Global Analysis of the Human RNA Degradome Reveals Widespread Decapped and Endonucleolytic Cleaved Transcripts


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
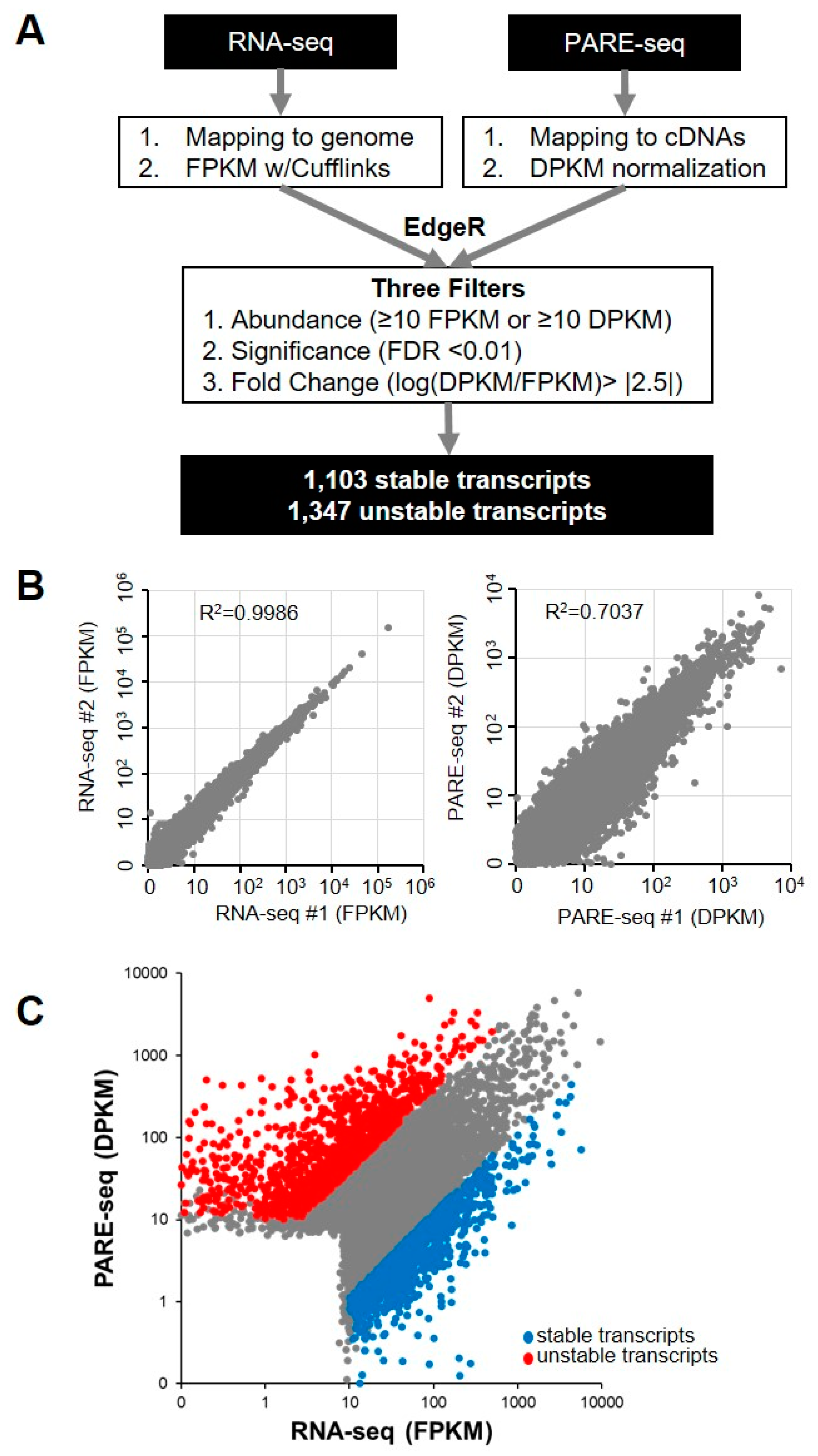
2.1. Global Analysis of mRNA Stability in XRN1-Knockdown HeLa Cells
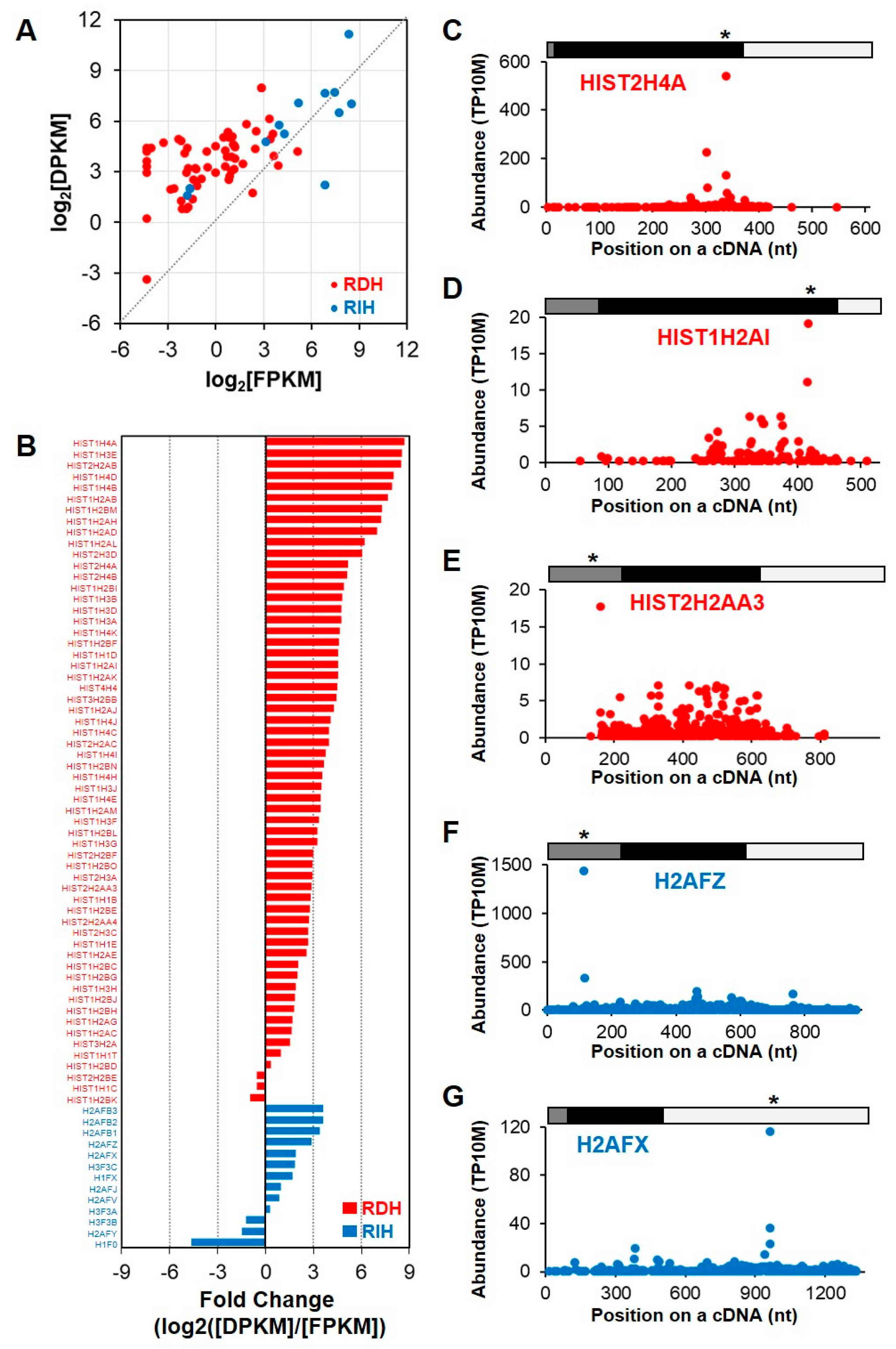
2.2. A Subset of the Replication-Dependent Histone Transcripts Is Polyadenylated and Rapidly Degraded
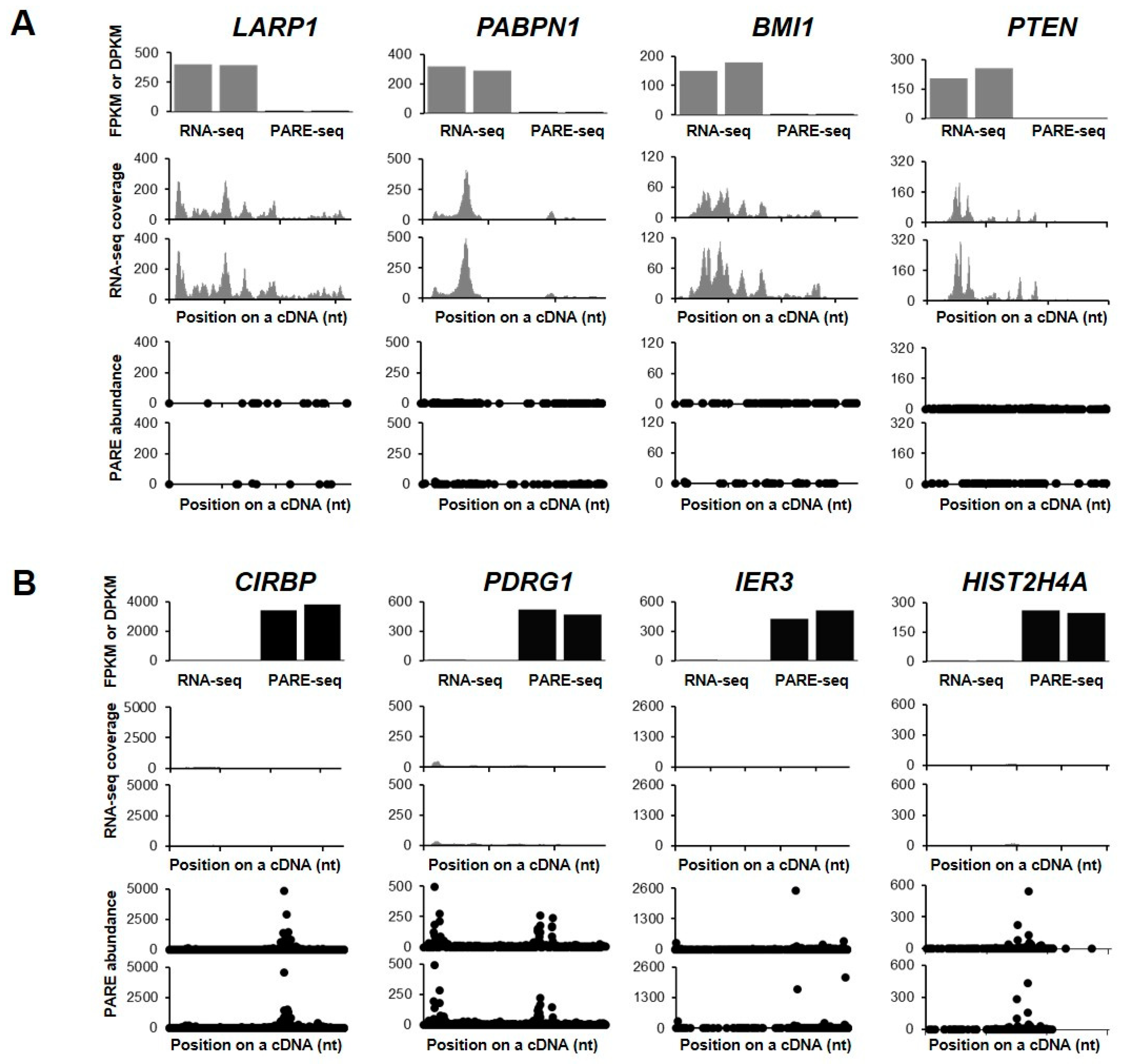
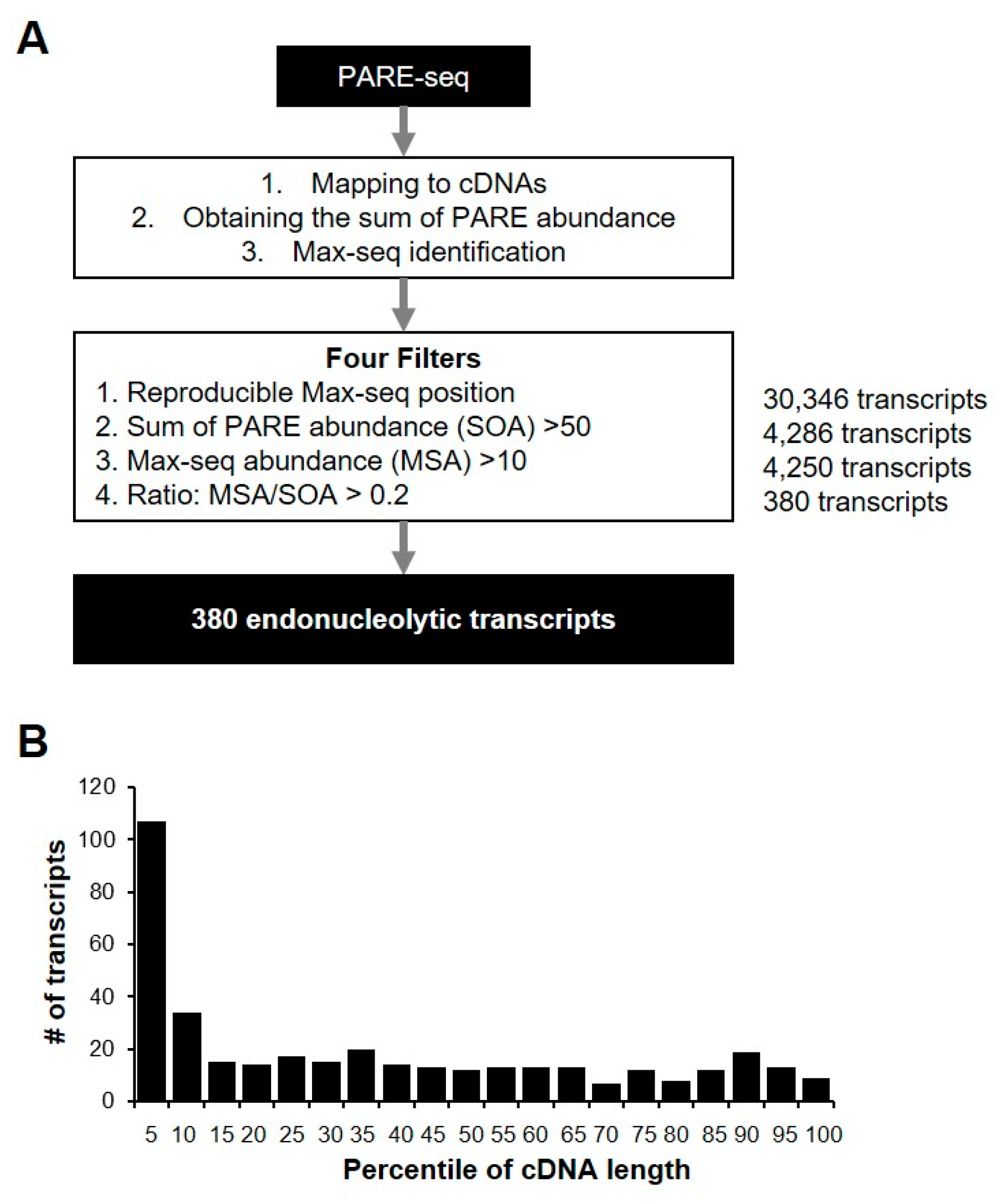
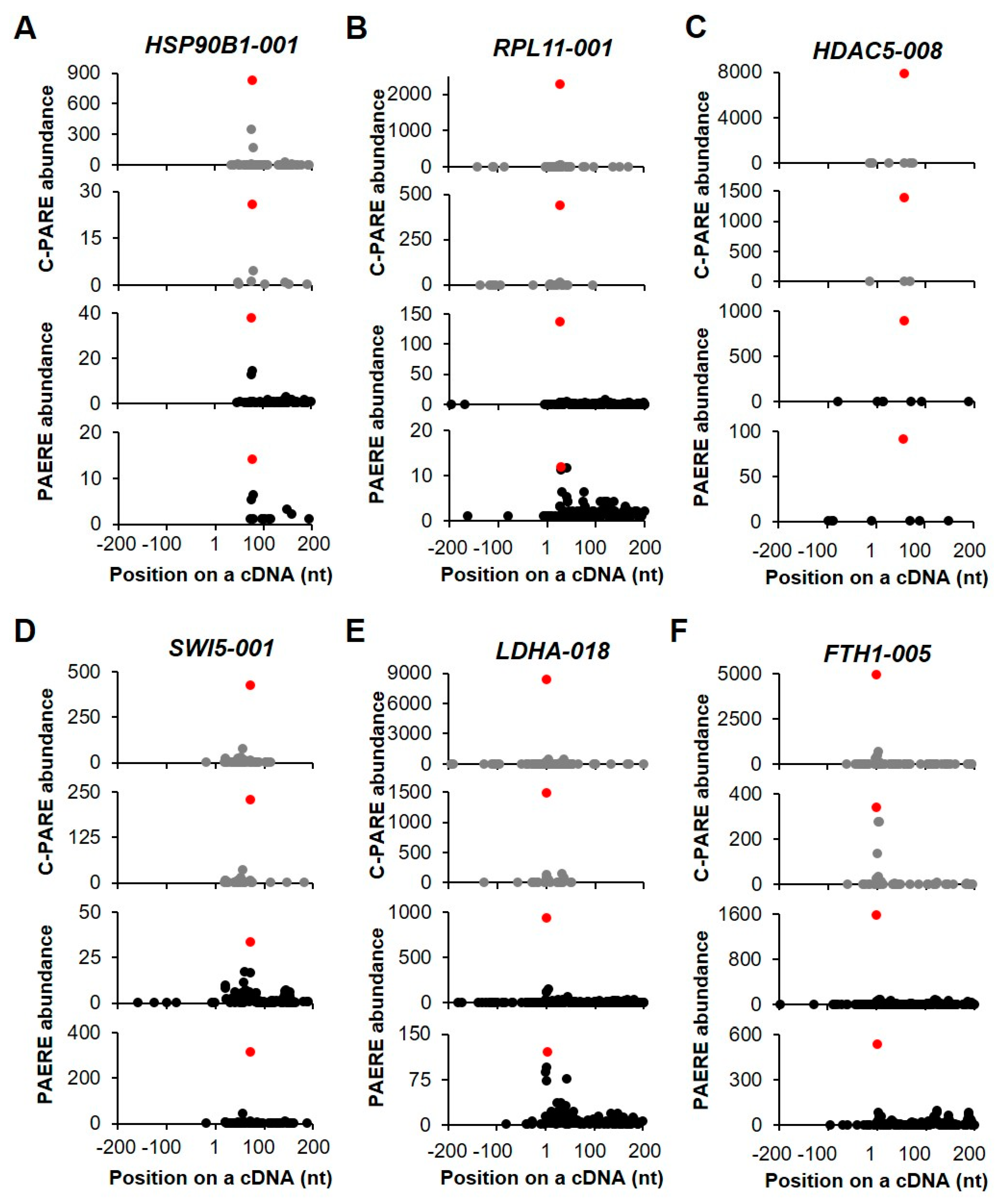
2.3. Identification of Endonucleolytically Cleaved Transcripts
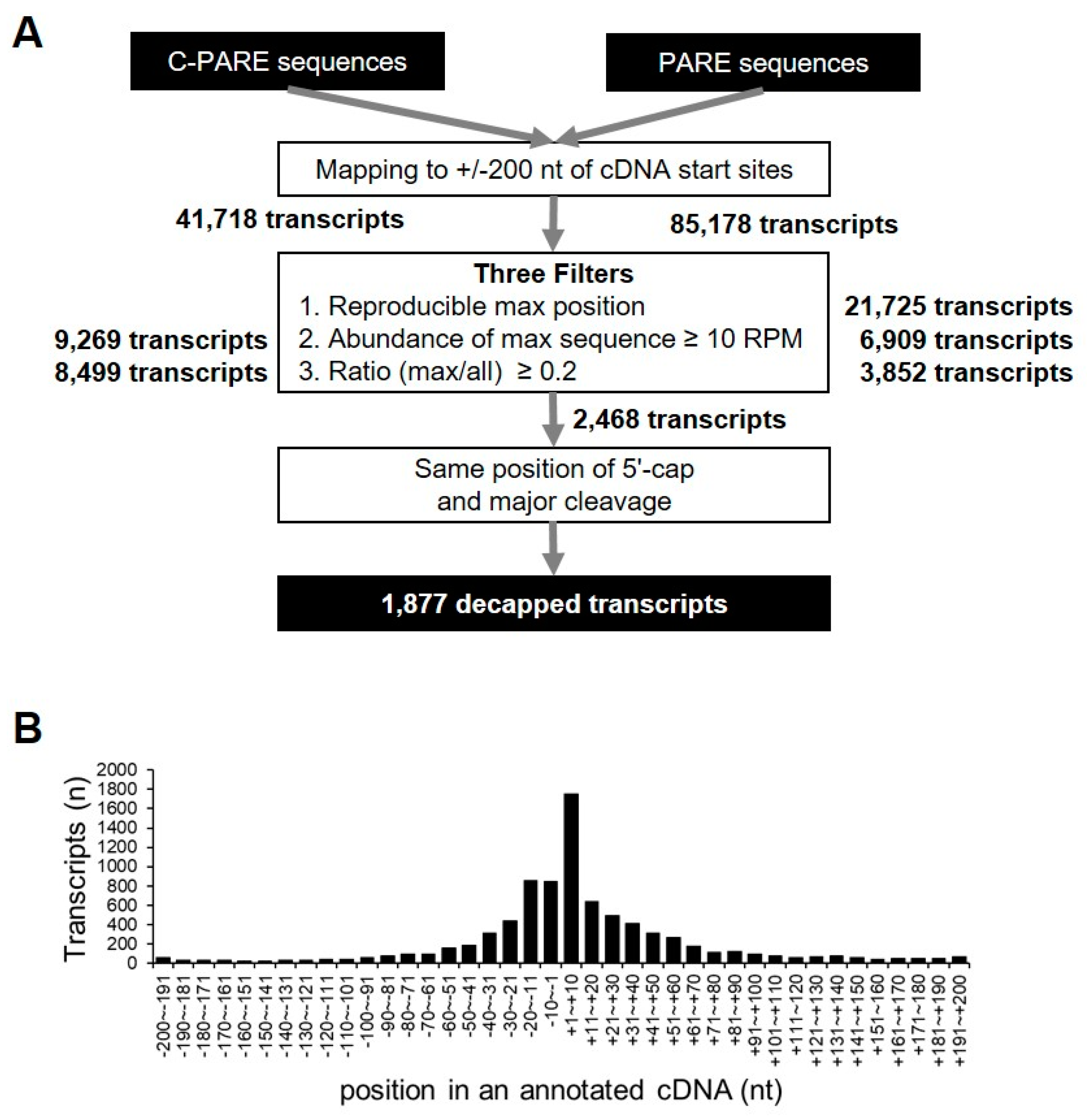
2.4. Identification of Decapped Transcripts
3. Discussion
4. Materials and Methods
4.1. PARE-Seq and C-PARE-Seq Data Processing
4.2. RNA-Seq Data Analysis
4.3. Comparative Analysis of PARE-Seq and RNA-Seq Data
4.4. Computational Analysis of Endonucleolytically Cleaved and Decapped Transcripts
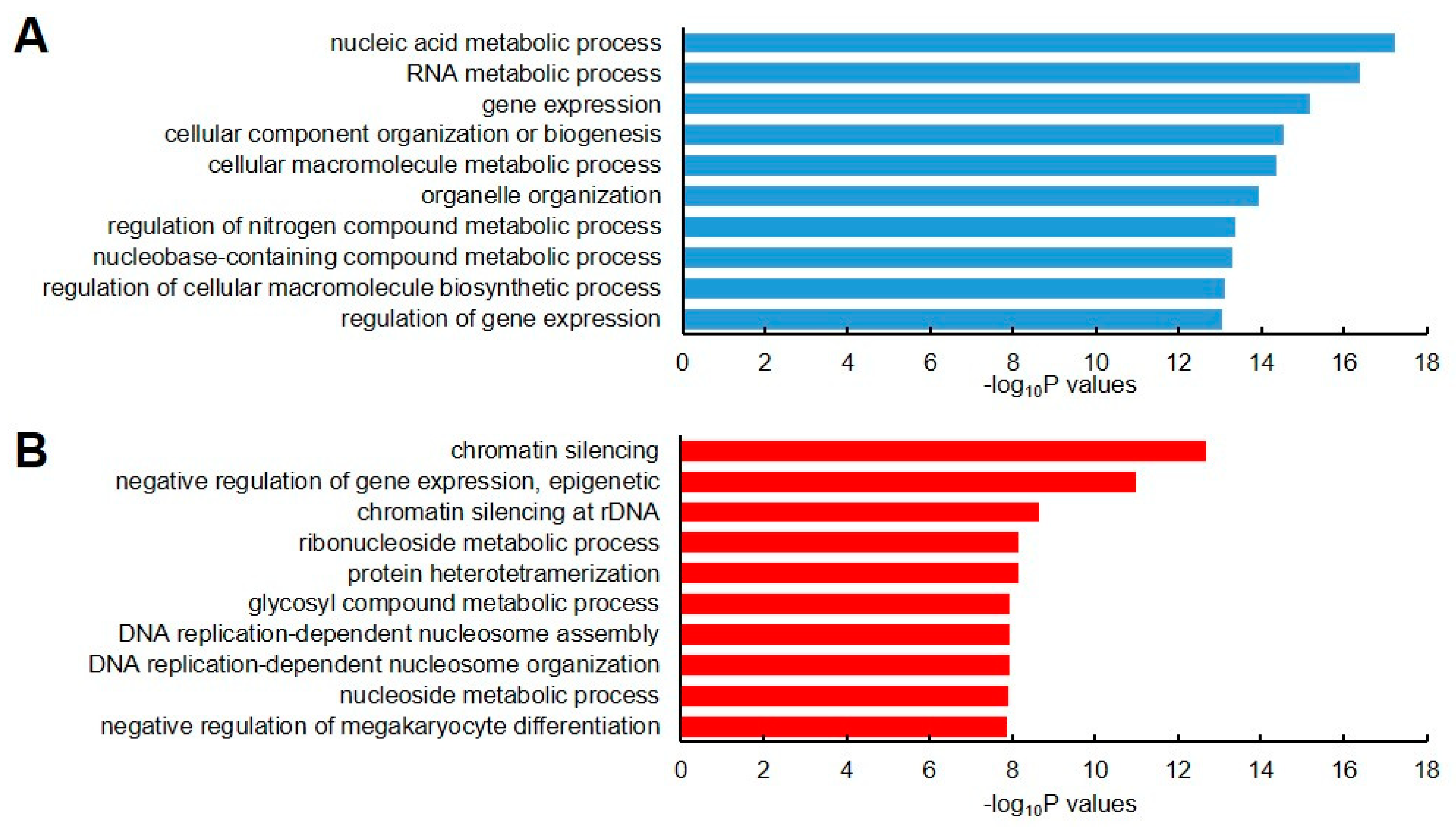
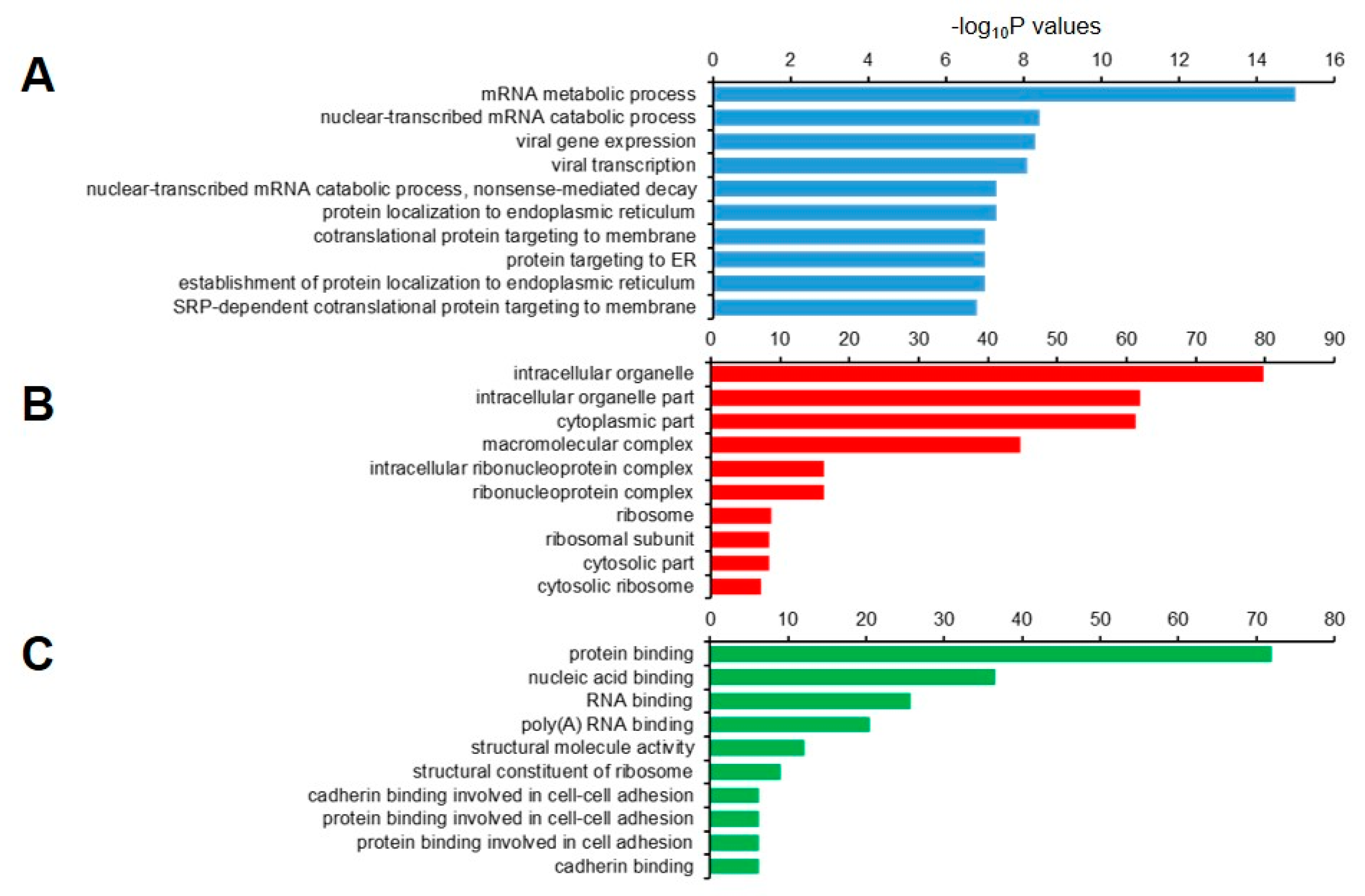
4.5. GO Annotation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Schoenberg, D.R.; Maquat, L.E. Regulation of cytoplasmic mRNA decay. Nat. Rev. Genet. 2012, 13, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-Y.A.; Shyu, A.-B. Mechanisms of deadenylation-dependent decay. Wiley Interdiscip. Rev. RNA 2011, 2, 167–183. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kiledjian, M. Regulation of mRNA decapping. Wiley Interdiscip. Rev. RNA 2010, 1, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Schoenberg, D.R. Mechanisms of endonuclease-mediated mRNA decay. Wiley Interdiscip. Rev. RNA 2011, 2, 582–600. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.; Song, H. The enzymes and control of eukaryotic mRNA turnover. Nat. Struct. Mol. Biol. 2004, 11, 121–127. [Google Scholar] [CrossRef]
- Goldstrohm, A.C.; Wickens, M. Multifunctional deadenylase complexes diversify mRNA control. Nat. Rev. Mol. Cell Biol. 2008, 9, 337–344. [Google Scholar] [CrossRef]
- Nagarajan, V.K.; Jones, C.I.; Newbury, S.F.; Green, P.J. XRN 5’→3’ exoribonucleases: Structure, mechanisms and functions. Biochim. Biophys. Acta 2013, 1829, 590–603. [Google Scholar] [CrossRef]
- Garneau, N.L.; Wilusz, J.; Wilusz, C.J. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 2007, 8, 113–126. [Google Scholar] [CrossRef]
- Tomecki, R.; Dziembowski, A. Novel endoribonucleases as central players in various pathways of eukaryotic RNA metabolism. RNA 2010, 16, 1692–1724. [Google Scholar] [CrossRef]
- Conti, E.; Izaurralde, E. Nonsense-mediated mRNA decay: Molecular insights and mechanistic variations across species. Curr. Opin. Cell Biol. 2005, 17, 316–325. [Google Scholar] [CrossRef]
- Lykke-Andersen, S.; Jensen, T.H. Nonsense-mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015, 16, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Ezzeddine, N.; Shyu, A.B. Messenger RNA half-life measurements in mammalian cells. Methods Enzymol. 2008, 448, 335–357. [Google Scholar] [PubMed]
- Elkon, R.; Zlotorynski, E.; Zeller, K.I.; Agami, R. Major role for mRNA stability in shaping the kinetics of gene induction. BMC Genom. 2010, 11, 1–8. [Google Scholar] [CrossRef]
- Hao, S.; Baltimore, D. The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat. Immunol. 2009, 10, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Barenco, M.; Brewer, D.; Papouli, E.; Tomescu, D.; Callard, R.; Stark, J.; Hubank, M. Dissection of a complex transcriptional response using genome-wide transcriptional modelling. Mol. Syst. Biol. 2009, 5, 327. [Google Scholar] [CrossRef]
- Jao, C.Y.; Salic, A. Exploring RNA transcription and turnover in vivo by using click chemistry. Proc. Natl. Acad. Sci. USA 2008, 105, 15779–15784. [Google Scholar] [CrossRef]
- Eidinoff, M.L.; Cheong, L.; Rich, M.A. Incorporation of Unnatural Pyrimidine Bases into Deoxyribonucleic Acid of Mammalian Cells. Science 1959, 129, 1550–1551. [Google Scholar] [CrossRef]
- Melvin, W.T.; Milne, H.B.; Slater, A.A.; Allen, H.J.; Keir, H.M. Incorporation of 6-Thioguanosine and 4-Thiouridine into RNA. Application to Isolation of Newly Synthesised RNA by Affinity Chromatography. Eur. J. Biochem. 1978, 92, 373–379. [Google Scholar] [CrossRef]
- Munchel, S.E.; Shultzaberger, R.K.; Takizawa, N.; Weis, K. Dynamic profiling of mRNA turnover reveals gene-specific and system-wide regulation of mRNA decay. Mol. Biol. Cell 2011, 22, 2787–2795. [Google Scholar] [CrossRef]
- German, M.A.; Pillay, M.; Jeong, D.-H.; Hetawal, A.; Luo, S.; Janardhanan, P.; Kannan, V.; Rymarquis, L.A.; Nobuta, K.; German, R.; et al. Global identification of microRNA–target RNA pairs by parallel analysis of RNA ends. Nat. Biotechnol. 2008, 26, 941–946. [Google Scholar] [CrossRef]
- Addo-Quaye, C.; Eshoo, T.W.; Bartel, D.P.; Axtell, M.J. Endogenous siRNA and miRNA Targets Identified by Sequencing of the Arabidopsis Degradome. Curr. Biol. 2008, 18, 758–762. [Google Scholar] [CrossRef]
- Gregory, B.D.; O’Malley, R.C.; Lister, R.; Urich, M.A.; Tonti-Filippini, J.; Chen, H.; Millar, A.H.; Ecker, J.R. A Link between RNA Metabolism and Silencing Affecting Arabidopsis Development. Dev. Cell 2008, 14, 854–866. [Google Scholar] [CrossRef] [PubMed]
- Bracken, C.P.; Szubert, J.M.; Mercer, T.R.; Dinger, M.E.; Thomson, D.W.; Mattick, J.S.; Michael, M.Z.; Goodall, G.J. Global analysis of the mammalian RNA degradome reveals widespread miRNA-dependent and miRNA-independent endonucleolytic cleavage. Nucleic Acids Res. 2011, 39, 5658–5668. [Google Scholar] [CrossRef] [PubMed]
- Karginov, F.V.; Cheloufi, S.; Chong, M.M.W.; Stark, A.; Smith, A.D.; Hannon, G.J. Diverse Endonucleolytic Cleavage Sites in the Mammalian Transcriptome Depend upon MicroRNAs, Drosha, and Additional Nucleases. Mol. Cell 2010, 38, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Shin, C.; Nam, J.-W.; Farh, K.K.-H.; Chiang, H.R.; Shkumatava, A.; Bartel, B. Expanding the MicroRNA Targeting Code: Functional Sites with Centered Pairing. Mol. Cell 2010, 38, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Ahn, S.; Kim, S.; Lee, J.; Nam, J.-W.; Shin, C. Degradome sequencing reveals an endogenous microRNA target inC. elegans. FEBS Lett. 2013, 587, 964–969. [Google Scholar] [CrossRef] [PubMed]
- Park, O.H.; Ha, H.; Lee, Y.; Boo, S.H.; Kwon, D.H.; Song, H.K.; Kim, Y.K. Endoribonucleolytic Cleavage of m6A-Containing RNAs by RNase P/MRP Complex. Mol. Cell 2019, 74, 494–507.e8. [Google Scholar] [CrossRef]
- Schmidt, S.A.; Foley, P.L.; Jeong, N.-H.; Rymarquis, L.A.; Doyle, F.; Tenenbaum, S.A.; Belasco, J.G.; Green, P.J. Identification of SMG6 cleavage sites and a preferred RNA cleavage motif by global analysis of endogenous NMD targets in human cells. Nucleic Acids Res. 2014, 43, 309–323. [Google Scholar] [CrossRef]
- Rymarquis, L.A.; Souret, F.F.; Green, P.J. Evidence that XRN4, an Arabidopsis homolog of exoribonuclease XRN1, preferentially impacts transcripts with certain sequences or in particular functional categories. RNA 2011, 17, 501–511. [Google Scholar] [CrossRef]
- Marzluff, W.F.; Koreski, K.P. Birth and Death of Histone mRNAs. Trends Genet. 2017, 33, 745–759. [Google Scholar] [CrossRef]
- Ryu, I.; Park, Y.; Seo, J.-W.; Park, O.H.; Ha, H.; Nam, J.-W.; Kim, Y.K. HuR stabilizes a polyadenylated form of replication-dependent histone mRNAs under stress conditions. FASEB J. 2018, 33, 2680–2693. [Google Scholar] [CrossRef]
- Saldi, T.; Fong, N.; Bentley, D.L. Transcription elongation rate affects nascent histone pre-mRNA folding and 3’ end processing. Genes Dev. 2018, 32, 297–308. [Google Scholar] [CrossRef]
- Jeong, D.-H.; Schmidt, S.A.; Rymarquis, L.A.; Park, S.; Ganssmann, M.; German, M.A.; Accerbi, M.; Zhai, J.X.; Fahlgren, N.; Fox, S.E.; et al. Parallel analysis of RNA ends enhances global investigation of microRNAs and target RNAs of Brachypodium distachyon. Genome Biol. 2013, 14, R145. [Google Scholar] [CrossRef]
- Zhang, J.; Mao, Z.; Chong, K. A global profiling of uncapped mRNAs under cold stress reveals specific decay patterns and endonucleolytic cleavages in Brachypodium distachyon. Genome Biol. 2013, 14, R92. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Chen, J.; Yu, J. Global analysis of uncapped mRNA changes under drought stress and microRNA-dependent endonucleolytic cleavages in foxtail millet. BMC Plant Biol. 2015, 15, 241. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Chen, L.; Yuan, X.; Luo, Q.; Liu, Y.; Xie, G.; Ma, Y.; Shen, L. Exosomal transfer of tumor-associated macrophage-derived miR-21 confers cisplatin resistance in gastric cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 53. [Google Scholar] [CrossRef] [PubMed]
- Egawa, H.; Jingushi, K.; Hirono, T.; Ueda, Y.; Kitae, K.; Nakata, W.; Fujita, K.; Uemura, M.; Nonomura, N.; Tsujikawa, K. The miR-130 family promotes cell migration and invasion in bladder cancer through FAK and Akt phosphorylation by regulating PTEN. Sci. Rep. 2016, 6, 20574. [Google Scholar] [CrossRef]
- Morf, J.; Rey, G.; Schneider, K.; Stratmann, M.; Fujita, J.; Naef, F.; Schibler, U. Cold-Inducible RNA-Binding Protein Modulates Circadian Gene Expression Posttranscriptionally. Science 2012, 338, 379–383. [Google Scholar] [CrossRef]
- Eckner, R.; Birnstiel, M.L. Evolutionary conserved multiprotein complexes interact with the 3’untranslated region of histone transcripts. Nucleic Acids Res. 1992, 20, 1023–1030. [Google Scholar] [CrossRef][Green Version]
- Ryu, I.; Kim, Y.K. AU-rich element–mediated mRNA decay via the butyrate response factor 1 controls cellular levels of polyadenylated replication-dependent histone mRNAs. J. Biol. Chem. 2019, 294, 7558–7565. [Google Scholar] [CrossRef]
- Li, Y.; Song, M.-G.; Kiledjian, M. Transcript-Specific Decapping and Regulated Stability by the Human Dcp2 Decapping Protein. Mol. Cell. Biol. 2007, 28, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Bönisch, C.; Temme, C.; Moritz, B.; Wahle, E. Degradation ofhsp70and Other mRNAs inDrosophilavia the 5′–3′ Pathway and Its Regulation by Heat Shock. J. Biol. Chem. 2007, 282, 21818–21828. [Google Scholar] [CrossRef] [PubMed]
- Goeres, D.C.; Van Norman, J.M.; Zhang, W.; Fauver, N.A.; Spencer, M.L.; Sieburth, L.E. Components of the Arabidopsis mRNA Decapping Complex Are Required for Early Seedling Development. Plant Cell 2007, 19, 1549–1564. [Google Scholar] [CrossRef] [PubMed]
- Trcek, T.; Larson, D.R.; Moldón, A.; Query, C.C.; Singer, R.H. Single-Molecule mRNA Decay Measurements Reveal Promoter-Regulated mRNA Stability in Yeast. Cell 2011, 147, 1484–1497. [Google Scholar] [CrossRef] [PubMed]
- Won, J.-I.; Lee, J.B.; Lee, H.W.; Shin, J.M.; Yoon, J.H.; Jeong, D.-H. WebPORD: A web-based pipeline of RNA degradome. Int. J. Data Min. Bioinform. 2019, 22, 216–230. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Brandon, M.C.; Lott, M.T.; Nguyen, K.C.; Spolim, S.; Navathe, S.B.; Baldi, P.; Wallace, U.C. MITOMAP: A human mitochondrial genome database—2004 update. Nucleic Acids Res. 2004, 33, D611–D613. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- da Huang, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Van Nimwegen, E.; Zavolan, M.; Rajewsky, N.; Schroeder, M.; Magnasco, M.; Darnell, J.E. Decay Rates of Human mRNAs: Correlation with Functional Characteristics and Sequence Attributes. Genome Res. 2003, 13, 1863–1872. [Google Scholar] [PubMed]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Won, J.-I.; Shin, J.; Park, S.Y.; Yoon, J.; Jeong, D.-H. Global Analysis of the Human RNA Degradome Reveals Widespread Decapped and Endonucleolytic Cleaved Transcripts. Int. J. Mol. Sci. 2020, 21, 6452. https://doi.org/10.3390/ijms21186452
Won J-I, Shin J, Park SY, Yoon J, Jeong D-H. Global Analysis of the Human RNA Degradome Reveals Widespread Decapped and Endonucleolytic Cleaved Transcripts. International Journal of Molecular Sciences. 2020; 21(18):6452. https://doi.org/10.3390/ijms21186452
Chicago/Turabian StyleWon, Jung-Im, JaeMoon Shin, So Young Park, JeeHee Yoon, and Dong-Hoon Jeong. 2020. "Global Analysis of the Human RNA Degradome Reveals Widespread Decapped and Endonucleolytic Cleaved Transcripts" International Journal of Molecular Sciences 21, no. 18: 6452. https://doi.org/10.3390/ijms21186452
APA StyleWon, J.-I., Shin, J., Park, S. Y., Yoon, J., & Jeong, D.-H. (2020). Global Analysis of the Human RNA Degradome Reveals Widespread Decapped and Endonucleolytic Cleaved Transcripts. International Journal of Molecular Sciences, 21(18), 6452. https://doi.org/10.3390/ijms21186452