrome in Community-Dwelling Older Adults. Diabetes Care 2007, 30, 1549–1555. [Google Scholar] [CrossRef] [PubMed]
- Krause, R.; Bühring, M.; Hopfenmüller, W.; Holick, M.F.; Sharma, A.M. Ultraviolet B and blood pressure. Lancet 1998, 352, 709–710. [Google Scholar] [CrossRef]
- Pfeifer, M.; Begerow, B.; Minne, H.W.; Nachtigall, D.; Hansen, C. Effects of a Short-Term Vitamin D3 and Calcium Supplementation on Blood Pressure and Parathyroid Hormone Levels in Elderly Women1. J. Clin. Endocrinol. Metab. 2001, 86, 1633–1637. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Forman, J.P.; Scott, J.B.; Ng, K.; Drake, B.F.; Suarez, E.G.; Hayden, D.L.; Bennett, G.G.; Chandler, P.D.; Hollis, B.W.; Emmons, K.M.; et al. Effect of Vitamin D Supplementation on Blood Pressure in Blacks. Hypertension 2013, 61, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Witham, M.D.; Nadir, M.A.; Struthers, A.D. Effect of vitamin D on blood pressure: A systematic review and meta-analysis. J. Hypertens. 2009, 27, 1948–1954. [Google Scholar] [CrossRef]
- Kunutsor, S.K.; Burgess, S.; Munroe, P.B.; Khan, H. Vitamin D and high blood pressure: Causal association or epiphenomenon? Eur. J. Epidemiol. 2014, 29, 1–14. [Google Scholar] [CrossRef]
- Arora, P.; Song, Y.; Dusek, J.; Plotnikoff, G.; Sabatine, M.S.; Cheng, S.; Valcour, A.; Swales, H.; Taylor, B.; Carney, E.; et al. Vitamin D Therapy in Individuals With Prehypertension or Hypertension. Circulation 2015, 131, 254–262. [Google Scholar] [CrossRef]
- Jorde, R.; Sneve, M.; Torjesen, P.; Figenschau, Y. No improvement in cardiovascular risk factors in overweight and obese subjects after supplementation with vitamin D 3 for 1 year. J. Intern. Med. 2010, 267, 462–472. [Google Scholar] [CrossRef]
- Nagpal, J.; Pande, J.N.; Bhartia, A. A double-blind, randomized, placebo-controlled trial of the short-term effect of vitamin D3 supplementation on insulin sensitivity in apparently healthy, middle-aged, centrally obese men. Diabet. Med. 2009, 26, 19–27. [Google Scholar] [CrossRef]
- Salehpour, A.; Shidfar, F.; Hosseinpanah, F.; Vafa, M.; Razaghi, M.; Hoshiarrad, A.; Gohari, M. Vitamin D3 and the risk of CVD in overweight and obese women: A randomised controlled trial. Br. J. Nutr. 2012, 108, 1866–1873. [Google Scholar] [CrossRef] [PubMed]
- Witte, K.K.; Byrom, R.; Gierula, J.; Paton, M.F.; Jamil, H.A.; Lowry, J.E.; Gillott, R.G.; Barnes, S.A.; Chumun, H.; Kearney, L.C.; et al. Effects of Vitamin D on Cardiac Function in Patients With Chronic HF: The VINDICATE Study. J. Am. Coll. Cardiol. 2016, 67, 2593–2603. [Google Scholar] [CrossRef] [PubMed]
- Giulietti, A.; van Etten, E.; Overbergh, L.; Stoffels, K.; Bouillon, R.; Mathieu, C. Monocytes from type 2 diabetic patients have a pro-inflammatory profile: 1,25-Dihydroxyvitamin D works as anti-inflammatory. Diabetes Res. Clin. Pr. 2007, 77, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, A.K.; Singh, H.V.; Raizada, A.; Singh, S.K. C-reactive protein, inflammation and coronary heart disease. Egypt Heart J. 2015, 67, 89–97. [Google Scholar] [CrossRef]
- Chen, S.; Swier, V.J.; Boosani, C.S.; Radwan, M.M.; Agrawal, D.K. Vitamin D Deficiency Accelerates Coronary Artery Disease Progression in Swine. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1651–1659. [Google Scholar] [CrossRef]
- Sharma, G.; She, Z.-G.; Valenta, D.T.; Stallcup, W.B.; Smith, J.W. Scavenger Receptor-Mediated Targeting of Macrophage Foam Cells in Atherosclerotic Plaque Using Oligonucleotide-Functionalized Nanoparticles. Nano Life 2010, 1, 207–214. [Google Scholar] [CrossRef]
- Das, R.; Ganapathy, S.; Mahabeleshwar, G.H.; Drumm, C.; Febbraio, M.; Jain, M.K.; Plow, E.F. Macrophage Gene Expression and Foam Cell Formation Are Regulated by Plasminogen. Circulation 2013, 127, 1209–1218. [Google Scholar] [CrossRef]
- Yin, K.; You, Y.; Swier, V.; Tang, L.; Radwan, M.M.; Pandya, A.N.; Agrawal, D.K. Vitamin D Protects Against Atherosclerosis via Regulation of Cholesterol Efflux and Macrophage Polarization in Hypercholesterolemic Swine. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2432–2442. [Google Scholar] [CrossRef]
- Koyama, T.; Shibakura, M.; Ohsawa, M.; Kamiyama, R.; Hirosawa, S. Anticoagulant Effects of 1α,25-Dihydroxyvitamin D3 on Human Myelogenous Leukemia Cells and Monocytes. Blood 1998, 92, 160–167. [Google Scholar] [CrossRef]
- Nakagawa, K.; Sasaki, Y.; Kato, S.; Kubodera, N.; Okano, T. 22-Oxa-1α,25-dihydroxyvitamin D3 inhibits metastasis and angiogenesis in lung cancer. Carcinogenesis 2005, 26, 1044–1054. [Google Scholar] [CrossRef]
- Oh, J.; Weng, S.; Felton, S.K.; Bhandare, S.; Riek, A.; Butler, B.; Proctor, B.M.; Petty, M.; Chen, Z.; Schechtman, K.B.; et al. 1,25(OH)2 Vitamin D Inhibits Foam Cell Formation and Suppresses Macrophage Cholesterol Uptake in Patients With Type 2 Diabetes Mellitus. Circulation 2009, 120, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Singh, S.S.; Yu, H.; Lee, J.Y.; Cho, B.R.; Kang, P.M. Vitamin D signaling pathway plays an important role in the development of heart failure after myocardial infarction. J. Appl. Physiol. 2013, 114, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Ford, K.; Latic, N.; Slavic, S.; Zeitz, U.; Dolezal, M.; Andrukhov, O.; Erben, R.G.; Andrukhova, O. Lack of vitamin D signalling per se does not aggravate cardiac functional impairment induced by myocardial infarction in mice. PLoS ONE 2018, 13, e0204803. [Google Scholar] [CrossRef] [PubMed]
- Melamed, M.L.; Muntner, P.; Michos, E.D.; Uribarri, J.; Weber, C.; Sharma, J.; Raggi, P. Serum 25-Hydroxyvitamin D Levels and the Prevalence of Peripheral Arterial Disease. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1179–1185. [Google Scholar] [CrossRef]
- Fahrleitner, A.; Dobnig, H.; Obernosterer, A.; Pilger, E.; Leb, G.; Weber, K.; Kudlacek, S.; Obermayer-Pietsch, B.M. Vitamin D deficiency and secondary hyperparathyroidism are common complications in patients with peripheral arterial disease. J. Gen. Intern. Med. 2002, 17, 663–669. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fahrleitner-Pammer, A.; Obernosterer, A.; Pilger, E.; Dobnig, H.; Dimai, H.P.; Leb, G.; Kudlacek, S.; Obermayer-Pietsch, B.M. Hypovitaminosis D, impaired bone turnover and low bone mass are common in patients with peripheral arterial disease. Osteoporos. Int. 2005, 16, 319–324. [Google Scholar] [CrossRef]
- Zhang, Q.-Y.; Jiang, C.-M.; Sun, C.; Tang, T.-F.; Jin, B.; Cao, D.-W.; He, J.-S.; Zhang, M. Hypovitaminosis D is associated with endothelial dysfunction in patients with non-dialysis chronic kidney disease. J. Nephrol. 2015, 28, 471–476. [Google Scholar] [CrossRef]
- Farrokhian, A.; Raygan, F.; Bahmani, F.; Talari, H.R.; Esfandiari, R.; Esmaillzadeh, A.; Asemi, Z. Long-Term Vitamin D Supplementation Affects Metabolic Status in Vitamin D–Deficient Type 2 Diabetic Patients with Coronary Artery Disease. J. Nutr. 2017, 147, 384–389. [Google Scholar] [CrossRef]
- Forouhi, N.G.; Menon, R.K.; Sharp, S.J.; Mannan, N.; Timms, P.M.; Martineau, A.R.; Rickard, A.P.; Boucher, B.J.; Chowdhury, T.A.; Griffiths, C.J.; et al. Effects of vitamin D2 or D3 supplementation on glycaemic control and cardiometabolic risk among people at risk of type 2 diabetes: Results of a randomized double-blind placebo-controlled trial. Diabetes Obes. Metab. 2016, 18, 392–400. [Google Scholar] [CrossRef]
- Hin, H.; Tomson, J.; Newman, C.; Kurien, R.; Lay, M.; Cox, J.; Sayer, J.; Hill, M.; Emberson, J.; Armitage, J.; et al. Optimum dose of vitamin D for disease prevention in older people: BEST-D trial of vitamin D in primary care. Osteoporos. Int. 2017, 28, 841–851. [Google Scholar] [CrossRef]
- Sokol, S.I.; Srinivas, V.; Crandall, J.P.; Kim, M.; Tellides, G.; Lebastchi, A.; Yu, Y.; Gupta, A.K.; Alderman, M.H. The effects of vitamin D repletion on endothelial function and inflammation in patients with coronary artery disease. Vasc. Med. 2012, 17, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Witham, M.D.; Dove, F.J.; Khan, F.; Lang, C.C.; Belch, J.J.F.; Struthers, A.D. Effects of Vitamin D supplementation on markers of vascular function after myocardial infarction—A randomised controlled trial. Int. J. Cardiol. 2013, 167, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Wang, T.; Zhu, S.; Li, L. Effects of vitamin D supplementation as an adjuvant therapy in coronary artery disease patients. Scand. Cardiovasc. J. 2016, 50, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Wöbke, T.K.; Sorg, B.L.; Steinhilber, D. Vitamin D in inflammatory diseases. Front. Physiol. 2014, 5, 244. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, A.; Frangogiannis, N.G. Aging and Cardiac Fibrosis. Aging Dis. 2011, 2, 158–173. [Google Scholar] [PubMed]
- Qu, H.; Lin, K.; Wang, H.; Wei, H.; Ji, B.; Yang, Z.; Peng, C.; Xiao, X.; Deng, H. 1,25(OH)2D3 improves cardiac dysfunction, hypertrophy, and fibrosis through PARP1/SIRT1/mTOR-related mechanisms in type 1 diabetes. Mol. Nutr. Food Res. 2017, 61, 201600338. [Google Scholar] [CrossRef]
- Chen, S.; Law, C.S.; Grigsby, C.L.; Olsen, K.; Hong, T.-T.; Zhang, Y.; Yeghiazarians, Y.; Gardner, D.G. Cardiomyocyte-specific deletion of the vitamin D receptor gene results in cardiac hypertrophy. Circulation 2011, 124, 1838–1847. [Google Scholar] [CrossRef]
- Artaza, J.N.; Norris, K.C. Vitamin D reduces the expression of collagen and key profibrotic factors by inducing an antifibrotic phenotype in mesenchymal multipotent cells. J. Endocrinol. 2009, 200, 207–221. [Google Scholar] [CrossRef]
- Bodyak, N.; Ayus, J.C.; Achinger, S.; Shivalingappa, V.; Ke, Q.; Chen, Y.S.; Rigor, D.L.; Stillman, I.; Tamez, H.; Kroeger, P.E.; et al. Activated vitamin D attenuates left ventricular abnormalities induced by dietary sodium in Dahl salt-sensitive animals. Proc. Natl. Acad. Sci. USA 2007, 104, 16810–16815. [Google Scholar] [CrossRef]
- Gotsman, I.; Shauer, A.; Zwas, D.R.; Hellman, Y.M.; Keren, A.; Lotan, C.; Admon, D. Vitamin D deficiency is a predictor of reduced survival in patients with heart failure; vitamin D supplementation improves outcome. Eur. J. Heart Fail. 2012, 14, 357–366. [Google Scholar] [CrossRef]
- Kim, D.H.; Sabour, S.; Sagar, U.N.; Adams, S.; Whellan, D.J. Prevalence of hypovitaminosis D in cardiovascular diseases (from the National Health and Nutrition Examination Survey 2001 to 2004). Am. J. Cardiol. 2008, 102, 1540–1544. [Google Scholar] [CrossRef] [PubMed]
- Zittermann, A.; Schleithoff, S.S.; Koerfer, R. Vitamin D insufficiency in congestive heart failure: Why and what to do about it? Heart Fail. Rev. 2006, 11, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Cubbon, R.M.; Lowry, J.E.; Drozd, M.; Hall, M.; Gierula, J.; Paton, M.F.; Byrom, R.; Kearney, L.C.; Barth, J.H.; Kearney, M.T.; et al. Vitamin D deficiency is an independent predictor of mortality in patients with chronic heart failure. Eur. J. Nutr. 2019, 58, 2535–2543. [Google Scholar] [CrossRef] [PubMed]
- Brøndum-Jacobsen, P.; Benn, M.; Jensen, G.B.; Nordestgaard, B.G. 25-Hydroxyvitamin D Levels and Risk of Ischemic Heart Disease, Myocardial Infarction, and Early Death. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2794–2802. [Google Scholar] [CrossRef] [PubMed]
- Melamed, M.L.; Michos, E.D.; Post, W.; Astor, B. 25-hydroxyvitamin D levels and the risk of mortality in the general population. Arch. Intern. Med. 2008, 168, 1629–1637. [Google Scholar] [CrossRef]
- Mahmood, S.S.; Levy, D.; Vasan, R.S.; Wang, T.J. The Framingham Heart Study and the epidemiology of cardiovascular disease: A historical perspective. Lancet 2014, 383, 999–1008. [Google Scholar] [CrossRef]
- Ameri, P.; Canepa, M.; Milaneschi, Y.; Spallarossa, P.; Leoncini, G.; Giallauria, F.; Strait, J.B.; Lakatta, E.G.; Brunelli, C.; Murialdo, G.; et al. Relationship between vitamin D status and left ventricular geometry in a healthy population: Results from the Baltimore Longitudinal Study of Aging. J. Intern. Med. 2013, 273, 253–262. [Google Scholar] [CrossRef]
- Seker, T.; Gur, M.; Ucar, H.; Turkoglu, C.; Baykan, A.O.; Özaltun, B.; Harbalioglu, H.; Kalkan, G.Y.; Kaypakli, O.; Kuloglu, O.; et al. Lower serum 25-hydroxyvitamin D level is associated with impaired myocardial performance and left ventricle hypertrophy in newly diagnosed hypertensive patients. Anatol. J. Cardiol. 2015, 15, 744–750. [Google Scholar] [CrossRef]
- Pandit, A.; Mookadam, F.; Boddu, S.; Aryal Pandit, A.; Tandar, A.; Chaliki, H.; Cha, S.; Lee, H.R. Vitamin D levels and left ventricular diastolic function. Open Heart 2014, 1, e000011. [Google Scholar] [CrossRef]
- Maggio, M.; De Vita, F.; Lauretani, F.; Ceda, G.P.; Volpi, E.; Giallauria, F.; De Cicco, G.; Cattabiani, C.; Melhus, H.; Michaëlsson, K.; et al. Vitamin D and Endothelial Vasodilation in Older Individuals: Data From the PIVUS Study. J. Clin. Endocrinol. Metab. 2014, 99, 3382–3389. [Google Scholar] [CrossRef]
- Van Ballegooijen, A.J.; Visser, M.; Cotch, M.F.; Arai, A.E.; Garcia, M.; Harris, T.B.; Launer, L.J.; Eiríksdóttir, G.; Gudnason, V.; Brouwer, I.A. Serum Vitamin D and Parathyroid Hormone in Relation to Cardiac Structure and Function: The ICELAND-MI Substudy of AGES-Reykjavik. J. Clin. Endocrinol. Metab. 2013, 98, 2544–2552. [Google Scholar] [CrossRef] [PubMed]
- Christensson, T.; Hellström, K.; Wengle, B. Blood pressure in subjects with hypercalcaemia and primary hyperparathyroidism detected in a health screening programme. Eur. J. Clin. Investig. 1977, 7, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Nainby-Luxmoore, J.C.; Langford, H.G.; Nelson, N.C.; Watson, R.L.; Barnes, T.Y. A case-comparison study of hypertension and hyperparathyroidism. J. Clin. Endocrinol. Metab. 1982, 55, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Piovesan, A.; Molineri, N.; Casasso, F.; Emmolo, I.; Ugliengo, G.; Cesario, F.; Borretta, G. Left ventricular hypertrophy in primary hyperparathyroidism. Effects of successful parathyroidectomy. Clin. Endocrinol. (Oxf.) 1999, 50, 321–328. [Google Scholar] [CrossRef]
- Almqvist, E.G.; Bondeson, A.G.; Bondeson, L.; Nissborg, A.; Smedgård, P.; Svensson, S.E. Cardiac dysfunction in mild primary hyperparathyroidism assessed by radionuclide angiography and echocardiography before and after parathyroidectomy. Surgery 2002, 132, 1126–1132. [Google Scholar] [CrossRef]
- Shoji, T.; Shinohara, K.; Kimoto, E.; Emoto, M.; Tahara, H.; Koyama, H.; Inaba, M.; Fukumoto, S.; Ishimura, E.; Miki, T.; et al. Lower risk for cardiovascular mortality in oral 1 -hydroxy vitamin D3 users in a haemodialysis population. Nephrol. Dial. Transpl. 2004, 19, 179–184. [Google Scholar] [CrossRef]
- Van Ballegooijen, A.J.; Snijder, M.B.; Visser, M.; van den Hurk, K.; Kamp, O.; Dekker, J.M.; Nijpels, G.; Stehouwer, C.D.; Henry, R.M.; Paulus, W.J.; et al. Vitamin D in relation to myocardial structure and function after eight years of follow-up: The Hoorn study. Ann. Nutr. Metab. 2012, 60, 69–77. [Google Scholar] [CrossRef]
- Avenell, A.; Maclennan, G.S.; Jenkinson, D.J.; McPherson, G.C.; McDonald, A.M.; Pant, P.R.; Grant, A.M.; Campbell, M.K.; Anderson, F.H.; Cooper, C.; et al. Long-Term Follow-Up for Mortality and Cancer in a Randomized Placebo-Controlled Trial of Vitamin D3and/or Calcium (RECORD Trial). J. Clin. Endocrinol. Metab. 2012, 97, 614–622. [Google Scholar] [CrossRef]
- Schleithoff, S.S.; Zittermann, A.; Tenderich, G.; Berthold, H.K.; Stehle, P.; Koerfer, R. Vitamin D supplementation improves cytokine profiles in patients with congestive heart failure: A double-blind, randomized, placebo-controlled trial. Am. J. Clin. Nutr. 2006, 83, 754–759. [Google Scholar] [CrossRef]
- Dalbeni, A.; Scaturro, G.; Degan, M.; Minuz, P.; Delva, P. Effects of six months of vitamin D supplementation in patients with heart failure: A randomized double-blind controlled trial. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 861–868. [Google Scholar] [CrossRef]
- Zittermann, A.; Ernst, J.B.; Prokop, S.; Fuchs, U.; Dreier, J.; Kuhn, J.; Knabbe, C.; Birschmann, I.; Schulz, U.; Berthold, H.K.; et al. Effect of vitamin D on all-cause mortality in heart failure (EVITA): A 3-year randomized clinical trial with 4000 IU vitamin D daily. Eur. Heart J. 2017, 38, 2279–2286. [Google Scholar] [CrossRef] [PubMed]
- Bouillon, R.; Suda, T. Vitamin D: Calcium and bone homeostasis during evolution. BoneKEy Rep. 2014, 3, 480. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Latic, N.; Erben, R.G. Vitamin D and Cardiovascular Disease, with Emphasis on Hypertension, Atherosclerosis, and Heart Failure. Int. J. Mol. Sci. 2020, 21, 6483. https://doi.org/10.3390/ijms21186483
Latic N, Erben RG. Vitamin D and Cardiovascular Disease, with Emphasis on Hypertension, Atherosclerosis, and Heart Failure. International Journal of Molecular Sciences. 2020; 21(18):6483. https://doi.org/10.3390/ijms21186483
Chicago/Turabian StyleLatic, Nejla, and Reinhold G. Erben. 2020. "Vitamin D and Cardiovascular Disease, with Emphasis on Hypertension, Atherosclerosis, and Heart Failure" International Journal of Molecular Sciences 21, no. 18: 6483. https://doi.org/10.3390/ijms21186483
APA StyleLatic, N., & Erben, R. G. (2020). Vitamin D and Cardiovascular Disease, with Emphasis on Hypertension, Atherosclerosis, and Heart Failure. International Journal of Molecular Sciences, 21(18), 6483. https://doi.org/10.3390/ijms21186483